

Significance
The fragile X mental retardation protein (FMRP) is most highly expressed in neurons, and is critical for proper synaptic functioning. Fragile X syndrome, a common cause of intellectual disability, is the result of absent or dysfunctional FMRP, highlighting its importance to the processes underlying learning and memory. A rapid upregulation of FMRP synthesis at the synapse in response to specific neuronal signals is a key step in maintaining a dynamic synapse, although the mechanisms governing this up-regulation are not well-understood. We show that a variant in the 3′UTR of fragile X mental retardation 1 (FMR1) causes the loss of this characteristic increase in synaptic FMRP synthesis, which may lead to developmental delay in patients. These data identify several mechanisms and molecules modulating activity-dependent translation of FMRP.
Keywords: fragile X syndrome, FMR1, FMRP, HuR, autism
Abstract
Fragile X syndrome is a common cause of intellectual disability and autism spectrum disorder. The gene underlying the disorder, fragile X mental retardation 1 (FMR1), is silenced in most cases by a CGG-repeat expansion mutation in the 5′ untranslated region (UTR). Recently, we identified a variant located in the 3′UTR of FMR1 enriched among developmentally delayed males with normal repeat lengths. A patient-derived cell line revealed reduced levels of endogenous fragile X mental retardation protein (FMRP), and a reporter containing a patient 3′UTR caused a decrease in expression. A control reporter expressed in cultured mouse cortical neurons showed an expected increase following synaptic stimulation that was absent when expressing the patient reporter, suggesting an impaired response to neuronal activity. Mobility-shift assays using a control RNA detected an RNA–protein interaction that is lost with the patient RNA, and HuR was subsequently identified as an associated protein. Cross-linking immunoprecipitation experiments identified the locus as an in vivo target of HuR, supporting our in vitro findings. These data suggest that the disrupted interaction of HuR impairs activity-dependent translation of FMRP, which may hinder synaptic plasticity in a clinically significant fashion.
Fragile X syndrome (FXS) is one of the most common forms of inherited intellectual disability, and represents a well-known genetic cause of autism spectrum disorder. FXS is a monogenic disorder characterized by the loss or dysfunction of the fragile X mental retardation protein (FMRP), the product of the fragile X mental retardation 1 (FMR1) gene (1). In a vast majority of FXS patients, FMRP expression is absent due to the expansion of an unstable CGG repeat in the promoter region of the FMR1 gene. This repeat tract is polymorphic in the population, where 5–45 CGG repeats are typical. FXS patients have in excess of 200 repeats, referred to as the full mutation (2), usually inherited via an unstable maternal premutation allele (55–200 repeats). At the full mutation length threshold of ∼200 repeats, an epigenetic event manifests that results in hypermethylation of the FMR1 promoter region and subsequent silencing of the transcript and protein expression (3–5).
Studies of FMRP function suggest that it is a selective RNA-binding protein (RBP) that primarily acts as a negative regulator of translation (6, 7), and is estimated to associate with about 4–5% of mRNA messages expressed in the brain, including its own transcript (8–10). FMRP is also a key regulator of translation downstream of glutamate receptor-mediated signaling in neurons, where it is rapidly inactivated by dephosphorylation upon receptor activation, thereby allowing protein synthesis of its targets to occur in response to the stimulus (11–14). The absence of FMRP uncouples glutamate receptor stimulation from the protein synthesis typically required for proper signal transduction at the synapse (15). These molecular defects are associated with impaired synaptic plasticity, widely believed to underlie the processes of learning and memory, which requires tightly controlled synaptic protein synthesis (16, 17) and is thought to be a principal cause of the cognitive disabilities in FXS patients (12, 18, 19). Whereas the effects of lacking FMRP entirely have been extensively investigated, few studies have focused on the consequences of still-present but dysregulated translation of FMRP.
Despite significant analysis of the FMR1 gene, only a small number of conventional genetic mutations, such as point mutations and insertions/deletions, have been reported to be associated with FXS or developmental delay (20–27). To identify causes of developmental delay attributable to FMR1 variants other than the repeat expansion, our group sequenced the FMR1 gene in 963 developmentally delayed males, each of whom tested negative for the CGG expansion mutation, and discovered a number of previously unreported variants (28). However, the molecular consequences, if any, of most of these variants remain unknown.
In this study, we describe the functional impact of a variant in the 3′UTR of FMR1 (c.*746T>C) using genetic, biochemical, and cell biological approaches. The variant is associated with reduced basal FMRP levels and impairs the normal response to activity-dependent synaptic translation in cultured primary neurons. Our data suggest that the RNA-binding protein HuR binds the locus normally but that this association is lost when the variant is present, leading to destabilized and rapidly degraded FMR1 transcript. These findings indicate that the c.*746C variant allele, detected at a frequency of 1 in 160 developmentally delayed male patients tested (28), may represent an unrecognized genetic basis of developmental delay via FMR1 dysregulation.
Results
Clinical Assessment of a Patient with the c.*746T>C Variant.
One of the patients identified by Collins et al. (28) as harboring the c.*746T>C variant was clinically evaluated by the Emory University Medical Genetics Clinic. The patient (pictured in Fig. 1A) was ∼10.5 y old at evaluation and was born full-term at 10 lb, 1 oz without complication. He was reported to have sat independently at 17 mo [typically achieved by 9 mo of age (29)] and first walked at 24 mo [typically achieved by 18 mo of age (29)]. At examination, he was nonverbal and exhibited stereotypic behavior consisting of rocking, spinning, rubbing his fingers, and repetitively touching his shirt collar. The patient had previously been diagnosed with autism spectrum disorder and attention deficit hyperactivity disorder (ADHD), and he attends special education classes. Cognitive abilities were assessed using the Stanford–Binet Intelligence Scales (5th Ed) (30), revealing moderate intellectual disability (IQ score 47). In terms of his growth parameters, the patient is within the 50–75th percentiles for weight (88 lb), height (60 in), and head circumference (55 cm). A physical examination was performed where bilateral flat feet with inversion were noted but no other significant findings. The patient’s half-brother was also evaluated, and genotyping revealed that he also possesses the c.*746C variant. He was reported to have sat independently at 17 mo old, taken his first steps at 24 mo, and spoken his first word at 5 y of age. Approximately 17 y old at examination, he speaks in short sentences, attends some regular 10th-grade classes with specialized classes for reading and math, is mildly intellectually disabled (IQ score 67), and has previously been diagnosed with ADHD.
Fig. 1.

Patient 3′UTR is associated with a significant reduction in endogenous FMRP and reporter activity in multiple cell types. (A) Photograph of a patient harboring the FMR1 c.*746C allele. (B) (Top) Representative Western blot of FMRP and eIF4e from two unrelated control male lymphoblastoid cell lines and the patient-derived lymphoblastoid cell line. (Bottom) The band density on the Western blot of three independent protein preparations was digitally quantified by ImageJ software (NIH). Data shown are the mean ± SD. (C) Luciferase assay in HEK293 and Neuro2a cell lines using vectors with a control 3′UTR or the patient 3′UTR. Each of three independent experiments was normalized to cotransfected Renilla luciferase activity; data shown are the mean ± SD. (D) Luciferase assay results of three independent experiments in HEK293 cells using vectors of the control, patient, and the patient’s half-brother. Data shown are the mean ± SD. Unpaired two-tailed t test, *P < 0.05; a.u., arbitrary units; n.s., not significant.
Patient 3′UTR Reduces Translation of FMRP and a Reporter.
In addition to the developmental delay and intellectual disability in the patient, several noteworthy lines of evidence led us to investigate the molecular impact of the FMR1 c.*746C allele. First, the variant was significantly enriched in unrelated developmentally delayed male patients compared with gender-matched controls [found in 6 of 963 patients and 0 of 1,260 controls; P = 0.007 (28)]. Additionally, the c.*746 position and broader genetic element are highly conserved evolutionarily, as indicated by PhyloP (31) (2.76), GERP (32) (5.52), and PhastCons (33) (1.00) scores. Last, the locus is thymine- and uridine-rich at the RNA level, which we hypothesized could serve as a site of interaction for a class of U-rich RBPs (Fig. S1). Based on these data, we explored whether the variant had any effect on FMRP expression in the patient. A lymphoblastoid cell line was established from the patient’s blood and Western blot analysis revealed a modest, but significant, reduction of the patient’s endogenous FMRP level compared with two healthy lymphoblastoid control lines (Fig. 1B). To corroborate these results, luciferase reporter vectors were constructed to include the full-length FMR1 3′UTR of the patient or a healthy control downstream of the firefly luciferase gene. We observed a significant decrease in normalized luciferase signal with the patient reporter compared with the control in two different cell lines (P = 0.007 in HEK293FT; P = 0.004 in Neuro2a; Fig. 1C). Additionally, the 3′UTR from the patient’s affected half-sibling brother, who harbored the c.*746C allele as well, showed a similar reduction in luciferase activity compared with the control (P < 0.001; Fig. 1D). Steady-state levels of luciferase transcript were equivalent between the patient and control vectors (Fig. S2), suggesting a posttranscriptional mechanism underlying the reduction in patient reporter activity. However, other mechanisms, such as mRNA instability, may be responsible for the observed decrease in luciferase activity, which these quantitative (q)RT-PCR assays cannot evaluate. Together, these data indicate that the patient 3′UTR is associated with a decrease in endogenous FMRP and reporter expression, and that the FMR1 gene may be regulated posttranscriptionally via the 3′UTR at the c.*746 locus.
Fig. S1.

Multiple-species DNA alignment at the FMR1 c.*746 locus. The c.*746 nucleotide (indicated by the red box) and the broader T-rich element (U-rich at the RNA level) are both highly conserved. PhyloP conservation scores are shown as bars (Top); these were generated by the UCSC Genome Browser (https://genome.ucsc.edu/). The pink boxes show that the PhyloP score for that particular nucleotide.
Fig. S2.
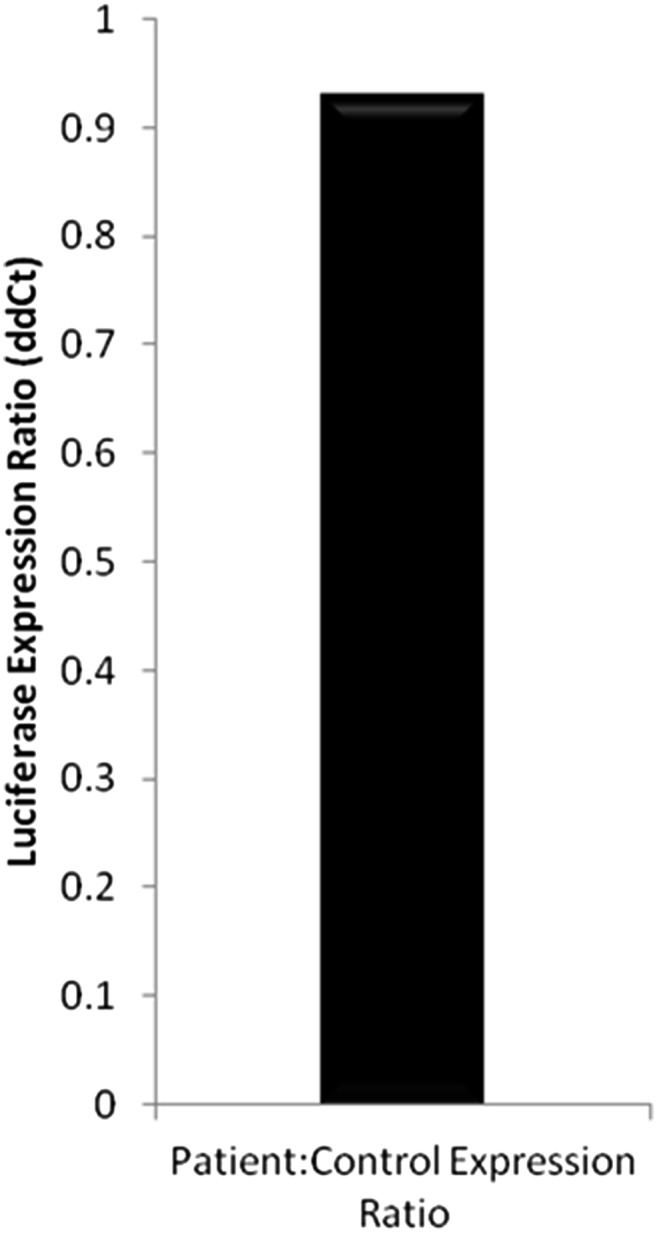
Luciferase mRNA was isolated from HEK293 cells that were transfected with the control or patient vectors. The data shown are the mean ∆∆Ct expression ratio of firefly luciferase normalized to Renilla luciferase; n = 3 independent experiments ± SD.
The c.*746 Locus Is Important for Translational Regulation.
To identify whether the c.*746 locus is the specific site responsible for the observed decrease in reporter activity because the 3′UTR of FMR1 exhibits frequent sequence variation, we used site-directed mutagenesis to change the variant 746C nucleotide in the patient reporter to the reference thymine. This single-nucleotide change restored luciferase activity to the level of the control. Conversely, mutagenesis of the control vector to the patient allele caused a reduction in reporter signal, indicating that the variant is specific in causing the diminished translation (Fig. 2A). Given that the variant does not completely abolish reporter activity, we examined whether mutagenesis of the broader U-rich motif would cause a more pronounced effect to determine the magnitude of translational misregulation caused directly by the c.*746C variant. We deleted 17 bp from the locus, including the variant site, in both the control and patient vectors (Fig. 2B). When expressing the control vector with the deletion, we observed a significant decrease in reporter activity compared with the control (P = 0.004) but not a more severe reduction than that observed with the patient reporter. The same deletion in the patient vector did not significantly alter expression from the unchanged patient reporter (P = 0.27). Because the patient vector with the deletion did not show significantly altered expression compared with the unchanged patient vector, the c.*746 position itself is likely a critical nucleotide in the motif. Taken together, these data implicate the locus, and the c.*746C variant in particular, as causative of the diminished reporter expression.
Fig. 2.

FMR1 c.*746 site modulates steady-state and activity-dependent reporter activity. (A) Results of three independent luciferase assays using mutagenized vectors in HEK293 cells. Reference allele at c.*746, T; patient allele, C. Data shown are the mean ± SD. (B) A 17-bp deletion in the vectors that deletes the U-rich locus including the 746 site (indicated by a horizontal bracket and red lettering, respectively) was used in luciferase assays. Results are the mean of three independent experiments ± SD. (C) Luciferase assay in WT mouse primary cortical neurons (E17.5) using various vectors with and without DHPG treatment (100 μM, 5 min) ∼24 h posttransfection. Data shown are the mean of four or five independent experiments ± SD. (D) Luciferase assay in Fmr1 KO mouse primary cortical neurons (E17.5) using various vectors with and without DHPG treatment (100 μM, 5 min) ∼24 h posttransfection. Data shown are the mean of four or five independent experiments ± SD. Unpaired two-tailed t test, *P < 0.05. C, control; Del, deletion; P, patient.
The c.*746C Variant Allele Is Refractory to Glutamate Receptor Signaling in Primary Neurons.
Although the patient’s lymphoblastoid cell line showed reduced levels of FMRP, ∼80% of normal levels were still present. We questioned whether this minimal reduction in FMRP could cause the developmental and cognitive disabilities displayed by the patient, and wondered whether there could be a neuron-specific mechanism of dysfunction that may not be apparent in other tissues. To explore this, we tested whether the variant allele affected activity-dependent protein synthesis in primary neurons. C57BL/6 (WT) and Fmr1 knockout (KO) mouse cortical neurons were isolated, cultured, and transfected with the described luciferase reporters to evaluate translation when treated with (RS)-3,5-dihydroxyphenylglycine (DHPG), a glutamate analog that stimulates synaptic activity via the group I metabotropic glutamate receptors (mGluRs) (12, 34, 35). The addition of DHPG to cultures expressing the control reporter caused an increase in translation in WT neurons, but not in Fmr1 KO neurons, as expected (Fig. 2 C and D, respectively). However, when expressing the patient reporter in WT neurons, there was no increase in translation after DHPG treatment in addition to the steady-state reduction (Fig. 2C). When the mutagenized control reporter was expressed in WT neurons, the normal up-regulation of translation elicited by DHPG treatment was lost (Fig. 2C). These data reveal a loss of activity-dependent translation in neurons in response to glutamate signaling by the patient reporter, and identify the c.*746 locus as associated with this deficit.
Loss of RNA-Binding Protein Association Caused by the c.*746C Variant.
Several potential mechanisms underlying these data were considered, including disruption of microRNA or RBP interactions caused by the variant allele. To determine whether an RBP targets the locus, we used electromobility-shift assays (EMSAs) to evaluate whether a 42-nt biotinylated RNA probe encompassing the c.*746 site interacts with a protein or protein complex from mouse whole-brain lysate. We detected a band shift using the control probe that was dosage-sensitive and -specific, because an excess amount of unlabeled competitor probe was able to outcompete the labeled probe for the interaction (Fig. 3 A and B; see also Fig. S3 and Table S1). To determine whether the c.*746C variant disrupted this interaction, we generated a biotinylated patient probe that differed only at the 746 position and compared this with the control probe binding. The patient probe lacked the band shift, suggesting a loss of association with the protein or protein complex due to the 746C variant (Fig. 3C and Table S1). Additionally, an unlabeled competitor patient probe could not compete away the interaction from the control, even at concentrations of 40× molar excess, confirming the inability of the protein to bind the patient sequence (Fig. 3D). These results demonstrate that the locus is a target of an RBP or protein complex and that the variant disrupts this association, identifying a potential mechanism underlying the observed changes in the reporter assays.
Fig. 3.

RNA EMSAs reveal a specific and dose-dependent interaction between a protein and the c.*746 locus that is absent with the patient sequence. (A) A biotinylated control RNA probe incubated with increasing amounts of mouse whole-brain lysate and resolved by 5% native TBE gel. (B) An unlabeled version of the control RNA probe was added in increasing amounts (up to 40× excess by molar concentration) to the whole-brain lysate binding reaction and resolved on a 5% native TBE gel. (C) A biotinylated patient RNA probe containing the c.*746C nucleotide was incubated with increasing amounts of mouse whole-brain lysate in the same manner as the control probe and resolved on a 5% native TBE gel. (D) An unlabeled version of the patient RNA probe was added in increasing amounts, up to 40× molar excess concentrations, to the binding reactions with the labeled control probe and resolved on a 5% native TBE gel.
Fig. S3.

Full image of the RNA EMSA from Fig. 3A is shown. The high molecular-weight bands likely represent endogenously biotinylated proteins, because this band is also present in the lysate-only lane and not the probe-only lane.
Table S1.
Sequences of the biotinylated RNA probes used in the RNA gel-shift assays
| Probe | Probe sequence, 5′ to 3′ |
| Control | GGACUUGUUUUUGUUUUUGUUUUGUUGCACUGAAGUUUGAUA |
| Patient | GGACUUGUUUUUGUUCUUGUUUUGUUGCACUGAAGUUUGAUA |
The single difference between the two probes at the c.*746 position is underlined.
The RNA-Binding Protein HuR Associates with the c.*746 Locus.
To identify the protein(s) that interacts with this RNA sequence, we used two approaches, both of which made use of tandem mass spectrometry (MS-MS) for protein identification. First, we performed two biological replicates of a coimmunoprecipitation (co-IP) assay where streptavidin-coated magnetic beads were fixed with the biotinylated control RNA probe and then incubated with mouse whole-brain lysate. We eluted bound proteins and subjected the sample to MS-MS peptide sequencing to identify enriched proteins compared with a bead-only control. Second, we performed the EMSAs as described and then excised the shifted band region from the gel and subjected the eluted protein sample to MS-MS analysis to directly identify proteins in the band-shift region (Table S2). Three candidates were detected by both screening methods: HuR (also known as ELAVL1), PUR-α, and PUR-β. Whereas the PUR proteins typically bind purine-rich motifs, HuR is known to target U-rich elements in the 3′UTR of genes. Given the U-rich nature and location of the FMR1 c.*746 locus, we pursued HuR as the top RBP candidate.
Table S2.
Rank aggregation analysis of proteins identified by MS-MS in two biological replicates of the RNA pull-down assay
| Rank | Gene symbol |
| 1 | PRKRA |
| 2 | KHSRP |
| 3 | ILF3 |
| 4 | FUBP1 |
| 5 | PURB |
| 6 | PURA |
| 7 | ELAV1 |
| 8 | SFPQ |
| 9 | RFC4 |
| 10 | ILF2 |
| 11 | HNRNPD |
| 12 | TARBP2 |
| 13 | AIMP1 |
| 14 | EEF1A1 |
| 15 | FUS |
| 16 | FKBP1A |
| 17 | TNIK |
| 18 | ALYREF |
| 19 | HNRNPU |
| 20 | FUBP3 |
The underlining indicates the proteins that were also identified by gel extraction of the band shift in the RNA EMSAs. Note: This list excludes two KRT (keratin) genes, because these are typically considered background in mass spectrometry protein ID experiments due to their abundance in the environment.
To determine whether the band shift we observed in the EMSAs was indeed HuR, we first performed a gel-supershift assay using an antibody to HuR. As shown in Fig. 4A, the shifted band disappears with the addition of HuR antibody to the binding reaction, although the supershifted band was not visible due to the high background signal inherent to using a whole-brain lysate. Next, we used a HuR antibody to immunopurify HuR-bound mRNAs from HEK293 cell lysates and then assayed for FMR1 as well as an established target of HuR (β-actin) and a gene not known to interact with HuR (GAPDH). FMR1 was enriched by HuR IP ∼400-fold over an IgG control, similar to the level of β-actin enrichment, whereas GAPDH showed no enrichment (Fig. 4B), indicating that HuR indeed associates with FMR1. To localize this interaction specifically to the 746 locus, we performed gel-shift, competition, and supershift assays with purified HuR protein rather than whole-brain lysate. These assays revealed an association between HuR and the control probe, and confirmed the lack of interaction between the patient probe and HuR (Fig. 4C). Altogether, FMR1 is bound by HuR at the c.*746 locus, and this interaction is disrupted by the variant allele.
Fig. 4.

HuR binds FMR1 at the c.*746 locus and is present at the synapse. (A) Supershift assays were performed using 32P-labeled control probe and HuR antibody. Binding reactions were carried out as described previously with the addition of 0.5–1 μg of antibody per 10 μg of mouse whole-brain lysate in each reaction. (B) Immunoprecipitation of HuR protein from HEK293 cell lysate was performed, and copurified mRNAs were assayed by real-time RT-PCR. The results show the fold enrichment of mRNA levels of β-actin (a known target of HuR), GAPDH (not known to interact with HuR), and FMR1 mRNA normalized to IP with IgG alone. All data are displayed as the ratio of IP mRNA levels to input mRNA levels. Data shows the mean of three independent experiments ± SD. (C) RNA EMSA using purified HuR protein with the control or patient probe, unlabeled competitor control probe, and HuR antibody. (Bottom) Free probe bands from a shorter exposure of the same blot to ensure equal probe loading. (D) Image of immunofluorescence staining for HuR (green), PSD-95 (red), and nuclei (blue) in primary cortical neurons. A merged image of all colors is shown (Top Left). (Scale bar, 10 μm.) The white boxes in the merged image are enlarged in the Bottom two rows, with the Top row representing the Left box and the Bottom row representing the Right box. The merged, HuR (green), and PSD-95 (red) images are shown in both Bottom rows from Left to Right, respectively. (Scale bars, 5 μm.) (E) Western blot showing the presence of HuR in a whole-cortical lysate and synaptoneurosomes. GFAP, a glial marker, was used as an indicator of synaptoneurosome purity. An antibody against eiF4e was used as a loading control. C1 and S1, cortical lysate and synaptoneurosomes, respectively, of preparation 1; C2 and S2, cortical lysate and synaptoneurosomes, respectively, of preparation 2. (F) Synaptoneurosome preparations were subjected to HuR immunoprecipitation, and the level of associated Fmr1 was assayed by qRT-PCR. The levels of GAPDH and β-actin mRNA were also assayed in the immunoprecipitate as a negative and positive control, respectively. Data shown are the mean of three independent synaptoneurosome preparations ± SD. (*P < 0.05 using a two-tailed unpaired t test.) (G) Actinomycin D (5 μg/mL) was added to control and patient lymphoblastoid cells to block transcription, and qRT-PCR was performed for FMR1 (Right) and PSD-95 (Left) after 1, 2, 4, and 6 h of drug treatment as well as an untreated control (0 h). Each point shows the mean percentage of mRNA remaining relative to the 0-h control ± SEM in six independent experiments. Nonlinear regression lines were fit to each set of data points, and the slopes were calculated and compared. For FMR1, the difference in slope was significantly different between the control and patient (P = 0.008); for PSD-95, the difference in slope was not significantly different (P = 0.582).
HuR Localizes to Dendrites and Synapses.
HuR is typically most abundant in the nucleus of cells, but is known to translocate to the cytoplasm under certain conditions to facilitate mRNA stability and translation (36, 37). If HuR is involved in the activity-dependent translation of FMR1, it must be present at the synapse, because previous work has shown that endogenous FMR1 mRNA localizes to dendrites (38) and can be locally translated in an activity-dependent manner (39). To determine whether HuR is present at the synapse, we used immunocytochemistry to visualize its location in primary cortical neurons. Although the majority of HuR is located in the nucleus, it is also present in the dendrites distal to the soma (Fig. 4D), a finding supported by other very recent data (40). To more specifically determine whether HuR localizes to the synapse, we performed immunoblot analyses from mouse synaptoneurosome preparations and found the protein to be present, suggesting that HuR is at the synapse (Fig. 4E). Previous work has shown that Fmr1 mRNA is present in synaptic fractions (18), so we investigated whether Fmr1 mRNA was in a complex with HuR in this compartment. Co-IP experiments in synaptoneurosomes revealed a significant enrichment of Fmr1 mRNA in HuR IP fractions, suggesting that these molecules interact at the synapse (Fig. 4F).
Patient FMR1 mRNA Decays Rapidly but Is Trafficked to Dendrites Normally.
HuR serves many functions, including translation regulation, transcript trafficking, and splicing. One of the best-studied functions of HuR is that of mRNA stabilization of transcripts that it binds via U-rich elements in the 3′UTR (36). Because HuR binding to the patient sequence appears to be impaired, we assessed the stability of endogenous FMR1 mRNA in the patient lymphoblastoid cell line. We performed mRNA decay assays using actinomycin D to block transcription and then tracked the longevity of the extant transcript over time. We observed rapid decay of FMR1 in the patient cell line, which was significantly faster than a control lymphoblastoid cell line (P = 0.008). Another gene, PSD-95, showed no difference in the rate of mRNA degradation after actinomycin D treatment between the patient and control (P = 0.582; Fig. 4G). These results suggest that the stability of endogenous FMR1 in the patient is diminished, potentially because of the inability of HuR to bind at the c.*746 locus and stabilize the transcript. This finding may explain the steady-state reduction in reporter assays and endogenous FMRP in the patient cell line, and potentially has implications for the activity-dependent defect observed in cultured neurons.
Another known function of HuR is trafficking of its mRNA targets to appropriate cellular compartments (41). Because HuR targets FMR1, we sought to determine whether the patient transcript was being properly transported to locations distal to the soma, as this may be controlled in part by HuR and may explain the defect in activity-dependent translation of the reporter in cultured neurons. Here we used fluorescently tagged FMR1 to track and compare the mutant transcript shuttling and localization in primary neurons compared with a control. Both versions of tagged transcript were observed in dendrites and colocalized with HuR and FMRP, suggesting that the patient message is being trafficked to the appropriate neuronal compartments (Fig. S4). We also tracked the movement of the FMR1 mRNA message over time to determine whether the rate of localization of the patient transcript was impaired relative to the control, and did not observe any significant difference (Fig. S5). Thus, the c.*746 locus likely does not play a role in FMR1 trafficking.
Fig. S4.

Fluorescently tagged FMR1 mRNAs were visualized in primary cortical neurons by immunocytochemistry. (Top) FMR1 mRNA localization with a control 3′UTR. (Bottom) FMR1 mRNA localization with the 746C variant allele included in the 3′UTR. The constructs used in these experiments are described in Fig. 4A. (Scale bars, 10 μm.)
Fig. S5.

(A) Schematic of the vectors used for immunofluorescence studies. Fmr1 mRNA harboring the reference 3′UTR or the 746C variant allele, in which four box B sequences were inserted, was visualized in primary hippocampal neurons by tagging it with mCherry fused to λN22 protein that binds to box B sequences, as previously described (61). (B) Fmr1 mRNA with a control 3′UTR colocalizes and is trafficked with FMRP. (Scale bar, 5 µm.) (C) Fmr1 with the 746C variant allele included in the 3′UTR colocalizes and is trafficked with FMRP. (Scale bar, 5 µm.) Empty arrowheads indicate FMRP/Fmr1 mRNA granules that are immobile. White arrowheads indicate dynamic FMRP/Fmr1 mRNA granules.
HuR Targets the c.*746 Locus in Vivo, and Knockdown of HuR Reduces FMR1 and FMRP Levels.
We next wanted to determine whether the HuR interaction at the c.*746 locus is recapitulated in vivo and whether disruption of the interaction has an effect on FMR1 and FMRP expression. To do this, we analyzed HuR binding profiles generated in three published datasets: two that used photoactivatable-ribonucleoside–enhanced cross-linking immunoprecipitation (PAR-CLIP) (42, 43) and one study comparing multiple CLIP protocols (44). CLIP assays capture in vivo interactions by UV-irradiating live cells or tissue at a wavelength that cross-links the RBP to the target RNA, thereby identifying specific sites or motifs of contact (45–47). All three studies identified the 3′UTR of FMR1 as a target of HuR. Although the sites of FMR1–HuR interaction differ slightly between the datasets, the Kishore et al. study (44) detected HuR association at the c.*746 locus through two different CLIP protocols, linking the patient variant site to HuR binding in vivo. The other two studies found HuR binding sites near the c.*746 locus, although the CLIP sequence tags do not directly overlap with the c.*746 site (Table S3). Taken together, there is evidence that the c.*746 site is targeted by HuR in vivo, although more studies will be necessary to confirm these findings.
Table S3.
PAR-CLIP data from Kishore et al. and Mukherjee et al. were analyzed for binding sites of HuR in FMR1
| Study | Assay | CLIP tag FMR1 3′UTR coordinate |
| Kishore et al. (44) | PAR-CLIP_Protocol 1 | c.*726–c.*765 |
| Kishore et al. (44) | PAR-CLIP_Protocol 2 | c.*727–c.*766 |
| Mukherjee et al. (43) | PAR-CLIP | c.*679–c.*710 |
| Lebedeva et al. (42) | PAR-CLIP | c.*977–c.*1002 |
From Kishore et al., protocol 1 used micrococcal nuclease to digest PAR-CLIP mRNA libraries and was performed in duplicate. Protocol 2 used RNase T1 to digest PAR-CLIP mRNA libraries and was performed once.
To examine the effect of HuR on its target genes, Lebedeva et al. (42) measured new protein synthesis and changes in mRNA levels using pulsed stable isotope labeling by amino acids in cell culture and RNA-sequencing experiments, respectively, in cells depleted of HuR and calculated the log2 fold change compared with a negative control for each target gene. Using these data, we plotted the distribution of changes in mRNA levels after HuR knockdown and discovered that FMR1 levels were significantly reduced compared with other genes assayed (log2 score −0.87; Fig. 5A). Additionally, FMRP was the third–least-abundant protein in the absence of HuR from over 2,000 proteins analyzed (log2 score −1.37; Fig. 5B). Even when normalizing protein synthesis by concomitant changes in mRNA levels, FMRP was still among the least-synthesized proteins (log2 score −0.51; Fig. 5B), indicating a role for HuR in translation regulation of FMR1. These data suggest that FMR1 is targeted by HuR at the c.*746 locus in vivo and is positively regulated by the protein, consistent with our in vitro results.
Fig. 5.

HuR depletion down-regulates FMR1 mRNA levels and FMRP synthesis. (A) The distribution of mRNA changes after knockdown (KD) of HuR reveals a significant decrease in FMR1 levels compared with over 2,000 other mRNAs assayed (log2 value −0.87). The red arrow indicates the bin in which FMR1 falls. (B) The distribution of protein-level changes shows a significant decrease in FMRP synthesis (log2 value −1.37). The red arrow indicates the bin in which FMRP falls. (C) The distribution of protein synthesis changes when accounting for concomitant mRNA changes indicates that the decrease in protein synthesis is not only the result of a reduction in mRNA levels (i.e., −log2 difference between protein and mRNA levels −0.51). The red arrow indicates the bin in which FMRP falls. All histograms were generated using data from Lebedeva et al. (42).
Discussion
The variant c.*746T>C in the FMR1 3′UTR reduces the basal level of FMRP in patient-derived cell lines and in multiple other cell types using reporter constructs and, perhaps more importantly, eliminates the normal response to glutamate signaling by reporter assay in primary cultured neurons. Our data suggest that both defects are the consequence of a disrupted RNA–protein interaction between HuR and FMR1 caused by the c.*746C variant. HuR is known to bind U-rich motifs in 3′UTRs and introns, particularly stretches of uridines with interspersed adenines or guanines (42, 43, 48). The change from uridine to cytosine at the c.*746 position, which interrupts a multiuridine stretch, presumably hinders the normal interaction of HuR at the locus. In support of this hypothesis, mutagenesis of the variant locus modulated the steady-state expression of the patient and control vectors as well as the activity-dependent translation. The c.*746C patient allele resulted in a molecular phenotype similar to that observed in Fmr1 KO neurons after glutamate receptor activation, where glutamate-driven protein synthesis was absent. Although these molecular findings are compelling, they do not definitively link the c.*746C variant to the patient’s phenotype. Consequently, it will be important to evaluate more families and individuals with the c.*746C allele to determine whether the variant is truly pathological and observe the range of severity and disabilities caused by the variant. The patient’s half-brother is also developmentally delayed/intellectually disabled, and his FMR1 3′UTR showed a reduction in expression by reporter assay, which provides a second case (albeit familial) that may be attributable to the c.*746C allele. However, he is affected to a lesser degree compared with the proband, which may indicate that other genetic or environmental factors specific to the proband play a role in the relative severity of the phenotype.
The role of synaptically expressed FMRP is hypothesized to function as a negative feedback loop, where existing FMRP is dephosphorylated and degraded after glutamate receptor stimulation and newly synthesized FMRP reins in the burst of translation after an appropriate amount of time (14, 34, 49). However, the importance of locally translated FMRP had not been directly addressed until recently, when it was discovered that a premutation FXS mouse model exhibited impaired activity-dependent FMRP synthesis likely due to the presence of expanded CGG repeats in the 5′UTR (50). Iliff et al. (50) exploited a well-characterized neuronal phenotype in Fmr1 KO mice, enhanced mGluR-mediated long-term depression (LTD), to evaluate the impact of dysfunctional local FMRP translation on synaptic plasticity in premutation brain tissue. Their results suggest that the reduced levels of pre-existing FMRP were not sufficient to suppress the enhanced mGluR-LTD occurring in the premutation neurons; locally synthesized FMRP was necessary for proper LTD. Whereas most studies of the premutation allele have focused on late-onset neurodegenerative phenotypes, accumulating evidence suggests that the premutation may have a demonstrable effect on neurodevelopment as well (51–53). Although the genetic defect in mGluR-mediated translation studied by Iliff et al. (i.e., the premutation in the 5′UTR) differs from the 3′UTR defect investigated here, it suggests that the c.*746C variant could hinder neurodevelopment and synaptic plasticity through the impairment of activity-dependent translation, and is a possible reason for the observed developmental delay and cognitive disability in the patient.
These data implicate the c.*746 locus in the basal and activity-dependent expression of FMRP. However, the exact mechanism(s) by which this deficit occurs remains to be determined. The translational insufficiencies could be a direct effect of the inability of HuR to bind FMR1 or an indirect consequence of this perturbed interaction, such as rapidly decaying transcript at the synapse that is insufficient to support activity-regulated protein synthesis. Another potential mechanism is based on the observation that HuR competes with other binding proteins, of which there are several that recognize a similar U-rich motif (36), to modulate the stability of a given target transcript. For example, the binding factor AUF1 (also known as hnRNP D), which was also pulled down in our co-IP/MS screen (Table S2), is associated with rapid degradation of bound mRNAs, opposing the function of HuR. Indeed, this type of competitive binding has recently been postulated to occur in vivo between HuR and another U-rich binding protein that negatively regulates transcript stability, ZFP36 (54). This presents the possibility that each is required as an antagonist to the other, and each performs their specific functions when cued by the appropriate cellular signals. If HuR binding is impaired at the c.*746 site, AUF1 or other similar transcript-destabilizing proteins may have an increased opportunity to degrade FMR1, leading to a reduced mRNA half-life, which we observed in the patient cell line.
A recently recognized function of HuR is that it can act as an “anti-RISC” (RNA-induced silencing complex); that is, HuR binds near microRNA (miRNA) sites, oligomerizes along the mRNA, and removes or blocks the miRNA machinery from reaching its target, thereby relieving the down-regulation typically imposed by miRNAs (43, 55, 56). One study has shown that miR-130b, an miRNA that is expressed in the brain and is highly conserved, interacts at the 746 variant locus (seed sequence binds at FMR1 c.*755–761), negatively regulates the expression of FMR1 in neural progenitor cells, and affects cell-fate specification (57). These findings present an alternate mode of dysfunction whereby the inability of HuR to bind and/or oligomerize adjacent to the miR-130b site hinders the removal of the miRNA, which then continues to repress FMRP translation unchecked and perhaps alters the response to mGluR stimulation. There is evidence of an miRNA-mediated response to glutamate signaling, which requires the removal of miR-125a to allow the normal increase in PSD-95 expression following glutamate receptor activation (58). If miR-130b is involved in activity-dependent translation of FMRP, failure to block or eliminate its association with FMR1 may prevent the typical up-regulation of FMRP synthesis following glutamate signaling. Although the disrupted FMR1–HuR interaction may be the basis of the observed dysfunctions, more research is required to identify the process(es) that is perturbed.
Multiple different approaches in this study demonstrate that HuR binds the FMR1 c.*746 locus in vitro. To investigate in vivo binding of HuR to the locus, we analyzed three published datasets that identified mRNA targets and binding locations of HuR by PAR-CLIP assays in human cell lines (HEK293 and HeLa) (42–44). All datasets showed that multiple 3′UTR and intronic sites of FMR1 are targeted by HuR. Because the specific binding locations identified by each dataset were inconsistent, it will be important to confirm these findings with more targeted studies. Additionally, it would be of interest to determine whether the interactions are recapitulated in different cell types, including neuronal cells or brain tissue. To determine the functional effect of HuR on FMR1, we analyzed data from experiments that tracked the translation of new proteins after knockdown of HuR, which revealed a drastic reduction in FMR1 mRNA levels and FMRP synthesis. In fact, FMRP levels were reduced more than all other proteins assayed except for two, one of which was HuR itself. Together, these results suggest that the locus is targeted by HuR and that FMRP expression is significantly down-regulated in the absence of HuR. If HuR-mediated regulation of FMR1 is via the 746 locus, the lack of HuR binding to the patient allele may underlie the reduction in expression and transcript stability observed in our data.
Because HuR is a member of a small protein family (HuR, HuB, HuC, and HuD) and the other Hu paralogs also bind a similar U-rich sequence motif (59), we were interested in whether these other Hu proteins associated with FMR1 at the c.*746 locus. Our co-IP followed by MS-MS assay did identify HuC as being copurified with the biotinylated probe, although it was not detected in the gel slice/MS-MS assay and, therefore, was not included in our top candidates. However, these data do suggest at least some level of interaction between the neuronal Hu and the c.*746 locus. Additionally, the neuronal Hu proteins were found to bind the c.*746 locus in vivo in mouse brain on actively translating polyribosomes (10). Taken together, the locus is a target of posttranscriptional regulation, and may be targeted by different Hu proteins under specific temporal, spatial, and environmental conditions, a possibility that should be examined further.
The findings presented here illustrate the impact of a single-nucleotide variant in the regulatory region of a gene, which can have significant molecular consequences and may be causative of a clinical phenotype. As whole-genome sequencing becomes more commonplace, sequence data in the UTRs and other regulatory regions will be available for analysis and should be explored for functional variants like the one described here when studying the genetic defects underlying certain diseases. Together, these data identify the FMR1 c.*746 locus as an important site of posttranscriptional regulation and shed new light on the mechanisms governing activity-dependent translation of FMRP.
Materials and Methods
Human Subject and Animal Research.
All experimental procedures requiring mouse models was approved by the Emory University Institutional Review Board (IRB). Informed consent was obtained from all family members described in the study and was approved by the Emory University IRB.
Luciferase Constructs and Site-Directed Mutagenesis.
The Dual-Luciferase System from Promega was used for reporter assays. In short, the control and patient 3′UTRs were amplified with a forward primer that included a 5′ XbaI site and a reverse primer that included a 5′ BamHI site. These amplicons were cloned using the TOPO TA Cloning System (Invitrogen), excised using BamHI and XbaI restriction enzymes, and ligated into a pGL3-promoter vector downstream of the luciferase gene. All constructs were sequenced to verify proper orientation and sequence. The pRL-SV40 vector was used as a transfection normalization control. The control and patient 3′UTR vectors were used as templates for all mutagenesis experiments using the Agilent QuikChange Lightning Kit as recommended.
Cell Culture and Transfection.
Neuro2a cells were grown in DMEM supplemented with 10% (vol/vol) FBS and 1× penicillin/streptomycin antibiotics; HEK293FT cells were grown in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 1× penicillin/streptomycin, 0.1 mM nonessential amino acids, and geneticin-selective antibiotic. Both cell lines were grown at 37 °C with 5% CO2.
Transfections in HEK293FT and Neuro2a cells were performed using Lipofectamine 2000 or Lipofectamine LTX (Invitrogen) in 6- or 12-well plates according to the manufacturer’s instructions. Lipofection complexes were left in the medium for 4–6 h and then removed and replaced with fresh medium. Cells were allowed to express the firefly and Renilla luciferase for ∼24 h before harvesting.
Primary mouse cortical neuron [embryonic day (E)17.5] cultures were isolated from C57BL/6 and Fmr1 KO embryos and plated in 12-well plates coated with 0.2 mg/mL poly–l-lysine. Transfections were carried out at 13 d in vitro using NeuroMag Transfection Reagent (OZ Biosciences) following the recommended protocol. Neurons were stimulated with 100 μM (RS)-3,5-DHPG (Tocris Biosciences) for 5 min before lysis with passive lysis buffer (Promega).
Luciferase Assay.
Lysis of the cells was performed using the passive lysis protocol from the Dual-Luciferase Assay Kit (Promega). To detect luciferase signal, 10–20 μL cell lysate and 50 μL Luciferase Assay Reagent II were mixed thoroughly by pipetting and placed in the luminometer, and firefly luminescence readings were collected. Immediately after, 50 μL Stop & Glo Reagent (Promega) was added and mixed thoroughly by pipetting, and the Renilla luminescence signal was detected. All values reported are the ratio of firefly:Renilla reporter signal.
Quantitative RT-PCR.
Reverse-transcription reactions were carried out using iScript One-Step RT-PCR or iTaq Universal SYBR Green Kits (Bio-Rad). All qPCR reactions were performed in duplicate or triplicate as technical replicates. All reactions were cycled for 40 cycles using the Bio-Rad CFX96 real-time system, followed by a melt curve cycle, and then analyzed with the Bio-Rad CFX96 software package.
Immunoprecipitation.
Dynabeads MyOne Streptavidin T1 (Thermo Fisher Scientific) was used for biotinylated RNA probe IP following the manufacturer’s recommended protocol. In each of two independent assays, 50 μg of biotinylated RNA probe was immobilized on 200 μL washed Dynabeads supplemented with cOmplete, Mini, EDTA-free (Roche) and SUPERase⋅In (Life Technologies) and then incubated with 1 mg whole-brain lysate and a 10× excess of Torula yeast RNA (Sigma-Aldrich) in binding buffer (1 M NaCl, 1 mM EDTA, 10 mM Tris⋅HCl, pH 7.5) for 1 h at room temperature with rotation. After three washes, coimmunoprecipitated proteins were eluted in 30 μL binding buffer with 0.1% (wt/vol) SDS at 95 °C for 5 min and subjected to mass spectrometry.
HuR antibody (MBL; RN004P) was immobilized on Dynabeads Protein G (Immunoprecipitation Kit; Life Technologies) and incubated with HEK293 cell lysates at room temperature for 12 min by rotation, along with a protein G-only control. After four washes, the coimmunoprecipitated RNA was eluted with 50 μL elution buffer at 95 °C for 5 min and then purified with TRIzol LS (Life Technologies). qRT-PCR was performed using FMR1, β-actin, and GAPDH primers for an input sample as well as the total amount of IP sample, and the ratio of calculated starting quantity of IP:input was used to determine the enrichment of each mRNA target.
Western Blotting.
Lysates from lymphoblast cell lines were generated by resuspending cells in lysis buffer [50 mM Tris⋅HCl, 300 mM NaCl, 30 mM EDTA, 0.5% Triton X-100, pH 7.6; cOmplete, Mini, Protease Inhibitor Tablet (Roche)] and incubated on ice for 15 min, followed by pelleting of cell debris at ˜23,000 × g for 15 min at 4 °C. SDS/PAGE (4–15% gradient) was performed with 10–20 μg total lysate as determined by the Bradford assay. Proteins were transferred to PVDF, blocked with T20 PBS blocking buffer (Thermo Scientific) for 15–30 min, and incubated with primary antibodies [anti-FMRP (Millipore; MAB2160) at 1:2,000 dilution and anti-eIF4e (BD Biosciences; 610270) at 1:2,000 dilution] overnight at 4 °C with gentle agitation. Next, the membrane was incubated with HRP-conjugated anti-mouse and anti-rabbit secondary antibodies at 1:10,000 dilution in 1% (wt/vol) milk blotto for 1 h at room temperature, washed three times with 1% milk blotto, and incubated with HyGLO ECL solution (Denville Scientific) for 1 min with agitation.
RNA Probe Generation by in Vitro Transcription.
DNA templates containing the T7 promoter and probe template sequence were generated by a modified overlap extension PCR strategy (60). Each oligonucleotide (2 μM) was added to a PCR mix consisting of 1× Herculase II reaction buffer, 0.25 mM each dNTP, 0.5 μL Herculase II fusion enzyme (Agilent Technologies), 3 μL 5 M betaine solution (Sigma-Aldrich), and MilliQ water up to 50 μL. The reaction was cycled under the following parameters: 95 °C for 2 min, followed by 35 cycles of 95 °C for 20 s, 45 °C for 30 s, and 72 °C for 30 s. A final extension cycle of 3 min at 72 °C was added. The resulting double-stranded PCR products were run on a 1% (wt/vol) agarose gel, extracted from the gel (Qiagen MinElute Gel Extraction Kit), and resuspended in MilliQ water. DNA templates were used at a concentration of 2 pM in the in vitro transcription reaction for 3–4 h at 37 °C (Ambion T7 MEGAshortscript Kit) in the presence of biotin-17-ATP (Enzo Biosciences) or biotin-14-CTP (Life Technologies). The resulting 42-nt RNA transcripts were purified using TRIzol LS (Ambion). The same in vitro transcription procedure was used to generate unlabeled RNA probes for 32P-end labeling and competition gel-shift assays.
Electromobility-Shift and -Supershift Assays.
Gel-shift assays were carried out using the LightShift Chemiluminescent RNA EMSA Kit (Thermo Scientific) according to the manufacturer’s protocol. Briefly, biotin-labeled RNA probes were mixed with C57BL/6 (WT) or FMR1 KO mouse whole-brain lysates and incubated at room temperature for 20–30 min and then electrophoresed through a 5% nondenaturing Tris-borate-EDTA (TBE) gel. The RNA–protein complexes were transferred to a positively charged nylon membrane and developed to film following the RNA EMSA protocol. Supershift assays were carried out in a similar manner with the following exceptions: addition of ∼1 μg of antibody and incubation at room temperature for 20 min before the addition of the radiolabeled or biotin-labeled RNA probe in a total of 20 μL reaction volume.
mRNA Decay Assay.
Lymphoblastoid cells were plated in six-well plates at a density of 5 × 106 cells per well in 2 mL medium consisting of RPMI-1640, 10% FBS, 2 mM l-glutamine, and 1× penicillin/streptomycin antibiotic. Cells were treated with 5 μg/mL actinomycin D (Sigma) for various amounts of time and then harvested by TRIzol LS (Ambion) extraction according to the manufacturer’s recommended protocol. qRT-PCR was used to assess the amount of FMR1 transcript remaining compared with an untreated control. All qRT-PCRs were performed in duplicate, and the mean level of FMR1 remaining at each time point was normalized to the mean level of β-actin remaining at each time point.
Acknowledgments
We thank Heather Clark for contacting the patient and recording the family history; Duc Duong, Nicholas Seyfried, and the Emory University Proteomics Core Facility for mass spectrometry data and advice; Tamika Malone for mouse colony maintenance and cortical tissue dissections; Pankaj Chopra for rank aggregation analysis of mass spectrometry assays; Mika Kinoshita, Leila Myrick, and Michael Santoro, among others in the S.T.W. and G.J.B. laboratories, for helpful insight and discussion. We also thank Robert Darnell and Jennifer Darnell for helpful discussions and data sharing. This work was supported by NIH Award NS091859 from the National Institute of Neurological Disorders and Stroke; Eunice Kennedy Shriver National Institute of Child Health and Human Development in support of the Emory National Fragile X Research Center (S.T.W.); NIH Award 1R21NS091038 (to G.J.B.); and a FRAXA Research Foundation fellowship (to J.A.S.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1514260112/-/DCSupplemental.
References
- 1.Verkerk AJ, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 2.Fu YH, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell. 1991;67(6):1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 3.Pieretti M, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66(4):817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 4.Sutcliffe JS, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1(6):397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 5.Alisch RS, et al. Genome-wide analysis validates aberrant methylation in fragile X syndrome is specific to the FMR1 locus. BMC Med Genet. 2013;14:18. doi: 10.1186/1471-2350-14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10(4):329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, et al. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29(11):2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashley CT, Jr, Wilkinson KD, Reines D, Warren ST. FMR1 protein: Conserved RNP family domains and selective RNA binding. Science. 1993;262(5133):563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 9.Brown V, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107(4):477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 10.Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99(11):7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27(20):5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci USA. 2003;100(24):14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nalavadi VC, Muddashetty RS, Gross C, Bassell GJ. Dephosphorylation-induced ubiquitination and degradation of FMRP in dendrites: A role in immediate early mGluR-stimulated translation. J Neurosci. 2012;32(8):2582–2587. doi: 10.1523/JNEUROSCI.5057-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Richter JD, Klann E. Making synaptic plasticity and memory last: Mechanisms of translational regulation. Genes Dev. 2009;23(1):1–11. doi: 10.1101/gad.1735809. [DOI] [PubMed] [Google Scholar]
- 17.Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127(1):49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 18.Weiler IJ, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA. 1997;94(10):5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiler IJ, et al. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA. 2004;101(50):17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Boulle K, et al. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993;3(1):31–35. doi: 10.1038/ng0193-31. [DOI] [PubMed] [Google Scholar]
- 21.Gedeon AK, et al. Fragile X syndrome without CCG amplification has an FMR1 deletion. Nat Genet. 1992;1(5):341–344. doi: 10.1038/ng0892-341. [DOI] [PubMed] [Google Scholar]
- 22.Hirst M, et al. Two new cases of FMR1 deletion associated with mental impairment. Am J Hum Genet. 1995;56(1):67–74. [PMC free article] [PubMed] [Google Scholar]
- 23.Wöhrle D, et al. A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am J Hum Genet. 1992;51(2):299–306. [PMC free article] [PubMed] [Google Scholar]
- 24.Lugenbeel KA, Peier AM, Carson NL, Chudley AE, Nelson DL. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat Genet. 1995;10(4):483–485. doi: 10.1038/ng0895-483. [DOI] [PubMed] [Google Scholar]
- 25.Myrick LK, et al. Fragile X syndrome due to a missense mutation. Eur J Hum Genet. 2014;22(10):1185–1189. doi: 10.1038/ejhg.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffee B, et al. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: A case report and review of the literature. Am J Med Genet A. 2008;146A(10):1358–1367. doi: 10.1002/ajmg.a.32261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myrick LK, et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc Natl Acad Sci USA. 2015;112(4):949–956. doi: 10.1073/pnas.1423094112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins SC, et al. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A. 2010;152A(10):2512–2520. doi: 10.1002/ajmg.a.33626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerber RJ, Wilks T, Erdie-Lalena C. Developmental milestones: Motor development. Pediatr Rev. 2010;31(7):267–276, quiz 277. doi: 10.1542/pir.31-7-267. [DOI] [PubMed] [Google Scholar]
- 30. Roid GH (2003) Stanford-Binet Intelligence Scales (SB5). Riverpubcom. Available at www.riverpub.com/products/sb5/details.html.
- 31. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20(1):110–121. [DOI] [PMC free article] [PubMed]
- 32. Cooper GM, et al. (2005) Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15(7):901–913. [DOI] [PMC free article] [PubMed]
- 33. Siepel A, et al. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15(8):1034–1050. [DOI] [PMC free article] [PubMed]
- 34.Hou L, et al. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51(4):441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 35.Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis–dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86(1):321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- 36.Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58(2):266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meisner NC, Filipowicz W. Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression. Adv Exp Med Biol. 2010;700:106–123. [PubMed] [Google Scholar]
- 38.Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tatavarty V, et al. Single-molecule imaging of translational output from individual RNA granules in neurons. Mol Biol Cell. 2012;23(5):918–929. doi: 10.1091/mbc.E11-07-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández E, et al. FXR2P exerts a positive translational control and is required for the activity-dependent increase of PSD95 expression. J Neurosci. 2015;35(25):9402–9408. doi: 10.1523/JNEUROSCI.4800-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keene JD. Why is Hu where? Shuttling of early-response-gene messenger RNA subsets. Proc Natl Acad Sci USA. 1999;96(1):5–7. doi: 10.1073/pnas.96.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lebedeva S, et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43(3):340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 43.Mukherjee N, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43(3):327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kishore S, et al. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011;8(7):559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- 45.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141(1):129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ule J, Jensen K, Mele A, Darnell RB. CLIP: A method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37(4):376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 47.Ule J, et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302(5648):1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 48.López de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci USA. 2004;101(9):2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao W, Chuang SC, Bianchi R, Wong RK. Dual regulation of fragile X mental retardation protein by group I metabotropic glutamate receptors controls translation-dependent epileptogenesis in the hippocampus. J Neurosci. 2011;31(2):725–734. doi: 10.1523/JNEUROSCI.2915-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iliff AJ, et al. Impaired activity-dependent FMRP translation and enhanced mGluR-dependent LTD in fragile X premutation mice. Hum Mol Genet. 2013;22(6):1180–1192. doi: 10.1093/hmg/dds525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y, et al. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2010;19(1):196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cunningham CL, et al. Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum Mol Genet. 2011;20(1):64–79. doi: 10.1093/hmg/ddq432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Besterman AD, et al. Towards an understanding of neuropsychiatric manifestations in fragile X premutation carriers. Future Neurol. 2014;9(2):227–239. doi: 10.2217/fnl.14.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukherjee N, et al. Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol. 2014;15(1):R12. doi: 10.1186/gb-2014-15-1-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125(6):1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 56.Kundu P, Fabian MR, Sonenberg N, Bhattacharyya SN, Filipowicz W. HuR protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res. 2012;40(11):5088–5100. doi: 10.1093/nar/gks148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gong X, et al. MicroRNA-130b targets Fmr1 and regulates embryonic neural progenitor cell proliferation and differentiation. Biochem Biophys Res Commun. 2013;439(4):493–500. doi: 10.1016/j.bbrc.2013.08.096. [DOI] [PubMed] [Google Scholar]
- 58.Muddashetty RS, et al. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol Cell. 2011;42(5):673–688. doi: 10.1016/j.molcel.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ince-Dunn G, et al. Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron. 2012;75(6):1067–1080. doi: 10.1016/j.neuron.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 61.Daigle N, Ellenberg J. LambdaN-GFP: An RNA reporter system for live-cell imaging. Nat Methods. 2007;4(8):633–636. doi: 10.1038/nmeth1065. [DOI] [PubMed] [Google Scholar]