

Abstract
Myopathy is a group of muscle diseases that can be induced or exacerbated by drug–drug interactions (DDIs). We sought to identify clinically important myopathic DDIs and elucidate their underlying mechanisms. Five DDIs were found to increase the risk of myopathy based on analysis of observational data from the Indiana Network of Patient Care. Loratadine interacted with simvastatin (relative risk 95% confidence interval [CI] = [1.39, 2.06]), alprazolam (1.50, 2.31), ropinirole (2.06, 5.00), and omeprazole (1.15, 1.38). Promethazine interacted with tegaserod (1.94, 4.64). In vitro investigation showed that these DDIs were unlikely to result from inhibition of drug metabolism by CYP450 enzymes or from inhibition of hepatic uptake via the membrane transporter OATP1B1/1B3. However, we did observe in vitro synergistic myotoxicity of simvastatin and desloratadine, suggesting a role in loratadine–simvastatin interaction. This interaction was epidemiologically confirmed (odds ratio 95% CI = [2.02, 3.65]) using the data from the US Food and Drug Administration Adverse Event Reporting System.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
-
1
Drug‐induced myopathy can be exacerbated by DDIs. No study to date has attempted to identify and investigate myopathic DDIs systematically.
WHAT QUESTION DID THIS STUDY ADDRESS?
-
1
This study identified DDIs that increased risk of myopathy and investigated their underlying mechanisms using a high‐throughput, translational approach.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
-
1
Five previously unknown DDIs were identified to increase the risk of myopathy, none of which appeared to result from inhibition of drug metabolism or hepatic uptake via OATP1B1/1B3. Synergistic myotoxicity may contribute to the interaction between loratadine and simvastatin.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS
-
1
Pharmacoepidemiologic screening followed by mechanistic investigations proved to be an efficient approach to identify clinically important DDIs.
Drug‐induced myopathy, among the most common causes of muscle disease,1 has clinical presentations ranging from asymptomatic muscle enzyme elevation to massive rhabdomyolysis with acute renal failure.2 Among 7 million case reports in the US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) from 2001–2010, about 100,000 cases involved myopathy as a suspected adverse drug reaction (ADR).3 Among various drug classes associated with myopathy, statins have received extensive public and scientific attention. Statin‐induced myopathy occurs in 5–20% of patients and is a significant barrier to maximizing the benefits of statin therapy.4 Considering that more than 18% of Americans aged ≥45 (∼127 million) took statins in 2012, 1.1 to 4.6 million patients might have experienced myopathy in 2012 alone.
Drug‐induced myopathy can be exacerbated by pharmacokinetic and/or pharmacodynamic drug–drug interactions (DDIs). In a pharmacokinetic myopathic DDI, the object drug induces myopathy, and the precipitant drug modifies the object drug's myopathic effects by changing its pharmacokinetics. One such example is the interaction between cerivastatin and gemfibrozil that contributed to the withdrawal of cerivastatin from the market.5 The risk of cerivastatin‐induced rhabdomyolysis is 10‐fold higher than that of other statins; with concurrent use of gemfibrozil, a drug that substantially inhibits the metabolism of cerivastatin, the risk is 50‐fold higher.6
Although drug‐induced myopathy and the role of DDIs as risk factors have been well documented, to our knowledge no study has attempted to identify and investigate unknown myopathic DDIs systematically. Research on DDIs has been mostly limited to pharmacokinetic DDIs with identifiable mechanisms, a small scope, a relatively low efficiency, and often a low clinical relevance. Recognizing the need for a translational approach for the study of DDIs,7 a promising new strategy involves pairing epidemiological studies with mechanistic investigations such as in vitro screening for metabolism‐based DDIs. This approach was recently successfully applied to the study of interactions between sulfonylureas and statins/fibrates.8 Our previous study predicted 13,197 potentially interacting drug pairs using data mined from PubMed abstracts,9 and narrowed down to 3,670 clinically prescribed drug pairs using data derived from electronic medical records.9 In the current study, by applying a large‐scale, translational approach, we sought to identify interacting drug pairs associated with myopathy and to elucidate their underlying pharmacokinetic and pharmacodynamic mechanisms.
RESULTS
DDIs associated with increased risk of myopathy
We applied the myopathy concept definition (Supplementary Table S1) to a subset (n = 828,905) of the Indiana Network for Patient Care (INPC) database (2004–2009) formatted in the Observational Medical Outcomes Partnership10 Common Data Model. We identified 59,572 myopathy cases, of which 48,877 (5.9%) had myalgia and myositis, 12,720 (1.5%) had muscle weakness, and 53 (0.0064%) had rhabdomyolysis. For each of the 3,670 drug pairs that we previously predicted to interact,9 we performed a simple cohort study. The demographics of the patient population were described previously9 and are shown in Supplementary Table S2. Since race information was missing for 65.8% of the patients, it was not included in the analyses. For each drug pair, we estimated a risk ratio (RR) adjusted for age and sex, both known risk factors of myopathy.11 An RR greater than 1 indicated that the incidence of myopathy following the prescriptions for both drugs was greater than the additive incidence following a prescription for either drug alone. Drug pairs with RRs greater than 1 were therefore considered to be interacting and associated with an increased risk of myopathy. As a small sample size may yield an unreliable estimate of risk ratio, drug pairs with counts of myopathy cases less than 100 were excluded. We identified five DDIs associated with an increased risk of myopathy (Table 1), four of which involved the widely used antihistamine loratadine. The risk of myopathy increased with age at 1.0015 (95% confidence interval [CI] = 1.00148, 1.00152) per year, and was 1.64‐fold (95% CI = 1.63, 1.65) higher in females (8.6%) than in males (5.4%) (Supplementary Table S3). Since sicker patients tend to take more medications, we used the number of prescribed medications, including the relevant drug pair, within drug exposure windows to adjust for confounding by morbidity. The average number of prescribed medications was 3.8 ± 2.5. The five DDIs remained significant after adjusting for the number of coprescribed medications (Supplementary Table S4).
Table 1.
Drug–drug interactions associated with increased risk of myopathy after adjusting for age and sex
| Drug 1 | Drug 2 | Risk1 | Risk2 | Risk12 | Risk ratio (95% CI) | M1 | N1 | M2 | N2 | M12 | N12 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Loratadine | Simvastatin | 0.022 | 0.033 | 0.093 | 1.69 (1.39, 2.06) | 1,264 | 44,245 | 4,197 | 102,345 | 137 | 1,223 |
| Loratadine | Alprazolam | 0.022 | 0.029 | 0.095 | 1.86 (1.50, 2.31) | 1,257 | 43,341 | 2,251 | 52,341 | 176 | 1,448 |
| Loratadine | Ropinirole | 0.020 | 0.018 | 0.122 | 3.21 (2.06, 5.00) | 1,218 | 43,491 | 164 | 6,531 | 17 | 123 |
| Promethazine | Tegaserod | 0.011 | 0.020 | 0.093 | 3.00 (1.94, 4.64) | 1,332 | 78,334 | 109 | 3,745 | 23 | 224 |
| Loratadine | Omeprazole | 0.022 | 0.059 | 0.102 | 1.26 (1.15, 1.38) | 1,260 | 44,207 | 4,339 | 70,345 | 304 | 2,734 |
Risk1 and risk2 are myopathy risks for drug 1 and drug 2, respectively. The risk ratios were calculated as risk12/(risk1+risk2). 95% CIs were calculated using multivariate logistic regression adjusted for age and sex. N1, N2, and N12 is the number of patients who had prescription for drug 1 only, drug 2 only, and both drugs, respectively; and M1, M2, and M3 is the number of myopathy cases who had prescription for drug 1 only, drug 2 only, and both drugs, respectively.
Inhibition of CYP‐mediated drug metabolism
Cytochrome P450s (CYPs) are responsible for about 75% of drug metabolism,12 and their inhibition is a common mechanism of pharmacokinetic DDIs.12 Since each drug in the five DDIs relies on CYPs for elimination, we examined whether the DDIs were possibly caused by inhibition of CYP drug metabolism. Using fluorometric CYP inhibition screening assays, we assessed the potential of the drugs, and their pharmacologically active metabolites, to inhibit the enzymatic activities of the major human CYPs isoforms CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. The half maximal inhibitory concentration (IC50s) are presented in Supplementary Table S6. It is commonly accepted that a dissociation constant (Ki) is more relevant than an IC50 when predicting the clinical risk of metabolism‐based DDIs. We therefore determined Kis for 11 drug‐enzyme pairs (Table 2) that showed relatively strong CYP inhibitions (IC50 ≤20 μM).
Table 2.
Predicting potential of CYP‐based drug–drug interaction
| Inhibitor | Pathway | Dissociation constant (Ki, µM) | Fraction of unbound (fu,inc) | Unbound dissociation constant (Ki,u, µM) | Peak plasma concentration (Cmax, ng/ml) | Inhibitor concentration ([I], µM) | Predicted R‐values |
|---|---|---|---|---|---|---|---|
| Simvastatin | CYP3A4 | 0.51 | 0.93 | 0.47 | — | 0.764 | 2.61 |
| Promethazine | CYP2D6 | 0.25 | 0.88 | 0.22 | 19.3 (36) | 0.068 | 1.31* |
| Tegaserod | CYP3A4 | 5 | 0.92 | 4.61 | — | 0.796 | 1.17 |
| Ropinirole | CYP2D6 | 0.85 | 0.84 | 0.71 | 26.9 (37) | 0.103 | 1.15* |
| Loratadine | CYP2D6 | 0.5 | 0.93 | 0.47 | 4.12 (38) | 0.011 | 1.02 |
| Tegaserod | CYP2D6 | 0.51 | 0.92 | 0.47 | 2.7 (39) | 0.009 | 1.02 |
| Loratadine | CYP2B6 | 2 | 0.93 | 1.86 | 4.12 (38) | 0.011 | 1.01 |
| Simvastatin | CYP2C9 | 18.3 | 0.93 | 17.03 | 25.4 (40) | 0.061 | 1.00 |
| Loratadine | CYP2C9 | 7.6 | 0.93 | 7.07 | 4.12 (38) | 0.011 | 1.00 |
| Tegaserod | CYP2C19 | 9.2 | 0.92 | 8.48 | 2.7 (39) | 0.009 | 1.00 |
| Tegaserod | CYP2C9 | 11.4 | 0.92 | 10.51 | 2.7 (39) | 0.009 | 1.00 |
Ki is the dissociation constant determined in vitro; fu,inc is the fraction of unbound drug in the incubation mixture and was predicted using the Hallifax‐Houston model41; Ki,u is the unbound dissociation constant estimated as Ki * fu,inc; Cmax is the peak total plasma concentration at the highest clinical dose; [I] is the inhibitor concentration used to predict R values and is equal to Cmax, except for CYP3A4 inhibitors administered orally. For simvastatin and tegaserod with CYP3A4, [I] is the estimated gut concentration at the highest proposed clinical dose, 80 mg (191 µM) and 6 mg (19.9 µM), respectively, divided by 250 mL (approximate gut volume); R values were estimated as 1 + [I]/Ki,inc; *R values ≥1.1 (or ≥11 for simvastatin and tegaserod with CYP3A4), indicating a probable clinical CYP450‐based DDI.
Following FDA guidelines for drug interaction studies,13 we applied a stepwise approach to evaluate the risk of clinical DDIs resulting from inhibition of drug metabolism by CYPs. For each of the 11 drug‐enzyme pairs for which a Ki was observed, we first used a conservative R‐value approach to evaluate each drug's potential to act as a hypothetical precipitant. An R‐value represents the predicted ratio of the area under concentration–time curve (AUC) of a hypothetical object drug that is exclusively metabolized by the inhibited CYP in the presence vs. absence of an inhibitor. Table 2 shows the predicted R‐values. Consistent with FDA guidelines,13 an R value ≥1.1 (or ≥11 for CYP3A4 inhibitors administered orally) indicates that the drug could act as a precipitant. With R‐values of 1.31 and 1.15, respectively, promethazine and ropinirole could potentially interact with drugs exclusively metabolized by CYP2D6, the isoform most strongly inhibited by both drugs. The predicted potential of the other inhibitor‐enzyme pairs was negligible. These determinations suggest that the DDIs not involving promethazine and ropinirole were unlikely to result from inhibition of drug metabolism by CYPs.
A limitation of R values is that they only account for inhibition of a single metabolic pathway without regard to object drugs.14 In cases where multiple pathways are responsible for the metabolism of an object drug, an AUC ratio (AUCR) taking into account the fractional contribution of inhibited pathways to the overall metabolism is preferred. We thus predicted AUCRs for the interaction between ropinirole and loratadine, and that between promethazine and tegaserod. Accounting for 10% of the hepatic metabolism of loratadine by CYP2D6 that would be inhibited by ropinirole,15 the AUCR of loratadine in the presence vs. absence of ropinirole was predicted to be 1.01. Consistent with the FDA guidelines,13 it indicates that loratadine and ropinirole are unlikely to have CYP‐based interactions. Because CYP2D6 is insignificant in tegaserod's elimination,16 the inhibition of CYP2D6 by promethazine was considered to have no clinical effect on the pharmacokinetics of tegaserod. Overall, our data suggest that CYP inhibition is unlikely the major mechanism underlying the significant DDIs identified previously.
Inhibition of OATP1B1/1B3‐mediated hepatic uptake
It has been increasingly recognized that organic anion‐transporting polypeptides (OATPs) represent an important site of DDIs. Particular attention has been paid to OATP1B1 and 1B3, the transporters of the OATP family demonstrated as most engaged in drug disposition.17 Among their substrates are many clinically important drugs including simvastatin acid,18 the active metabolite of simvastatin. The risk of simvastatin‐induced myopathy was 4.5‐fold higher in individuals with a genetic variant of SLCO1B1 (the OATP1B1 gene), compared to those with the wildtype allele.19
We hypothesized that the DDIs identified previously may result from, at least in part, the inhibition of OATP1B1/1B3 that leads to impaired hepatic uptake and compromised hepatic clearance. We first evaluated the potential of the drugs, as well as their pharmacologically active metabolites, to inhibit the active uptake of β‐estradiol 17‐β‐D‐glucuronide (E217βDG) in cryopreserved rat hepatocytes. E217βDG is a relatively specific substrate of OATP1B2, a functional homolog of human OATP1B1/1B3 with very similar substrate specificity.17, 20 At 100 μM, simvastatin acid, omeprazole, alprazolam, desloratadine (the active metabolite of loratadine), simvastatin, tegaserod, ropinirole, loratadine, and promethazine inhibited E217βDG uptake by 103.3 ± 0.5%, 60.1 ± 4.8%, 54.5 ± 0.3%, 44.9 ± 14.2%, 36.3 ± 6.0%, 24.6 ± 15.3%, 23.7 ± 2.7%, 18.1 ± 10.9%, and 17.7 ± 7.7%, respectively. We then determined the inhibitory potencies of the drugs showing ≥45% inhibition. The IC50s (95% CI) of simvastatin acid, omeprazole, alprazolam, and desloratadine were 4.3 μM (3.5, 5.3), 84.3 μM (49.8, 142.9), 99.5 μM (79.5, 124.6), and 140.5 (111.4, 177.1) μM, respectively (inhibition curves are shown in Supplementary Figure 1).
Figure 1.
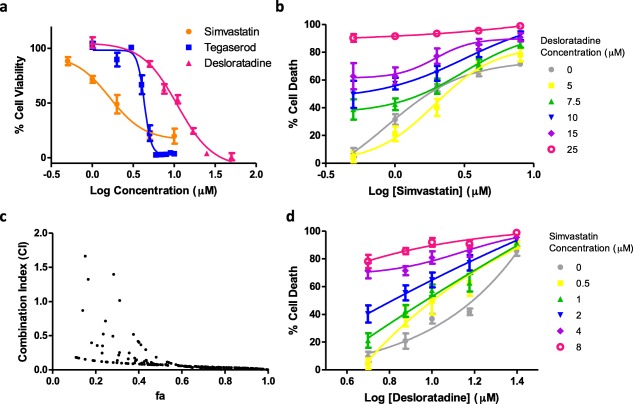
(a) Dose–response curves of simvastatin (orange), tegaserod (blue), and desloratadine (pink). Healthy, fully differentiated rat L6 myotubes were treated with individual drugs at various concentrations for 5 days, and myotube viability was determined using MTS/PMS assays. (b) Concentration–effect curves of simvastatin in the presence of various fixed concentrations of desloratadine at various concentrations. (d) Concentration–effect curves of desloratadine in the presence of various fixed concentrations of simvastatin. (c) Combination index (CI) – fractional myotube death (fa) plot. CI = 1 indicates additivity (no interaction). The points above 1 indicate antagonism and those below indicate synergism. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Following a similar strategy for evaluating CYP‐based DDIs, we estimated R‐values (from IC50s) to evaluate the drugs' potential to interact clinically with OATP1B1/1B3 substrates. The R‐values of simvastatin acid, omeprazole, alprazolam, and desloratadine were 3.85, 1.23, 1.01, and 1.01, respectively (Table 3). Consistent with the FDA guidelines,13 simvastatin acid and omeprazole (R value ≥1.1) might interact with drugs relying on OATP1B1/1B3 for hepatic uptake. The potential of alprazolam and desloratadine as precipitants was negligible.
Table 3.
Predicting potential of OATP1B1‐based drug–drug interaction
| Drug | Half maximum inhibition concentration (IC50, μM) | Dose (mg/mmol) | Molecular weight (g/mol) | Peak plasma concentration (Cmax, μM) | Maximal hepatic inlet concentration ([I]inlet,max, μM) | Predicted R value |
|---|---|---|---|---|---|---|
| Simvastatin Acid | 4.3 | 80/0.183 | 436.6 | 0.058 (40) | 12.274 | 3.85 |
| Omeprazole | 84.3 | 80/0.232 | 345.42 | 4.146 (42) | 19.586 | 1.23 |
| Alprazolam | 99.5 | 3/0.01 | 308.76 | 0.333 (43) | 0.981 | 1.01 |
| Desloratadine | 140.5 | 5/0.16 | 310.8 | 0.015 (44) | 1.088 | 1.01 |
Dose is the highest proposed clinical dose; Cmax is the peak plasma concentration at the highest proposed clinical dose; [I]inlet,max was estimated as Cmax + (ka x Dose x FaFg/Qh) (13), where Qh is the hepatic blood flow (1,500 mL/min), ka is the absorption rate constant, and FaFg is the fraction of oral dose that reaches the liver. Because the values of ka and FaFg were not available, for conservative predictions, they were assumed equal to the theoretical maxima of 0.1 min−1 and 1,13 respectively. R values were estimated as 1 + [I]inlet,max/Ki. Because the concentration of E217βDG (1 μM) was well below its Km,45, 46 the Kis were approximated by the IC50s based on Ki = IC50 / (1 + [S]/Km).47 For simvastatin acid, the Cmax and dose were assumed equal to those of simvastatin. For desloratadine, R values were estimated with the Cmax following the highest clinical dose of desloratadine since it is higher than the Cmax following that of loratadine.48, 49
Direct myotoxicity
Although all the drugs involved in the DDIs have known muscle‐related side effects, their direct myotoxicity, except that of simvastatin, has not been examined. We tested whether the DDIs resulted from the direct toxicity of the individual drugs, or their combinations, to muscle cells. We first evaluated the myotoxicity of each individual drug to rat L6 myotubes, a commonly used in vitro skeletal muscle model previously used to study mechanisms of statin‐induced myopathy.21, 22 After treatment of healthy, fully differentiated myotubes with each drug individually at 10 μM for 5 days, tegaserod, simvastatin, desloratadine, and simvastatin acid induced 97.9 ± 0.4%, 73.7 ± 2.6%, 73.3 ± 1.1%, and 33.0 ± 2.1% myotube death, respectively, compared to dimethyl sulfoxide (DMSO) control. The remaining drugs were not myotoxic. We then determined the concentration–effect curves of tegaserod, simvastatin, and desloratadine since they induced >50% myotube death. The IC50s (95% CI) of tegaserod, simvastatin, and desloratadine were 4.32 μM (4.15, 4.49), 1.64 μM (1.05, 2.56), and 10.94 μM (9.24, 12.96), respectively (Figure 1 a).
The myotoxicity of simvastatin and desloratadine led us to suspect a synergistic interaction that increases risk of myotoxicity when used in combination. We treated myotubes with simvastatin and desloratadine in combination at various concentrations to evaluate their combined toxic effect. The dose–response curves of simvastatin shifted leftward with increasing concentration of desloratadine (Figure 1 b). The same trend was observed for desloratadine in the presence of simvastatin (Figure 1 d). Using the method of Chou,23 combination index (CI) values were calculated and plotted against fractional myotube death (fa) (Figure 1 c). Most CI values were less than unity; a few CI values greater than unity near the region fa = 0 likely resulted from methodological flaw,24 indicating that the interaction between simvastatin and desloratadine was synergistic, such that the drugs notably increased each other's myotoxic effect. This synergistic myotoxicity may contribute to the interaction between simvastatin and loratadine. Direct toxicity to muscle cells, however, was unlikely to explain the other DDIs we identified.
Validation of loratadine‐simvastatin interaction
The interaction between loratadine and simvastatin was further validated using an independent dataset, the FAERS. A distinct feature of the FAERS is that it only includes patients who experience suspected ADRs. As a case‐only design was considered more appropriate using the FARES, we performed a similar study using the INPC dataset to compare the results. An odds ratio (OR), estimated from a case‐only study, is equivalent to a relative risk estimated from a cohort study.25 The ORs are presented in Table 4. Consistent with the RRs presented previously, the concomitant use of loratadine and simvastatin was significantly associated with increased risk of myopathy, with ORs of 2.20 (95% CI = 2.02, 3.65) and 1.53 (95% CI = 1.28, 1.82) in the FAERS and INPC databases, respectively. In additional subgroup analyses stratified by sex, age, or myopathy type (muscle weakness or myalgia), the interaction between loratadine and simvastatin remained significant in specific subgroups of patients (Supplementary Table S8).
Table 4.
Testing and validation of the loratadine/simvastatin interaction using case‐only datasets
| Datasets | Odds ratio | 95% CI | N12 | N1 | N2 | N00 |
|---|---|---|---|---|---|---|
| INPC CDM | 1.53 | (1.28, 1.82) | 37 | 1,264 | 4,197 | 5,572 |
| FAERS | 2.20 | (2.02, 3.65) | 37 | 276 | 6,116 | 100,531 |
N12, N1, N2, and N00 is the number of myopathy cases with prescription for both simvastatin and loratadine, simvastatin only, loratadine only, and neither drug, respectively. INPC CDM stands for Indiana Network of Patient Care Common Data Model. FAERS stands for the FDA adverse event reporting system.
DISCUSSION
Research on pharmacokinetic DDIs traditionally involves prediction of potential DDIs based on molecular mechanistic understanding of the interaction between a drug and its relevant drug‐metabolizing enzymes or drug transporters. The clinical importance of hypothesized DDIs is then examined in clinical trials or pharmacoepidemiologic studies. This approach is often limited to a small scope and a relatively low efficiency when used to identify unknown, clinically important DDIs. We sought to overcome these limitations by applying a translational and systematic approach involving pharmacoepidemiologic screening followed by mechanistic investigations.
Our study identified a synergistic myotoxic interaction between simvastatin and loratadine that has never been reported. As simvastatin is one of the most widely prescribed statins, this myopathic interaction could potentially affect a large population. We suggest further studies to confirm this interaction and its myopathic effects. Simvastatin is known to interact clinically with a number of drugs that may further increase its risk of myopathy, including CYP3A inhibitors, such as verapamil, ketoconazole, itraconazole, tacrolimus, erythromycin, clarithromycin, and amiodarone26, 27 and OATP1B1 inhibitors (e.g., gemfibrozil).28 Our study, however, did not identify any known DDIs with statins that would increase the risk of myopathy, except for amiodarone (Supplementary Table S5). One possible explanation is that our predefined 1‐month drug exposure window cannot well capture the concomitance of statins with many CYP3A inhibitors that typically have short exposure. Amiodarone, however, is used chronically and its concomitance with statins is easier to capture. The other explanation is the underpowered interaction analyses between statins and CYP3A inhibitors. Power analysis for these reported DDIs in Supplementary Table S5 showed that almost all of them had less than 10% power, except for the interactions between amiodarone and statins, which had power higher than 70%. Referring back to our initial drug interaction study design, a requested minimum sample size of 100 for two‐committed drugs and a minimum of 1.5 risk ratio would give us 65% power in testing the drug interaction effect.
Four out of five DDIs identified in our study involved a commonly used antihistamine, loratadine. Myalgia is one of the side effects of both loratadine and desloratadine.29, 30 Our results suggest that loratadine and desloratadine may be more myotoxic than previously recognized, and can pose even higher risk of myopathy with concomitant use of other drugs.
The IC50s and Kis that we reported provide a comprehensive view of the potential of these drugs to cause CYP‐based DDIs. These data are consistent with those published previously.9 To our knowledge, we are the first to describe the potential of these drugs (except simvastatin) to inhibit OATP1B2 in rat hepatocytes and assess their potential OATP‐mediated DDIs in humans. We are also likely the first to report myotoxicity of desloratadine and tegaserod, which may underlie their muscle‐related side effects. Of note, simvastatin was much more toxic than simvastatin acid to myotubes in vitro, an observation previously reported,31 suggesting that simvastatin‐induced myopathy is due primarily to simvastatin rather than simvastatin acid. Similarly, the in vitro myotoxicity of desloratadine suggests that myalgia associated with loratadine may be primarily due to its metabolite, desloratadine.
Although inhibition of drug metabolism by CYPs and inhibition of OATP1B1/1b3 are the most common mechanisms underlying pharmacokinetic DDIs, they are unlikely the major mechanisms for the DDIs that we observed. The results from the R‐value approach suggest that simvastatin acid and omeprazole may interact with drugs that rely on OATP1B1/1B3 for hepatic uptake. We suggest that such data be interpreted with caution, as the R‐value approach, for both CYPs and transporters, is known to overpredict the risk of clinical DDIs and lead to spurious conclusions that a drug is a precipitant when it is not.32 It implies, however, that the drug pairs predicted not to interact using this approach in our study are very unlikely to have real interactions.
There are a few limitations to our study. We used a simple cohort design that may be subject to residual confounding and misclassification. The use of the FAERS may not provide a definitive validation for a simvastatin–loratadine interaction. The CYP450 inhibition assays involve fluorogenic substrates and recombinant CYP enzymes that occasionally generate inhibitory potencies very different from those using conventional approaches. Both the R value and AUCR approaches use a single static in vivo concentration of an inhibitor drug, which may overestimate the risk of DDI for drugs, such as simvastatin, with relatively short half‐lives and whose circulating concentrations drop rapidly following a dose. We did not evaluate the drugs as direct substrates of OATP1B1/1B3 or other transporters, limiting our understanding of the role of drug transporters in the DDIs. We also used cryopreserved rat hepatocytes and rat L6 myotubes, which are less clinically relevant than human‐derived cell models. Future studies are warranted to further evaluate the underlying mechanisms of these DDIs.
METHODS
Evaluation of CYP450 inhibition
Fluorometric cytochrome P450 inhibition kits (BD Biosciences/Gentest, San Jose, CA) were used to determine the IC50s of the drugs for the major CYPs. The assays were performed following the manufacturer's instructions under the conditions in Supplementary Table S7.33 Data were analyzed using GraphPad Prism 5 software (La Jolla, CA).
R values were estimated as 1+ [I]/Ki,u, where [I] is the peak total plasma inhibitor concentration (Cmax) at the highest proposed clinical dose obtained from the published literature, and Ki,u is the unbound dissociation constant of the inhibitor. For drugs that inhibited CYP3A4 administered orally, [I] was estimated as [I] = Igut = molar dose/250 mL. AUCRs were predicted using the mechanistic static model in Eq. 1,34
| (1) |
where fm,CYPj is the fractional metabolism of the object drug through the jth inhibited CYP pathway, Finhibited and F are the bioavailabilities of the object drug in the presence and absence of the inhibitor, respectively. Because Finhibited and F were not available, for conservative prediction they were assumed to be unity.
Evaluation of inhibition of OATP1B1/1B3
The drugs (100 μM) were incubated with cryopreserved rat hepatocytes (1 × 106 cells/mL) and [3H] E217βDG (1 μM, 0.1 μCi) for 3 minutes at 37°C and 0°C in triplicate. Uptake was stopped with addition of 1 mL ice‐cold PBS and immediate centrifugation at 4500 rpm for 1 minute at 4°C. Cells were resuspended in 1 mL ice‐cold PBS and centrifuged again. After removing supernatant, cell pellets were lysed with 200 μL of 50% acetonitrile in H2O, followed by vigorous vortexing. The fraction of uptake was the ratio of the radioactivity in hepatocyte lysate to the total radioactivity in both lysate and supernatants. The fraction of active uptake was the difference between the total uptake at 37°C and that at 0°C.
Evaluation of myotoxicity
Rat L6 muscle cells were cultured as previously detailed by Klip and colleagues35 with slight modifications. Cells were maintained in monolayer culture in α‐MEM containing 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic solution (10,000 U/ml penicillin G, 10 mg/ml streptomycin, and 25 mg/ml amphotericin B) in an atmosphere of 5% CO2 at 37°C. Five days after seeding, myoblasts were differentiated into multinucleated myotubes with 2% FBS. All drug treatments were initiated 5 days after the initiation of differentiation and continued for 5 days. The CellTiter 96 aqueous nonradioactive cell proliferation (MTS/PMS) assay (Promega, Madison, WI) was used to measure cell viability after drug treatment.
Combination index (CI) values were calculated as described by Chou.23 The fraction of unaffected (fu), in this case equivalent to cell viability, was calculated as described above. Fractional inhibition (fa) was calculated as 1 – fu. The slope factor m and IC50 of simvastatin and desloratadine were estimated by fitting the data of each drug when applied alone to Eq. 2,
| (2) |
CI values were then calculated using Eq. 3,
| (3) |
A CI ‐ fa plot was constructed by plotting CI values and fa on y and x axes, respectively.
ACKNOWLEDGMENTS
This research work is supported by the following grants: DK102694, GM10448301, and LM011945.
AUTHOR CONTRIBUTIONS
L.L. and X.H. wrote the article; L.L., S.K.Q., J.S.E., and D.F. designed the research; L.L., X.H., Z.W., and J.S.E. performed the research; L.L., X.H., and P.Z. analyzed the data; J.D., Z.D., and D.F. contributed new reagents/analytical tools.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- 1. Miller, M.L. Drug‐Induced Myopathies <http://www.uptodate.com/contents/drug-induced-myopathiess> (2013).
- 2. Kuncl, R.W. Agents and mechanisms of toxic myopathy. Curr. Opin. Neurol. 22, 506–515 (2009). [DOI] [PubMed] [Google Scholar]
- 3. Hart, G.W. Dynamic O‐linked glycosylation of nuclear and cytoskeletal proteins. Annu. Rev. Biochem. 66, 315–335. (1997). [DOI] [PubMed] [Google Scholar]
- 4. Sathasivam, S. Statin induced myotoxicity. Eur. J. Intern. Med. 23, 317–324 (2012). [DOI] [PubMed] [Google Scholar]
- 5. Furberg, C.D. & Pitt, B. Withdrawal of cerivastatin from the world market. Curr. Control. Trials Cardiovasc. Med. 2, 205–207 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Graham, D.J. et al Incidence of hospitalized rhabdomyolysis in patients treated with lipid‐lowering drugs. JAMA 292, 2585–2590 (2004). [DOI] [PubMed] [Google Scholar]
- 7. Hennessy, S. & Flockhart, D.A. The need for translational research on drug‐drug interactions. Clin. Pharmacol. Ther. 91, 771–773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schelleman, H. et al Pharmacoepidemiologic and in vitro evaluation of potential drug‐drug interactions of sulfonylureas with fibrates and statins. Br. J. Clin. Pharmacol. 78, 639–648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duke, J.D. et al Literature based drug interaction prediction with clinical assessment using electronic medical records: novel myopathy associated drug interactions. PLoS Comput. Biol. 8, e1002614 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stang, P.E. et al Advancing the science for active surveillance: rationale and design for the Observational Medical Outcomes Partnership. Ann. Intern. Med. 153, 600–606 (2010). [DOI] [PubMed] [Google Scholar]
- 11. Schech, S. et al Risk factors for statin‐associated rhabdomyolysis. Pharmacoepidemiol . Drug Saf. 16, 352–358 (2007). [DOI] [PubMed] [Google Scholar]
- 12. Wienkers, L.C. & Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 4, 825–833 (2005). [DOI] [PubMed] [Google Scholar]
- 13. FDA . Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. (2012).
- 14. Einolf, H.J. Comparison of different approaches to predict metabolic drug‐drug interactions. Xenobiotica 37, 1257–1294 (2007). [DOI] [PubMed] [Google Scholar]
- 15. Ghosal, A. et al Metabolism of loratadine and further characterization of its in vitro metabolites. Drug Metab. Lett. 3, 162–170 (2009). [DOI] [PubMed] [Google Scholar]
- 16. Vickers, A.E. , Zollinger, M. , Dannecker, R. , Tynes, R. , Heitz, F. & Fischer, V. In vitro metabolism of tegaserod in human liver and intestine: assessment of drug interactions. Drug Metab. Dispos. 29, 1269–1276 (2001). [PubMed] [Google Scholar]
- 17. Niemi, M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics 8, 787–802 (2007). [DOI] [PubMed] [Google Scholar]
- 18. Shitara, Y. , Maeda, K. , Ikejiri, K. , Yoshida, K. , Horie, T. & Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 34, 45–78 (2013). [DOI] [PubMed] [Google Scholar]
- 19. Link, E. et al SLCO1B1 variants and statin‐induced myopathy—a genomewide study. N. Engl. J. Med. 359, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 20. Zaher, H. et al Targeted disruption of murine organic anion‐transporting polypeptide 1b2 (Oatp1b2/Slco1b2) significantly alters disposition of prototypical drug substrates pravastatin and rifampin. Mol. Pharmacol. 74, 320–329 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Itagaki, M. , Takaguri, A. , Kano, S. , Kaneta, S. , Ichihara, K. & Satoh, K. Possible mechanisms underlying statin‐induced skeletal muscle toxicity in L6 fibroblasts and in rats. J. Pharmacol. Sci. 109, 94–101 (2009). [DOI] [PubMed] [Google Scholar]
- 22. Matzno, S. et al Statin‐induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J. Pharm. Pharmacol. 57, 1475–1484 (2005). [DOI] [PubMed] [Google Scholar]
- 23. Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58, 621–681 (2006). [DOI] [PubMed] [Google Scholar]
- 24. Greco, W.R. , Bravo, G. & Parsons, J.C. The search for synergy: a critical review from a response surface perspective. Pharmacol. Rev. 47, 331–385 (1995). [PubMed] [Google Scholar]
- 25. Yang, Q. et al Case‐only design to measure gene‐gene interaction. Epidemiology 10, 167–170. (1999). [PubMed] [Google Scholar]
- 26. Jacobson, T.A. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am. J. Cardiol. 94, 1140–1146 (2004). [DOI] [PubMed] [Google Scholar]
- 27. Kantola, T. , Kivisto, K.T. & Neuvonen, P.J. Erythromycin and verapamil considerably increase serum simvastatin and simvastatin acid concentrations. Clin. Pharmacol. Ther. 64, 177–182 (1998). [DOI] [PubMed] [Google Scholar]
- 28. Backman, J.T. , Kyrklund, C. , Kivisto, K.T. , Wang, J.S. & Neuvonen, P.J. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin. Pharmacol. Ther. 68, 122–129 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Merck. CLARITIN® Product Information.
- 30.Merck. CLARINEX® Product Information.
- 31. Skottheim, I.B. , Gedde‐Dahl, A. , Hejazifar, S. , Hoel, K. & Asberg, A. Statin induced myotoxicity: the lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur. J. Pharm. Sci. 33, 317–325 (2008). [DOI] [PubMed] [Google Scholar]
- 32. Zamek‐Gliszczynski, M.J. et al ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport‐mediated PK and DDIs in humans. Clin. Pharmacol. Ther. 94, 64–79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller, V.P. , Stresser, D.M. , Blanchard, A.P. , Turner, S. & Crespi, C.L. Fluorometric high‐throughput screening for inhibitors of cytochrome P450. Ann. N. Y. Acad. Sci. 919, 26–32 (2000). [DOI] [PubMed] [Google Scholar]
- 34. Rostami‐Hodjegan, A. & Tucker, G. ‘In silico' simulations to assess the ‘in vivo' consequences of ‘in vitro' metabolic drug‐drug interactions. Drug Discov. Today Technol. 1, 441–448 (2004). [DOI] [PubMed] [Google Scholar]
- 35. Khayat, Z.A. , Tsakiridis, T. , Ueyama, A. , Somwar, R. , Ebina, Y. & Klip, A. Rapid stimulation of glucose transport by mitochondrial uncoupling depends in part on cytosolic Ca2+ and cPKC. Am. J. Physiol. 275, C1487–C1497 (1998). [DOI] [PubMed] [Google Scholar]
- 36. Strenkoski‐Nix, L.C. , Ermer, J. , DeCleene, S. , Cevallos, W. & Mayer, P.R. Pharmacokinetics of promethazine hydrochloride after administration of rectal suppositories and oral syrup to healthy subjects. Am. J. Health Syst. Pharm. 57, 1499–1505 (2000). [DOI] [PubMed] [Google Scholar]
- 37. Hubble, J. et al Linear pharmacokinetic behavior of ropinirole during multiple dosing in patients with Parkinson's disease. J. Clin. Pharmacol. 40, 641–646 (2000). [PubMed] [Google Scholar]
- 38. Carr, R.A. et al Steady‐state pharmacokinetics and electrocardiographic pharmacodynamics of clarithromycin and loratadine after individual or concomitant administration. Antimicrob. Agents Chemother. 42, 1176–1180 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Appel‐Dingemanse, S. , Hirschberg, Y. , Osborne, S. , Pommier, F. & McLeod, J. Multiple‐dose pharmacokinetics confirm no accumulation and dose proportionality of the novel promotile drug tegaserod (HTF 919). Eur. J. Clin. Pharmacol. 56, 889–891 (2001). [DOI] [PubMed] [Google Scholar]
- 40. Bergman, A.J. et al Simvastatin does not have a clinically significant pharmacokinetic interaction with fenofibrate in humans. J. Clin. Pharmacol. 44, 1054–1062 (2004). [DOI] [PubMed] [Google Scholar]
- 41. Hallifax, D. & Houston, J.B. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab. Dispos. 34, 724–726; author reply 7 (2006). [DOI] [PubMed] [Google Scholar]
- 42. Kovacs, P. , Edwards, D.J. , Lalka, D. , Scheiwe, W.M. & Stoeckel, K. High‐dose omeprazole: use of a multiple‐dose study design to assess bioequivalence and accuracy of CYP2C19 phenotyping. Ther. Drug Monit. 21, 526–531 (1999). [DOI] [PubMed] [Google Scholar]
- 43. Greenblatt, D.J. , von Moltke, L.L. , Harmatz, J.S. , Ciraulo, D.A. & Shader, R.I. Alprazolam pharmacokinetics, metabolism, and plasma levels: clinical implications. J. Clin. Psychiatry 54 (suppl), 4–11; discussion 2–4 (1993). [PubMed] [Google Scholar]
- 44. Affrime, M. , Gupta, S. , Banfield, C. & Cohen, A. A pharmacokinetic profile of desloratadine in healthy adults, including elderly. Clin. Pharmacokinet. 41 (suppl.1), 13–19 (2002). [DOI] [PubMed] [Google Scholar]
- 45. Kouzuki, H. , Suzuki, H. , Ito, K. , Ohashi, R. & Sugiyama, Y. Contribution of sodium taurocholate co‐transporting polypeptide to the uptake of its possible substrates into rat hepatocytes. J. Pharmacol. Exp. Ther. 286, 1043–1050 (1998). [PubMed] [Google Scholar]
- 46. Shitara, Y. et al Function of uptake transporters for taurocholate and estradiol 17beta‐D‐glucuronide in cryopreserved human hepatocytes. Drug Metab. Pharmacokinet. 18, 33–41 (2003). [DOI] [PubMed] [Google Scholar]
- 47. Cheng, Y. & Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 (1973). [DOI] [PubMed] [Google Scholar]
- 48. Ramanathan, R. et al Disposition of loratadine in healthy volunteers. Xenobiotica 37, 753–769 (2007). [DOI] [PubMed] [Google Scholar]
- 49. Ramanathan, R. et al Disposition of desloratadine in healthy volunteers. Xenobiotica 37, 770–787 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information