

Abstract
Pain sensing neurons in the periphery (called nociceptors) and the central neurons that receive their projections show remarkable plasticity following injury. This plasticity results in amplification of pain signaling that is now understood to be crucial for the recovery and survival of organisms following injury. These same plasticity mechanisms may drive a transition to a non-adaptive chronic pain state if they fail to resolve following the termination of the healing process. Remarkable advances have been achieved in the past two decades in understanding the molecular mechanisms that underlie pain plasticity following injury. The mechanisms bear a striking resemblance to molecular mechanisms involved in learning and memory processes in other brain regions, including the hippocampus and cerebral cortex. Here those mechanisms, their commonalities and subtle differences, will be highlighted and their role in causing chronic pain will be discussed. Arising from these data is the striking argument that chronic pain is a disease of the nervous system, which distinguishes this phenomena from acute pain that is frequently a symptom alerting the organism to injury. This argument has important implications for the development of disease modifying therapeutics.
Introduction: pain plasticity and “pain memory”
A core feature of all nervous systems is an ability to adapt to sensory information. This adaptive process is referred to as plasticity and the study of neuronal plasticity has led to some of the most exciting advances in modern biological research. The pain system, comprised of peripheral neurons responsible for detecting damaging or potentially damaging peripheral stimuli, called nociceptors, and the neurons of the CNS that receive direct or indirect inputs from these neurons, rapidly change upon injury. In almost all studied cases this adaptation results in an amplification of signaling (Woolf and Walters, 1991). This pain amplification is thought to underlie some important psychophysical aspects of pain such as an enhanced response to a normally noxious stimulus (hyperalgesia) and a noxious response to a normally innocuous stimulus (allodynia, (Cervero, 1996)). Plasticity may also lead to changes in nociceptors or other neurons in the pain pathway that cause them to fire action potentials without any direct stimulation (ectopic activity) or fire continuously following stimulation (afterdischarge) both of which likely contribute to what is commonly called spontaneous pain that is a common feature of chronic neuropathic pain (Lisney and Devor, 1987; Devor et al., 1994). While all of these states can exist acutely following an injury, they are also prominent features of chronic pain disorders.
On the most general level, plasticity in the pain system occurs at two locations, the primary afferent nociceptor (Reichling et al., 2013) and at synapses receiving nociceptive input throughout the CNS (Ji et al., 2003; Woolf, 2007; Latremoliere and Woolf, 2009). Preclinical models of acute and chronic inflammatory pain as well as models of neuropathic pain have revealed a plethora of molecular targets that have advanced our understanding of how chronic pain develops as well as revealing important potential therapeutic intervention points. These experimental studies have also revealed a striking similarity in mechanisms underlying pain amplification and learning and memory in areas of the brain such as hippocampus and cerebral cortex (Ji et al., 2003; Sandkuhler, 2007; Ruscheweyh et al., 2011). These findings have given rise to the idea that a “pain memory” is encoded within the nervous system and that reversing this pain memory may be the key to terminating chronic pain disorders (Reichling and Levine, 2009; Price and Ghosh, 2013; Reichling et al., 2013). In other words, reversing plasticity in pain circuits may provide the opportunity to permanently alleviate chronic pain.
While the term “pain memory” has been used in a variety of forms for decades, the first specific uses in the scientific literature, to our knowledge, can be attributed to Ronald Melzack, one of the experimental pioneers widely credited with advancing pain science into the modern age of neuroscience. In 1979 Dennis and Melzack described a series of experiments where painful irritation of the rat paw prior to a denervation injury led to an exacerbation of neuropathic pain (Dennis and Melzack, 1979). They hypothesized that this pre-irritation led to the generation of a “pain memory” that could not be repressed by descending pain modulation centers, due to the subsequent nerve injury, and therefore persisted unabated after the nerve injury. Subsequently, in 1990, Katz and Melzack presented this same term in the context of phantom limb pain (Katz and Melzack, 1990), pain arising in a limb that has been amputated. They postulated that this sort of pain occurs due to the “memory” of pain that was caused by damage to the limb that was subsequently amputated. Since many amputations occur due to injury to a limb that is irreparable, this could explain this common clinical finding in amputees. While this idea is now widely disputed in the pain field (Flor et al., 2006), it remains an area of intense investigation. Very recently clinical evidence was published indicating that this “pain memory” causing phantom limb pain is very likely to reside in the peripheral nervous system (Vaso et al., 2014). These investigators found that local nerve block applied to the dorsal root ganglion (DRG) innervating the limb with phantom pain led to an immediate resolution of pain in 31 out of 31 patients. Strikingly, this finding is directly paralleled by preclinical (a term used to describe animal model experiments in the neuroscience area) work from Coderre and Melzack in 1987 where they concluded that neuropathic “pain memory” almost certainly resides in the peripheral nervous system (Coderre and Melzack, 1987).
Since the first experiments into “pain memory” in the Melzack lab at McGill University, many animal models of acute and chronic pain have been developed. For the purposes of the present chapter we will focus on a particular model developed in the late 1990s and early 2000s by Jon Levine and colleagues called “hyperalgesic priming” (for extensive review see (Reichling and Levine, 2009)). This model provides unique insight into plasticity in the nociceptive system because it allows for molecular dissection of pain states in two distinct phases. These models involve a priming stimulus, aimed at causing an acute sensitization of peripheral nociceptors and their central inputs. Next, the initial sensitization is allowed to resolve and a second, normally sub-threshold, stimulus is delivered. This second stimulus, which has only a transient effect in naïve animals, leads to a prolonged state of nociceptive hypersensitivity that allows for investigation of molecular mechanisms that define the primed nociceptor and/or the primed nociceptive system. Here we will describe how models of hyperalgesic priming give unique insight into how acute pain states lead to reorganization of molecular machinery throughout the pain system rendering animals, and almost certainly humans, susceptible to prolonged pain states provoked by insults that would have little effect in unprimed individuals. This primed state, therefore, represents a kind of “pain memory”. Hence, our goals in this chapter will be to highlight mechanisms underlying this “pain memory”. This will include three major themes 1) molecular signaling in the peripheral nociceptor and their parallels to memory mechanisms (local translation), 2) mechanisms controlling plasticity of synapses in the spinal dorsal horn and their relation to memory mechanisms (long-term potentiation) and 3) signaling mechanisms in the spinal dorsal horn that parallel findings from the learning and memory area (atypical PKCs (aPKC) and brain derived neurotrophic factor (BDNF)).
The adaptive nature of pain plasticity
Before delving into molecular mechanisms of pain plasticity and pain memory, it is useful to first consider the evolutionary relevance of nociception and nociceptive plasticity. First, nearly all organisms with nervous systems have sensory neurons that can be considered nociceptors. That is, they have sensory detectors that are capable of sensing damaging chemicals, temperatures or tissue insult and whose action can lead to avoidance of real or potential damage to the organism. Investigators interested in nociceptor biology have employed model organisms such as Drosophila melanogaster (fruit flies), Caenorhabditis elegans (transparent nematodes) and Aplysia californica (sea hares) due to the ease of manipulation of their genome, their transparency (for imaging purposes), rapid life cycle or their stereotypic behaviors and large, easily accessible neurons. Some of the most relevant work has been done using Aplysia. The gill withdrawal reflex in these animals has been studied in great detail and involves a simple circuit made up of a sensory neuron, an interneuron and a motor neuron. Plasticity in this circuit has been widely studied as a simple model for learning and memory (Kandel, 2004) and molecular mechanisms discovered in these neurons led to the awarding of the Nobel Prize for Physiology or Medicine to Eric Kandel in 2000 (De Camilli and Carew, 2000). Importantly, the sensory neuron that plays a critical role in this reflex shares many properties with nociceptors and has been extensively investigated as a model for nociceptive plasticity (Woolf and Walters, 1991; Clatworthy and Walters, 1993; Illich and Walters, 1997). Studying the response of this neuron to injury led to the first demonstration of local translation contributing to the increased excitability of nociceptors after injury (Weragoda et al., 2004). Hence, there is strong evidence that nociceptive plasticity is evolutionarily ancient and that the basic mechanisms of this plasticity are conserved across a broad variety of organisms (Woolf and Walters, 1991; Walters and Moroz, 2009). This has profound implications for understanding the crucial nature of nociceptive plasticity for organisms to survive in their natural environments and also suggests that mechanisms of neuronal plasticity in general may have first evolved in the nociceptive system.
Does nociceptive plasticity provide an adaptive advantage to organisms? On the surface, the answer to this question seems obvious. Of course it does. It is paramount for animals to protect an injured area until the healing process has run its course. This idea, however, is difficult to demonstrate experimentally. In an elegant set of experiments, Crook and colleagues used squid to address this question directly (Crook et al., 2014). When the squid arm is injured it causes nociceptive neurons that innervate the squid arm to become sensitized. The sensitization results in reduced thresholds for mechanical stimulation similar to observations of hyperalgesia in rodents and humans. Local anesthetic applied at the time of squid arm injury completely blocks nociceptor sensitization and amplified nociceptive behavioral responses. Hence, it is possible to experimentally injury squid and have two distinct outcomes: squid with injury and hyperalgesia and squid with identical injury but no hyperalgesia. When these squid are exposed to a natural predator a remarkable difference in behavior is observed. Squid with injury and hyperalgesia orient to the predator more quickly and are able to escape attack. On the other hand, squid with injury and no hyperalgesia fail to mount this response to the predator resulting in markedly increased predation. Hence, hyperalgesia, or pain amplification, holds a distinct survival advantage, placing pain plasticity in a new evolutionary light. Moreover, these findings highlight the evolutionarily ancient nature of plasticity in peripheral nociceptors as a mechanism to drive persistent pain (Crook et al., 2014; Price and Dussor, 2014).
The peripheral nociceptor as the locus of “pain memory”, focus on translation control
In 1982 Steward and Levy published a landmark study describing the preferential localization of ribosomes at the base of dendritic spines (Steward and Levy, 1982). While the potential importance of dendritic spines for nervous system function had been postulated since the time of Santiago Ramon y Cajal, this finding came less than a decade after the original description of LTP by Bliss and Lomo in 1973 (Bliss and Lomo, 1973). At the time of the work of Steward and Levy, the mechanisms underlying LTP were still poorly understood but their finding led to a striking conclusion, the localization of ribosomes at the base of dendritic spines places the translation machinery at the perfect location to control local changes that regulate postsynaptic plasticity. What has followed has been a veritable explosion of work on translation control and its contribution to synaptic plasticity and LTP (Costa-Mattioli et al., 2009). While other aspects of LTP will be covered in more detail below, it is now known that LTP decays if local translation is blocked and signaling molecules that control activity-dependent plasticity are enriched in and around dendritic spines. Moreover, RNA binding proteins anchor mRNAs at dendritic spines and play critical roles in brain function, for instance, they are mutated in several neurodevelopmental disorders (e.g. fragile X mental retardation protein (FMRP) and fragile X syndrome, (Bassell and Warren, 2008)). Therefore, local, activity-dependent control of gene expression at the level of translation is at the very foundation of plasticity in the brain, including the most widely studied learning and memory mechanism, LTP (Abraham and Williams, 2008b).
Why is local translation control so important for neuronal plasticity? Here it is important to remember the basic anatomy/organization of neurons in relation to gene expression demands and the correct sorting of protein localization given this complexity. Individual neurons can have tens of thousands of synapses and plasticity can occur at any of these individual synapses depending on afferent input to the neuron. This creates a difficult problem if changes in gene expression are required for the full expression of plasticity, as is now well accepted. If new proteins required for this plasticity were all contributed from the cell body, the neuron would need a mechanism in place to correctly sort all of these plasticity-related proteins to their correct location. A more parsimonious solution is for the neuron to traffic mRNAs to dendritic spines and hold them in a translationally dormant state until an appropriate signal is received (e.g. strong synaptic input). Changes in gene expression can then be achieved locally via activity-dependent translation. Overwhelming evidence, from a broad variety of brain regions, including the dorsal horn of the spinal cord, supports the preceding statements (Abraham and Williams, 2008b; Costa-Mattioli et al., 2009; Price and Geranton, 2009; Sacktor, 2011; Price and Ghosh, 2013).
But which mRNAs are trafficked to these distal sites in neurons? This has likewise been an area of intense investigation. Investigators have focused on finding mRNAs that associate with mRNA binding proteins, such as FMRP (Brown et al., 2001; Darnell et al., 2001) but these experiments have been technically demanding and have led to disparate results depending on the technique. Another approach has been to use multi-chambered devices where neuronal extensions, be they dendrites or axons, can extend into an isolated chamber where these neuronal components can be isolated and mRNA can be extracted (Willis et al., 2005; Willis et al., 2007) (Figure 1). While these approaches have identified important candidate mRNAs it has only been recently that bioinformatics approaches have led to insights into the species of mRNAs that are found at distal sites in neurons and whether these mRNAs differ from those that are preferentially translated in the neuronal soma (Weatheritt et al., 2014). These investigations found several distinguishing factors in the proteins encoded by these distally localized mRNAs, most prominently an enrichment of sites for post-translational modification (e.g. phosphorylation) and increased intrinsic disorder. These protein properties indicate that the protein is highly prone to changes in conformation upon interaction with other proteins or signaling factors (Tantos et al., 2012). Consistent with this, proteins encoded by distally localized mRNAs have more protein-protein interaction partners and a broader interaction network. They also show large changes in protein expression after cellular stimulation, likely because their mRNAs are targets for signaling mechanisms to the translation machinery, and the proteins have shorter half-lives, consistent with tight temporal control of distally localized signaling processes that are integrated by translation control (Weatheritt et al., 2014). This study reached several conclusions yielding important insights into distally localized mRNAs and their localized translation: 1) local translation of these genes likely minimizes off-target protein-protein interactions that might otherwise be expected amongst proteins that have wide interaction networks, 2) it decentralizes protein expression for efficient processing of plasticity-related signaling, 3) it permits strong amplification of spatially localized signaling, an important property of proteins with high intrinsic disorder and 4) it facilitates the organization of localized assemblies such as the post-synaptic density (Figure 1). Interestingly, these features share a variety of similarities with mRNAs and proteins that are highly regulated in cancer and the upstream mechanisms that are thought to regulate these genes are likewise shared (Boussemart et al., 2014; Wolfe et al., 2014).
Figure 1. Use of Microfluidic Devices to elucidate properties of distally localized mRNAs.

The left panel shows a schematic of a microfluidic device while the middle panel shows an immunocytochemical image of DRG neurons in culture labeled with βIII-tubulin staining. DRG somas are found on the bottom side of the chamber and extend axons through the microfluidic barrier where they then elaborate extensive axonal arobrations on the axonal side. Properties of distally (e.g. axonal) localized mRNAs vs. those found restricted to cell bodies are listed on the right.
Local translation is a key mediator of nociceptor priming
In hyperalgesic priming models, there is now clear evidence that persistent plasticity in peripheral nociceptors is critical to the initiation and maintenance of the primed state (Reichling and Levine, 2009). A broad variety of signaling mechanisms are altered in this state including switches in kinase and G protein coupled receptor (GPCR) signaling cascades (Dina et al., 2009; Joseph and Levine, 2010; Bogen et al., 2012; Ferrari et al., 2012; Wang et al., 2013) but a key feature of this form of plasticity is changes in gene expression regulated at the level of translation. Translation can be controlled, in an activity-dependent fashion, by extracellular factors signaling via kinase cascades offering rapid, locally-mediated control of gene expression. Two important kinases for translation control are the mechanistic target of rapamycin complex 1 (mTORC1) and extracellular signal regulated kinase (ERK, (Topisirovic and Sonenberg, 2011)). Both of these kinases signal to proteins that bind to the 5′ cap structure of mRNAs. In sensory neurons, nerve growth factor (NGF) and interleukin 6 (IL-6), two factors known to induce priming, induce an increase in ERK and mTORC1 signaling leading to a local, axonal increase in protein synthesis (Melemedjian et al., 2010; Melemedjian et al., 2013a). Blockade of these kinases, or blockade of eIF4F complex formation with the eIF4F inhibitor compound 4EGI1, inhibits mechanical hypersensitivity induced by these factors and abrogates precipitation of priming by a normally subthreshold stimulus (Melemedjian et al., 2010; Asiedu et al., 2011) (Figure 2). Hence, axonal translation is required for the induction of priming.
Figure 2. Translational control pathways involved in hyperalgesic priming.

mTORC1 phosphorylates 4EBPs, negative regulators of eIF4F formation. This results in its dissociation from eIF4E, allowing the binding of eIF4E to eIF4G. Phosphorylation of eIF4E (via ERK/MNK1/2) or eIF4G (via mTORC1) enhances the formation of the eIF4F complex, promoting translation. Phosphorylation of CPEB by CamKIIα enhances translation efficiency by increasing the length of the poly A tail in mRNAs containing a CPE sequence. Taken together, eIF4F complex formation enhances cap-dependent translation, which is necessary for the induction of priming via translational control of gene expression in sensory afferents.
One mechanism to decrease ERK and mTORC1 signaling is via stimulation of adenosine monophosphate activated protein kinase (AMPK). AMPK is a widely expressed kinase well known to inhibit mTORC1 (Inoki et al., 2003; Carling et al., 2012) and ERK signaling (Jakobsen et al., 2001; Shen et al., 2013) (Figure 3). In sensory neurons AMPK activation with pharmacological stimulators (for review see (Price and Dussor, 2013)) leads to decreased ERK and mTORC1 activity (Melemedjian et al., 2011; Tillu et al., 2012), decreased eIF4F complex formation (Melemedjian et al., 2011; Tillu et al., 2012) and inhibition of axonal protein synthesis (Melemedjian et al., 2013a). AMPK activators also decrease peripheral nerve injury- and inflammation-induced mechanical hyperalgesia (Melemedjian et al., 2011; Russe et al., 2013) suggesting an important role for this kinase in peripheral pain plasticity across pain models. In the context of hyperalgesic priming, AMPK activation decreases mechanical hypersensitivity caused by incision or IL-6 exposure and completely blocks the development of priming when given locally around the time of incision (Tillu et al., 2012).
Figure 3. AMPK activation pathway.
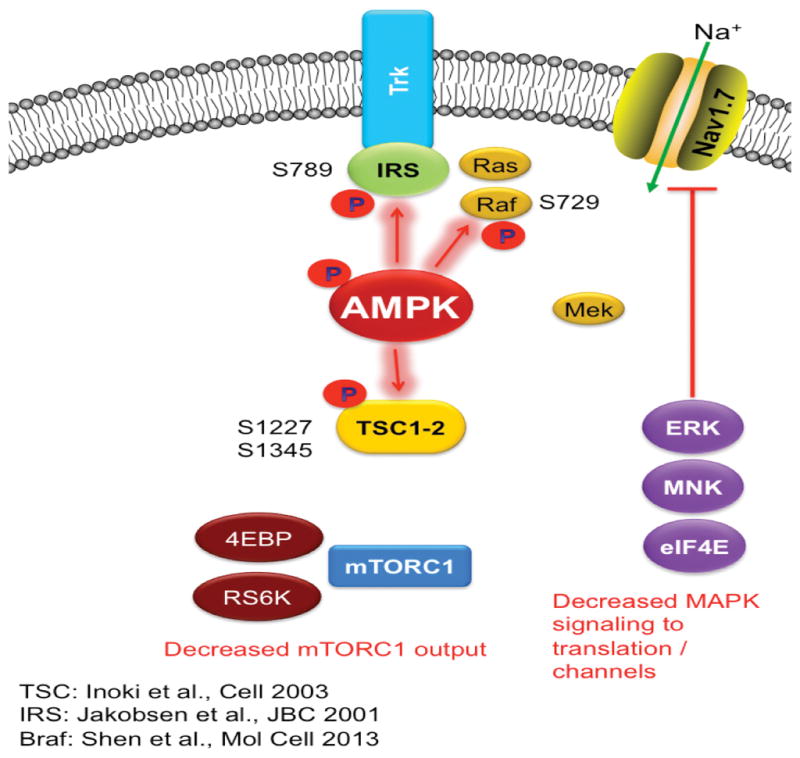
AMPK activation phosphorylates TSCs at Ser 1227 and 1345 leading to the inhibition of mTORC1. This is shown in the figure as an uncoupling of mTORC1 from Trk signaling via phosphorylation of TSC1/2 by AMPK. AMPK activation also phosphorylates Braf (Raf) at Ser 729 leading to inhibition of ERK signaling. Again, this is shown in the figure by an uncoupling of Raf/Mek signaling to ERK, MNK and eIF4E via phosphorylation of Raf at Ser 729. Finally, AMPK phosphorylates IRS1 at Ser 789 leading to further inhibition of tyrosine kinase receptor signaling.
The regulation of translation via 5′ cap binding proteins (the eIFs) and their upstream kinases clearly comprise an important mechanism for the priming of nociceptors. Translation is also regulated by RNA binding proteins that bind to either 5′ or 3′ untranslated regions (UTRs) of mRNAs. For instance, The fragile X mental retardation protein (FMRP) is a key RNA binding protein regulating plasticity in the PNS and CNS (Bassell and Warren, 2008). As such, FMRP knockout mice fail to sensitize in several preclinical pain models (Price et al., 2007; Price and Melemedjian, 2012) and these mice also have deficits in priming induced by NGF and IL-6 (Asiedu et al., 2011). Cytoplasmic polyadenylation element binding protein (CPEB) binds preferentially to mRNAs containing a CPE sequence in their 3′ UTR near the polyadenylation sequence. These mRNAs contain short poly A tails and CPEB acts to enhance poly A tail length leading to enhanced translation efficiency in an activity-dependent fashion (Richter, 2007). This process is linked to LTP in the CNS (Udagawa et al., 2012) and plays a central role in nociceptor priming (Bogen et al., 2012; Ferrari et al., 2012; Ferrari et al., 2013a) as evidenced by inhibition of the initiation of priming via CPEB knockdown in the DRG (Bogen et al., 2012). CPEB is phosphorylated by the aurora family kinases and by Ca2+ / calmodulin activated protein kinase II α (CaMKIIα, (Atkins et al., 2005)). Importantly, in priming induced by peripheral inflammation, CPEB may act downstream of CaMKIIα to initiate and maintain a primed state (Ferrari et al., 2013a). Since CPEB is thought to have prion-like properties that are linked to its role in memory maintenance (Si et al., 2003b; Si et al., 2003a), these findings highlight a potential role for CPEB in creating a permanently primed state in peripheral nociceptors. This could occur due to the self-perpetuating, prion-like properties of activated CPEB.
Which mRNAs are locally translated in the setting of hyperalgesic priming? One candidate is the cyclic AMP response element binding protein (CREB) transcription factor (Melemedjian et al., 2014). Interestingly, CREB was one of the first memory genes identified using the aplysia gill reflex model system (Dash et al., 1990; Kaang et al., 1993). Subsequently, a broad variety of studies have shown a key role for CREB in memory throughout the brain and spinal cord (Rahn et al., 2013). More recently CREB mRNA was identified in developing DRG axons where its local translation is regulated by NGF to control the survival of developing neurons (Cox et al., 2008). In adult DRG axons, stimulation with IL-6 leads to local, nascent synthesis of CREB that is then retrogradely transported to the DRG nucleus where it regulates changes in transcription that are crucial for the establishment of hyperalgesic priming. One gene that is regulated by this retrograde signaling transcription factor is BDNF (Melemedjian et al., 2014).
While it is clear that translation regulation is required to initiate a primed state in the periphery, an important question is whether continuous local translation is required to maintain priming once it has been established. An experimental paradigm to test this translation dependency is to induce priming with a locally administered stimulus and then allow the initial mechanical hypersensitivity to resolve. Then, prior to injection of the stimulus to precipitate the second episode of mechanical hypersensitivity in primed animals, translation inhibitors can be administered locally to test whether continuous translation is required to express a primed state (Asiedu et al., 2011; Ferrari et al., 2013a). In this regard, following injection of IL-6 and resolution of mechanical hypersensitivity in mice, injection of anisomycin or rapamycin (at doses that block the initiation of priming) fail to reverse a primed state when given two days prior to PGE2 injection (Asiedu et al., 2011). In contrast, in rats, injection of carrageenan causes priming that is disrupted both at the time of carrageenan injection and during the maintenance phase by either the mTORC1 inhibitor rapamycin or the polyadenylation inhibitor cordycepin (Ferrari et al., 2013b). Similar effects with rapamycin and cordycepin are observed in rats primed with paw injection of CaMKIIα. Since CaMKIIα phosphorylates CPEB and CPEB regulates CaMKIIα translation this raises the intriguing possibility that CaMKIIα/CPEB signaling could represent a positive feedback mechanism to maintain pain memory in the peripheral nociceptor (Ferrari et al., 2013a). Hence, while there are conflicting results in different models, it is possible that brief disruption of local translation in primed nociceptors is capable of resolving a “pain memory” stored in the peripheral nociceptor.
“Pain memory” in the spinal dorsal horn
LTP in the spinal dorsal horn, a neurophysiological correlate of pain memory?
LTP has been described at synapses throughout the CNS and is widely believed to be a core mechanism of plasticity for the nervous system. In the hippocampus LTP occurs during learning and its persistence is correlated with memory acquisition and consolidation (Whitlock et al., 2006). Likewise, learning and LTP induce changes in dendritic spines in the hippocampus and cortex and these changes in spine shape are thought to be critical for the maintenance of potentiation of postsynaptic responses (De Roo et al., 2008). Interestingly, changes in spine shape also occur in the spinal dorsal horn after injury providing a potential structural change associated with pain memory (Tan et al., 2008; Tan et al., 2009; Tan et al., 2011). Likewise, and similar to memory circuits, LTP can be observed in synapses activated by C-fiber afferent activity (Sandkuhler, 2007; Ruscheweyh et al., 2011). Key sites for nociceptor activity-dependent LTP are the outer lamina of the spinal dorsal horn (Ikeda et al., 2006) where projection neurons expressing the neurokinin receptor type 1 (NK1, substance P receptor) are found and in the deep dorsal horn where many wide dynamic range neurons (neurons that receive both Aβ- and C-fiber input) reside. LTP at these synapses shares many molecular and electrophysiological mechanisms with hippocampal and cortical LTP (Sandkuhler, 2007; Ruscheweyh et al., 2011), with a notable and very important distinction. Whereas low frequency afferent stimulation causes long term depression (LTD) at most synapses in the brain, low frequency stimulation of C-fibers, which largely matches their normal firing frequency, is sufficient to evoke LTP at a subset of dorsal horn neurons receiving direct C-fiber input (Ikeda et al., 2006).
LTP has been equated with an important term used to describe activity-dependent plasticity in the dorsal horn: central sensitization. This topic has been covered by recent reviews (Latremoliere and Woolf, 2009) and editorial comments (Latremoliere and Woolf, 2010; Sandkuhler, 2010) and it is still controversial as to whether spinal LTP and central sensitization are parallel processes. What is less controversial is that LTP correlates well with hyperalgesia and, from a neurophysiological perspective, provides a parsimonious explanation for this form of pain amplification, at least at lamina I synapses in the dorsal horn. This is because homosynaptic LTP is observed at these synapses and hyperalgesia, an enhanced response to a normally noxious stimulus, can be explained by a monosynaptic amplification of C-fiber input onto projection neurons (Sandkuhler, 2007; Ruscheweyh et al., 2011). Allodynia, a noxious response to a normally innocuous stimulus, is more difficult to explain in terms of LTP because it would require a heterosynaptic form of plasticity. This is because Aβ-fibers that are stimulated by light touch do not have a monosynaptic connection to lamina I projection neurons. Interestingly, heterosynaptic LTP has been described at GABAergic synapses in the dorsal horn (Fenselau et al., 2011) but a potential role for this form of plasticity in allodynia has not been established.
Critically, LTP can be induced by natural stimulation of C-fibers with algogens such as capsaicin and formalin (Ikeda et al., 2006). This form of LTP is consolidated into late-LTP and shares mechanisms with hippocampal and cortical LTP (Ikeda et al., 2006; Sandkuhler, 2007; Drdla et al., 2009; Ruscheweyh et al., 2011; Drdla-Schutting et al., 2012). Once spinal dorsal horn LTP reaches the late-phase it does not readily reverse over a several hour time course. However, hyperalgesia induced by both capsaicin and formalin eventually reverses after several days. Does this mean that late phase spinal LTP eventually decays? One possibility is that endogenous analgesic mechanisms mask the hyperalgesic state that would otherwise be evident as a result of a persistent form of LTP. Hyperalgesic priming models may be capable of revealing such a mechanism. In this scenario, the precipitation of hyperalgesia in primed animals would override these endogenous inhibitory mechanisms leading to the reemergence of a hyperalgesic state revealed by a normally sub-threshold stimulus. One candidate for endogenous analgesia overriding late-phase LTP and hyperalgesia is the endogenous opioid system. This system is robust in the dorsal horn with interneurons capable of releasing peptides that act on μ opioid receptors (MORs) expressed throughout the dorsal horn (Ribeiro-da-Silva et al., 1992; Ma et al., 1997), including presynaptic nociceptor nerve endings (Schroeder et al., 1991; Schroeder and McCleskey, 1993). In support of this idea, infusion of MOR inverse agonists immediately precipitates a reinstatement of hyperalgesia in animals that have been primed with an inflammatory stimulus that is known to induce spinal LTP after the resolution of hyperalgesia (Corder et al., 2013). This effect is absent in sham animals and is analogous to precipitation of hyperalgesia in primed animals with a sub-threshold peripheral stimulus. What governs this effect? Peripheral inflammation, and presumably other nociceptive stimuli, induces a change in spinal MORs such that they now acquire constitutive activity (signaling through G proteins in the absence of agonist). This MOR constitutive activity then causes a tonic inhibition of pain signaling that masks a hyperalgesic state that would otherwise persist following the initial insult (Corder et al., 2013).
These findings have several important implications for understanding central mechanisms governing hyperalgesic priming. They provide an elegant solution to why initial hyperalgesia resolves despite the persistence of a primed state and the potential durability of spinal LTP. This evidence also provides links to priming and late-LTP maintenance that potentially solve questions stated above. Opioid-dependent mechanisms play an important role in regulating spinal LTP. While there is evidence that high dose opioids can stimulate LTP at certain synapses after their abrupt removal (Drdla et al., 2009), there is likewise evidence that MOR activation can resolve even late-LTP at spinal synapses (Drdla-Schutting et al., 2012). Based on this, it is possible that the initial priming stimulus, or induction of LTP, leads to late-LTP consolidation but this is subsequently resolved by endogenous opioid-mediated mechanisms. Does this mean that the previous establishment of late-LTP at central synapses causes a drop in threshold for establishment of subsequent LTP? If the mechanisms governing the MOR-dependent reversal of spinal late-LTP are constitutively expressed, as appears to be the case (Corder et al., 2013), then this may lead to a tonic reversal of late LTP with underlying mechanisms (e.g., aPKC and BDNF/trkB signaling, discussed below) still in place. While this idea obviously requires experimental testing, it could represent an important mechanism linking changes in peripheral sensitivity to CNS plasticity responsible for the maintenance of priming. Reversing these mechanisms could lead to revolutionary new therapeutics with disease modifying effects on chronic pain.
Atypical PKCs and Brain Derived Neurotropic Factor (BDNF)
Early-LTP requires the activation of CaMKIIα, PKA and conventional PKC leading to the phosphorylation of AMPA receptors (Huganir and Nicoll, 2013). Early-LTP also leads to changes in gene expression that occur both at the level of transcription and translation. These changes in gene expression are needed for the consolidation of early-LTP into late-LTP (Abraham and Williams, 2008a). Mechanisms involved in the maintenance of late-LTP have been more difficult to clearly elucidate but are thought to involve an atypical PKC (aPKC) isoform called PKMζ (Sacktor, 2011). Late-LTP can be reversed by inhibition of aPKCs with a peptide-based, pseudo substrate inhibitor called ZIP (Pastalkova et al., 2006). Intrathecal injection of ZIP either at the time of priming induction or following the resolution of the initial hyperalgesia leads to a complete reversal of hyperalgesic priming (Asiedu et al., 2011; Melemedjian et al., 2013b). ZIP also reverses established pain states that have become dependent on central plasticity following sustained afferent input (Laferriere et al., 2011). These findings are consistent with a role for PKMζ in the maintenance of late-LTP, memory retention and the maintenance of a chronic pain state. On the other hand, recent experiments using genetic models to dissect the role of PKMζ in late-LTP and memory maintenance have called the specificity of ZIP and the role of PKMζ in these effects into question (Lee et al., 2013; Volk et al., 2013). It remains to be seen if PKMζ plays a specific role in the maintenance of hyperalgesic priming in the dorsal horn of the spinal cord (for review on this topic see (Price and Ghosh, 2013)).
An important component of the proposed role of PKMζ in LTP and memory is the trafficking of AMPA receptors to synaptic sites leading to a persistent augmentation of postsynaptic glutamate-mediated signaling (Sacktor, 2011). This trafficking can be disrupted with a peptide called pep2m that blocks AMPA receptor association with trafficking molecules (Migues et al., 2010). Similar to experiments in other CNS regions, intrathecal injection of pep2m disrupts the maintenance of hyperalgesic priming (Asiedu et al., 2011) suggesting that aPKC-mediated regulation of AMPA receptor trafficking may play a central role in chronic pain states. This is consistent with a wide variety of experimental findings indicating that AMPA receptor trafficking plays a central role in mediating pain plasticity induced by peripheral injury (Tao, 2012) and that an increase in AMPA receptors at the postsynaptic density is required for LTP consolidation and maintenance.
As mentioned above, while it is clear that ZIP is capable of permanently reversing a primed state in a variety of experimental models (Asiedu et al., 2011; Laferriere et al., 2011; Melemedjian et al., 2013b), the molecular mechanisms engaged by ZIP are less clear based on evidence from transgenic mice (Lee et al., 2013; Volk et al., 2013). One possibility is that aPKC isoforms play a redundant role in synaptic plasticity and therefore another aPKC, PKCλ, may be involved in maintenance mechanisms of hyperalgesic priming (Price and Ghosh, 2013). This isoform is also inhibited by ZIP (Melemedjian et al., 2013b; Volk et al., 2013), therefore, this provides an explanation for the discrepancy between pharmacological effects of ZIP and findings from mice lacking aPKCs derived from the Prckz locus (PKMζ and PKCζ). If this were the case, upstream mechanisms that regulate all aPKCs isoforms would represent alternative targets to reverse hyperalgesic priming. A candidate molecule fitting this description is BDNF.
BDNF is well recognized as an important mediator of pain plasticity. BDNF is expressed by DRG neurons and released in the spinal dorsal horn (Balkowiec and Katz, 2000), where it can act on pre- and post-synaptic trkB receptors to regulate plasticity of pre-synaptic afferent fibers (Matayoshi et al., 2005) and post-synaptic dorsal horn neurons (Kerr et al., 1999; Pezet et al., 2002; Garraway et al., 2003). BDNF expression increases following peripheral injury (Mannion et al., 1999), nociceptor-specific knockout of BDNF leads to a profound reduction in many forms of injury-induced pain plasticity (Zhao et al., 2006) and microglial BDNF expression is increased by nerve injury (Trang et al., 2011). BDNF is also a key factor in LTP. In hippocampus, BDNF is required for the induction of LTP and dendritic-expressed BDNF is postulated to play an autocrine role in maintenance of late phase LTP (Lu et al., 2008). Likewise, BDNF is sufficient to induce LTP in dorsal horn neurons (Zhou et al., 2008) linking BDNF-induced pain plasticity to memory-like mechanisms that may be involved in the maintenance of hyperalgesic priming. Indeed, intrathecal injection of compounds that interfere with BDNF action, blocks hyperalgesia induced by priming agents and prevents the precipitation of a primed state by subsequent stimulation. Significantly, interruption of BDNF/trkB signaling after the establishment of a primed state leads to a resolution of priming (Melemedjian et al., 2013b) suggesting a key role of BDNF/trkB signaling in the maintenance of a primed state. At spinal synapses, BDNF induces phosphorylation and translation of the two major aPKC isoforms found in the CNS, PKMζ and PKCλ indicating a link between BDNF/trkB and aPKCs (Melemedjian et al., 2013b) (Figure 4). Therefore, these findings point to BDNF/trkB signaling as a therapeutic target for the reversal of established chronic pain states. Therapeutics aimed at this signaling axis could lead to disease modification in chronic pain patients.
Figure 4. The role of aPKCs and BDNF in hyperalgesic priming initiation and maintenance.
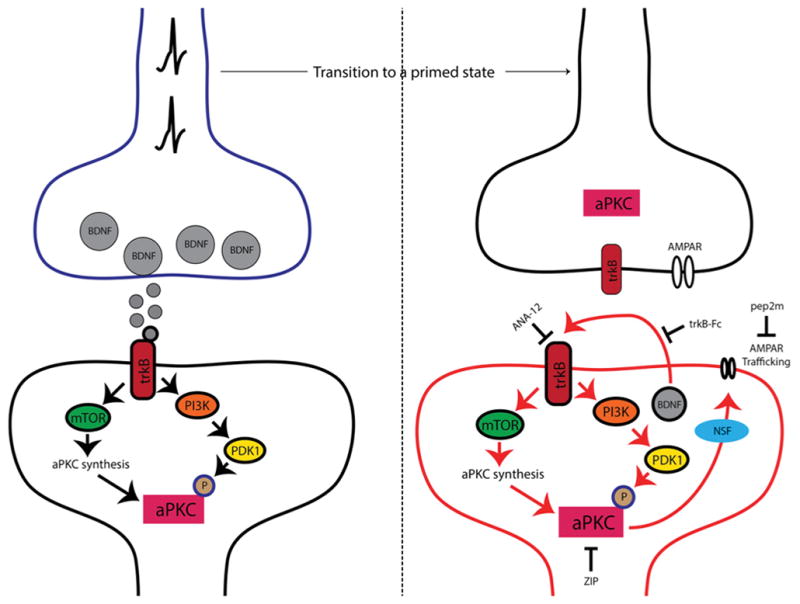
Nociceptor activation leads to spinal BDNF release and a postsynaptic mTORC1-dependent translation of aPKC protein. These newly synthesized aPKCs are then phosphorylated by PDK1. Increased levels and phosphorylation of aPKCs are thought to be involved in initiating priming. Once priming is established (right panel), increased aPKC protein and phosphorylation leads to a constitutive increase in AMPAR trafficking to the postsynaptic membrane. This appears to be regulated by BDNF signaling via trkB with BDNF potentially being released from postsynaptic dendrites in the maintenance stage of priming. Presynaptic trkB may also be activated by increased BDNF action in primed animals. Once established, hyperalgesic priming can be permanently reversed by inhibition of aPKCs with ZIP, disruption of AMPAR trafficking with pep2M or via inhibition of trkB/BDNF signaling with ANA-12 or trkB-Fc, respectively.
Reconsolidation of pain memory
The mechanisms discussed above provide a foundation for understanding spinal mechanisms that encode a memory trace for pain. These mechanisms primarily tie into the concept that spinal dorsal horn plasticity shares molecular, neurophysiological and structural similarities with plasticity in other regions of the brain that are involved in learning and memory. Do spinal memory traces share other features with hippocampal or cortical memory mechanisms? An important concept in the learning and memory literature is the idea of reconsolidation. Reconsolidation is a memory updating mechanism during which a memory trace can become labile and susceptible to revision with behavioral intervention and/or pharmacological manipulation (Nader and Hardt, 2009). Classical reconsolidation experiments involve fear conditioning. Here an animal is trained to fear a neutral stimulus through pairing with a foot shock. This creates a long-lasting memory that can be revealed by freezing induced by presentation of the neutral stimulus that has been paired with shock. Infusion of a protein synthesis inhibitor at the time of training blocks the acquisition of the fear memory, however, after the memory has consolidated (e.g. within 6 hours after training) protein synthesis inhibition no longer alters the memory trace, so long as the protein synthesis inhibitor is given outside of the training context. However, if the animal is placed back in the training context and exposed to the neutral stimulus to trigger recall of the memory, infusion of a protein synthesis inhibitor will again lead to a reversal of fear conditioning (Nader et al., 2000). Hence, during reconsolidation, a memory trace is made labile and can be reversed by pharmacological mechanisms that were only effective prior to consolidation of the long-term memory. Importantly, these pharmacological mechanisms share strong similarities with early- and late-LTP, as described above (Figure 5A).
Figure 5. Consolidation of late phase LTP (late-LTP) and reconsolidation.

A) Following high frequency stimulation of afferent input (3 upward arrows), early-LTP (e-LTP in the figure) is induced and this consolidates to late-LTP (l-LTP in the figure) over the course of 30 – 60 minutes. Application of translation control inhibitors, such as anisomycin (red line), during early-LTP cause a failure of late-LTP consolidation. Likewise, aPKC inhibition with ZIP (green line) blocks consolidation of late-LTP. Vehicle application (blue line) has no impact on consolidation of late-LTP B) Once late-LTP is established administering translation inhibitors (e.g. anisomycin, red line) in the absence of high frequency stimulation of afferents fails to reverse late-LTP while ZIP application (green line) does induce late-LTP decay. Restimulation of afferents at high frequency during late-LTP (upward arrows) opens a reconsolidation window. Application of translation inhibitors such as anisomycin (red line) during this reconsolidation period leads to late-LTP decay, an effect that is presumably linked to reversal of a chronic pain state in similar behavioral pharmacology experiments.
Bonin and De Koninck recently showed that a mechanism akin to reconsolidation is engaged in the spinal dorsal horn. Using capsaicin as a noxious stimulus to induce pain plasticity they showed that inhibition of spinal protein synthesis paired with either a second capsaicin injection or optogenetic stimulation of C-fibers led to a reversal of pain plasticity (Bonin and De Koninck, 2014). In parallel experiments they examined spinal LTP and its dependence on protein synthesis following the consolidation of late-LTP (Figure 5B). They found that after late-LTP consolidation spinal LTP was not reversed by protein synthesis inhibition unless C-fibers were tetanized at the time of protein synthesis inhibitor application. This crucial experiment parallels reconsolidation of pain memory in a behavioral paradigm providing a strong link between behavioral manifestations of pain plasticity (hyperalgesia) and spinal LTP. Similar experiments using the hyperalgesic priming paradigm indicate that reconsolidation of pain memory can be engaged even at late stages after the initiation of a pain memory trace (Kim and Price unpublished observations) suggesting that opening of a reconsolidation window in chronic pain patients may provide an opportunity for reversal of pain plasticity and a resolution of chronic pain.
Clinical implications of pain memory
In order to fully grasp the importance of the research findings discussed herein, it is important to reflect on the utility of using experimental models of pain memory to gain better insight into human pain plasticity. Along these lines it is must be noted that human experimental models of perceptual LTP, and even LTD, exist and involve afferent stimulation protocols that are similar if not identical to those used in preclinical studies. There is also evidence that LTP stimulation protocols in humans can lead to a transition to late-LTP in certain individuals. Another important point is that the experimental framework of the hyperalgesic priming model, which is often used to study pain memory, provides important insight into clinical chronic pain because it captures the recurrent nature of some of the most common pathological pain conditions (Reichling and Levine, 2009). In 1921, Wilfred Harris described his clinical experience treating patients with presumed injuries to peripheral nerves. He described pain in these patients as episodic with pain episodes provoked by acute exacerbation (Harris, 1921). Hence, from some of the earliest descriptions of pain as a disease, the notion of priming followed by sub-threshold provocation of long-lived pain episodes has been apparent.
Population-based studies in prevalent chronic pain conditions have directly demonstrated the episodic yet progressive nature of chronic pain. Perhaps the best-known example is headache and, in the case of migraine, frequency of attacks is the best predictor of a transition to chronic migraine (Lipton, 2009). In fact, the vast majority of migraineurs move from a low-frequency episodic headache stage to a high-frequency stage and eventually into chronic migraine (Bigal and Lipton, 2008). Moreover, migraines can frequently be provoked by migraine triggers. These are, by definition, sub-threshold stimuli because they fail to provoke migraines in the non-migraineur population. This situation is not unique to migraine. Acute episodes of low back pain generally resolve (Bartleson, 2001; Cassidy et al., 2005) but recurrence rates over 5 years are as high as 70% (Von Korff and Saunders, 1996; Carey et al., 1999; Cassidy et al., 2005; Kolb et al., 2011) and lifetime recurrence is estimated at 85% (Andersson, 1999; Tamcan et al., 2010). The probability of low back pain recurrence increases with previous episodes of low back pain (Kolb et al., 2011). A similar clinical picture has been found for chronic neck pain (Croft et al., 2001; Nolet et al., 2010). Finally, in the case of surgery and chronic post-surgical pain, there is evidence that pre-existing pain is a major risk factor for chronic post-incision pain suggesting that a pain memory trace might already be in place in some patients causing precipitation of a very long-lasting pain state induced by incision (Althaus et al., 2012; Pinto et al., 2013). Hence, the “priming” event in the hyperaglesic priming model may be viewed as an induction of a pain memory with important clinical parallels that help to explain mechanisms of disease and potential pathways to resolution of plasticity that drives chronicity.
Conclusions
The concept of pain memory, first proposed by Ronald Melzack and colleagues nearly 40 years ago, has inspired remarkable progress into our understanding of mechanisms that cause pain to become chronic. Here we have described two major potential mechanisms for pain memory: 1) changes in gene expression in peripheral nociceptors that permanently alter the phenotype and function of these crucial neurons for pain plasticity and 2) changes in synaptic strength in the dorsal horn governed by mechanisms that play an active role in maintaining a chronic pain state. We have highlighted several intervention points that have potential to lead to disease modifying therapeutics for the permanent reversal of chronic pain states. While these mechanisms ultimately await testing in human patients, there is little question that the concept of pain memory will continue to provoke new research questions and provide novel insight into how plasticity leads to the neurological disorder that is chronic pain.
Acknowledgments
This work was supported by NIH grants NS065926 (TJP) and GM102575 (TJP and GD) and The University of Texas STARS program (TJP).
References
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem. 2008a;89:260–268. doi: 10.1016/j.nlm.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Williams JM. LTP maintenance and its protein synthesis-dependence. Neurobiology of learning and memory. 2008b;89:260–268. doi: 10.1016/j.nlm.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Althaus A, Hinrichs-Rocker A, Chapman R, Arranz Becker O, Lefering R, Simanski C, Weber F, Moser KH, Joppich R, Trojan S, Gutzeit N, Neugebauer E. Development of a risk index for the prediction of chronic post-surgical pain. Eur J Pain. 2012;16:901–910. doi: 10.1002/j.1532-2149.2011.00090.x. [DOI] [PubMed] [Google Scholar]
- Andersson GB. Epidemiological features of chronic low-back pain. Lancet. 1999;354:581–585. doi: 10.1016/S0140-6736(99)01312-4. [DOI] [PubMed] [Google Scholar]
- Asiedu MN, Tillu DV, Melemedjian OK, Shy A, Sanoja R, Bodell B, Ghosh S, Porreca F, Price TJ. Spinal protein kinase M zeta underlies the maintenance mechanism of persistent nociceptive sensitization. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:6646–6653. doi: 10.1523/JNEUROSCI.6286-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Davare MA, Oh MC, Derkach V, Soderling TR. Bidirectional regulation of cytoplasmic polyadenylation element-binding protein phosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase 1 during hippocampal long-term potentiation. J Neurosci. 2005;25:5604–5610. doi: 10.1523/JNEUROSCI.5051-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci. 2000;20:7417–7423. doi: 10.1523/JNEUROSCI.20-19-07417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartleson JD. Low Back Pain. Curr Treat Options Neurol. 2001;3:159–168. doi: 10.1007/s11940-001-0051-4. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal ME, Lipton RB. Clinical course in migraine: conceptualizing migraine transformation. Neurology. 2008;71:848–855. doi: 10.1212/01.wnl.0000325565.63526.d2. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCepsilon activation of CPEB. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:2018–2026. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin RP, De Koninck Y. A spinal analog of memory reconsolidation enables reversal of hyperalgesia. Nat Neurosci. 2014;17:1043–1045. doi: 10.1038/nn.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F, Mateus C, Routier E, Kamsu-Kom N, Agoussi S, Eggermont AM, Desaubry L, Robert C, Vagner S. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014 doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Carey TS, Garrett JM, Jackman A, Hadler N. Recurrence and care seeking after acute back pain: results of a long-term follow-up study. North Carolina Back Pain Project. Med Care. 1999;37:157–164. doi: 10.1097/00005650-199902000-00006. [DOI] [PubMed] [Google Scholar]
- Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- Cassidy JD, Cote P, Carroll LJ, Kristman V. Incidence and course of low back pain episodes in the general population. Spine (Phila Pa 1976) 2005;30:2817–2823. doi: 10.1097/01.brs.0000190448.69091.53. [DOI] [PubMed] [Google Scholar]
- Cervero F. Spinal cord mechanisms of hyperalgesia and allodynia: role of peripheral input from nociceptors. Prog Brain Res. 1996;113:413–422. doi: 10.1016/s0079-6123(08)61101-2. [DOI] [PubMed] [Google Scholar]
- Clatworthy AL, Walters ET. Rapid amplification and facilitation of mechanosensory discharge in Aplysia by noxious stimulation. J Neurophysiol. 1993;70:1181–1194. doi: 10.1152/jn.1993.70.3.1181. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Melzack R. Cutaneous hyperalgesia: contributions of the peripheral and central nervous systems to the increase in pain sensitivity after injury. Brain Res. 1987;404:95–106. doi: 10.1016/0006-8993(87)91359-x. [DOI] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK. Constitutive mu-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science. 2013;341:1394–1399. doi: 10.1126/science.1239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol. 2008;10:149–159. doi: 10.1038/ncb1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft PR, Lewis M, Papageorgiou AC, Thomas E, Jayson MI, Macfarlane GJ, Silman AJ. Risk factors for neck pain: a longitudinal study in the general population. Pain. 2001;93:317–325. doi: 10.1016/S0304-3959(01)00334-7. [DOI] [PubMed] [Google Scholar]
- Crook RJ, Dickson K, Hanlon RT, Walters ET. Nociceptive sensitization reduces predation risk. Current Biology. 2014 doi: 10.1016/j.cub.2014.03.043. in press. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Carew TJ. Nobel celebrates the neurosciences. Modulatory signaling in the brain. Cell. 2000;103:829–833. doi: 10.1016/s0092-8674(00)00185-9. [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Garcia PM, Poglia L, Muller D. Spine dynamics and synapse remodeling during LTP and memory processes. Prog Brain Res. 2008;169:199–207. doi: 10.1016/S0079-6123(07)00011-8. [DOI] [PubMed] [Google Scholar]
- Dennis SG, Melzack R. Self-mutilation after dorsal rhizotomy in rats: effects of prior pain and pattern of root lesions. Exp Neurol. 1979;65:412–421. doi: 10.1016/0014-4886(79)90108-0. [DOI] [PubMed] [Google Scholar]
- Devor M, Janig W, Michaelis M. Modulation of activity in dorsal root ganglion neurons by sympathetic activation in nerve-injured rats. J Neurophysiol. 1994;71:38–47. doi: 10.1152/jn.1994.71.1.38. [DOI] [PubMed] [Google Scholar]
- Dina OA, Khasar SG, Gear RW, Levine JD. Activation of Gi induces mechanical hyperalgesia poststress or inflammation. Neuroscience. 2009;160:501–507. doi: 10.1016/j.neuroscience.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drdla R, Gassner M, Gingl E, Sandkuhler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210. doi: 10.1126/science.1171759. [DOI] [PubMed] [Google Scholar]
- Drdla-Schutting R, Benrath J, Wunderbaldinger G, Sandkuhler J. Erasure of a spinal memory trace of pain by a brief, high-dose opioid administration. Science. 2012;335:235–238. doi: 10.1126/science.1211726. [DOI] [PubMed] [Google Scholar]
- Fenselau H, Heinke B, Sandkuhler J. Heterosynaptic long-term potentiation at GABAergic synapses of spinal lamina I neurons. J Neurosci. 2011;31:17383–17391. doi: 10.1523/JNEUROSCI.3076-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Role of nociceptor alphaCaMKII in transition from acute to chronic pain (hyperalgesic priming) in male and female rats. J Neurosci. 2013a;33:11002–11011. doi: 10.1523/JNEUROSCI.1785-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Chu C, Levine JD. Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain. 2013b;14:731–738. doi: 10.1016/j.jpain.2013.01.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Alessandri-Haber N, Levine E, Gear RW, Levine JD. Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain. Neuroscience. 2012;222:392–403. doi: 10.1016/j.neuroscience.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor H, Nikolajsen L, Staehelin Jensen T. Phantom limb pain: a case of maladaptive CNS plasticity? Nat Rev Neurosci. 2006;7:873–881. doi: 10.1038/nrn1991. [DOI] [PubMed] [Google Scholar]
- Garraway SM, Petruska JC, Mendell LM. BDNF sensitizes the response of lamina II neurons to high threshold primary afferent inputs. Eur J Neurosci. 2003;18:2467–2476. doi: 10.1046/j.1460-9568.2003.02982.x. [DOI] [PubMed] [Google Scholar]
- Harris W. Persistent Pain in Lesions of the Peripheral and Central Nervous System. Br Med J. 1921;2:896–900. doi: 10.1136/bmj.2.3178.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and Synaptic Plasticity: The Last 25 Years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jager T, Sandkuhler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Illich PA, Walters ET. Mechanosensory neurons innervating Aplysia siphon encode noxious stimuli and display nociceptive sensitization. J Neurosci. 1997;17:459–469. doi: 10.1523/JNEUROSCI.17-01-00459.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE. 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem. 2001;276:46912–46916. doi: 10.1074/jbc.C100483200. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Multiple PKCepsilon-dependent mechanisms mediating mechanical hyperalgesia. Pain. 2010;150:17–21. doi: 10.1016/j.pain.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaang BK, Kandel ER, Grant SG. Activation of cAMP-responsive genes by stimuli that produce long-term facilitation in Aplysia sensory neurons. Neuron. 1993;10:427–435. doi: 10.1016/0896-6273(93)90331-k. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialog between genes and synapses. Biosci Rep. 2004;24:475–522. doi: 10.1007/s10540-005-2742-7. [DOI] [PubMed] [Google Scholar]
- Katz J, Melzack R. Pain ‘memories’ in phantom limbs: review and clinical observations. Pain. 1990;43:319–336. doi: 10.1016/0304-3959(90)90029-D. [DOI] [PubMed] [Google Scholar]
- Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci. 1999;19:5138–5148. doi: 10.1523/JNEUROSCI.19-12-05138.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb E, Canjuga M, Bauer GF, Laubli T. Course of back pain across 5 years: a retrospective cohort study in the general population of Switzerland. Spine (Phila Pa 1976) 2011;36:E268–273. doi: 10.1097/BRS.0b013e3181f324b5. [DOI] [PubMed] [Google Scholar]
- Laferriere A, Pitcher MH, Haldane A, Huang Y, Cornea V, Kumar N, Sacktor TC, Cervero F, Coderre TJ. PKMzeta is essential for spinal plasticity underlying the maintenance of persistent pain. Mol Pain. 2011;7:99. doi: 10.1186/1744-8069-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Synaptic plasticity and central sensitization: author reply. J Pain. 2010;11:801–803. doi: 10.1016/j.jpain.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO. Prkcz null mice show normal learning and memory. Nature. 2013;493:416–419. doi: 10.1038/nature11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton RB. Tracing transformation: chronic migraine classification, progression, and epidemiology. Neurology. 2009;72:S3–7. doi: 10.1212/WNL.0b013e3181974b19. [DOI] [PubMed] [Google Scholar]
- Lisney SJ, Devor M. Afterdischarge and interactions among fibers in damaged peripheral nerve in the rat. Brain Res. 1987;415:122–136. doi: 10.1016/0006-8993(87)90275-7. [DOI] [PubMed] [Google Scholar]
- Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89:312–323. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Ribeiro-da-Silva A, De Koninck Y, Radhakrishnan V, Cuello AC, Henry JL. Substance P and enkephalin immunoreactivities in axonal boutons presynaptic to physiologically identified dorsal horn neurons. An ultrastructural multiple-labelling study in the cat. Neuroscience. 1997;77:793–811. doi: 10.1016/s0306-4522(96)00510-6. [DOI] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matayoshi S, Jiang N, Katafuchi T, Koga K, Furue H, Yasaka T, Nakatsuka T, Zhou XF, Kawasaki Y, Tanaka N, Yoshimura M. Actions of brain-derived neurotrophic factor on spinal nociceptive transmission during inflammation in the rat. J Physiol. 2005;569:685–695. doi: 10.1113/jphysiol.2005.095331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Mejia GL, Lepow TS, Zoph OK, Price TJ. Bidirectional regulation of P body formation mediated by eIF4F complex formation in sensory neurons. Neurosci Lett. 2013a doi: 10.1016/j.neulet.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. IL-6- and NGF-Induced Rapid Control of Protein Synthesis and Nociceptive Plasticity via Convergent Signaling to the eIF4F Complex. J Neurosci. 2010;30:15113–15123. doi: 10.1523/JNEUROSCI.3947-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Tillu DV, Moy JK, Asiedu MN, Mandell EK, Ghosh S, Dussor G, Price TJ. Local translation and retrograde axonal transport of CREB regulates IL-6-induced nociceptive plasticity. Mol Pain. 2014;10:45. doi: 10.1186/1744-8069-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Tillu DV, Asiedu MN, Mandell EK, Moy JK, Blute VM, Taylor CJ, Ghosh S, Price TJ. BDNF regulates atypical PKC at spinal synapses to initiate and maintain a centralized chronic pain state. Mol Pain. 2013b;9:12. doi: 10.1186/1744-8069-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melemedjian OK, Asiedu MN, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T, Sonenberg N, Dussor G, Price TJ. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain. 2011;7:70. doi: 10.1186/1744-8069-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. 2010;13:630–634. doi: 10.1038/nn.2531. [DOI] [PubMed] [Google Scholar]
- Nader K, Hardt O. A single standard for memory: the case for reconsolidation. Nat Rev Neurosci. 2009;10:224–234. doi: 10.1038/nrn2590. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nolet PS, Cote P, Cassidy JD, Carroll LJ. The association between a lifetime history of a neck injury in a motor vehicle collision and future neck pain: a population-based cohort study. Eur Spine J. 2010;19:972–981. doi: 10.1007/s00586-010-1344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW, Williams RJ, McMahon SB. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Molecular and cellular neurosciences. 2002;21:684–695. doi: 10.1006/mcne.2002.1205. [DOI] [PubMed] [Google Scholar]
- Pinto PR, McIntyre T, Ferrero R, Almeida A, Araujo-Soares V. Risk factors for moderate and severe persistent pain in patients undergoing total knee and hip arthroplasty: a prospective predictive study. PLoS One. 2013;8:e73917. doi: 10.1371/journal.pone.0073917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Geranton SM. Translating nociceptor sensitivity: the role of axonal protein synthesis in nociceptor physiology. Eur J Neurosci. 2009;29:2253–2263. doi: 10.1111/j.1460-9568.2009.06786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Melemedjian OK. Fragile X mental retardation protein (FMRP) and the spinal sensory system. Results Probl Cell Differ. 2012;54:41–59. doi: 10.1007/978-3-642-21649-7_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Dussor G. AMPK: An emerging target for modification of injury-induced pain plasticity. Neurosci Lett. 2013 doi: 10.1016/j.neulet.2013.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Ghosh S. ZIPping to pain relief: the role (or not) of PKMzeta in chronic pain. Mol Pain. 2013;9:6. doi: 10.1186/1744-8069-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Dussor G. Evolution: the advantage of ‘maladaptive’ pain plasticity. Curr Biol. 2014;24:R384–386. doi: 10.1016/j.cub.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007;27:13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn EJ, Guzman-Karlsson MC, David Sweatt J. Cellular, molecular, and epigenetic mechanisms in non-associative conditioning: implications for pain and memory. Neurobiol Learn Mem. 2013;105:133–150. doi: 10.1016/j.nlm.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB, Green PG, Levine JD. The fundamental unit of pain is the cell. Pain. 2013 doi: 10.1016/j.pain.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro-da-Silva A, De Koninck Y, Cuello AC, Henry JL. Enkephalin-immunoreactive nociceptive neurons in the cat spinal cord. Neuroreport. 1992;3:25–28. doi: 10.1097/00001756-199201000-00006. [DOI] [PubMed] [Google Scholar]
- Richter JD. CPEB: a life in translation. Trends Biochem Sci. 2007;32:279–285. doi: 10.1016/j.tibs.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Ruscheweyh R, Wilder-Smith O, Drdla R, Liu XG, Sandkuhler J. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Mol Pain. 2011;7:20. doi: 10.1186/1744-8069-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russe OQ, Moser CV, Kynast KL, King TS, Stephan H, Geisslinger G, Niederberger E. Activation of the AMP-Activated Protein Kinase Reduces Inflammatory Nociception. J Pain. 2013;14:1330–1340. doi: 10.1016/j.jpain.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Sacktor TC. How does PKMzeta maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- Sandkuhler J. Understanding LTP in pain pathways. Mol Pain. 2007;3:9. doi: 10.1186/1744-8069-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkuhler J. Central sensitization versus synaptic long-term potentiation (LTP): a critical comment. J Pain. 2010;11:798–800. doi: 10.1016/j.jpain.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Schroeder JE, McCleskey EW. Inhibition of Ca2+ currents by a mu-opioid in a defined subset of rat sensory neurons. J Neurosci. 1993;13:867–873. doi: 10.1523/JNEUROSCI.13-02-00867.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JE, Fischbach PS, Zheng D, McCleskey EW. Activation of mu opioid receptors inhibits transient high- and low-threshold Ca2+ currents, but spares a sustained current. Neuron. 1991;6:13–20. doi: 10.1016/0896-6273(91)90117-i. [DOI] [PubMed] [Google Scholar]
- Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, Zheng B. Phosphorylation of BRAF by AMPK Impairs BRAF-KSR1 Association and Cell Proliferation. Mol Cell. 2013;52:161–172. doi: 10.1016/j.molcel.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell. 2003a;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell. 2003b;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- Steward O, Levy WB. Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci. 1982;2:284–291. doi: 10.1523/JNEUROSCI.02-03-00284.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamcan O, Mannion AF, Eisenring C, Horisberger B, Elfering A, Muller U. The course of chronic and recurrent low back pain in the general population. Pain. 2010;150:451–457. doi: 10.1016/j.pain.2010.05.019. [DOI] [PubMed] [Google Scholar]
- Tan AM, Choi JS, Waxman SG, Hains BC. Dendritic spine remodeling after spinal cord injury alters neuronal signal processing. J Neurophysiol. 2009;102:2396–2409. doi: 10.1152/jn.00095.2009. [DOI] [PubMed] [Google Scholar]
- Tan AM, Chang YW, Zhao P, Hains BC, Waxman SG. Rac1-regulated dendritic spine remodeling contributes to neuropathic pain after peripheral nerve injury. Exp Neurol. 2011;232:222–233. doi: 10.1016/j.expneurol.2011.08.028. [DOI] [PubMed] [Google Scholar]
- Tan AM, Stamboulian S, Chang YW, Zhao P, Hains AB, Waxman SG, Hains BC. Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J Neurosci. 2008;28:13173–13183. doi: 10.1523/JNEUROSCI.3142-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantos A, Han KH, Tompa P. Intrinsic disorder in cell signaling and gene transcription. Mol Cell Endocrinol. 2012;348:457–465. doi: 10.1016/j.mce.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Tao YX. AMPA receptor trafficking in inflammation-induced dorsal horn central sensitization. Neuroscience bulletin. 2012;28:111–120. doi: 10.1007/s12264-012-1204-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillu DV, Melemedjian OK, Asiedu MN, Qu N, De Felice M, Dussor G, Price TJ. Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Molecular Pain. 2012;8:5. doi: 10.1186/1744-8069-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Sonenberg N. mRNA translation and energy metabolism in cancer: the role of the MAPK and mTORC1 pathways. Cold Spring Harb Symp Quant Biol. 2011;76:355–367. doi: 10.1101/sqb.2011.76.010785. [DOI] [PubMed] [Google Scholar]
- Trang T, Beggs S, Salter MW. Brain-derived neurotrophic factor from microglia: a molecular substrate for neuropathic pain. Neuron Glia Biol. 2011;7:99–108. doi: 10.1017/S1740925X12000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa T, Swanger SA, Takeuchi K, Kim JH, Nalavadi V, Shin J, Lorenz LJ, Zukin RS, Bassell GJ, Richter JD. Bidirectional control of mRNA translation and synaptic plasticity by the cytoplasmic polyadenylation complex. Mol Cell. 2012;47:253–266. doi: 10.1016/j.molcel.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, Devor M. Peripheral nervous system origin of phantom limb pain. Pain. 2014;155:1384–1391. doi: 10.1016/j.pain.2014.04.018. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Korff M, Saunders K. The course of back pain in primary care. Spine (Phila Pa 1976) 1996;21:2833–2837. doi: 10.1097/00007632-199612150-00004. discussion 2838–2839. [DOI] [PubMed] [Google Scholar]
- Walters ET, Moroz LL. Molluscan memory of injury: evolutionary insights into chronic pain and neurological disorders. Brain Behav Evol. 2009;74:206–218. doi: 10.1159/000258667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Heijnen CJ, van Velthoven CT, Willemen HL, Ishikawa Y, Zhang X, Sood AK, Vroon A, Eijkelkamp N, Kavelaars A. Balancing GRK2 and EPAC1 levels prevents and relieves chronic pain. J Clin Invest. 2013 doi: 10.1172/JCI66241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatheritt RJ, Gibson TJ, Babu MM. Asymmetric mRNA localization contributes to fidelity and sensitivity of spatially localized systems. Nat Struct Mol Biol. 2014;21:833–839. doi: 10.1038/nsmb.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weragoda RM, Ferrer E, Walters ET. Memory-like alterations in Aplysia axons after nerve injury or localized depolarization. J Neurosci. 2004;24:10393–10401. doi: 10.1523/JNEUROSCI.2329-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Willis D, Li KW, Zheng JQ, Chang JH, Smit A, Kelly T, Merianda TT, Sylvester J, van Minnen J, Twiss JL. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–791. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis DE, van Niekerk EA, Sasaki Y, Mesngon M, Merianda TT, Williams GG, Kendall M, Smith DS, Bassell GJ, Twiss JL. Extracellular stimuli specifically regulate localized levels of individual neuronal mRNAs. J Cell Biol. 2007;178:965–980. doi: 10.1083/jcb.200703209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AL, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature. 2014 doi: 10.1038/nature13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. Central sensitization: uncovering the relation between pain and plasticity. Anesthesiology. 2007;106:864–867. doi: 10.1097/01.anes.0000264769.87038.55. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Walters ET. Common patterns of plasticity contributing to nociceptive sensitization in mammals and Aplysia. Trends in neurosciences. 1991;14:74–78. doi: 10.1016/0166-2236(91)90024-o. [DOI] [PubMed] [Google Scholar]
- Zhao J, Seereeram A, Nassar MA, Levato A, Pezet S, Hathaway G, Morenilla-Palao C, Stirling C, Fitzgerald M, McMahon SB, Rios M, Wood JN. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci. 2006;31:539–548. doi: 10.1016/j.mcn.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Zhou LJ, Zhong Y, Ren WJ, Li YY, Zhang T, Liu XG. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Exp Neurol. 2008;212:507–514. doi: 10.1016/j.expneurol.2008.04.034. [DOI] [PubMed] [Google Scholar]