

Introduction
This reports aims to describe a unique case of rapidly progressive neurodegenerative disease in an individual with both a valosin containing protein (VCP) mutation (R155H) and a CAG repeat expansion in the gene encoding Huntingtin (HTT) (44 CAG repeats).
Mutations in the VCP gene are the most frequent cause of multisystem proteinopathy (MSP), a pleiotropic degenerative disorder affecting brain, muscle and bone(1, 2). Patients with MSP may present with familial amyotrophic lateral sclerosis (fALS)(3), frontotemporal dementia (FTD), inclusion body myopathy, Paget's disease of bone, or a combination of these disorders. VCP is an ubiquitin segregase involved in multiple cellular activities. Disease mutations in VCP impair multiple cellular processes, including autophagosome maturation, mitochondrial quality control, endocytosis, and regulation RNA granule dynamics(2). Huntington Disease (HD) results from polyglutamine expansion in the protein Huntingtin, resulting in a toxic gain of function characterized by mitochondrial dysfunction and accumulation of ubiquitin-positive protein inclusions(4).
Case report
A 47 year old man with a history of Paget's disease and a family history of both HD (maternal) and ALS (paternal) presented to the HD clinic for initial evaluation, four years after developing vocal tics, followed by chorea, then a one year history of rapidly progressive cognitive deterioration. He began to develop progressive weakness 6 months earlier and could no longer walk unassisted. He was found to have dementia, slow saccades, chorea and profound weakness with widespread muscle atrophy and fasciculations together with upper motor neuron signs. A clinical diagnosis of Shoulson and Fahn Stage III HD was confirmed with genetic testing, and he was seen at the ALS clinic one month later. At presentation, his ALS functional rating scale was 26/48. He was awake, alert and oriented to self, place and year, but had limited insight and was a poor autobiographic historian. His Montreal cognitive assessment score was 13/30. Abnormal findings on examination included spastic dysarthria, bilateral trapezius weakness, mild chorea, increased tone with both rigidity and spasticity of all limbs, with marked atrophy of his shoulder girdle, biceps and quadriceps muscles and frequent fasciculations throughout his body. He had severe, mildly asymmetrical weakness (Medical Research Council 0 to 4-), worse in proximal musculature. His deep tendon reflexes were brisk and he had Babinski's sign bilaterally. His sensory exam was grossly normal, but his cognition limited detailed testing.
An electrodiagnostic evaluation demonstrated mildly reduced motor amplitudes with mild generalized conduction slowing. F-waves were markedly prolonged. Sensory nerve conductions were normal. Needle EMG testing was performed of the right deltoid, biceps, triceps, first dorsal interosseus, flexor carpi ulnaris, and left vastus medialis quadriceps, tibialis anterior, gastrocnemius as well as the right T5 paraspinals. All muscles demonstrated fibrillation and positive sharp wave potentials, and the motor units recruited in a reduced neurogenic pattern. Motor unit remodeling was seen in most muscles with large polyphasic units of long duration being present. The findings were felt to be consistent with ALS, without apparent myopathy.
A clinical diagnosis of fALS with FTD was made and a mutation in the VCP gene (R155H) was identified.
The patient died 6 months later - 4.5 years after his first symptom and 12 months after the onset of weakness - from advanced dementia and respiratory muscle weakness.
A post mortem exam showed that the unfixed brain weighed 1090g. The right hemisphere had focal atrophy of the superior temporal gyrus, pre-central gyrus, and the pre-frontal area. The ventricles were minimally dilated. The substantia nigra appeared pale. The caudate nucleus was normal on gross examination with microscopic evidence of mild to moderate focal gliosis (Figure 1a), thus corresponding to the grade 1 (out of 0-4) Huntington's pathology according to the classification proposed by Vonsattel et al. (5). Microscopic analysis also revealed bilateral cortico-spinal tract degeneration and depletion of anterior horn cells with degeneration of many of the remaining motor neurons.
Figure1.
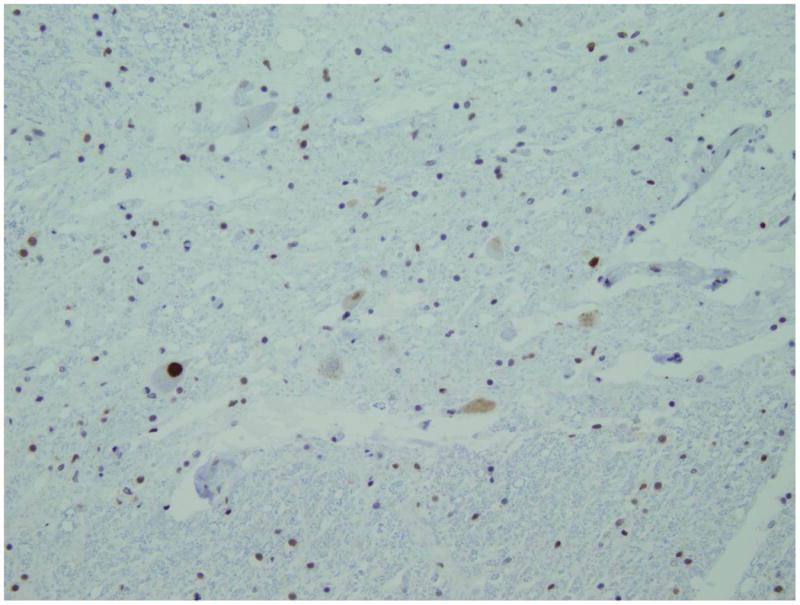
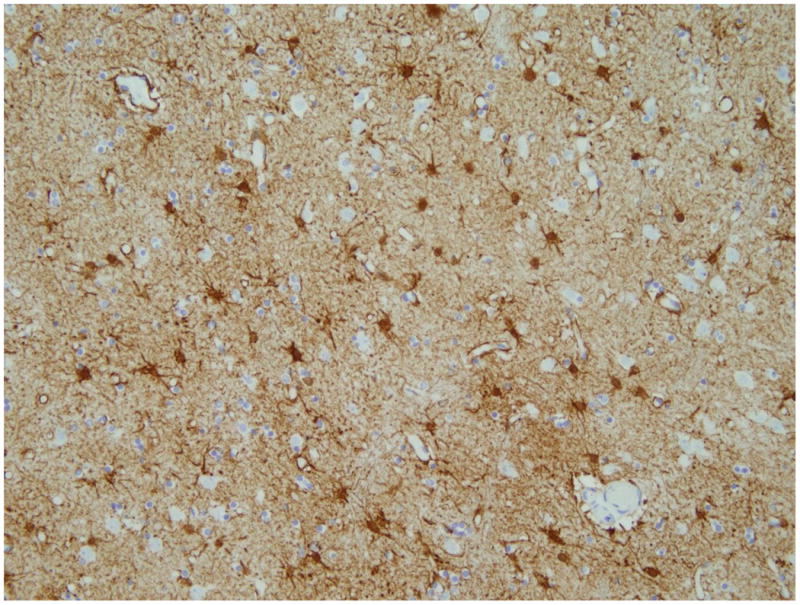
Histopathology a) GFAP immunostain of the head of caudate nucleus, which shows moderate astrogliosis. b) TDP-43 immunostain shows few anterior horn cells in the level of section (lumbar). The neurons show reduced TDP-43 nuclear immunoreactivity or with an intensely stained cytoplasmic inclusion (arrow). Both original magnifications × 200.
Anterior horn cells, more in the lumbar cord, showed intracytoplasmic inclusions, including ubiquitin negative and TDP-43-positive inclusions in the form of skeins and occasional large ovoid inclusions (Figure 1b). Additionally, there were ubiquitin-positive stubby neurites seen throughout the neocortex and hippocampal pyramidal cell layer, as well as occasional TDP-43-positive intracytoplasmic inclusions. Muscle tissue was analyzed revealing grouped atrophy and increased internal nucleii without red rimmed vacuoles or other definitively myopathic changes.
Discussion
There have been previous reports of patients with co-existent HD and fALS (6), but this is the first case in which a (VCP) mutation (R155H) has been identified. We propose that the presence of both mutations led to synergistic neurodegeneration. The resulting phenotype was one of very rapid neurodegeneration with death occurring 12 months after the first symptom of neurogenic weakness. This sharply contrasts with the median survival in HD which is about two decades (7). The patient's father, who died from ALS had slowly progressive weakness over nearly two decades before death. The MSP ALS phenotype is varied but only rarely does it culminate in rapidly progressive motor neuron disease lasting less than one year(1). Only two of the other 16 MSP patients with MND our case series had upper motor neuron signs(1). The most prominent cellular defects caused by VCP mutations are impaired protein and mitochondrial quality control(8). Meanwhile, the two most prominent features of HD pathology are accumulation of proteinaceous inclusions and mitochondrial dysfunction. Thus we suggest that the rapid disease course experienced by the patient described here reflects synergy between the pathological processes at work in MSP and HD as has been suggested in in vitro studies(9). Genetic testing of the offspring of this patient with two dominant neurodegenerative diseases has not been performed. When they reach adulthood they will be confronted with the choice of being tested for the two disease genes. Whether to undergo pre-symptomatic testing is a difficult decision for the individual with potential social and legal implications. The information can only be used for family and career planning until effective treatments have been developed.
Acknowledgments
The authors thank the participant in this study as well as his family members for their commitment to and engagement in the research process.
The UC Davis Alzheimer's Disease center
HDSA Center of Excellence at UC Davis
UC Davis Multidisciplinary ALS clinic, an ALS Association Certified Center of Excellence
Funding: NIH UL1 TR 000002 and KL2 TR 000134
NIA P30 AG10129
ALS Association, grant 1862; ALS Recovery Fund; Robert Packard Center for ALS Research.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Contributor Information
Björn Oskarsson, University of California, Davis.
Victoria Wheelock, University of California, Davis.
Michael Benatar, University of Miami.
J. Paul Taylor, St. Jude Children's Research Hospital.
Nanette Joyce, University of California, Davis.
David Chesak, Sutter Center for Neuroscience Medical Group.
Lee-Way Jin, University of California, Davis.
References
- 1.Benatar M, Wuu J, Fernandez C, Weihl CC, Katzen H, Steele J, et al. Motor neuron involvement in multisystem proteinopathy: implications for ALS. Neurology. 2013;80:1874–80. doi: 10.1212/WNL.0b013e3182929fc3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron. 2013;78:65–80. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–64. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.La Spada AR, Weydt P, Pineda VV. Huntington's Disease Pathogenesis Mechanisms and Pathways. 2011 [PubMed] [Google Scholar]
- 5.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. Journal of neuropathology and experimental neurology. 1985;44:559–77. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Rubio A, Steinberg K, Figlewicz DA, MacDonald ME, Greenamyre T, Hamill R, et al. Coexistence of Huntington's disease and familial amyotrophic lateral sclerosis: case presentation. Acta neuropathologica. 1996;92:421–7. doi: 10.1007/s004010050539. [DOI] [PubMed] [Google Scholar]
- 7.Rinaldi C, Salvatore E, Giordano I, De Matteis S, Tucci T, Cinzia VR, et al. Predictors of survival in a Huntington's disease population from southern Italy. Can J Neurol Sci. 2012;39:48–51. doi: 10.1017/s0317167100012671. [DOI] [PubMed] [Google Scholar]
- 8.Ju JS, Miller SE, Hanson PI, Weihl CC. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J Biol Chem. 2008;283:30289–99. doi: 10.1074/jbc.M805517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poksay KS, Madden DT, Peter AK, Niazi K, Banwait S, Crippen D, et al. Valosin-containing protein gene mutations: cellular phenotypes relevant to neurodegeneration. J Mol Neurosci. 2011;44:91–102. doi: 10.1007/s12031-010-9489-8. [DOI] [PMC free article] [PubMed] [Google Scholar]