

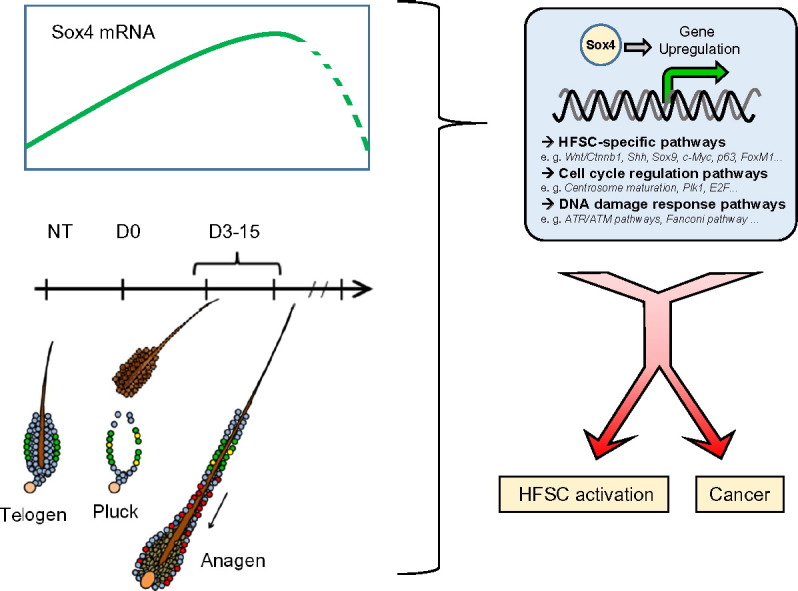
Abstract
Adult stem cells (ASCs) reside in specific niches in a quiescent state in adult mammals. Upon specific cues they become activated and respond by self-renewing and differentiating into newly generated specialised cells that ensure appropriate tissue fitness. ASC quiescence also serves as a tumour suppression mechanism by hampering cellular transformation and expansion (White AC et al., 2014). Some genes restricted to early embryonic development and adult stem cell niches are often potent modulators of stem cell quiescence, and derailed expression of these is commonly associated to cancer (Vervoort SJ et al., 2013). Among them, it has been shown that recommissioned Sox4 expression facilitates proliferation, survival and migration of malignant cells. By generating a conditional Knockout mouse model in stratified epithelia (Sox4cKO mice), we demonstrated a delayed plucking-induced Anagen in the absence of Sox4. Skin global transcriptome analysis revealed a prominent defect in the induction of transcriptional networks that control hair follicle stem cell (HFSC) activation such as those regulated by Wnt/Ctnnb1, Shh, Myc or Sox9, cell cycle and DNA damage response-associated pathways. Besides, Sox4cKO mice are resistant to skin carcinogenesis, thus linking Sox4 to both normal and pathological HFSC activation (Foronda M et al., 2014). Here we provide additional details on the analysis of Sox4-regulated transcriptome in Telogen and Anagen skin. The raw and processed microarray data is deposited in GEO under GSE58155.
Keywords: Adult stem cells, Sox4, Skin, Cancer, Microarray
Graphical abstract
| Specifications | |
|---|---|
| Organism/cell line/tissue | Mus musculus/keratinocytes/back skin epidermis |
| Sex | Male |
| Sequencer or array type | Whole Mouse Genome DNA Microarray (Agilent) |
| Data format | Raw and analysed |
| Experimental factors | Telogen vs Anagen. Sox4WT vs Sox4cKO mouse skin. |
| Experimental features | Telogen (resting) and Anagen (proliferative; 12 days post-plucking) skin from adult Sox4WT vs Sox4cKO mice was collected and total RNA was extracted to study global transcriptome changes, in the absence of Sox4, in active HFSCs. |
| Consent | Mice were maintained under specific pathogen-free conditions at the CNIO, and mouse experimentation was performed in agreement with the recommendations of FELASA. |
| Sample source location | N.A. |
1. Direct link to deposited data
The deposited data can be found at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58155.
2. Experimental design, materials and methods
2.1. Experimental groups and conditions
To understand the links between cancer and stem cell activation [1], [2], we generated Sox4cKO mice in stratified epithelia by crossing Sox4lox/lox with Keratin5-Cre transgenic mice [3], [4]. Sox4cKO mice display reduced Sox4 expression in the whole organism and undetectable levels in back and tail skin epidermis [3]. As Sox4 expression is restricted to active HFSCs, its depletion should principally affect this compartment during skin and hair regeneration [5], [6]. First, male mice were selected to avoid potential hormonal-dependent hair cycle alterations [7]. We initially performed Telogen skin analysis on Sox4WT vs Sox4cKO mice of 4–5 months of age (n = 3 mice per genotype). At this stage mouse skin enters into Anagen at a slower pace and asynchronously in small discrete areas, instead of happening in sagittal coordinated waves [7]. During sampling, we therefore avoided collecting Anagen skin patches arising from dominant behaviour among male mice or spontaneous Anagen induction patches [7], [8]. In order to study the contribution of Sox4 to HFSC activation, we performed hair plucking, a bona-fide method for assessing hair regeneration [5], [7], [8]. In this setting, we used young adult (4–5 months, 3 mice per genotype) and aged mice (17–18 months, 3 mice per genotype). We did not observe any differences in Anagen induction or in the contribution of Sox4 to modulating HFSC pathways in young versus aged mice [3], therefore all these mice were grouped in this study based solely on their genotype. To induce Anagen, 1 cm2 hair skin patch was plucked with tweezers and tape-stripped from the lower dorsal part of anaesthetized mice. 12 days later, the plucked patch is in full Anagen and can be identified macroscopically as a darkly pigmented skin area [7], [8].
2.2. Skin collection, RNA extraction and microarray hybridization
A critical point to obtain high quality RNA from skin is to preserve the samples immediately after collection by immersing them on RNAlater (QIAGEN); this method works for skin RNA extraction even more efficiently than liquid nitrogen flash-freezing, in our experience. For skin collection we first hair-clipped the mice and then culled them by cervical dislocation to minimize the time elapsed before tissue harvesting. Dorsal back skin was then cut and pulled with forceps and placed dermis side up in a Petri dish; then, the dermal fat was thoroughly removed by scrapping with a scalpel, immediately immersed in RNAlater in a new Petri dish and cut into 3 × 3 small pieces. RNAlater-soaked samples were preserved in 1.5 mL of fresh RNAlater solution at 4 °C overnight and − 80 °C thereafter until RNA extraction. After removing RNAlater completely, we then performed tissue disruption and RNA extraction. For this, tissue explants were placed on a 2 mL homogenisation tube containing 1 mL of TRIzol reagent (Life Technologies) and 5 zirconium oxide beads per 3 × 3mm2 tissue bits. After 2 runs (4 cycles) on programme 2 in a Precellys-24 Tissue disruptor (Precellys), we proceeded to TRIzol-based RNA extraction; we adjusted the hydrophobicity of TRIzol's chloroform-extracted aqueous phase by adding ethanol up to a final concentration of 36%, and applied it directly to a silica spin column for RNA purification (RNeasy, QIAGEN). If RNA integrity number (RIN) is not greater than 7.5, additional RNA extraction from the original sample might be required to fulfil this Quality Control (QC). Nevertheless, a sample with RIN 6.7 was successfully analysed in this study. As an alternative approach we recommend using the Fibrous Tissue RNeasy minikit (QIAGEN) that includes a Proteinase K step, and gives high RNA yields and good RIN values. RNA integrity numbers were obtained on an Agilent 2100 Bioanalyzer. A final amount of 100 ng of total RNA was labelled using a Low Input Quick Amp Labelling kit (“One-color Microarray-based Gene Expression analysis kit” v6.5, Agilent) following the manufacturer's instructions. Labelled samples were further purified on silica spin columns (RNeasy, QIAGEN). Finally, samples were hybridized to a 60K Whole Mouse Genome DNA Microarray (Agilent design ID 028005 SurePrint G3 Mouse GE 8x60K Microarray) in a SureHyb chamber (Agilent) in a final volume of 50 μL during 17 h at 65 °C. Subsequently, arrays were washed and images obtained with a G2505C DNA microarray scanner (Agilent).
2.3. Gene expression analysis
Images were analysed with Agilent Feature Extraction (FE) Software (ver. 10.1.1). FE performs spot quantitation and systematic gradient correction by spatial de-trending algorithms. QC reports provided by FE (which inform on signal qualities, background level and overall sensitivity within well-established acceptance thresholds) were examined for the absence of anomalies. We then performed background subtraction using the normexp method [9]. To normalize the dataset, we performed loess within-array normalization and quantile approach for between-array normalization. Differentially expressed genes were obtained by applying linear models with R limma package [10] (Bioconductor project, http://www.bioconductor.org). To account for multiple hypothesis testing, the estimated significance level (p value) was adjusted using Benjamini & Hochberg false discovery rate (FDR) correction [11]. Those genes with FDR < 0.05 were selected as differentially expressed genes (DEGs). Gene set enrichment analysis (GSEA) was studied using annotations from NCI [12], KEGG [13] and Reactome [14] databases. We additionally studied some custom-made gene lists obtained from genomic datasets publicly available [3], [5]. Genes were ranked according to their limma moderated t statistic. After Kolmogorov–Smirnov testing, those gene sets showing FDR < 0.25, a well-established cut-off for the identification of biologically relevant gene sets [15], were considered enriched between Sox4WT vs Sox4cKO mice (Fig. 1A and B).
Fig. 1.

A) Venn's diagram showing the overlap between the significantly enriched pathways in Sox4WT vs Sox4cKO mouse skin in Telogen (green) and Anagen (yellow). Only pathways showing FDR < 0.05 were included for this comparison. Note that most deregulated pathways belong to the Anagen group, reinforcing a role for Sox4 in this condition, and that the overlap is minimal between both categories.
B) Enrichment plots for selected pathways, obtained from the NCI repository at FDR < 0.150, where N indicates the number of genes included in each list, and FDR means false discovery rate (q value). The red to blue bar represents the ranked list of genes (red = Sox4WT, blue = Sox4cKO). Genes showing differential expression between genotypes are located at the edges of the bar, and similarly-expressed genes are in the centre. The Y axis represents the enrichment score (ES). Plk1, ATM, nuclear β-catenin, E2F, FoxM1 and TAP63 pathways are shown from left to right and top to bottom.
C) Comparison of the values obtained by qPCR and microarray in Sox4WT vs Sox4cKO mouse skin, for the selected DEGs. Fold change relative to Sox4WT (set to 1) is shown. N = 6 mice per genotype.
2.4. Microarray validation
To validate the DEGs and the significantly changed pathways, we extracted RNA from similar groups of mice following RNA later and RNeasy protocols (described above). We retrotranscribed 500 ng of total RNA using iSCRIPT Advanced (Bio-Rad) following the manufacturer's instructions. We used 1/10th dilutions of the resulting cDNA for subsequent quantitative-real time PCR (qPCR) in a 7900HT 384-well real-time fast thermocycler (Applied Biosystems) using SYBR 2 × qPCR master mix (Life Technologies). Fold change (FC) of genes of interest was further validated by comparing the results obtained in the microarray. Most DEGs displayed consistent FC in microarray versus qPCR values (Fig. 1C).
3. Discussion
Adult tissues are composed by specialised cells that need to be constantly fuelled up with fresh cells upon tissue damage or during normal homeostatic turnover. In one of the most paradigmatic examples, skin replenishment is carried out by its tissue stem cells, which have a high proliferative potential when compared to their progeny, but remain in a relatively quiescent state until the appropriate signals pull them out of their niches to migrate, differentiate and specialise [16]. On one hand, it is acknowledged that stem cell function decline is one of the hallmarks of ageing, and molecular ageing is linked to decreased stem cell activation potential [17], [18]. On the other hand, adult stem cell activation constitutes a prerequisite to license carcinogenesis initiation and progression, at least in a two-step chemically-induced mouse skin carcinogenesis model [1]. Therefore, this leads to the speculation that there might be common signals that link stem cell activation with ageing and cancer. We proved that Sox4 is a key factor that warrants normal hair follicle stem cell activation during hair regeneration and wound healing. Interestingly, abrogation of Sox4 expression in skin prevented chemically-induced tumorigenesis and globally reduced mRNA levels correlated with diminished incidence of spontaneous cancer in mice [3]. By analysing the transcriptome of Sox4-depleted skin during hair regeneration we observed deficiencies in the induction of pathways that modulate HFSC activation, but we also detected strong deregulation of groups of genes that are typically associated to cancer. In summary, our results demonstrate that understanding the Sox4-regulated transcriptome might be an interesting subject of study for effective cancer therapies and controlled regenerative medicine approaches, and thus we hope that this data resource constitutes a valuable tool for the research community.
Author contributions
MF performed most of the experiments. LM-P helped with microarray validation. GG-L and DG-P performed Bioinformatics analysis. OD performed microarray hybridization and scanning. MF and MAB conceived and designed the experiments, interpreted results and wrote the paper. MAB supervised the project and secured funding. All the authors commented and approved the final manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We are indebted to G. Luengo and the CNIO Technical Units for technical support and the Blasco and Serrano Lab members for scientific discussion. MF was funded by a Long-Term EMBO Fellowship (ALTF 836-2014) and a Marie Sklowdoska-Curie IF (661656). The Blasco Lab is funded by the Spanish Ministry of Economy and Competitiveness Projects SAF2008-05384 and CSD2007-00017, the European Union FP7 Projects 2007-A-201630 (GENICA) and 2007-A-200950 (TELOMARKER), the European Research Council (ERC) Project TEL STEM CELL (GA # 232854), the Körber Foundation, the AXA Research Fund, Fundación Botín and Fundación Lilly (Spain).
Contributor Information
Miguel Foronda, Email: miguel.foronda@ed.ac.uk.
Maria A. Blasco, Email: mblasco@cnio.es.
References
- 1.White A.C., Khuu J.K., Dang C.Y., Hu J., Tran K.V., Liu A., Gomez S., Zhang Z., Yi R., Scumpia P., Grigorian M., Lowry W.E. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat. Cell Biol. 2014;16(1):99–107. doi: 10.1038/ncb2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vervoort S.J., van Boxtel R., Coffer P.J. The role of SRY-related HMG box transcription factor 4 (SOX4) in tumorigenesis and metastasis: friend or foe? Oncogene. 2013;32:3397–3409. doi: 10.1038/onc.2012.506. [DOI] [PubMed] [Google Scholar]
- 3.Foronda M., Martinez P., Schoeftner S., Gomez-López G., Schneider R.P., Flores J.M., Pisano D.G., Blasco M.A. Sox4 links tumor suppression to accelerated aging in mice by modulating stem cell activation. Cell Rep. 2014;8(2):487–500. doi: 10.1016/j.celrep.2014.06.031. (24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramirez A., Page A., Gandarillas A., Zanet J., Pibre S., Vidal M., Tusell L., Genesca A., Whitaker D.A., Melton D.W., Jorcano J.L. A keratin K5Cre transgenic line appropriate for tissue-specific or generalized Cre-mediated recombination. Genesis. 2004;39(1):52–57. doi: 10.1002/gene.20025. [DOI] [PubMed] [Google Scholar]
- 5.Lien W.H., Polak L., Lin M., Lay K., Zheng D., Fuchs E. In vivo transcriptional governance of hair follicle stem cells by canonical Wnt regulators. Nat. Cell Biol. 2014;16(2):179–190. doi: 10.1038/ncb2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greco V., Chen T., Rendl M., Schober M., Pasolli H.A., Stokes N., Dela Cruz-Racelis J., Fuchs E. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell. 2009;4(2):155–169. doi: 10.1016/j.stem.2008.12.009. (6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller-Rover S., Handjiski B., van der Veen C., Eichmuller S., Foitzik K., McKay I.A., Stenn K.S., Paus R. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Investig. Dermatol. 2001;117(1):3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- 8.Jensen K.B., Driskell R.R., Watt F.M. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat. Protoc. 2010;5(5):898–911. doi: 10.1038/nprot.2010.39. [DOI] [PubMed] [Google Scholar]
- 9.Ritchie M.E., Silver J., Oshlack A., Holmes M., Diyagama D., Holloway A., Smyth G.K. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23(20):2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- 10.Smyth G.K., Michaud J., Scott H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21(9):2067–2075. doi: 10.1093/bioinformatics/bti270. (1) [DOI] [PubMed] [Google Scholar]
- 11.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995;57(1):289–300. [Google Scholar]
- 12.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., Yefanov A., Lee H., Zhang N., Robertson C.L., Serova N., Davis S., Soboleva A. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41(Database issue):D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R., Jassal B., Jupe S., Matthews L., May B., Palatnik S., Rothfels K., Shamovsky V., Song H., Williams M., Birney E., Hermjakob H., Stein L., D'Eustachio P. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42(Database issue):D472–D477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solanas G., Benitah S.A. Regenerating the skin: a task for the heterogeneous stem cell pool and surrounding niche. Nat. Rev. Mol. Cell Biol. 2013;14(11):737–748. doi: 10.1038/nrm3675. [DOI] [PubMed] [Google Scholar]
- 17.Sharpless N.E., DePinho R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 18.López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–11217. doi: 10.1016/j.cell.2013.05.039. (6) [DOI] [PMC free article] [PubMed] [Google Scholar]