

Abstract
2-O-Propargyl ethers are shown to be advantageous in the 4,6-O-benzylidene acetal directed β-mannosylation reaction. The effect is most pronounced when the 3-O-protecting group is bulky silyl ether or a glycosidic bond, however, even with a 3-O-benzyl ether the use of a 2-O-propargyl ether results in a significant increase in diastereoselectivity. The beneficial effect of the propargyl ether is thought to be a combination of its minimal steric bulk, as determined by measurement of the steric A-value, and of its moderately disarming nature, as reflected in the pKa of propargyl alcohol. Conversely, the application of a 3-O-propargyl ether in the benzylidene acetal directed mannosylation has a detrimental effect on stereoselectivity, for which no explanation is at present available. Deprotection is achieved by base-catalyzed isomerization of the propargyl ether group to the corresponding allenyl ether, followed by oxidative cleavage with N-methylmorpholine N-oxide and catalytic osmium tetroxide.
Graphical Abstract
Introduction
Protecting groups play a central role in carbohydrate chemistry,1 with applications extending beyond the simple blocking of hydroxyl groups to the modulation of reactivity of both glycosyl donors and acceptors, and, critically, the control of anomeric stereochemistry. Indeed, the development of new protecting groups capable of rendering enhanced control of regioselectivity,2 reactivity,3 and stereoselectivity,4 can be said to be one of the current frontiers of the discipline.
The influence of even remote protecting groups on the control of anomeric stereochemistry is illustrated by the 4,6-O-benzylidene protected β-mannosyl donors developed in this laboratory,5 in which the benzylidene acetal, or its surrogate,4d,6 is now understood to function by restricting the C5-C6 bond to the more disarming7 tg conformer,8,9 thereby limiting the lifetime of the transient contact ion pair10 that is in equilibrium with the covalent glycosyl triflate intermediate.5c,11
However powerful this method may be in the synthesis of complex oligosaccharides containing the β-mannopyranoside and related linkages,12,13 it is not without limitations. Thus, the use of donors bearing bulky groups on O-3, either silyl ethers or glycosidic bonds, diminishes the selectivity of the mannosylation.14
Although the effect of the O-3 protecting group on anomeric stereoselectivity is not yet fully understood, we introduced the use of 2-O-propargyl ether as a means of overcoming the loss of selectivity as a result of the use of bulky groups at O-3.15 In this Article we examine in greater detail the potential of the readily cleavable, minimally sterically intrusive propargyl ether protecting group and show how, in conjunction with the correct choice of other protecting groups, it can lead to considerable enhancements in the stereoselectivity of mannopyranosylation reactions and even the very challenging rhamnopyranosylations.
Results and Discussion
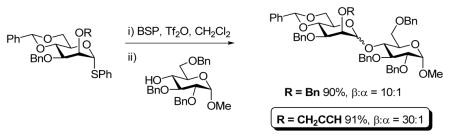
The problem of diminished selectivity caused by bulky groups on O-3 was initially encountered in the synthesis of the common core pentasaccharide of the N-linked glycans,13a when coupling of the 2-O-benzyl-3-O-TBDMS mannosyl donor 2 with pentenyl glycoside acceptor 1 exhibited poor selectivity (77%, α:β = 1.8:1). In contrast, with the 2-O-TBDMS-3-O-benzyl donor 3, the selectivity was significantly better (72%, α:β = 1:3), albeit still not at the high levels typically experienced with more standard 2,3-di-O-benzyl protected donors.16
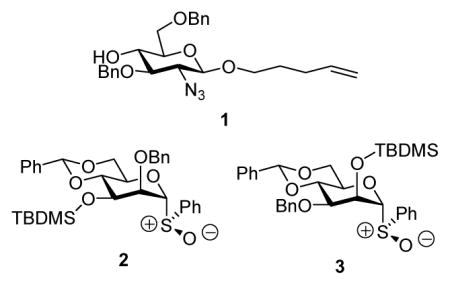
A more critical manifestation of this problem presented itself during the synthesis of the alternating β-(1→3)-β-(1→4)-mannan common to Rhodotorula glutinis, Rhodotorula mucilaginosa and Leptospira biflexa.14b Donors 4 and 5, both displaying very bulky glycosyl substituents on O-3, showed unusually poor β-selectivity in coupling reactions, thereby reducing the efficiency of the convergent synthesis of the target polysaccharide.
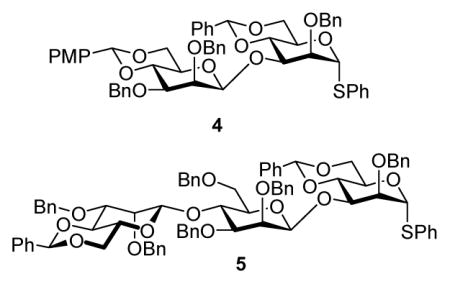
We hypothesized that the poor selectivity seen with donors 2, 4, and 5 was the result of steric buttressing between the O-2 and O-3 protecting groups, resulting in unusually high shielding of the β-face of the glycosyl donor.14a,15 Thus, as illustrated for the triflate derived from 2, we reason that of the three possible staggered conformations around the O-3-substituent bond, (A) is disfavored by the steric interaction with the rigid benzylidene ring leading to the preferential population of conformers (B) and (C) in which the bulky silyl group is gauche to C-2 and its substituent (Fig. 1).
Figure 1.
Staggered Conformations about the C3-O3 Bond
Viewed from the perspective of the O-2-substituent bond, the population of conformer (D) is likely extremely small due to high steric congestion. The bulky group on O-3 presumably destabilizes conformation (E) thus leaving (F) as the most populous state (Fig. 2). In conformer F the 2-O-benzyl ether is in close proximity to the β-face of the α-mannosyl triflate. This enhanced steric shielding retards attack on the β-face, either on the covalent triflate itself or on the transient contact ion pair arising from the covalent triflate, thereby resulting in the observed loss of β-selectivity.
Figure 2.
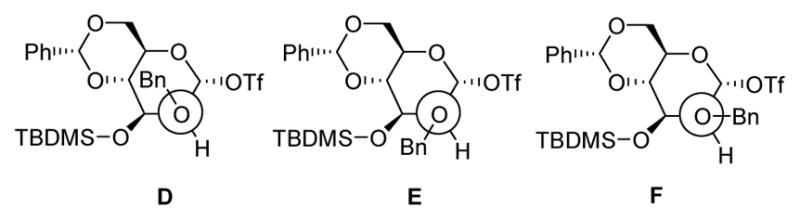
Staggered Conformations about the C2-O2 Bond
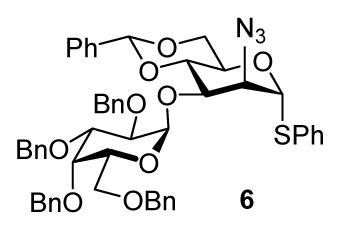
In systems such as 2 this problem can be circumvented by the simple ruse of switching to a less bulky O-3 protecting group, however in target-directed convergent oligosaccharide synthesis there is no way to avoid the use of donors such as 4 and 5. We reasoned that the unfavorable steric interaction in conformer E could be reduced by minimizing the size of the O-2 protecting group, which should have the effect of increasing the population of E at the expense of F. At the same time, the use of a protecting group with a low steric demand on O-2, should serve to minimize the detrimental effect of any residual population of conformer F. We were encouraged in this line of thinking, by van Boom’s work on the successful β-glycosylation of several acceptors by donor 6 with the relatively small 2-azido group.17 However, the size of the azido group cannot be viewed independently of its strongly disarming properties, thereby complicating the interpretation of this precedent. For similar reasons, we decided not to pursue the very small but also moderately disarming cyanate esters,4e and focused instead on the allyl and propargyl ethers.
We began with the synthesis of the 3-O-silyl compounds 10 and 13 (Scheme 1) by standard means from the known thioglycoside 7.14b In these syntheses the 3-O-silyl group was introduced after the allyl or propargyl ethers to preempt problems of silyl migration that were anticipated in the reverse protocol.
Scheme 1.
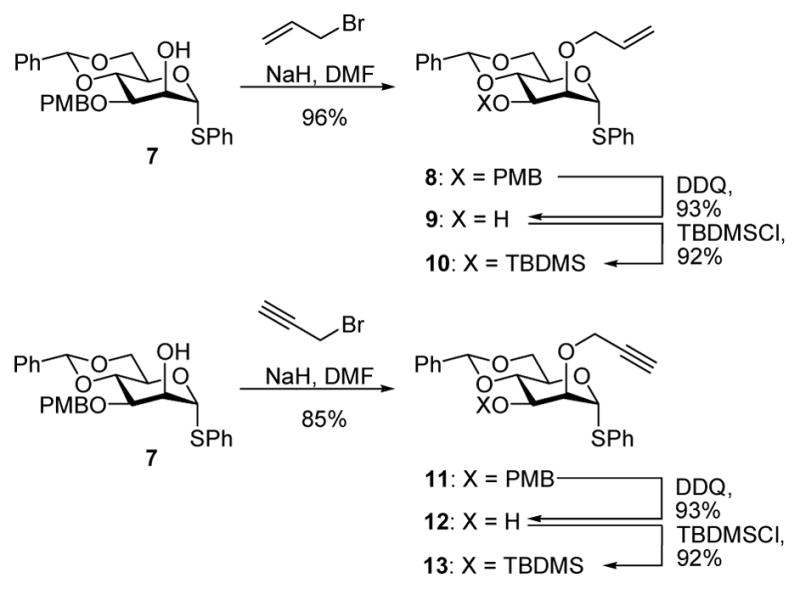
Synthesis of Donors 10 and 13
Donors 10 and 13 were then coupled to the acceptor 14 by our standard BSP/TTBP/Tf2O protocol5c,16 leading to the yields and selectivities outlined in Table 1. Included in Table 1 for comparison is the previous coupling14a of donor 2 to acceptor 1, by the directly analogous sulfoxide method.16
Table 1.
Influence of the O2 Protecting Group on Selectivity

|

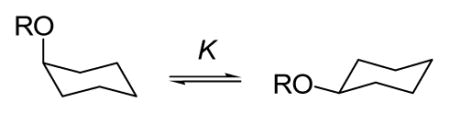
These results strongly support the hypothesis of the beneficial effect of reducing the bulk of the 2-O protecting group on the stereochemical outcome of the reaction, with the best anomeric ratio obtained with the smallest O-2 protecting group. To put the inverse relationship between the steric bulk of the O-2 protecting group and anomeric selectivity on a more secure footing we measured steric A-values for the propargyloxy, allyloxy, benzyloxy, and tert-butyldimethylsiloxy groups by the classical 1H VT-NMR method (Table 2).18 The observed trend in A-values fully supports the initial hypothesis, with the propargyl ether being significantly smaller than the allyl ether, which in turn is smaller than the benzyl ether. The A-value for the tert-butyldimethylsiloxy group determined here, and included for comparison purposes, is significantly greater than that previously measured by Eliel for the same group using an alternative 13C-NMR method,19 but is consistent with the general trend of coupling selectivities observed in this entire study.
Table 2.
Steric A-Values and pKa’s
| ||||
|---|---|---|---|---|
| A-Values, R’ = C6H11 (18–21) | pKa (R’ = H) | |||
| Tempa (K) | Kb | Ac | ||
|
|
193 | 17.7 | 1.10 | 13.6 |
|
|
193 | 26.2 | 1.25 | 15.5 |
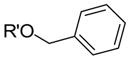
|
203 | 31.5 | 1.39 | 15.4 |
| R’O TBDMS | 193 | 49.4 | 1.50 | - |
) Measurement temp.
) Equilbrium const.
) A = RTlnK
In addition to the smaller size of the propargyl ether we also considered the possibility that it might exhibit an electron-withdrawing effect. Indeed, the sp-hybridization of the alkyne carbon renders the propargyloxy group moderately electron-withdrawing with respect to the other ethers studied, as seen from the pKa’s of the corresponding alcohols (Table 2),20 and it is likely that the beneficial effect of the 2-O-propargyl ethers arises from a combination of the minimal steric bulk and its moderately disarming property.
The coupling of donor 13 to two further substrates, again with excellent results (Table 3), confirmed the ability of the 2-O-propargyl ether protecting group to overcome the deleterious effects of a 3-O-silyl ether.
Table 3.
Further Couplings to Donor 13

|
Attention was next focused on donors bearing a glycosidic bond at O-3, analogous to the problematic 4 and 5. Furthermore, bearing in mind the potential for the eventual use in mannan synthesis, a glycosyl acceptor carrying a 2-O-propargyl ether was also prepared (Scheme 2).
Scheme 2.
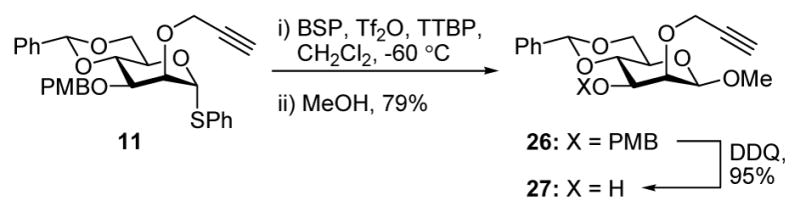
Synthesis of Acceptor 27
Acceptor 12 was successfully coupled to the known donor 285c with α:β selectivity of 1:16 in 88% yield (Scheme 3). Per the protocol of van Boom,14b,17,21 triethyl phosphate was added after addition of the acceptor 12 to limit premature activation its thioglycoside functionality by any extraneous thiophiles. Coupling of disaccharide donor 29 to acceptor 27 then gave the mannotriose in 80% yield with an α:β ratio of 1:5, presenting a very significant improvement over the approximately 1:1 α:β ratio observed with donor 4 and a related acceptor.14b Additionally, the successful couplings employing compounds 12 and 27 illustrate that propargyl ethers are also suitable for protection of acceptors.
Scheme 3.
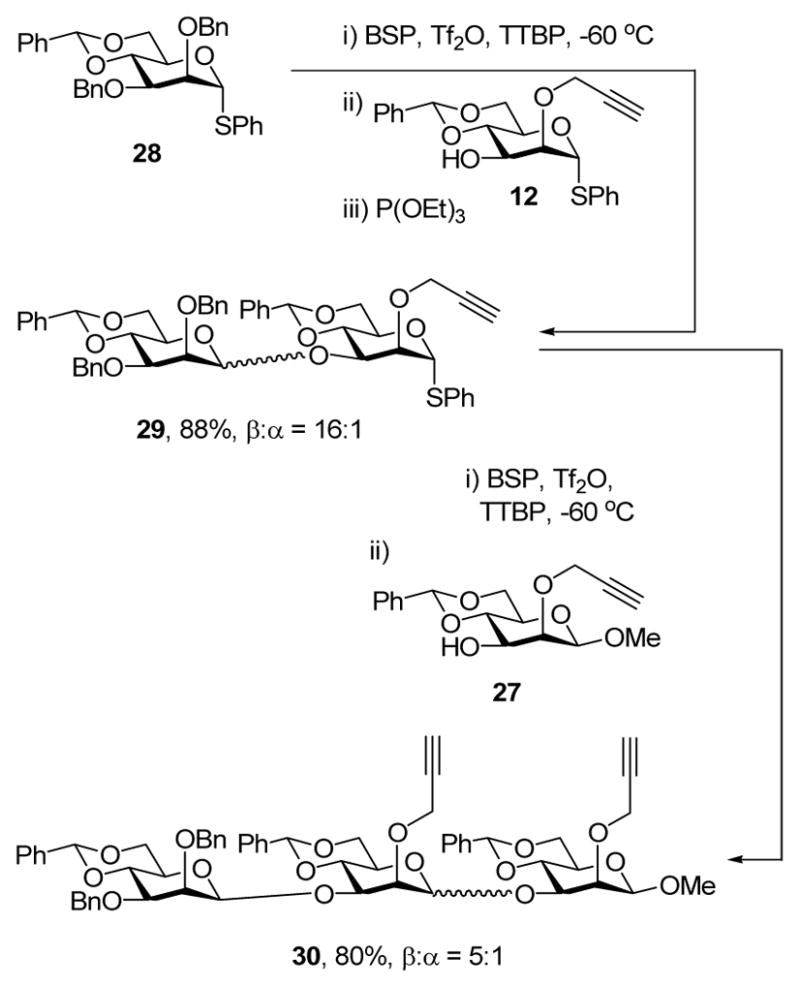
Synthesis of a Mannotriose using 2-O-Propargyl Ethers
With a means to overcome the unfavorable effect of a bulky 3-O substituent in hand, we proceeded to undertake a broader investigation into the general effects of propargyl ethers on stereoselectivity in 4,6-O-benzylidene-directed β-mannosylation reactions. Specifically, we reasoned that while the steric buttressing effect discussed above and illustrated in Figs 1 and 3 will be maximized with a large group on O-3, it will necessarily be present with more common protecting groups on O-3, albeit to a lesser extent. Accordingly, the use of a 2-O-propargyl even in conjunction with a 3-O-benzyl ether should lead to enhanced selectivity over the more typical 2,3-di-O-benzyl protected donors. To probe this idea, a series of four donors was prepared, using standard techniques from diol 3114b via the known monobenzyl ethers 32 and 3414b (Scheme 4).
Scheme 4.
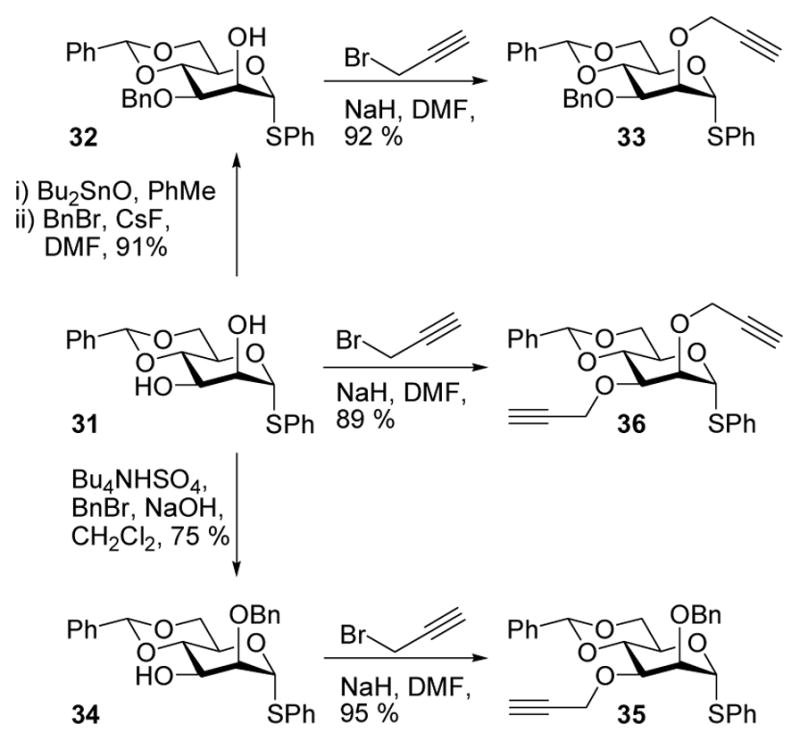
Synthesis of Donors 33, 35, and 36
Subsequent coupling of this series of donors to a standard acceptor 14 gave the results presented in Table 4. Comparison of entries 1 and 2 in Table 4 clearly demonstrates a 2-O-propargyl ether lead to enhanced β-selectivity even with the 3-O-benzyl protected system, in accordance with the above stated hypothesis. The 3-O-propargyl donor 35 (Table 4, entry 3) gave surprisingly poor but reproducible results, for which we have no satisfactory explanation at the present time. It is clear, however, that the O-3 group plays a major role in these 4,6-O-benzylidene protected β-mannosylation reactions, and that the issue of steric bulk and buttressing discussed here is only one facet of the problem.22 Taking into account the selectivities obtained with donors 33 and 35, it is clear that the modest 10:1 β:α selectivity obtained with the 2,3-di-O-propargyl protected donor 36 (Table 4, entry 4) is a compromise between the excellent selectivity obtained with a 2-O-propargyl group alone, and the obviously harmful effect of the 3-O-propargyl ether.
Table 4.
Coupling of Mono and Di-O-proparyl Protected Donors to 14

|
The very encouraging results obtained with donor 33, featuring the combination of the 2-O-propargyl and 3-O-benzyl ether protecting groups were then extended to encompass a broader range of typical acceptor alcohols (Table 5). In each excellent yields and β:α selectivies surpassing 20:1 were obtained.
Table 5.
Coupling of 33 to Further Acceptors

|
While the use of allyl ethers as protecting groups is extremely widespread,1a, 23 that of propargyl ethers is novel, and required the investigation of suitable deprotection conditions. It has been reported that propargyl ethers may be cleaved with benzyltriethylammonium tetrathiomolybdate, 24 with low valent titanium in hot THF, 25 and by a nickel-catalyzed electrochemical protocol.26 However, based on experience in our laboratory with allyl ethers in oligosaccharide synthesis,12c,d, 27 we have preferred a method involving base-catalyzed isomerization to the corresponding allenyl ether followed by oxidative cleavage with catalytic osmium tetroxide in the presence of N-methyl morpholine N-oxide (NMNO) as reported by Mereyala and co-workers,28 albeit under somewhat milder conditions. Thus, a representative series of propargyl ether protected saccharides was treated with potassium tert-butoxide in THF at room temperature, followed by exposure to catalytic OsO4 in the presence of NMNO, also at room temperature, resulting in hydrolysis to the corresponding alcohols (Table 6).
Table 6.
Cleavage of Propargyl Ethers.

|
Finally, we have briefly investigated the potential of the 2-O-propargyl ether protecting group in the synthesis of β-L-rhamnopyranosides, a cognate problem to that of the β-D-mannopyranosides but one which does not allow the use of the stereodirecting 4,6-O-benzylidene acetal function in the donor. As part of our ongoing effort in this area,4e,29 we reported that the 3,4-O-carbonate protected rhamnosyl donor 60 gave moderate to good β:α selectivity (1.5:1 to β only) on coupling to various acceptors under the standard BSP/Tf2O/TTBP conditions, depending on the reactivity of the acceptor.4f
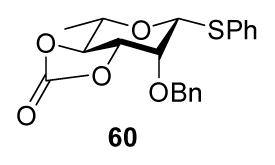
It was reasonable, therefore, to investigate the analogous 2-O-carbonate 64, which was prepared as set out in Scheme 5 from the known4f bisacetal 61.
Scheme 5.
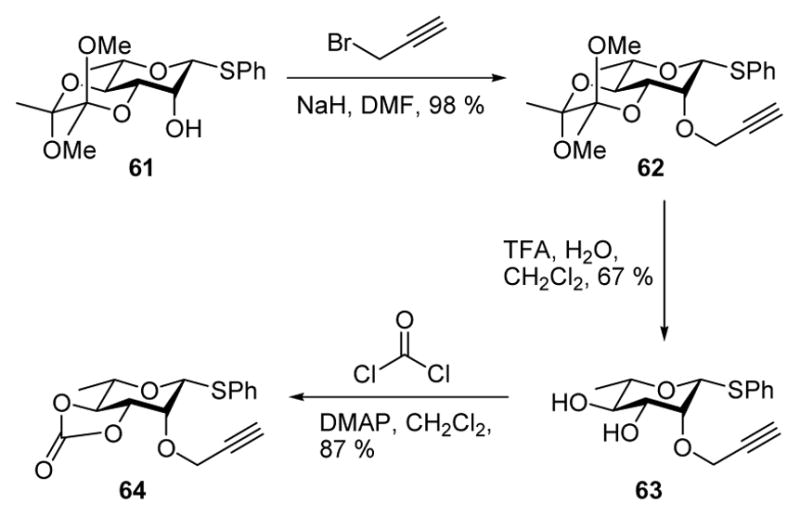
Preparation of Rhamnosyl Donor 64.
Activation of 64, which proceeded smoothly under the standard conditions, was followed by the addition of methyl 2,3,6-tri-O-benzyl-α-D-glucopyranoside 14, a member of the glucose 4-OH derivatives which are known to be relatively difficult to glycosylate, 30 giving the disaccharide 65 in 65% yield in the form of a 2:1 β:α mixture (Scheme 6). This represents only a modest improvement of selectivity over the 1.5:1 β:α ratio obtained on coupling of 60 with 14,4f and discouraged us from further work with this donor. Presumably, the there is very little buttressing interaction between the tied back carbonate and the protecting group on O-2, as such the effect on stereoselectivity of changing the O-2 protecting from the benzyl ether to the propargyl ether is very small.
Scheme 6.
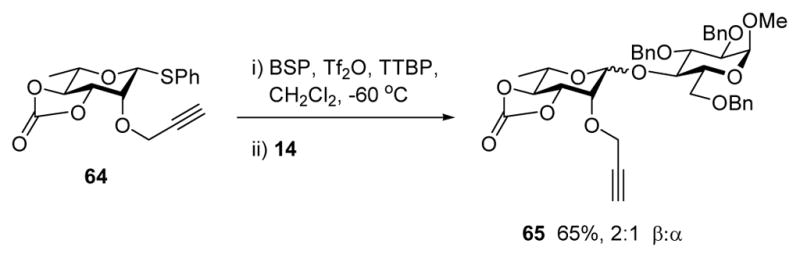
Glycosylation in the Rhamnopyranose Series
Overall, propargyl ethers are readily introduced and cleaved protecting groups for alcohols that bring about significant improvements in the diastereoselectivity of many mannosylation reactions, which we attribute to the combination of their minimal steric bulk and their modest disarming power. While we have focused on the application of this protecting group to the solution of current problems in our laboratory, we anticipate that it will find broader application in organic synthesis, especially in situations in which the steric bulk of a protecting group is a factor.
Experimental Section
Phenyl 4,6-O-benzylidene-2-O-(prop-2-ynyl)-3-O-p-methoxybenzyl-1-thio-α-D-mannopyranoside (11)
To a stirred solution of phenyl 4,6-O-benzylidene-3-O-p-methoxybenzyl-1-thio-α-D-mannopyranoside (2.5 g, 5.5 mmol) in dry dimethylformamide (15 mL) at 0 ºC was added NaH 60% in oil (0.33 g, 8.3 mmol) and stirred for 15 min. Propargyl bromide (0.93 mL, 8.3 mmol) was added dropwise to the above reaction mixture and continued stirring for 3 h. The rection mixture was quenched by addition of methanol, diluted with CH2Cl2 (25 mL) and washed with sat. NaHCO3. The organic layer was separated and dried over anhydrous Na2SO4 and concentrated under vaccum. The crude product was purified by flash column cromatography on silica gel (hexane:ethyl acetate; 8:1) to give 11 (2.46 g, 85%), [α]24.5D + 155.8 (c, 2.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 2.4 (t, J = 2.4 Hz, 1H), 3.82 (s, 3H), 3.87 (t, J = 11.0 Hz, 1H), 3.98 (dd, J = 3.0, 10.0 Hz, 1H), 4.19–4.24 (m, 3H), 4.26–4.31 (m, 1H), 4.4 (dd, J = 0.5, 2.0 Hz, 2H), 4.70 (d, J = 12.0 Hz, 1H), 4.81 (d, J = 12.0 Hz, 1H), 5.61 (d, J = 1.5 Hz, 1H), 5.63 (s, 1H), 6.9 (d, J = 8.6 Hz, 2H), 7.3–7.45 (m, 10H), 7.5–7.56 (m, 2H); 13C NMR (125 MHz, CDCl3) δ: 55.3, 58.8, 65.4, 68.5, 72.9, 75.2, 75.7, 77.6, 79.0, 79.4, 87.4, 101.5, 113.8, 126.1, 127.6, 128.2, 128.8, 129.1, 129.2, 129.4, 130.2, 131.6, 133.7, 134.5, 137.5, 159.3 ESIHRMS Calcd for C30H30O6S [M+Na]+: 541.1661. Found 541.1658.
Phenyl 4,6-O-benzylidene-2-O-(prop-2-ynyl)-1-thio-α-D-mannopyranoside (12)
To stirred solution of 11 (0.47 g, 0.91 mmol) in CH2Cl2 (8 mL) and water (0.4 mL) was added DDQ (0.3 g, 1.3 mmol) at room temp. After 3 h, sat. NaHCO3 was added, and the mixture was extracted with CH2Cl2. The extract was washed several times with sat NaHCO3, and dried over Na2SO4. Evaporation of the solvent in vacuo gave an oil, which was chromatographed on a flash silica gel column (hexane:ethyl acetate; 4:1) to give 12 (0.34 g, 93%) as a white solid MP 128 ºC [α]27D + 119 (c, 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 2.49 (t, J = 2.4 Hz, 1H), 2.5 (bs, 1H), 3.84 (t, J = 10.2 Hz, 1H), 3.9 (t, J = 9.6 Hz, 1H), 4.16 (dd, J = 3.6, 10.0 Hz, 1H), 4.21–4.24 (m, 2H), 4.27–4.32 (m, 1H), 4.34 (dd, J = 2.4, 16.1 Hz, 1H), 4.42 (dd, J = 2.4, 16.1 Hz, 1H), 5.59 (s, 1H), 5.68 (s, 1H), 7.32–7.53 (m, 10H); 13C NMR (125 MHz, CDCl3) δ: 58.6, 64.7, 68.4, 68.9, 75.7, 78.9, 79.3, 79.4, 86.4, 102.2, 126.3, 127.7, 128.3, 129.2, 131.7, 133.8, 137.2. ESIHRMS Calcd for C22H22O5S [M+Na]+: 421.1086. Found 421.1095.
Phenyl 4,6-O-benzylidene-2-O-(prop-2-ynyl)-3-O-(2,3-di-O-benzyl-4,6-O-benzylidene-β-D-mannopyranosyl)-1-thio-α-D-mannopyranoside 29β and the α-anomer 29α
To a stirred solution of donor 28 (480 mg, 0.88 mmol), BSP (223 mg 1.06 mmol), TTBP (331 mg, 1.33 mmol), and 4 Å molecular sieves in CH2Cl2 (5 mL), at −60 ºC under an Ar atmosphere, was added Tf2O (195 μL 1.15 mmol). After 30 min. the temperature was brought down to −78 ºC, and then acceptor 12 (424 mg 1.06 mmol) in CH2Cl2 (3 mL), was slowly added. The reaction mixture was stirred for 2h. at −78 ºC, and quenched by the addition of triethylphosphite (435 μL, 2.7 mmol), and continued stirring for 1 h at −78 ºC and allowed to reached room temperature. The reaction mixture was diluted with CH2Cl2 (10 mL) and molecular sieves were filtered off and washed with saturated NaHCO3. The organic layer was separated, dried and concentrated. The crude was purified by radial chromatography (hexane:ethyl acetate; 8:1) to give 29β and 29α in 83% and 5% yield respectively. 29β: [α]24D + 26.3 (c, 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ : 2.23 (t, J = 2.4 Hz, 1H), 3.29–3.34 (m, 1H), 3.6 (dd, J = 3.2, 9.7 Hz, 1H), 3.86 (t, J = 10.3 Hz, 1H), 3.93 (t, J = 10.3 Hz, 1H), 4.0 (d, J = 3.0 Hz, 1H), 4.13 (t, J = 9.7 Hz, 1H), 4.25–4.40 (m, 8H), 4.65 (d, J = 12.5, Hz, 1H), 4.76 (d, J = 12.5 Hz, 1H), 4.84 (s, 1H), 4.86 (d, J = 11.9 Hz, 1H), 4.98 (d, J = 11.8 Hz, 1H), 5.58 (s, 1H), 5.62 (s, 1H), 5.64 (s, 1H), 7.24–7.49 (m, 25H); 13C NMR (125 MHz, CDCl3) δ: 57.5, 65.3, 67.8, 68.5, 68.6, 72.3, 73.4, 74.8, 75.6, 75.7, 76.5, 77.5, 77.6, 78.6, 79.0, 86.0, 98.9, 101.3, 101.9, 126.0, 126.2, 127.4, 127.6, 127.7, 127.8, 128.0, 128.1, 128.2, 128.3, 128.4, 128.8, 129.0, 129.2, 131.6, 133.6, 137.3, 137.6, 138.4, 138.7. ESIHRMS Calcd for C49H48O10S [M+Na]+: 851.2866. Found 851.2875. 29α: [α]24D + 76.4 (c, 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ : 2.4 (t, J = 2.4 Hz, 1H), 3.8 (t, J = 10.5 Hz, 1H), 3.92–3.96 (m, 2H), 3.99–4.04 (m, 2H), 4.15 (t, J = 9.5 Hz, 1H), 4.2 (dd, J = 2.5, 16.1 Hz, 1H), 4.25–4.38 (m, 7H), 4.5 (d, J = 12.4 Hz, 1H), 4.62 (d, J = 12.4 Hz, 1H), 4.63 (d, J = 12.2 Hz, 1H), 4.7 (d, J = 12.2 Hz, 1H), 5.4 (d, J = 1.2 Hz, 1H), 5.57 (s, 1H), 5.59 (s, 1H), 5.67 (s, 1H), 7.15–7.52 (m, 25H); 13C NMR (125 MHz, CDCl3) δ: 58.3, 64.7, 65.1, 68.5, 68.8, 72.1, 72.7, 72.8, 75.3, 75.7, 78.5, 79.0, 79.2, 86.9, 99.6, 101.4, 101.9, 125.9, 126.1, 127.5, 127.6, 127.7, 127.8, 128.2, 128.4, 128.8, 129.2, 129.3, 131.6, 133.5, 137.3, 137.7, 137.8, 138.5. ESIHRMS Calcd for C49H48O10S [M+Na]+: 851.2866. Found 851.2874.
General procedure for deprotection of propargyl ethers
To a stirred solution of propargyl ether (1 mmol) in dry THF (5 mL) was added KOtBu (1.1 mmol), stirring was continued at room temperature for 3–12 h untill TLC indicated completion. The reaction mixture was diluted with CH2Cl2 (10 mL). The organic phase was separated, washed with water, dried (Na2SO4) and concentrated on a rotary evaporator to give the allenyl ethers in quantitative yields. A homogeneous solution of allenyl ethers (1 mmol) in acetone: water (4:1, 5 mL) was treated with OsO4 (0.1 mmol) and N-methyl morpholine N-oxide (2 mmol) and the mixture was stirred for 3 h at room temperature. After completion of the reaction, acetone was removed under vaccum and the residue was dissolved in CH2Cl2 (10 mL) and washed with sat. NaHSO3. The organic phase was separated, dried (Na2SO4), and concentrated on a rotary evaporator. The residues were purified by flash or radial chromatography on silica gel to yield deprotected di and trisaccharides in 80–91%.
Supplementary Material
Acknowledgments
We thank the NIH (GM57335) for support of our work in this area.
Footnotes
Supporting Information Available. Full experimental details and copies of nmr spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.a) Grindley TB. In: In Modern Methods in Carbohydrate Chemistry. Khan SH, O’Neill RA, editors. Harwood Academic; Amsterdam: 1996. pp. 225–250. [Google Scholar]; b) Green LG, Ley SV. In: In Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Wiley-VCH; Weinheim: 2000. pp. 427–448. [Google Scholar]; c) Fraser-Reid B, López JC, Gómez AM, Uriel C. Eur J Org Chem. 2004:1387–1395. [Google Scholar]
- 2.Ley SV, Baeschlin DK, Dixon DJ, Foster AC, Ince SJ, Priepke HWM, Reynolds DJ. Chem Rev. 2001;101:53–80. doi: 10.1021/cr990101j. [DOI] [PubMed] [Google Scholar]
- 3.a) Glaudemans CPJ, Fletcher HG. J Am Chem Soc. 1965;87:4636–4641. [Google Scholar]; b) Ishikawa T, Fletcher HG. J Org Chem. 1969;34:563–571. doi: 10.1021/jo01255a018. [DOI] [PubMed] [Google Scholar]; c) Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5584. [Google Scholar]; d) Fraser-Reid B, Madsen R. In: In Preparative Carbohydrate Chemistry. Hanessian S, editor. Dekker; New York: 1997. pp. 339–356. [Google Scholar]; e) Fraser-Reid B, Anilkumar G, Gilbert MR, Joshi S, Kraehmer R. In: In Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Wiley-VCH; Weinheim: 2000. pp. 135–154. [Google Scholar]; f) Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc Perkin Trans. 1998;1:51–65. [Google Scholar]; g) Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 4.a) Crich D, Vinod AU. J Org Chem. 2005;70:1291–1296. doi: 10.1021/jo0482559. [DOI] [PubMed] [Google Scholar]; b) Crich D, Dudkin V. J Am Chem Soc. 2001;121:6819–6825. doi: 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]; c) Crich D, Jayalath P. J Org Chem. 2005;70:7252–7259. doi: 10.1021/jo0508999. [DOI] [PubMed] [Google Scholar]; d) Crich D, Yao Q. J Am Chem Soc. 2004;126:8232–8236. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]; e) Crich D, Hutton TK, Banerjee A, Jayalath P, Picione J. Tetrahedron: Asym. 2005;16:105–119. [Google Scholar]; f) Crich D, Vinod AU, Picione J. J Org Chem. 2003;68:8453–8458. doi: 10.1021/jo035003j. [DOI] [PubMed] [Google Scholar]; g) Crich D, Vinod AU, Picione J, Wink DJ. ARKIVOC. 2005;(vi):339–344. [PMC free article] [PubMed] [Google Scholar]; h) Kim JH, Yang H, Boons GJ. Angew Chem Int Ed. 2005;44:947–949. doi: 10.1002/anie.200461745. [DOI] [PubMed] [Google Scholar]; i) Kim JH, Yang H, Park J, Boons GJ. J Am Chem Soc. 2005;127:12090–12097. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]; j) Smoot JT, Pornsuriyasak P, Demchenko AV. Angew Chem Int Ed. 2005;44:7123–7126. doi: 10.1002/anie.200502694. [DOI] [PubMed] [Google Scholar]; k) Bowers SG, Coe DM, Boons GJ. J Org Chem. 1998;63:4570–4571. [Google Scholar]; l) Jiao H, Hindsgaul O. Angew Chem Int Ed. 1999;38:346–348. doi: 10.1002/(SICI)1521-3773(19990201)38:3<346::AID-ANIE346>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; m) Debenham J, Rodebaugh R, Fraser-Reid B. Liebigs Ann/Recueil. 1997:791–802. [Google Scholar]; n) Yu H, Williams DL, Ensley HE. Tetrahedron Lett. 2005;46:3417–3421. doi: 10.1016/j.tetlet.2005.03.099. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Chéry F, Rollin P, De Lucchi O, Cossu S. Synthesis. 2003:286–292. [Google Scholar]; p) Imamura A, Ando H, Korogi S, Tanabe G, Muraoka O, Ishidaa H, Kiso M. Tetrahedron Lett. 2003;44:6725–6728. [Google Scholar]; q) Haberman JM, Gin DY. Org Lett. 2001;3:1665–1668. doi: 10.1021/ol015854i. [DOI] [PubMed] [Google Scholar]; r) Wei P, Kerns RJ. J Org Chem. 2005;70:4195–4198. doi: 10.1021/jo047812o. [DOI] [PubMed] [Google Scholar]; s) Benakli K, Zha C, Kerns RJ. J Am Chem Soc. 2001;123:9461–9462. doi: 10.1021/ja0162109. [DOI] [PubMed] [Google Scholar]
- 5.a) Crich D, Sun S. J Org Chem. 1997;62:1198–1199. [Google Scholar]; b) Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]; c) Crich D, Smith M. J Am Chem Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]; d) Crich D. In: In Glycochemistry: Principles, Synthesis, and Applications. Wang PG, Bertozzi CR, editors. Dekker; New York: 2001. pp. 53–75. [Google Scholar]; e) Crich D. J Carbohydr Chem. 2002;21:663–686. [Google Scholar]
- 6.a) Crich D, Smith M. J Am Chem Soc. 2002;124:8867–8869. doi: 10.1021/ja011406u. [DOI] [PubMed] [Google Scholar]; b) Crich D, de la Mora M, Vinod AU. J Org Chem. 2003;68:8142–8148. doi: 10.1021/jo0349882. [DOI] [PubMed] [Google Scholar]
- 7.In the terminology of Fraser-Reid a disarming protecting group is one that deactivates a glycosyl donor, whereas an arming protecting group is one that activates a glycosyl donor.1b,3c
- 8.Jensen HH, Nordstrom M, Bols M. J Am Chem Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 9.For earlier studies on the disarming influence of acetal protecting groups see: Andrews CW, Rodebaugh R, Fraser-Reid B. J Org Chem. 1996;61:5280–5289. doi: 10.1021/jo961115h.Fraser-Reid B, Wu ZC, Andrews W, Skowronski E. J Am Chem Soc. 1991;113:1434–1435.
- 10.Crich D, Chandrasekera NS. Angew Chem Int Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 11.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 12.for examples from this laboratory see: ref 4d and Crich D, Li H, Yao Q, Wink DJ, Sommer RD, Rheingold AL. J Am Chem Soc. 2001;121:5826–5828. doi: 10.1021/ja015985e.Crich D, Li H. J Org Chem. 2002;67:4640–4646. doi: 10.1021/jo0108818.Crich D, Dai Z. Tetrahedron. 1999;55:1569–1580.Crich D, Barba GR. Tetrahedron Lett. 1998;39:9339–9342.Crich D, de la Mora MA, Cruz R. Tetrahedron. 2002;58:35–44.Crich D, Banerjee A, Yao Q. J Am Chem Soc. 2004;126:14930–14934. doi: 10.1021/ja047194t.Crich D, Banerjee A. Org Lett. 2005;7:1935–1938. doi: 10.1021/ol050224s.Crich D, Dudkin V. J Am Chem Soc. 2002;124:2263–2266. doi: 10.1021/ja0123958.Dudkin VY, Crich D. Tetrahedron Lett. 2003;44:1787–1789.
- 13.For examples from other laboratories see: Dudkin VK, Miller JS, Danishefsky SJ. J Am Chem Soc. 2004;126:736–738. doi: 10.1021/ja037988s.Miller JS, Dudkin VY, Lyon GJ, Muir TW, Danishefsky SJ. Angew Chem Int Ed. 2003;42:431–434. doi: 10.1002/anie.200390131.Nicolaou KC, Mitchell HJ, Rodriguez RM, Fylaktakidou KC, Suzuki H, Conley SR. Chem Eur J. 2000;6:3149–3165. doi: 10.1002/1521-3765(20000901)6:17<3149::aid-chem3149>3.0.co;2-l.Kim KS, Kang SS, Seo YS, Kim HJ, Jeong KS. Synlett. 2003:1311–1314.Wu X, Schmidt RR. J Org Chem. 2004;69:1853–1857. doi: 10.1021/jo0354239.Kwon YT, Lee YJ, Lee K, Kim KS. Org Lett. 2004;6:3901–3904. doi: 10.1021/ol048648u.Tanaka S-i, Takashina M, Tokimoto H, Fujimoto Y, Tanaka K, Fukase K. Synlett. 2005:2325–2328.
- 14.a) Crich D, Dudkin V. Tetrahedron Lett. 2000;41:5643–5646. [Google Scholar]; b) Crich D, Li W, Li H. J Am Chem Soc. 2004;126:15081–15086. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]; c) Codée JDC, Hossain LH, Seeberger PH. Org Lett. 2005;7:3251–3254. doi: 10.1021/ol051038p. [DOI] [PubMed] [Google Scholar]
- 15.Crich D, Jayalath P. Org Lett. 2005;7:2277–2280. doi: 10.1021/ol050680g. [DOI] [PubMed] [Google Scholar]
- 16.Crich D, Lim LBL. Org React. 2004;64:115–251. [Google Scholar]
- 17.a) Codée JDC, Litjens REJN, den Heeten R, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1519–1522. doi: 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]; b) Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Tetrahedron. 2004;60:1057–1064. [Google Scholar]
- 18.Jensen FR, Bushweller CH, Beck BH. J Am Chem Soc. 1969;91:344–351. [Google Scholar]
- 19.Eliel EL, Satici H. J Org Chem. 1994;59:688–689. [Google Scholar]
- 20.Dictionary of Organic Compounds. Chapman and Hall; London: 1996. [Google Scholar]
- 21.a) Codée JDC, van den Bos J, Litjens REJN, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1947–1950. doi: 10.1021/ol034528v. [DOI] [PubMed] [Google Scholar]; b) Sliedregt LAJM, van der Marel GA, van Boom JH. Tetrahedron Lett. 1994;35:4015–4018. [Google Scholar]; c) Alonso I, Khiar N, Martin-Lomas M. Tetrahedron Lett. 1996;37:1477–1480. [Google Scholar]
- 22.For example, we have previously discussed the highly α-selective couplings obtained in the presence of a 3-O-carboxylate ester. The origin of these effects is currently under active investigation in our laboratory. See ref 4d and Crich D, Cai W, Dai Z. J Org Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482.
- 23.a) Guibé F. Tetrahedron. 1997;53:13509–13556. [Google Scholar]; b) Guibé F. Tetrahedron. 1998;54:2967–3042. [Google Scholar]; c) Kocienski PJ. Protecting Groups. 3. Thieme; Stuttgart: 2005. [Google Scholar]; d) Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3. Wiley; New York: 1999. [Google Scholar]
- 24.Swamy VM, Ilankumaran P, Chandrasekaran S. Synlett. 1997:513–514. [Google Scholar]
- 25.Nayak SK, Kadam SM, Banerji A. Synlett. 1993:581–582. [Google Scholar]
- 26.Olivero S, Dunach E. Tetrahedron Lett. 1997;38:6193–6196. [Google Scholar]
- 27.Crich D, Hwang JT, Yuan H. J Org Chem. 1996;61:6189–6198. doi: 10.1021/jo960799q. [DOI] [PubMed] [Google Scholar]
- 28.Mereyala HB, Gurrala SR, Mohan SK. Tetrahedron. 1999;55:11331–11342. [Google Scholar]
- 29.Crich D, Picione J. Org Lett. 2003;5:781–784. doi: 10.1021/ol0340890. [DOI] [PubMed] [Google Scholar]
- 30.Paulsen H. Angew Chem Int Ed Engl. 1982;21:155–224. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.