

Abstract
The four stereoisomers of azetidine-2,3-dicaroxylic acid (L-trans-ADC, L-cis-ADC, D-trans-ADC, and D-cis-ADC) were synthesized in a stereocontrolled fashion following two distinct strategies; one providing the two cis-ADC enantiomers and one giving access to the two trans-ADC enantiomers. The four azetidinic amino acids were characterized in a radio ligand binding assay ([3H]CGP39653) at native NMDA receptors: L-trans-ADC showed the highest affinity (Ki = 10 μM) followed by the D-cis-ADC stereoisomer (21 μM). On the other hand, the two analogs L-cis-ADC and D-trans-ADC were low affinity ligands (>100 and 90 μM, respectively). Electrophysiological characterization of the ADC compounds at the four NMDA receptor subtypes NR1/NR2A, NR1/NR2B, NR1/NR2C, and NR1/NR2D expressed in Xenopus oocytes showed that L-trans-ADC displayed the highest agonist potency at NR1/NR2D (EC50 = 50 μM), which was 9.4-, 3.4- and 1.9-fold higher compared to the potencies at NR1/NR2A-C, respectively. D-cis-ADC was shown to be a partial agonist at NR1/NR2C and NR1/NR2D with medium-range micromolar potencies (EC50 = 720 μM and 230 μM, respectively). A subsequent in-silico ligand-protein docking study suggested an unusual binding mode for these amino acids in the agonist binding site.
Keywords: Conformational restriction, Enantioselective synthesis, Glutamate, NMDA, Subtype selectivity
Introduction
Fast excitatory neurotransmission is a neurological process during which (S)-glutamate (Glu, Fig. 1) is released from the presynaptic membrane, diffuses across the synaptic cleft, and activates ligand-gated ion channels located at the postsynaptic membrane. These ligand-gated ion channels are named the ionotropic glutamate receptors (iGluRs) and are further divided into three functional groups, named the N-methyl-D-aspartate (NMDA) receptors, the 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionate (AMPA) receptors, and the kainate receptors (Fig. 1).[1] The NMDA receptors play pivotal roles in fast glutamatergic neurotransmission and are critically involved in many important neuronal functions including frequency encoding of information,[2] synaptic plasticity,[3] and neuronal development.[4] Under a variety of acute conditions, such as ischemia, seizures or traumatic brain injury, the release of excess amounts of glutamate and the resultant NMDA receptor-mediated Ca2+ flux into the cell may be of sufficient magnitude as to promote neuronal death (excitotoxicity).[5] Under chronic conditions of enhanced neuronal susceptibility, as in Parkinson’s, Huntington’s, and Alzheimer’s diseases, the potential involvement of NMDA receptor-mediated excitotoxicity may be of a slower process.[6–9] For these reasons, there has been an extensive interest in understanding structure, function, localization, and regulation of NMDA receptors with the goal of designing new therapeutic strategies for a number of diseases.
Figure 1.
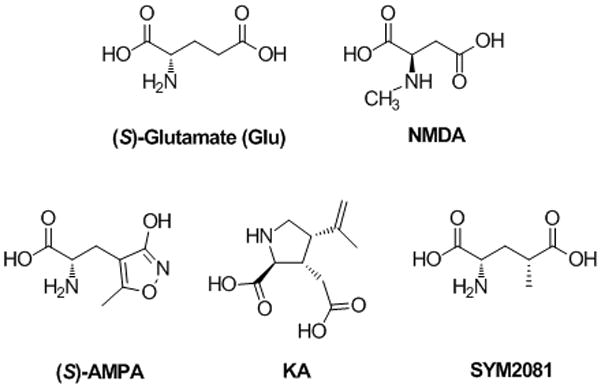
(S)-Glutamate (Glu) and ionotropic glutamate receptor (iGluR) ligands: N-methyl-D-aspartic acid (NMDA), 2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid (AMPA), kainic acid (KA), and (4R)-methyl Glu (SYM2081)
Functional NMDA receptors are assembled from two NR1 subunits and two NR2 subunits and are activated by the simultaneous binding of glycine and glutamate to the NR1 and NR2 subunits, respectively.[10] One NR1 subunit and four different NR2 subunits (NR2A, NR2B, NR2C, and NR2D) have been identified, and these different NR2 subunits determine the physiological role of the NMDA receptor subtype.[11] In response to agonist binding, NMDA receptors undergo conformational changes that open a cation-conducting channel pore. The time course of these conformational changes differs considerably among the NMDA receptor subtypes (NR1/NR2A, NR1/NR2B, NR1/NR2C, and NR1/NR2D), and these differences influence the amplitude and time course of excitatory postsynaptic currents at glutamatergic synapses. Progress towards understanding this functional variation among the NMDA receptor subtypes and the increasingly precise anatomical localization of NR2 subunits have strengthened the therapeutic interest in the development of subunit-selective NMDA receptor agonists of which only few exist.
In this paper, we present the stereoselective synthesis of the four stereoisomers of 2,3-ADC, subsequent radio ligand binding experiments and electrophysiological recordings at recombinant NMDA receptors. Finally, we have conducted an in-silico study and put forward new hypotheses as to how rational design of potentially subtype selective NMDA receptor agonists may be attained.
Design of Azetidines
Azetidine-2,3-dicarboxylic acids (ADC) (Figure 2) may be envisaged as highly conformationally restricted analogs of NMDA and are thus potential agonists at NMDA receptors. Whereas azetidinic α-amino acids have been prepared by means of several diverse strategies,[12] the class of azetidine 2,3-dicarboxylic acids has only been synthesized via a stereoselective strategy,[13] and by an approach in which an electrocylic reaction was the key step.[14] Furthermore, pharmacology of this class of amino acids has only been investigated briefly at native excitatory amino acid transporters (EAATs) at which only the L-trans-ADC stereoisomer showed affinity in the medium micromolar range.[13]
Figure 2.
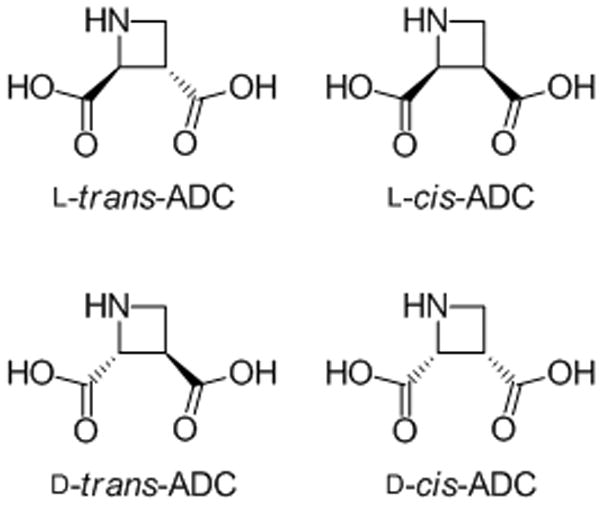
The four stereoisomers of azetidine-2,3-dicarboxylic acid: L-trans-ADC, L-cis-ADC, D-trans-ADC and D-cis-ADC.
Chemistry
We developed two distinct synthetic strategies for the synthesis of the cis-stereoisomers and the trans-stereoisomers (Scheme 1 and 2, respectively), The stereospecific synthesis of D-cis-ADC (Scheme 1) commenced with the reaction of (R)-O-Benzyl glycidol 1 with benzyl amine to give amino alcohol 2.[13] This compound was N-alkylated with t-butylbromoacetate, and the resulting alcohol 3 was chlorinated with thionyl chloride. As previously described with similar substrates,[12] this chlorination gave a mixture of regioisomeric chlorides 5 and 6 via aziridinium intermediate 4 that were next equilibrated by heating in DMF, to afford in good overall yield the more stable secondary chloride 5. This sequence has been shown to occur without racemization with similar substrates.[15] Upon treatment with LiHMDS, this chloride was closed to the corresponding 2,3-cis-azetidine 7 with a total diastereoselectivity and a fair yield of 60%. Azetidine 7 was subsequently N- and O-debenzylated and N-Boc protected in one step to afford 8 in very good yield. The free primary alcohol was next oxidized into the corresponding carboxylic acid[16] to give 9, which upon treatment with TFA, followed by ion-exchange chromatography, gave a good yield of the target amino acid 10. This same strategy was applied to the synthesis of ent-10 starting from ent-1, with similar yields
Scheme 1.
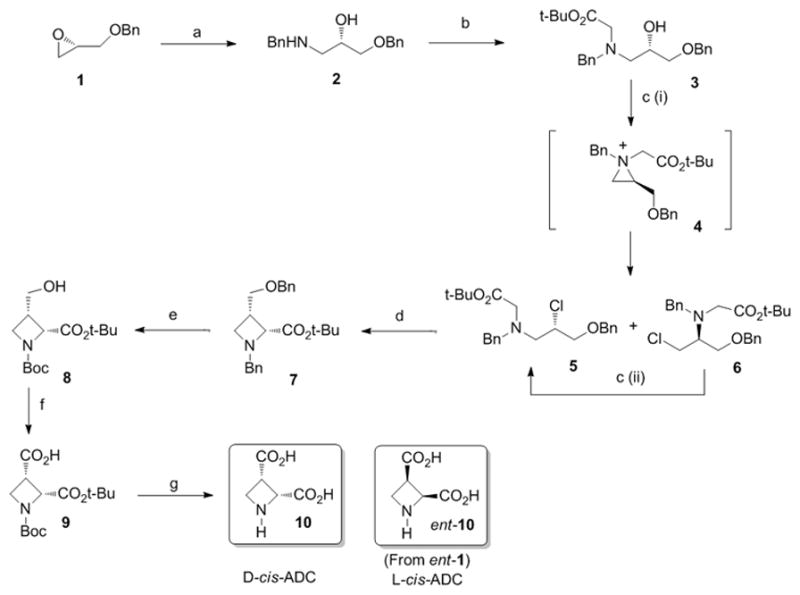
Reagents and conditions: (a) BnNH2, H2O cat., 60°C, 5h (80%) (b) tButylbromoacetate, NaHCO3, NaI, DMF, rt, 5h (70%) (c) (i) SOCl2, DCM, reflux, 3h (ii) DMF, 65°C (90%) (d) LiHMDS, THF/HMPA, −78°C to 0°C (60%) (e) H2, 15 bars, Pd(OH)2 cat., EtOH, Boc2O (96%) (f) RuCl3 cat., NaIO4, MeCN/CCl4/H2O (71%) (g) TFA/DCM, rt, 12h then Dowex 5WX8-200 (90%).
Scheme 2.
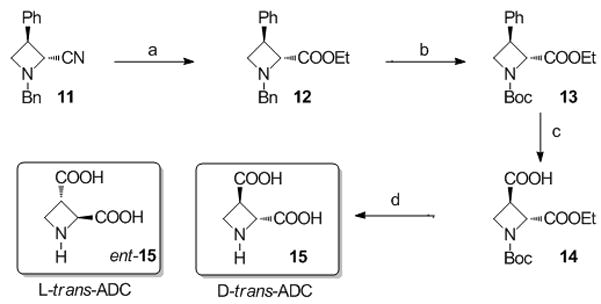
Reagents and conditions: (a) H2SO4, EtOH, reflux, 12h (89%) (b) H2, 1 bar, 10%Pd/C cat., Boc2O, EtOH, 6 days (quant.) (c) NaIO4, RuCl3, CH3CN/CCl4/H2O: 1/1/1, rt, 48h (75%) (d) NaOH, MeOH/H2O: 1/1, rt, 48h then HCl 1M, rt, 12h then Dowex 5WX8-200 (72%).
The D-trans-ADC isomer was synthesized following a different pathway, depicted in Scheme 2. The known cyano azetidine 11,[17] easily prepared from (R)-phenylglycinol as the source of chirality, was converted into the ethyl ester 12 by treatment with concentrated H2SO4 in ethanol, followed by aqueous workup. No epimerization was observed during these quite harsh conditions. Next, the N-benzyl protective group was cleaved, with in situ reprotection as a tert-butyl carbamate to give 13. The 3-carboxylic acid functionality was unveiled by oxidation of the phenyl ring with sodium periodate in the presence of a catalytic amount of ruthenium chloride[18] to give the expected acid 14 with a 75% yield. The final deprotection to give D-trans-ADC 15 were effected in one pot: Saponification of the ethyl ester with sodium hydroxide followed by acidification with hydrochloric acid 1M gave the amino diacid in its protonated form, and the zwitterionic amino acid was obtained in good yield after ion-exchange chromatography. The same sequence was conducted starting with ent-11 (prepared starting from (S)-phenylglycinol) and gave ent-15 with similar yields.
Pharmacology
First, the affinities of the four stereoisomeric azetidine-2,3-dicarboxylic acids (ADC) for the glutamate binding site of native NMDA receptors were determined in radio ligand binding assay (rat brain synaptosomes). Summarized in Table 1, L-trans-ADC and L-cis-ADC displayed affinities in the low micromolar range (10 and 21 μM, respectively), whereas D-trans-ADC and L-cis-ADC were essentially without affinity.
Table 1.
Binding affinities of azetidines at native NMDA receptors (rat brain synaptosomes)
| Azetidine | [3H]CGP39653 Ki (μM) |
|---|---|
| SYM2081 | 5.9 |
| NMDA | 6.2 |
| L-trans-ADC | 10 [5.04 ± 0.10] |
| L-cis-ADC | >100 |
| D-trans-ADC | 90 [4.05 ± 0.03] |
| D-cis-ADC | 21 [4.74 ± 0.13] |
Next, we evaluated the four ADC analogues in a functional assay for agonist activity at the four different heteromeric NMDA receptor subtypes (NR1/NR2A-D). The NMDA receptor subtypes were expressed in Xenopus oocytes and current responses to application of different concentrations of the compounds were recorded using two-electrode voltage-clamp electrophysiology. Of the four stereoisomeric ADC analogs, only L-trans-ADC was able to (partially) activate NR1/NR2A. Activation of subtypes NR1/NR2B, NR1/NR2C, and NR1/NR2D was also observed with highest potency at NR1/NR2D. With respect to subtype selectivity, L-trans-ADC showed a notable 9.4-fold preference for NR2D over NR2A, whereas the EC50 values at NR2B and NR2C were within the range of that of NR2D (170, 95 and 50 μM, respectively). In contrast, L-cis-ADC was unable to activate NR1/NR2A and NR1/NR2B, and only at high concentrations (1000 μM) some activation of NR1/NR2C and NR1/NR2D subtypes could be detected. D-trans-ADC and D-cis-ADC activated NR1/NR2B, NR1/NR2C, and NR1/NR2D subtypes with the highest potency at NR1/NR2D. All active compounds by which concentration-response data were obtained were partial agonists at the NMDA receptor subtypes, since they display sub-maximal activation relative to glutamate. The concentration-response data is shown in Figure 3 and the EC50-values and maximal responses relative to glutamate are summarized in Table 2.
Figure 3.

Mean concentration–response curves for ADC analogues: (A) L-trans-ADC, (B) D-cis-ADC, and (C) D-trans-ADC determined using two-electrode voltage-clamp recordings on Xenopus oocytes expressing NMDA receptor subtypes NR1/NR2A-D. The curves are normalized to the maximal current response (Imax) to glutamate in the same recording. Data points are represented as mean ± SEM. All EC50-values are listed in Table 2. (D) Comparison of maximal currents induced by D-trans-ADC, D-cis-ADC, and L-trans-ADC relative to glutamate at NR1/NR2A-D subtypes. All bars are represented as mean + SEM. All relative Imax-values are listed in Table 2.
Table 2.
Characterization of ADC analogues at recombinant NMDA receptor subtypes NR1/NR2A-D expressed in Xenopus oocytes using electrophysiological recordings.
| NR1/NR2A | NR1/NR2B | NR1/NR2C | NR1/NR2D | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Compound | EC50 (μM) [pEC50] | I/Imax | EC50 (μM) [pEC50] | I/Imax | EC50 (μM) [pEC50] | I/Imax | EC50 (μM) [pEC50] | I/Imax |
| NMDA[a] | 75 [4.13±0.07] | 0.90 ± 0.04 | 22 [4.66±0.03] | 0.77 ± 0.01 | 23 [4.63±0.02] | 0.73 ± 0.02 | 8.3 [5.08±0.04] | 0.80 ± 0.02 |
| L-trans-ADC | 470 [3.32±0.03] | 0.38 ± 0.02 | 170 [3.76±0.01] | 0.48 ± 0.01 | 95 [4.02±0.02] | 0.73 ± 0.03 | 50 [4.30±0.02] | 0.80 ± 0.01 |
| L-cis-ADC | NR | ND | NR | ND | >1000 | ND | >1000 | ND |
| D-trans-ADC | NR | ND | >1000 | ND | >1000 | ND | 660 [3.18±0.02] | 0.69 ± 0.01 |
| D-cis-ADC | NR | ND | >3000 | ND | 720 [3.15±0.06] | 0.47 ± 0.03 | 230 [3.63±0.01] | 0.67 ± 0.01 |
Mean pEC50 ± SEM (shown in brackets) and the corresponding EC50-values (in [μM]) as well as the mean relative maximal response (I/Imax ± SEM) (relative to maximal response to glutamate; 1.0) were calculated from full concentration-response curves. For all agonist data, the hill coefficients were between 1.1 and 2.1, and the number of oocytes was between 5 and 10. NR indicates no response up to 1000μM. ND indicates that the relative maximal response was not determined. [a] Data from reference [21].
In-silico docking study
In order to better understand the binding mode and subtype activity of these NMDA receptor agonists we performed a docking study in the recently published X-ray structure of the agonist binding domain (ABD) of NR2A.[22] In this structure (Fig. 3A), Glu connects the upper and lower domain of the ABD. A grid was created of the ABD around glutamate excluding all water molecules and the ligands displaying activity were docked (flexibly) in this grid. Subsequently, the best scoring binding modes was minimized with the ABD leaving the ligand and amino acid side chains flexible, but restricting the movements of the protein backbone. This minimization did not significantly alter the position of the side chains or the ligand. The binding modes are shown together with that of Glu in Fig. 4. In the X-ray structure, the α-carboxylic acid of Glu makes a bidentate electrostatic interaction with R518 (Fig. 4B). This is a general characteristic of all amino acid analogs that have been crystallized with ABDs of GluRs.[10] The distal carboxylic acid of Glu is bound by hydrogen bonds. Surprisingly, the three ligands docked inversely to Glu with the distal carboxylic acid group having the bidentate interaction with R518 (Fig. 4B–D) and the α-carboxylic acid of the ADC ligands overlaying with the distal carboxylic acid group of Glu. We also docked the ligands including two water molecules important for Glu binding.[22] However, the general binding mode with the amino group in the same position as that of Glu (Fig. 4E) was only observed for D-cis-ADC. An X-ray crystallographic study of these new ligands in the ABD of the NMDA receptors would further address the validity of suggested inverse binding modes.
Figure 4.

(A) X-ray structure of ABD of NR2A with Glu bound. (B–D) Proposed binding mode of L-trans-ACD (orange), D-cis-ADC (yellow) and D-trans-ADC (purple), respectively, in the NR2A agonist binding site compared to Glu (grey). (E) Alternative binding mode of D-cis-ADC (yellow) when docked with two water molecules in the binding site. (F) View of proposed binding mode of L-trans-ACD (orange), D-cis-ADC (yellow) rotated 90° to the right and emphasizing a possible interaction with Y730.
The inverse binding mode is probably due to a close contact (approx. 3 Å for D-cis-ADC and L-trans-ADC and 5 Å for D-trans-ADC) between the positively charged amino group of the ADC ligands and Y730 (Fig. 4F) that further mediates an interdomain contact to E413. Y730 is also close to V715 that is part of a short sequence containing several non-conserved residues among NR2A-D. Thus, altered conformation of Y730 could affect V715 and may explain the activity variance seen at these subtypes.
In a recent study, mutagenesis of Y730 to phenylalanine in NR2A decreased the potency of glutamate by 45-fold, thereby supporting the idea that an interdomain contact predicted from molecular dynamics simulations between E413 and Y730 in NR2A contributes to agonist potency.[23] In the same study, the reduction of glutamate potency was not observed when this potential interdomain contact was disrupted in NR2D (Y732F mutation in NR2D). In fact, the potency of glutamate was slightly increased on NR2D(Y732F). Furthermore, the glutamate analog (2S,4R)-4-methylglutamate (SYM2081) displays a 46-fold higher potency at NR1/NR2D over NR1/NR2A that can be explained by steric clash between the methyl group of SYM2081 and Y730 in NR2A.[23] These data argue that the E413:Y730 interdomain hydrogen bonds can stabilize the active conformation of the ABD and thus influence the energetics underlying agonist binding to the NR2A subunit. It is possible that the close contact between the positively charged amino group of the ADC ligands and Y730 in NR2A disrupts the conformation of Tyr730 and possibly also the predicted E413:E730 interdomain contact, thereby reducing ability of the ADC ligands to activate NR1/NR2A.
Conclusion
We have synthesized the four stereoisomers of ACD (L-trans-ADC, L-cis-ADC, D-trans-ADC, and D-cis-ADC) in a stereocontrolled fashion following distinct strategies for the cis-ADC and for the trans-ADC enantiomers. The four ACDs were investigated in a radio ligand binding assay ([3H]CGP39653) at native NMDA receptors and subsequently characterized as potential agonists at the four NMDA receptor subtypes NR1/NR2A, NR1/NR2B, NR1/NR2C, and NR1/NR2D. Most notable was L-trans-ADC, which showed the highest potency at NR1/NR2D (EC50 = 50 μM), with a 9.4-, 3.4 and 1.9-fold preference over NR1/NR2A-C, respectively. Subsequent in-silico ligand-protein docking suggested an unusual binding mode for these amino acids in the agonist binding site.
Experimental section
Chemistry
General comments: 1H and 13C spectra (CDCl3 solution unless otherwise stated) were recorded on a Bruker AC 200 or 300 spectrometer at 200, 300 (1H), 50.3 and 75.5 (13C) MHz; chemical shifts are reported in ppm from TMS. Optical rotations were determined with a Perkin Elmer 241 instrument. All reactions were carried out under argon. Column chromatography was performed on silica gel 230–400 mesh by using various mixtures of diethyl ether (E), ethyl acetate (AcOEt), cyclohexane (CyH) and petroleum ether (PE). TLC’s were run on Merck Kieselgel 60F254 plates. Melting points are uncorrected. THF was distilled from sodium/benzophenone ketyl. Dichloromethane and triethylamine were distilled from calcium hydride. HMPA was distilled before use. Other reagents were used as purchased. Mention of “usual workup” means: (i) decantation of the organic layer, (ii) extraction of the aqueous layer with ether, (iii) washing the combined organic layers with brine and drying of the combined organic phases over MgSO4, (iv) solvent evaporation under reduced pressure. Compositions of stereoisomeric mixtures were determined by NMR analysis on crude products before any purification. HRMS were performed at the “Service Central d’Analyses du CNRS” (Vernaison, France). Mass spectra were recorded on a GC/MS HP MS 5989B spectrometer at the University of Versailles.
(2R)-[Benzyl-(3-benzyloxy-2-hydroxy-propyl)-amino]-acetic acid tert-butyl ester 3 and ent-3
To a solution of the β-amino alcohol 2[24] (2.8 g, 10.33 mmol), NaI (3.1 g, 20.66 mmol), and NaHCO3 (1.73 g, 20.66 mmol) in DMF (50 mL) was added dropwise t-butylbromoacetate (3.1 mL, 20.66 mmol). The suspension was stirred at rt for 12h and it was poured into a 1/1 mixture of water and diethyl ether. Usual workup gave a residue that was purified by flash chromatography (PE/AcOEt : 85/15). Compound 3 was obtained as a colourless oil. Yield: 2.8g (70%); Rf = 0.40 (PE/AcOEt : 85/15); [α]D20 = −30.9 (c = 0.4, CHCl3); δH (300 MHz, CDCl3, Me4Si): 1.51 (s, 9H, t-Bu), 2.73 (dd, J = 13 and 9.3 Hz, 1H, NCHHCHOH), 2.94 (dd, J = 13 and 3.6 Hz, 1H, NCHHCHOH), 3.29 (s, 2H, NCH2CO2t-Bu), 3.56 (Apparent d, J = 4.9 Hz, 2H, CH2OBn), 3.81 (d, J = 13.5 Hz, 1H, NCHHPh), 3.94 (d, J = 13.5 Hz, 1H, NCHHPh), 4.02 (quint., J = 4.7 Hz, 1H, CHOH), 4.61 (s, 2H, OCH2Ph), 7.27–7.47 (m, 10H, Ar) ; δc (75 MHz, CDCl3, Me4Si): 28.2 (CH3), 55.8, 58.0, 58.9 (CH2), 67.6 (CH), 72.5, 73.5 (CH2), 81.3 (Cq), 127.6, 127.7, 127.9, 128.4, 128.5, 128.7, 128.8, 129.0 (CHAr), 138.3, 138.5 (CqAr), 171.1 (CO) ; MS (CI, NH3): m/z (%) 386 (100) (MH+), 330 (10), 284 (20), 234 (14), 178 (7); HRMS (ESI) calcd. For C23H32NO4 [MH+]: 386.2331; found 386.2328.
Starting from ent-2 and following the same procedure, ent-3 was obtained in 78% yield. [α]D20 = +30.4 (c = 0.4, CHCl3).
(2R)-[Benzyl-(3-benzyloxy-2-chloro-propyl)-amino]-acetic acid tert-butyl ester 5 and ent-5
To a solution of amino alcohol 3 (1 g, 2.6 mmol) in DCM (10 mL) cooled to 0°C was added dropwise thionyl chloride (415 μL, 5.33 mmol). The resulting mixture was warmed to rt and refluxed for 2h. The reaction was then cooled to 0°C and treated by dropwise addition of a saturated aqueous solution of NaHCO3 (10 mL). The aqueous layer was extracted with diethyl ether (2×15 mL) and usual workup gave a residue (mixture of regioisomeric chlorides) that were rapidly filtrated on a short pad of silica gel eluted with 1/1 : PE/AcOEt. After evaporation of the eluent, the residue (1 g) was taken up in DMF and heated under argon at 65°C for 64 hrs. At this time, DMF was removed in vacuo and the residue was purified by flash chromatography (PE/AcOEt : 9/1) to give chloride 5 as a thick oil. Yield: 960 mg (96%); Rf = 0.70 (PE/AcOEt : 9/1); [α]57820 = +1 (c = 0.4, CHCl3); δH (300 MHz, CDCl3, Me4Si): 1.54 (s, 9H, t-Bu), 3.13 (dd, J = 14.3 and 6.4 Hz, 1H, NCHHCHCl), 3.26 (dd, J = 14.3 and 6.9 Hz, 1H, NCHHCHCl), 3.35 (s, 2H, NCH2CO2t-Bu), 3.75 (dd, J = 10.4 and 5.6 Hz, 1H, CHHOBn), 3.83 (dd, J = 10.4 and 4.6 Hz, 1H, CHHOBn), 3.96 (s, 2H, NCH2Ph), 4.12 (quint., J = 6.6 Hz, 1H, CHCl), 4.62 (s, 2H, OCH2Ph), 7.27–7.48 (m, 10H, Ar) ; δc (75 MHz, CDCl3, Me4Si): 28.3 (CH3), 55.7, 57.9, 58.8 (CH2), 58.9 (CH), 71.9, 73.3 (CH2), 81.1 (Cq), 127.3, 127.7, 127.8, 128.4, 128.9 (CHAr), 137.9, 139.5 (CqAr), 170.7 (CO) ; MS (CI, NH3): m/z (%) 404 (100) (MH+), 348 (15), 312 (50), 303 (40); HRMS (ESI) calcd. For C23H31NO3Cl [MH+]: 404.1992; found 404.2014.
Starting from ent-3 and following the same procedure, ent-5 was obtained in 80% yield. [α]57820 = −0.9 (c = 0.4, CHCl3).
(2R,3S)-tert-Butyl-1-benzyl-3-[(benzyloxy)methyl]azetidine-2-carboxylate 7
To a solution of chloride 5 (403 mg, 1mmol) in a THF-HMPA mixture (5 mL + 0.5 mL respectively) was added dropwise at −78°C a solution of LiHMDS (1M solution in THF, 1.5 mL, 1.5 mmol). The reaction was monitored by TLC and was warmed gradually at 0°C (2h) and then quenched by addition of an aqueous saturated solution of NH4Cl (5 mL). Addition of diethyl ether and water was followed by usual workup. The crude residue was purified by flash chromatography (PE/AcOEt : 4/1) to give azetidine 7 as an oil. Yield: 220 mg (60%); Rf = 0.50 (PE/AcOEt : 4/1); [α]57820 = +23.1 (c = 0.13, CHCl3); δH (300 MHz, CDCl3, Me4Si) : 1.38 (s, 9H, t-Bu), 2.82–2.97 (m, 1H, H-3), 3.06 (Apparent t, J = 7.3 Hz, 1H, H-4), 3.26 (dd, J = 7.0 and 2.5 Hz, 1H, H-4′), 3.63–3.90 (m, 5H, H-2, NCH2Ph, OCH2CH), 4.54 (s, 2H, OCH2Ph), 7.24–7.42 (m, 10H, Ar) ; δc (75 MHz, CDCl3, Me4Si): 28.0 (CH3), 33.6 (C-3), 53.4 (C-4), 61.9 (CH2), 65.6 (CH), 69.8, 73.3 (CH2), 81.8 (Cq), 127.6, 127.8, 128.3, 128.4, 129.3 (CHAr), 137.2, 138.3 (CqAr), 170.2 (CO) ; MS (CI, NH3): m/z (%) 368 (100) (MH+), 312 (21), 266 (30), 91 (10); HRMS (ESI) calcd. For C23H30NO3 [MH+]: 368.2226; found 368.2239.
Starting from ent-5 and following the same procedure, ent-7 was obtained in 58% yield. [α]57820 = −22.9 (c = 0.1, CHCl3).
(2R,3S)-Di-tert-butyl-3-hydroxymethylazetidine-1,2-dicarboxylate 8
To a solution of azetidine 7 (2.25 g, 6.13 mmol) and di-tert-butyl-dicarbonate (2.67 g, 12.4 mmol) in absolute ethanol (15 mL) was added Pd(OH)2, 20%wt. on carbon (1.5 g). The suspension was hydrogenated at rt under 15 bar (218 psi) of hydrogen for 36h. The reaction mixture was then filtrated over Celite, concentrated and dried in vacuum. The residue was purified by flash chromatography (PE/AcOEt : 1/1) to give 8 as a thick oil. Yield: 1.7 g (96%); Rf = 0.35 (PE/AcOEt : 1/1); [α]D20 = +43.8 (c = 0.27, CHCl3); δH (300 MHz, CDCl3, Me4Si) : 1.36 (s, 9H, t-Bu), 1.44 (s, 9H, t-Bu), 2.54 (bs, 1H, OH), 2.97 ( Apparent sext., J = 7.1 Hz, 1H, H-3), 3.58–3.77 (m, 3H, H-4, CH2OH), 3.84 (Apparent. t., J = 8.3 Hz, 1H, H-4′), 4.52 (d, J = 9.1 Hz, H-2) ; δc (75 MHz, CDCl3, Me4Si): 27.9, 28.2 (CH3), 33.8 (C-3), 49.5 (C-4), 61.4 (CH2), 63 (C-2), 79.8, 82.2 (Cq), 155.3, 169.1 (CO) ; MS (ESI): m/z (%) 310 (100) (M+Na+); HRMS (ESI) calcd. For C14H25NO5Na [MH+]: 310.1630; found 310.1639.
Starting from ent-7 and following the same procedure, ent-8 was obtained in 92% yield. [α]D20 = −44.2 (c = 0.3, CHCl3)
(2R,3S)-1,2-Di-(tert-butoxycarbonyl)azetidine-3-carboxylic acid 9
To a suspension of NaIO4 (165 mg, 0.77 mmol) in a (1/1/1) mixture of MeCN/CCl4/H2O (5 mL) was added RuCl3.H2O (15 mg, 0.07 mmol) and the mixture was stirred at rt for 45 min. To this mixture was added alcohol 8 (200 mg, 0.7 mmol) dissolved in MeCN (3 mL), followed by the addition of a second portion of NaIO4 (150 mg, 0.7 mmol). The resulting mixture was stirred at rt for 0.5h, then filtered through a pad of celite and thoroughly washed with EtOAc. The combined filtrates were dried over MgSO4 and concentrated to give a residue which was purified by flash chromatography (CHCl3/MeOH : 9/1). Compound 9 was obtained as a low-viscous oil. Yield: 150 mg (71%); Rf = 0.25 (CHCl3/MeOH : 9/1) ; [α]D20 = +29.9 (c = 0.85, CHCl3); δH (300 MHz, CDCl3, Me4Si) : 1.36 (s, 9H, t-Bu), 1.39 (s, 9H, t-Bu), 3.57 (Apparent. quart., J = 9.0 Hz, 1H, H-3), 3.93 (Appt. t., J = 7.5 Hz, 1H, H-4), 4.21 (Apparent. t., J = 8.4 Hz, 1H, H-4′), 4.60 (d, J = 9.4 Hz, H-2) ; δc (75 MHz, CDCl3, Me4Si): 27.9, 28.3 (CH3), 35.3 (C-3), 49.5 (C-4), 63.6 (C-2), 80.7, 82.5 (Cq), 167.8, 174.8 (CO) ; MS (ESI): m/z (%) 324 (100) (M+Na+); HRMS (ESI) calcd. For C14H23NO6Na [MH+]: 324.1423; found 324.1433.
Starting from ent-8 and following the same procedure, ent-9 was obtained in 70% yield. [α]D20 = −30.5 (c = 0.8, CHCl3)
(2R,3S)-Azetidine-2,3-dicarboxylic acid 10
To a solution of N-Boc azetidine 9 (160 mg, 0.53 mmol) in dichloromethane (3 mL) was added TFA (3 mL) and the solution was stirred at rt overnight. Upon completion the reaction mixture was concentrated in vacuum and the residue was triturated with small portions of dry acetone which were removed in vacuum. The obtained trifluoroacetate salt was dissolved in the minimal quantity of water and deposited on an ion-exchange DOWEX resin (DOWEX 50WX-200, 7g) previously washed with water until neutrality. Elution with water was followed by elution with a 1% aqueous ammonia solution. The ninhydrin positive fractions were lyophilized to give the title compound as a hygroscopic foam. Yield: 70 mg (90%) ; Rf = 0.1 (EtOH/30% aqu. NH3/H2O : 9/3/1) ; Mp : 189–90°C (Litt6 : 187°C) [α]57820 = +736.6 (c = 0.06, H2O) ; [α]36520 +636.4 (c 0.06, H2O) δH (300 MHz, D2O) : 3.52 (td., J = 5.7 and 3.2 Hz, 1H, H-3), 3.84 (dd, J = 10.6 and 5.5 Hz, 1H, H-4), 4.031 (Apparent. t., J = 9.6 Hz, 1H, H-4′), 4.81 (d, J = 9.6 Hz, H-2) ; δc (75 MHz, D2O): 41.1 (C-3), 45.4 (C-4), 60.7 (C-2), 171.5, 175.6 (CO) ; MS (ESI): m/z (%) 168 (100) (M+Na+); HRMS (ESI) calcd. For C5H8NO4Na [MH+]: 146.0453; found 146.0488.
Starting from ent-9 and following the same procedure, ent-10 was obtained in 85% yield. [α]D20 [α]57820 = −727 (c = 0.08, H2O)
(2R,3R)-1-Benzyl-2-ethoxycarbonyl-3-phenyl-azetidine 12
To a solution of cyano azetidine 11 (1.1 g, 4.43 mmol) in absolute ethanol (150 mL) was added sulphuric acid (9.5 mL, 40 equiv.) at 0°C. The mixture was then heated to reflux for a period of 24h, when it was poured carefully in a large beaker containing a saturated solution of sodium bicarbonate (300 mL) precooled at 0°C. After the gas evolution had ceased and the pH was check to be below 7, the aqueous phase was extracted with CH2Cl2 (3 × 100 mL), the combined organic layers were dried over magnesium sulphate and concentrated in vacuo. The title compound was obtained as a clear oil which was used without further purification. An analytical sample was purified by flash chromatography using (Et2O/CyH: 20/80). Yield: 1.2 g (89%) ; Rf = 0.49 (Et2O/CyH: 20/80); [α]D20 = −6.0 (c = 1.00, CHCl3); δH (300 MHz, CDCl3) : 1.21 (t, J = 7.2 Hz, 3H, H-8), 3.14 (dd, J = 6.2 and 8.5 Hz, 1H, H-4), 3.70 (A of an AB syst., J = 12.5 Hz, 1H, H-5), 3.78 (dd, J = 6.2 and 7.9 Hz, 1H, H-4′), 3.81 (d, J = 7.9 Hz, 1H, H-2), 3.91 (B of an AB system., J = 12.5 Hz, 1H, H-5′), 3.87–3.95 (m, 1H, H-3), 4.05–4.22 (m, 2H, H-7), 7.22–7.36 (m, 5H, Ph); δc (75 MHz, CDCl3): 14.3 (C-8), 39.9 (C-3), 57.4 (C-4), 60.9 (C-7), 62.6 (C-5), 71.3 (C-2), 126.9–129.3 (CHAr), 137.0 and 140.4 (Cipso-Ph), 172.1 (C-6); Elemental analysis, calc. for C19H21NO2, C: 77.26, H: 7.17, N: 4.74, found, C: 77.14, H: 7.44, N: 4.55.
Starting from ent-11 and following the same procedure, ent-12 was obtained in 96% yield. [α]D20 = +6.4(c = 0.8, CHCl3).
(2R,3R)-3-Phenyl-azetidine-1,2-dicarboxylic acid 1-tert-butyl ester 2-ethyl ester 13
The N-benzyl protected azetidine 12 (1.09 g, 3.69 mmol) was dissolved in 50 mL of absolute ethanol. Boc2O was added (1.6 g, 7.39 mmol, 2 equiv.) followed by 300 mg of palladium on charcoal 10%wt. The suspension was stirred under a hydrogen atmosphere for six days, then filtered over celite and concentrated in vacuum. The residue was purified by flash chromatography (Et2O/CyH: 20/80), and gave 13 as a colorless oil. Yield: 1.13 g (quant.) ; Rf = 0.6 (Et2O/CyH: 20/80); [α]D20 = −13.8 (c = 9.35, CHCl3); δH (300 MHz, CDCl3) : 1.22 (t, J = 7.11 Hz, 3H, CH2CH3); 1.37 (s, 9H, t-Bu); 3.55–3.62 (m, 1H, H-3); 3.89 (dd, J = 5.8 and 8.1 Hz, 1H, H-4); 4.18 (q, J = 7.11 Hz, 2H, CH2CH3); 4.32 (t, J = 8.1 Hz, 1H, H-4′); 4.52 (d, J = 5.4 Hz, 1H, H-2); 7.19–7.28 (m, 5H, Ph); δc (75 MHz, CDCl3): 14.3 (CH3); 28.3 (CH3, tBu); 38.4 (C-3); 54.3 (C-4); 61.3 (CH2); 67.8 (C-2); 80.3 (Cq.tBu); 126.8, 127.5, 128.9 (CHAr); 140.2 (CqAr); 155.5 (CO); 170.7 (CO); HRMS (ESI) calcd. For C19H22NO4 [MH+]: 328.1549; found 328.1543.
Starting from ent-12 and following the same procedure, ent-13 was obtained in 92% yield. [α]D20 = +14.3(c = 1.5, CHCl3)
(2R,3R)-Azetidine-1,2,3-tricarboxylic acid 1-tert-butyl ester 2-ethyl ester 14
To a suspension of NaIO4 (823 mg, 3.85 mmol, 20 equiv.) in a (1/1/1) mixture of MeCN/CCl4/H2O (6 mL) was added RuCl3.H2O (7 mg) and the mixture was stirred at rt for 30min. To this suspension was added compound 13 (58.8 mg, 0.19 mmol) dissolved in MeCN (3 mL). The reaction was stirred at rt for 24h, it was then diluted with water and extracted with ethyl acetate (4 × 20 mL). The organic layers were dried over MgSO4 and concentrated to give a dark residue that was purified by flash chromatography (CHCl3/MeOH : 9/1) to give 14 as a clear oil. Yield: 39.3 mg (75%); Rf = 0.5 (CHCl3/MeOH : 9/1); [α]D20 = +4.3 (c = 1.1, CHCl3); δH (300 MHz, CDCl3) : 1.24 (t, J = 7.11 Hz, 3H, CH2CH3); 1.36 (s, 9H, t-Bu); 3.27 (dt, J = 5.6 and 10.4 Hz, 1H, H-3), 3.98 (dd, J = 5.6 and 8.1 Hz, 1H, H-4); 4.11–4.23 (m, 3H, H-4′ and CH2CH3); 4.73 (d, J = 5.4 Hz, 1H, H-2); 9.15 (broad s, 1H, COOH); δc (75 MHz, CDCl3): 14.1 (CH3); 28.2 (CH3, tBu); 36.2 (C-3); 50.1 (C-4); 61.7 (CH2); 62.8 (C-2); 81.1 (Cq tBu); 155.4 (CO); 169.8 (CO); 175.1 (CO2H); HRMS (ESI) calcd. For C10H21NO6Na [MH+]: 274.1267; found 274.1278.
Starting from ent-13 and following the same procedure, ent-14 was obtained in 74% yield. [α]D20 = −5 (c = 0.9, CHCl3)
(2R,3R)-Azetidine-2,3-dicarboxylic acid 15
To a solution of N-Boc azetidine 14 (52.1 mg, 0.19 mmol) in methanol (1 mL) was added 23 mg of sodium hydroxide dissolved in 1 mL of water. The solution was stirred at rt for 48h when 2 ml of hydrochloric acid 1M was added. The solution was stirred 5h at rt and it was concentrated in vacuo. The white residue was dissolved in the minimal quantity of water and deposited on an ion-exchange DOWEX resin (DOWEX 50WX-200, 7g) previously washed with water until neutrality. Elution with water was followed by elution with a 1% aqueous ammonia solution. The ninhydrin positive fractions were lyophilized to give of the title compound as a white powder. Yield: 72% (20 mg); [α]D20 = +45.6 (c = 0.09, H2O); δH (300 MHz, D2O) : 3.50 (dt, J = 7.7 and 9.2 Hz, 1H, H-3); 4.01–4.17 (m, 2H, H-4); 4.83 (d, J = 7.7 Hz, 1H, H-2) ; δc (75 MHz, D2O): 42.2 (C-3); 46.4 (C-4); 62.5 (C-2); 173.3 (COOH); 176.9 (COOH) ; HRMS (ESI) calcd. For C5H8NO4 [MH+]: 146.0453; found 146.0446.
Starting from ent-14 and following the same procedure, ent-15 was obtained in 40% yield. [α]D20 = −47 (c = 0.2, H2O)
Two-electrode voltage-clamp electrophysiology
cRNA for rat NR1-1a (hereafter NR1) and NR2A, B, C, and D were synthesized in vitro and injected (5–10 ng) into Xenopus laevis oocytes as previously described.[25] Rat cDNAs for NR1 and NR2 subunits (GenBank numbers NR1: U11418 and U08261; NR2A: D13211; NR2B: U11419; NR2C: M91563; NR2D: L31611 (modified according to Monyer et al. (1994)) were provided by Drs. S. Heinemann (Salk Institute), S. Nakanishi (Kyoto University), and P. Seeburg (University of Heidelberg). Two-electrode voltage-clamp current recordings were made 24–72 hours post injection as previously described.[23] Recordings from 5–10 oocytes from two different Xenopus laevis were performed for all compounds. Agonist concentration–response data were pooled among oocytes and composite dose-response data were fitted by the following equation:
Imax is the maximum current in response to the agonist, nH denotes the Hill coefficient, [A] is the agonist concentration, and EC50 is the agonist concentration that produces half-maximum response. Relative Imax were calculated from a full concentration–response measurement as Imax(agonist)/Imax(Glu), where Imax(agonist) is the fitted Imax according to the Hill equation and Imax(Glu) is the maximum current obtained from glutamate in the same recording.
NMDA receptor binding
The four stereoisomeric azetidines were evaluated for NMDA receptor binding affinity ([3H]CGP39653) in native rat synaptosomes in accordance with previously described experimental procedures.[26]
Molecular Modelling
The docking of the ligands was performed using essentially the same procedure as previously described,[27] the only difference being that the ligands are docked in NR2A rather than a homology model of NR2B.
Acknowledgments
We would like to thank the Carlsberg Foundation, the Alfred Benzon Foundation, the Villum Kann Rasmussen Foundation, the Lundbeck Foundation, the Danish Medical Research Council, NIH-NINDS (NS36654), and IFCPAR (Indo-French Center for the Promotion of Advanced Research) is gratefully acknowledged for funding (Project N°3005-1).
Contributor Information
Prof. François Couty, Email: couty@chimie.uvsq.fr.
Assoc. Prof. Lennart Bunch, Email: lebu@farma.ku.dk.
References and notes
- 1.Dingledine R, Borges K, Bowie D, Traynelis SF. Pharmacological Reviews. 1999;51:7–61. [PubMed] [Google Scholar]
- 2.Froemke RC, Poo MM, Dan Y. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Otano I, Ehlers MD. Trends in Neurosciences. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Nacher J, McEwen BS. Hippocampus. 2006;16:267–270. doi: 10.1002/hipo.20160. [DOI] [PubMed] [Google Scholar]
- 5.Dirnagl U, Iadecola C, Moskowitz MA. Trends in Neurosciences. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 6.Bräuner-Osborne H, Egebjerg J, Nielsen EO, Madsen U, Krogsgaard-Larsen P. Journal Of Medicinal Chemistry. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- 7.Danysz W, Parsons CG. Pharmacological Reviews. 1998;50:597–664. [PubMed] [Google Scholar]
- 8.Palmer GC. Current Drug Targets. 2001;2:241–271. doi: 10.2174/1389450013348335. [DOI] [PubMed] [Google Scholar]
- 9.Waxman EA, Lynch DR. Neuroscientist. 2005;11:37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- 10.Erreger K, Chen PE, Wyllie DJ, Traynelis SF. Crit Rev Neurobiol. 2004;16:187–224. doi: 10.1615/critrevneurobiol.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- 11.Cull-Candy SG, Leszkiewicz DN. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- 12.Couty F, Evano G. Organic Preparations and Procedures International. 2006;38:427–465. [Google Scholar]
- 13.Bridges RJ, Lovering FE, Humphrey JM, Stanley MS, Blakely TN, Cristofaro MF, Chamberlin AR. Bioorganic & Medicinal Chemistry Letters. 1993;3:115–121. [Google Scholar]
- 14.Arakawa Y, Murakami T, Arakawa Y, Yoshifuji S. Chem Pharm Bull (Tokyo) 2003;51:96–97. doi: 10.1248/cpb.51.96. [DOI] [PubMed] [Google Scholar]
- 15.Sivaprakasam M, Couty F, Evano G, Srinivas B, Sridhar R, Rao KR. Synlett. 2006:781–785. [Google Scholar]
- 16.Bräuner-Osborne H, Bunch L, Chopin N, Couty F, Evano G, Jensen AA, Kusk M, Nielsen B, Rabasso N. Organic and Biomolecular Chemistry. 2005;3:3926–3936. doi: 10.1039/b509514j. [DOI] [PubMed] [Google Scholar]
- 17.Agami C, Couty F, Evano G. Tetrahedron-Asymmetry. 2002;13:297–302. [Google Scholar]
- 18.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. Journal Of Organic Chemistry. 1981;46:3936–3938. [Google Scholar]
- 19.Bunch L, Johansen TH, Bräuner-Osborne H, Stensbol TB, Johansen TN, Krogsgaard-Larsen P, Madsen U. Bioorganic & Medicinal Chemistry. 2001;9:875–879. doi: 10.1016/s0968-0896(00)00304-7. [DOI] [PubMed] [Google Scholar]
- 20.Clausen RP, Hansen KB, Cali P, Nielsen B, Greenwood JR, Begtrup M, Egebjerg J, Brauner-Osborne H. European journal of pharmacology. 2004;499:35–44. doi: 10.1016/j.ejphar.2004.07.049. [DOI] [PubMed] [Google Scholar]
- 21.Hansen KB, Bräuner-Osborne H, Egebjerg J. Combinatorial Chemistry & High Throughput Screening. 2008;11:304–315. doi: 10.2174/138620708784246040. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa H, Singh SK, Mancusso R, Gouaux E. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 23.Erreger K, Geballe MT, Kristensen A, Chen PE, Hansen KB, Lee CJ, Yuan H, Le P, Lyuboslavsky PN, Micale N, Jorgensen L, Clausen RP, Wyllie DJ, Snyder JP, Traynelis SF. Mol Pharmacol. 2007;72:907–920. doi: 10.1124/mol.107.037333. [DOI] [PubMed] [Google Scholar]
- 24.Berg S, Larsson LG, Renyi L, Ross SB, Thorberg SO, Thorell-Svantesson G. Journal of Medicinal Chemistry. 1998;41:1934–1942. doi: 10.1021/jm970806i. [DOI] [PubMed] [Google Scholar]
- 25.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermit MB, Greenwood JR, Nielsen B, Bunch L, Jorgensen CG, Vestergaard HT, Stensbol TB, Sanchez C, Krogsgaard-Larsen P, Madsen U, Bräuner-Osborne H. European journal of pharmacology. 2004;486:241–250. doi: 10.1016/j.ejphar.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 27.Clausen RP, Christensen C, Hansen KB, Greenwood JR, Jørgensen L, Micale N, Madsen JC, Nielsen B, Egebjerg J, Bräuner-Osborne H, Traynelis SF, Kristensen JL. Journal of Medicinal Chemistry. 2008;51:4179–4187. doi: 10.1021/jm800025e. [DOI] [PMC free article] [PubMed] [Google Scholar]