

Abstract
Proper functioning of intracellular membranes is critical for many cellular processes. A key feature of membranes is their ability to adapt to changes in environmental conditions by adjusting their composition so as to maintain constant biophysical properties, including fluidity and flexibility. Similar changes in the biophysical properties of membranes likely occur when intracellular processes, such as vesicle formation and fusion, require dramatic changes in membrane curvature. Similar modifications must also be made when nuclear pore complexes (NPCs) are constructed within the existing nuclear membrane, as occurs during interphase in all eukaryotes. Here we report on the role of the essential nuclear envelope/endoplasmic reticulum (NE/ER) protein Brl1 in regulating the membrane composition of the NE/ER. We show that Brl1 and two other proteins characterized previously—Brr6, which is closely related to Brl1, and Apq12—function together and are required for lipid homeostasis. All three transmembrane proteins are localized to the NE and can be coprecipitated. As has been shown for mutations affecting Brr6 and Apq12, mutations in Brl1 lead to defects in lipid metabolism, increased sensitivity to drugs that inhibit enzymes involved in lipid synthesis, and strong genetic interactions with mutations affecting lipid metabolism. Mutations affecting Brl1 or Brr6 or the absence of Apq12 leads to hyperfluid membranes, because mutant cells are hypersensitive to agents that increase membrane fluidity. We suggest that the defects in nuclear pore complex biogenesis and mRNA export seen in these mutants are consequences of defects in maintaining the biophysical properties of the NE.
INTRODUCTION
The nuclear envelope (NE) of eukaryotic cells compartmentalizes the nuclear material and separates it from the cytoplasm. The double membrane of the NE consists of an outer and an inner nuclear membrane (ONM and INM) that differ in protein and lipid composition. The NE is structurally and functionally related to the endoplasmic reticulum (ER), and the ONM is contiguous with the ER (1, 2). Embedded in the NE are the nuclear pore complexes (NPCs) and, in budding yeast, the spindle pole body (SPB). NPCs are extremely large and are constructed from multiple copies of about 30 different nucleoporins (nups) (3, 4). NPCs mediate selective trafficking of proteins and other macromolecules between the nucleus and the cytoplasm but also serve other important functions, including gene activation and mRNA surveillance (5, 6). The biogenesis of NPCs and their distribution over the NE are highly regulated processes and are coordinated with the cell cycle (7). During interphase, the number of NPCs doubles. In budding yeast, the NE remains intact throughout the cell cycle, and all the formation of NPCs occurs through de novo construction within the NE.
In addition to the ONM and the INM, the NE contains a pore membrane domain (POM), formed by the fusion of the INM and ONM at sites where NPCs are assembled (8). The POM is a highly curved region of the NE that is intimately associated with nucleoporins, including multiple integral membrane nups. Early steps of the fusion of the inner and outer membrane leaflets of the NE that accompanies NPC formation require extensive changes in membrane curvature and thus depend directly on lipidic factors, such as the shape and size of lipid molecules. For example, in vitro reconstitution experiments using Xenopus egg extracts showed that pore formation was inhibited by lysophosphatidylcholine, and this inhibition was completely reversed by the concomitant addition of oleic acid or phosphatidylethanolamine (9). Interestingly, deletion of membrane-bending reticulons in yeast cells causes defects in NPC biogenesis. Reticulons have therefore been regarded as essential factors for proper NPC biogenesis and distribution (10). Additionally, many NPC proteins, including yeast Nup85, Nup120, and Nup133, contain amphipathic helical ALPS (ArfGAP1 lipid-packing sensor) motifs that sense lipid packaging at the curved membranes of the POM (11). Thus, a complex array of lipid-protein interactions is thought to stabilize the highly curved pore membranes, but the roles of these interactions in membrane fusion and curvature are not fully understood (12, 13).
Recent findings from our labs suggest that NPC biogenesis is very sensitive to alterations in the membrane composition and biophysical properties of the NE/ER. Two integral membrane proteins of the NE/ER, Apq12 and Brr6, are required for efficient NPC biogenesis and are implicated in mediating lipid homeostasis in budding yeast (14, 15). Apq12 was identified in multiple genetic screens that suggested a possible role in mRNA metabolism, since the absence of Apq12 led to defects in mRNA export and 3′ pre-mRNA processing (16, 17). Subsequent analysis showed that cells lacking Apq12 are cold sensitive for growth, exhibit changes in the membrane properties and morphology of the NE following a shift to a lower temperature, and are defective in the assembly of NPCs at nonpermissive temperatures (14).
BBR6 was identified initially in a screen for cold-sensitive mutants affecting mRNA export; depletion of BRR6 led to abnormalities in NPC distribution and NE morphology (18). We identified BRR6 as a dosage suppressor of the growth and mRNA export defects seen in apq12Δ cells and showed that brr6 mutant cells display defects in nuclear envelope structure and NPC assembly very similar to those seen in apq12Δ cells (15). Both the apq12Δ mutant and the brr6 conditional mutant are hypersensitive to drugs that inhibit lipid biosynthesis and show synthetically lethal interactions with mutations in various lipid biosynthetic pathways (14, 15). As in the budding yeast, mutations affecting the orthologous protein of Schizosaccharomyces pombe also lead to changes in membrane properties (19). These results support the hypothesis that Brr6 and Apq12 monitor the membrane environment and coordinate the changes in the lipid composition of the NE required to maintain optimal function, thereby facilitating the construction of supramolecular protein complexes such as NPCs and SPBs (14, 19, 20).
Many fungi, including Saccharomyces cerevisiae, contain a gene closely related to BRR6 called BRL1. BRL1 was initially identified in budding yeast as a suppressor of mutations of the nuclear exportin Crm1/Xpo1, which mediates the export of many proteins and some RNAs from the nucleus (21). Members of the Brr6/Brl1 family of proteins are found in all fungi and lower eukaryotes that undergo closed mitosis. Whereas S. cerevisiae and many closely related fungi have both genes, S. pombe, Pneumocystis carinii, and other fungi distantly related to S. cerevisiae have a unique gene that is more closely related to Brl1 than to Brr6 (19, 22). Brl1 is not a nucleoporin; its distribution in the NE is unaffected in strains carrying nucleoporin mutations that cause NPCs to cluster together (18, 21). Brr6 and Brl1 of budding yeast show synthetic genetic interactions. The two proteins have been shown to interact in a yeast two-hybrid analysis, although it was not determined how direct their interaction is (21). Interestingly, S. pombe Brr6 has been shown to be required for the insertion of SPBs into the NE and for NE integrity during the late stages of mitosis (19).
We report here the isolation and characterization of two conditional alleles of BRL1, brl1-1 and brl1-2. We show that these mutants behave similarly to apq12Δ and brr6 mutants in causing defects in mRNA export, NPC biogenesis, and lipid metabolism. brl1 mutant cells accumulate excess sterols and sterol precursors, and the synthesis of neutral lipids becomes essential. brl1 mutants show synthetic lethality with mutations affecting lipid biosynthetic pathways, and the cells are hypersensitive to drugs that inhibit these pathways and to agents that increase membrane fluidity.
Our earlier findings with brr6 and apq12Δ mutants led us to hypothesize that cells carrying mutations affecting Brr6 and Apq12 are able to sense a downshift in temperature, leading to modification of their membrane lipid composition, but are unable to sense when proper membrane properties have been restored, a defect that results in hyperfluidization (14, 15). Here we show that brl1 mutants display all of the phenotypes of brr6 and apq12Δ mutant cells. Importantly, results from fluidity adaptation experiments provide strong support for the hypothesis that brr6, brl1, and apq12Δ mutants overshoot in their compensatory changes. Upon a temperature downshift, mutants accumulate higher levels of monounsaturated versus saturated fatty acids, and display a greater shift to shorter fatty acid chain lengths, than wild-type (WT) cells. Together, these biochemical and genetic data suggest that Brl1, Brr6, and Apq12 are candidates for components of a sensory complex that plays a role in ensuring appropriate alterations in membrane composition needed to maintain the proper biophysical properties of ER and NE membranes. In support of the notion that Brl1, Brr6, and Apq12 are components of such a complex, we find that the proteins partially colocalize in punctate structures at the NE and can be coprecipitated. The interaction of Brl1 and Brr6 is reduced in cells lacking Apq12, suggesting that at least some of these interactions involve an Apq12–Brr6–Brl1 complex.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The strains used in this work are listed in Table S1 in the supplemental material. Deletion mutants were obtained by standard yeast transformation protocols using loxP marker deletion cassettes (23). Strains were grown in YPD rich medium (1% Bacto yeast extract, 2% Bacto peptone [US Biological, Swampscott, MA], 2% glucose [Reactolab, Servion, Switzerland]) or minimal medium (0.67% yeast nitrogen base without amino acids [US Biological], 2% glucose, 0.73 g/liter amino acids). To test the sensitivities of mutants to different drugs, strains were grown in minimal or YPD medium; the cultures were diluted back to a starting optical density at 600 nm (OD600) of 0.2; and then 1/10 serial dilutions were made. Serial dilutions were spotted onto plates, which were incubated for 3 to 5 days.
Microscopy.
The micrographs in Fig. 1 were recorded using a Nikon TE2000-E microscope (Nikon 100× Plan Apochromat oil objective; numerical aperture [NA], 1.4), an Orca-ER charge-coupled-device (CCD) camera (Hamamatsu), and Volocity software, version 5.3.2 (PerkinElmer). To stain lipid droplets (LDs) with 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene (BODIPY), cells were incubated with BODIPY 493/503 (Invitrogen, Life Technologies) at a final concentration of 1 μg/ml for 30 min in the dark. Cells were washed once with phosphate-buffered saline (PBS) containing 50 μM bovine serum albumin (BSA) (fatty acid free) and once with PBS, resuspended in residual PBS, and analyzed by fluorescence microscopy using a Carl Zeiss Axioplan 2 microscope (Carl Zeiss, Oberkochen, Germany) fitted with a AxioCam CCD camera and AxioVision software, version 3.1. The genomically tagged proteins shown in Fig. 6 and in Fig. S5 and S6 in the supplemental material were visualized using a DeltaVision Elite imaging system (GE Healthcare, Pittsburgh, PA), consisting of an Olympus 1X71 inverted microscope equipped with a CCD camera (CoolSNAP HQ2; Photometrics). Images were acquired with a UPlan S-Apo 100× (NA, 1.4) oil immersion objective (Olympus) and a green fluorescent protein (GFP)/mCherry filter set. Six to 10 0.2-μm-thick optical sections taken through the nucleus were deconvoluted using the iterative constrained deconvolution program in softWoRx (Applied Precision). Single sections are displayed.
FIG 1.
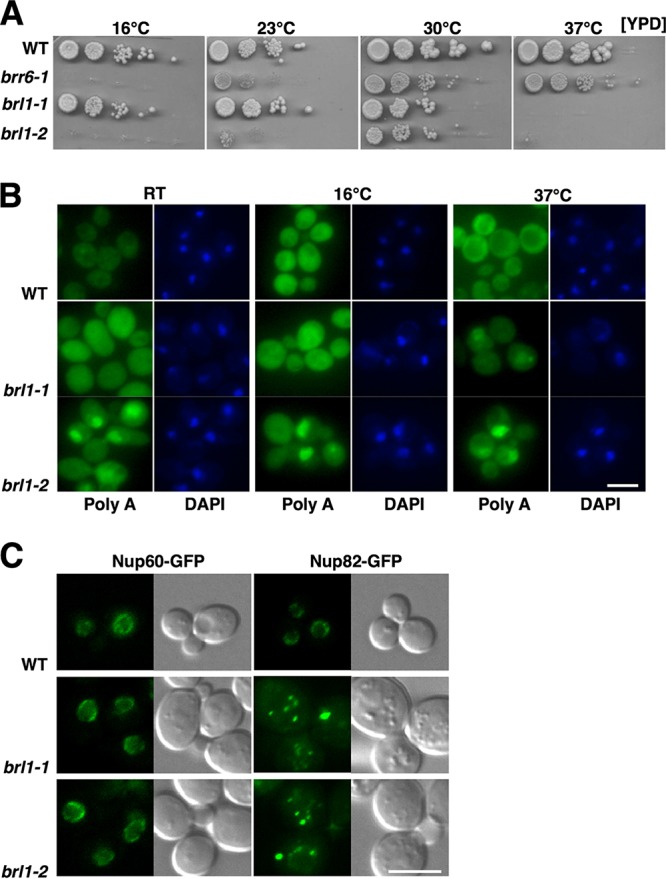
Mutants with mutations in Brl1 are temperature sensitive and affect mRNA export and NPC assembly and distribution. (A) Temperature sensitivity of brl1 mutant strains. Cells were grown at 30°C in YPD, and serial dilutions were spotted onto YPD plates. (B) Mutations in Brl1 affect mRNA export. Cells were incubated at the indicated temperatures for 18 h, and the subcellular distribution of poly(A)+ mRNA was analyzed by in situ hybridization. DNA was stained with DAPI. (C) Brl1 affects the distribution and assembly of the NPC. The distribution of the nuclear basket protein Nup60-GFP and that of Nup82-GFP, a component of the cytoplasmic filaments, were analyzed by fluorescence microscopy. Bar, 5 μm.
FIG 6.

Brl1 localizes in punctate structures within the NE and interacts with Brr6 and Apq12. (A) Brl1-GFP localizes to a few puncta within the NE. Cells of the indicated genotypes were cultivated to the mid-log phase (OD600, 1 to 2) at 30°C, except in the experiment performed with nup120Δ mutants (24°C), and were analyzed by fluorescence microcopy. All proteins visualized in this panel have been tagged at their carboxy termini. Colocalization is indicated by arrows. Bar, 5 μm. (B) Brl1 interacts with Brr6 and Apq12. Lysates from cells expressing Apq12-GFP, Brr6-HA, and Brl1-HA (load) were immunoprecipitated with antibodies against GFP. Immunopurified fractions were analyzed by Western blotting for the presence of Brr6-HA and Brl1-HA (IP). The yeast endoplasmic reticulum transmembrane protein 3 (Yet3) served as a loading control. (C) Lysates from cells expressing Brr6–13Myc (load) were immunoprecipitated (precipitate) with antibodies against the Myc epitope. Fractions were analyzed by Western blotting for the presence of Brr6–13Myc and Brl1-HA, in the presence and absence of Apq12. The presence of the 13Myc tag shifts the molecular mass of Brr6 by 15.6 kDa.
Fluorescence in situ hybridization.
Cells were grown overnight at room temperature (RT), back diluted, and allowed to regrow for 4 h before being shifted overnight to the nonpermissive temperatures (16°C and 37°C). Room temperature samples were again back diluted the following day and were allowed to recover for 2 h before all cells were processed for in situ hybridization as described previously (24).
Fatty acid analysis.
Fatty acid methyl esters (FAMEs) were prepared from 50 OD600 units of yeast cells. Wild-type and mutant cells were grown in synthetic complete (SC) medium at 24°C and were diluted to an OD600 of 1. Aliquots were either shifted to 16°C (overnight) or left at 24°C. Cells were washed once in cold water, and a fatty acid standard (C17:0; 50 μg; Sigma) was added. Cells were disrupted using glass beads, and lipids were extracted using chloroform-methanol (1:1). FAMEs were produced using boron trifluoride at 100°C for 45 min and were recovered by extraction with petrol ether. After evaporation, extracts were resuspended in hexane. FAMEs were separated with an Agilent 7890A gas chromatograph (GC) equipped with a DB-23 capillary column (30 m by 250 mm by 0.25 mm; Agilent Technologies, Santa Clara, CA). The temperature of the injection port was set to 250°C, its pressure to 26.24 lb/in2 (average velocity, 48.17 cm/s), and the septum purge flow to 3 ml/min. Split injections occurred through an Agilent 7693A automated liquid sampler. The initial oven temperature (100°C; held for 2 min) was increased to 160°C at the rate of 25°C/min and was then increased again to 250°C at 8°C/min. The final oven temperature was held for an additional 4 min. FAMEs were detected with a flame ionization detector (Agilent Technologies) set at 270°C with H2, air, and He flows set at 30, 400, and 27.7 ml/min, respectively. FAMEs were quantified relative to the internal standard, and the relative response factor for each FAME was determined from a 4-level calibration curve (r2, >0.999).
Immunoprecipitation (IP).
Two liters of YP medium supplemented with 2% galactose (YPG) was inoculated using a 20-ml YPG culture, and the mixture was shaken overnight at 24°C. Cells were harvested, resuspended in 10 mM HEPES (pH 7.4) and 1.2% polyvinylpyrrolidone, pelleted, and frozen in liquid nitrogen. Cells were then homogenized for 3 min in dry ice using commercial coffee grinders and were stored at −80°C. To prepare lysates for immunopurification, 2 g of the homogenized cell powder was added to 4 ml of lysis buffer composed of 20 mM HEPES (pH 7.4), 110 mM potassium acetate, 150 mM sodium chloride, 2 mM magnesium chloride, 1 mM EDTA, 1% filtered Triton X-100 (SLHV033RS filter; pore size, 0.45 μm; Millipore), and protease inhibitors (product no. 11836170001; Roche). The ground cells were then resuspended, chilled on ice for 10 min, and clarified with a low-speed spin at 3,200 relative centrifugal force (RCF). The soluble lysate was transferred to a fresh tube; load fractions were taken; and 40 μl of anti-GFP or anti-Myc magnetic beads was added to the remaining suspension (catalog no. D153-9 and M047-1; Medical & Biological Laboratories). Lysate bead slurries were rotated at 4°C for 2 h. The beads were then pelleted on ice using magnetic racks and were rinsed 3 times with 0.5 ml of the remaining cold lysis buffer. Proteins were eluted off beads with SDS loading buffer, and immunoblotting was performed by standard methods. GFP-tagged proteins were detected with a rabbit-raised anti-GFP antibody provided by Bill Wickner. Hemagglutinin (HA)-tagged proteins were detected with a mouse monoclonal antibody (H9658; Sigma). Myc epitopes were detected with an anti-c-Myc mouse monoclonal antibody (9E10; Santa Cruz Biotechnology). Yet3 was detected with a rabbit-raised anti-Yet3 antibody given to us by Charlie Barlowe.
RESULTS
Growth, mRNA export, and NPC assembly defects of brl1 mutants.
To study Brl1 functions, we isolated two brl1 alleles following error-prone PCR and screening for cold- and heat-sensitive growth. The brl1-1 mutant is temperature sensitive and fails to grow at 37°C. As seen previously with the brr6-1 mutant, the brl1-2 mutant exhibits limited growth at 23°C and fails to grow at either 16°C or 37°C, indicating that this allele is sensitive to both cold and heat. brr6-1, brl1-1, and brl1-2 cells grow best at 30°C, although none of them grows as well as the wild type at this temperature (Fig. 1A).
We next performed in situ hybridization analysis to localize poly(A)+ RNA in mutant cells. brl1-1 cells do not accumulate poly(A)+ RNA in nuclei when grown at either 16°C or room temperature, two conditions under which mutant cells can grow. However, when shifted to 37°C, brl1-1 cells showed moderate accumulation of poly(A)+ RNA in nuclei, indicating a defect in mRNA export. The brl1-2 mutant was more defective for mRNA export when grown at 16°C, 23°C, and 37°C, temperatures at which it either fails to grow or shows very limited growth (Fig. 1B).
The effects of brl1 mutations on NPC assembly and distribution were assessed by localizing two GFP-tagged nucleoporins, Nup60 and Nup82. Brl1 mutant cells displayed a near-normal distribution of Nup60-GFP, a component of the nuclear basket of NPCs, but may have a mild NPC clustering phenotype (Fig. 1C) (21). In contrast, Nup82-GFP, a component of the cytoplasmic filaments (CFs), lacked signal at the nuclear periphery and displayed aberrant localization in large punctate structures in the brl1 mutants, indicating that NPC assembly is defective in these mutants (Fig. 1C). Taken together, these results demonstrate that Brl1 is essential for growth, NPC assembly, and optimal mRNA export. In this respect, brl1 mutants closely resemble brr6-1 and apq12Δ mutants. These three types of mutants are referred to collectively below as BBA mutants and their protein products as BBA proteins.
Defects in sterol metabolism and genetic interactions in brl1 mutant cells.
Given the high degree of similarity between brl1 and brr6 mutants, including the shared defects in growth and mRNA export, as well as nucleoporin mislocalization, it was not surprising to find that brl1 mutants also have defects very similar to those we reported for brr6-1 and apq12Δ mutants with respect to lipid metabolism, sensitivity to drugs that inhibit lipid biosynthetic pathways, and genetic interactions with genes encoding enzymes involved in lipid biosynthesis (14, 15).
Mutants with mutations that directly affect ergosterol metabolism or indirectly disturb sterol homeostasis are often hypersensitive to inhibitors of sterol biosynthetic enzymes. brl1 mutants were hypersensitive to drugs that inhibit sterol biosynthesis (terbinafine, fenpropimorph, and fluconazole) even at 30°C, their optimal growth temperature (Fig. 2A). In accordance with the drug sensitivity observed, brl1 mutations exhibited strong aggravating genetic interactions with mutations in sterol biosynthesis and transport (erg5Δ, erg6Δ, arv1Δ) (Table 1).
FIG 2.
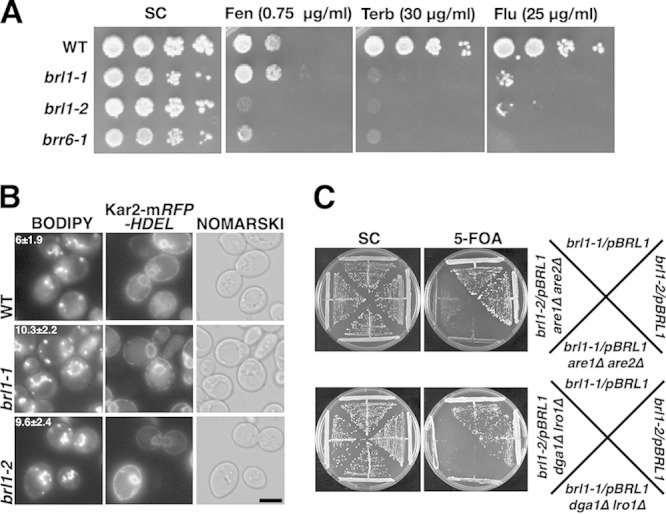
brl1 mutants have defects in sterol homeostasis. (A) Hypersensitivity of brl1 mutant cells to inhibitors of sterol biosynthesis. Serial dilutions of wild-type, brl1, and brr6 strains were spotted onto SC plates containing the indicated concentration of fenpropimorph (Fen), terbinafine (Terb), or fluconazole (Flu), and plates were incubated at 30°C. (B) brl1 cells accumulate lipid droplets (LDs) and have aberrant LD morphology. Cells of the indicated genotypes were grown in SC medium, and LDs were stained with BODIPY 493/503. Colocalization with the ER marker Kar2-mRFP-HDEL reveals perinuclear clustering of LDs in brl1 cells. The average number of LDs ± the standard deviation of the mean (n, 100 cells) is given for each genotype. Bar, 5 μm. (C) brl1 mutants are synthetically lethal with mutants that fail to synthesize TAG (dga1Δ lro1Δ mutants) or STE (are1Δ are2Δ mutants). brl1 mutant cells rescued by a plasmid-borne wild-type copy of BRL1 (URA3) were deleted for the indicated neutral-lipid biosynthetic gene, and loss of the wild-type copy of BRL1 was assessed on plates containing 5-fluoroorotic acid (5-FOA).
TABLE 1.
Genetic interactions of brl1 mutationsa with mutations affecting lipid biosynthesis
| Genotype of lipid mutant tested | Function of encoded proteinb | Interactionc |
|---|---|---|
| elo2Δ | Fatty acid elongation | SL |
| elo3Δ | Fatty acid elongation | SL |
| arv1Δ | Sterol homeostasis, GPI anchor synthesis | SL |
| erg5Δ | Ergosterol biosynthesis | SL |
| erg6Δ | Ergosterol biosynthesis | SL |
| mga2Δ | Transcription factor for OLE1 | SS |
| spt23Δ | Transcription factor for OLE1 | SS |
The brl1-1 and brl1-2 mutants show similar interactions with the mutants listed. The growth of the double mutants was assessed at 30°C.
GPI, glycosylphosphatidylinositol.
SL, synthetically lethal; SS, synthetically sick.
brl1 mutants also displayed much higher levels of the sterol ergosterol and its precursors than wild-type cells (see Fig. S1A and B in the supplemental material). We previously identified BRR6 as a dosage suppressor of the cold-sensitive growth and mRNA export defects of apq12Δ cells and, conversely, showed that overexpression of Apq12 partially suppressed the defects in mRNA export, NPC biogenesis, and aberrant sterol accumulation exhibited by brr6 cells (15). We found that overexpression of APQ12 or BRR6 restores wild-type levels of both ergosterol and episterol in both brl1 mutants (see Fig. S2A to D in the supplemental material). Likewise, overexpression of wild-type BRL1 or APQ12 decreased the levels of these sterols in a brr6 background (see Fig. S2E and F in the supplemental material). These data indicate that the three BBA proteins share a function in sterol homeostasis, since overexpression of any one of them can suppress the altered sterol phenotype induced by mutations in the other genes.
Neutral lipid synthesis is essential for brl1 mutants.
Yeast and mammalian cells esterify sterols to form steryl esters (STE), which are removed from membranes and packaged into lipid droplets (LDs). In addition to STE, LDs also contain high levels of triacylglycerols (TAG). Storage of STE and TAG in LDs allows cells to sequester both free sterols and free fatty acids and thus suppress the toxicity associated with the accumulation of these lipids. These neutral lipids can be mobilized rapidly when cells need to form new membranes, for example, upon the resumption of growth following dilution into fresh medium. STE levels were considerably higher in brl1 mutant cells than in wild-type cells (see Fig. S3 in the supplemental material).
The overproduction of STE in brl1 mutants prompted us to look for any alterations in the number or morphology of LDs in these cells. LDs were stained with the neutral lipid-staining dye BODIPY 493/503, and the average number of LDs per cell was determined. In wild-type cells, LDs remained connected to the ER membrane but appeared to be distributed throughout the cell (25). In wild-type cells, the average numbers of LDs differed depending on the growth phase, reaching 4 to 6 per cell in exponentially growing cells. The number of LDs in brl1 mutants increased about 1.5-fold to reach about 10 per cell (Fig. 2B). Strikingly, LDs in the brl1 mutants were often arranged in a circular fashion, coinciding with the NE/ER, as indicated by their colocalization with the ER marker Kar2-RFP-HDEL (Fig. 2B). These observations are similar to those reported for brr6-1 (15).
Since both brl1 mutants incorporate more fatty acids into STE, have higher numbers of LDs per cell, and display aberrant perinuclear accumulation of LDs, we wondered whether the increased synthesis of neutral lipids is important for the growth and survival of these mutants. The synthesis of these neutral lipids depends on the activities of four acyltransferases: two, Lro1 and Dga1, for TAG synthesis and two, Are1 and Are2, for STE synthesis (26). Neutral lipid synthesis and LD formation are nonessential in yeast under normal growth conditions. Indeed, yeast cells carrying double deletions of dga1 and lro1 or of are1 and are2 have very few or no LDs yet are viable. Similarly, cells carrying deletions of all four of these acyltransferases have no LDs and are also viable (27). We reported previously that the formation of neutral lipids is essential for the viability of brr6 cells (15). This is also the case for brl1, since triple mutant cells combining a brl1 mutation with either dga1Δ and lro1Δ mutations or are1Δ and are2Δ mutations can grow only if a wild-type copy of BRL1 (pBRL1) is present (Fig. 2C).
We also examined genetic interactions between brl1 mutants and mutations affecting genes involved in fatty acid metabolism. We found that the brl1 alleles are synthetically lethal with mutations of genes whose products are involved in fatty acid elongation (elo2Δ, elo3Δ) and synthetically sick with mutations affecting the regulation of fatty acid desaturation (mga2Δ, spt23Δ) (Table 1).
The ability to adapt rapidly to changes in membrane properties brought about by changes in temperature or by the addition of agents that fluidize membranes is essential for viability. We reported previously that both brr6-1 and apq12Δ mutants were hypersensitive to benzyl alcohol (BA) and to oleic acid, a major unsaturated fatty acid of yeast (15). We compared the growth of brl1 mutant cells with that of brr6-1and apq12Δ cells in the presence of BA or oleic acid. All strains were hypersensitive to both BA and oleic acid (15) (Fig. 3A). Interestingly, cells unable to make neutral lipids (STE and TAG) were also hypersensitive to BA, indicating that the ability to store neutral lipids in LDs is also essential when cells are adjusting to the increased fluidity caused by the addition of BA (Fig. 3B).
FIG 3.
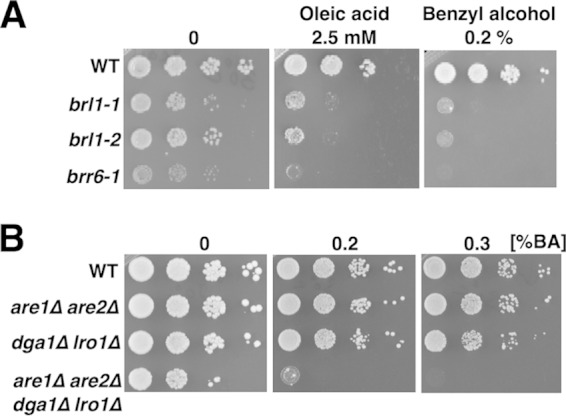
brl1 mutant cells are hypersensitive to the membrane-fluidizing agents oleic acid and benzyl alcohol. (A) Hypersensitivity of brl1 mutant cells to membrane-fluidizing agents. Strains were grown in YPD; 10-fold serial dilutions were spotted onto plates containing the indicated concentration of oleic acid or BA; and plates were incubated at 30°C. The hypersensitivity of brl1 and brr6 mutant strains to these agents is indicative of hyperfluid membranes and deregulation of membrane composition. (B) Mutants lacking the capacity to synthesize neutral lipids are hypersensitive to BA. The indicated strains were cultured in YPD, diluted, and spotted onto plates containing the indicated concentration of BA.
Analysis of membrane lipid composition in WT, brl1, brr6, and apq12Δ cells suggests that mutants are defective in the regulation of membrane properties.
We wanted to test the hypothesis that the hypersensitivity of BBA mutants to BA and oleic acid, the observed alterations in lipid metabolism, and the synthetic lethality of these mutants with mutations in genes needed for neutral lipid synthesis reflect misregulation of the adaptation of membranes to temperature or BA. When cells are shifted to a lower temperature, membranes become more rigid until changes in membrane lipid composition needed to restore membrane homeostasis take place. Because substantial changes in lipid composition occur after a temperature downshift of the BBA mutants, we conclude that the mutant cells sense the temperature change, leading to alterations in lipid metabolism. A proper response that would restore normal fluidity would be to increase the level of monounsaturated fatty acids and to shift the membrane fatty acid profile toward shorter chain lengths.
We analyzed the levels and composition of total fatty acids in BBA mutant cells at different temperatures. Exponentially growing cells were shifted from 24°C to 16°C for 16 h. Control cells were maintained at 24°C. Lipids were extracted, and fatty acid methyl esters (FAMEs) were analyzed by gas chromatography (GC) and quantified relative to internal standards. As expected, following a shift to 16°C, levels of the monounsaturated fatty acids palmitoleic acid (C16:1) and oleic acid (C18:1) increased in both wild-type and brl1 mutant cells, but to a much greater extent in mutant than in wild-type cells (Fig. 4A and B). Following this shift, the difference in the levels of oleic acid (C18:1) between the mutants and the wild type was most pronounced for brr6 cells but was also observed for the other mutants. Levels of the short-chain fatty acid myristic acid (C14:0) and its desaturated form, myristoleic acid (C14:1), were considerably higher in BBA mutant cells than in wild-type cells, particularly at 16°C (Fig. 4C and D). Levels of the saturated fatty acids palmitic acid (C16:0) and stearic acid (C18:0) were increased in brr6-1 cells at 16°C, but overall they displayed the least temperature-induced change (Fig. 4E and F).
FIG 4.

brl1, brr6, and apq12Δ mutant cells accumulate short-chain and unsaturated fatty acids. Cells were cultivated at the indicated temperatures in SC medium for 16 h; lipids were extracted; and fatty acid methyl esters were analyzed by GC using C17:0 as an internal standard. Palmitoleic acid (A) and oleic acid (B) levels are increased at 16°C, as are the levels of the C14 short-chain fatty acids myristic acid (C) and myristoleic acid (D). Palmitic acid (E) and stearic acid (F) levels are affected in the brr6-1 mutant. Values are means ± standard deviations for at least two independent experiments.
Taken together, these data show that cells carrying mutations affecting the BBA proteins have much higher levels of monounsaturated and shorter-chain fatty acids than wild-type cells. There was a 3-fold increase in the level of myristoleic acid, in addition to higher levels of myristic and palmitoleic acids. In addition, in mutant cells, there was a shift toward shorter-chain fatty acids, with the greatest increase occurring for myristic acid (C14:0). The increased levels of these fatty acids at 16°C confirm that mutant cells can sense the shift to a lower temperature and attempt to restore membrane homeostasis by altering membrane lipid composition. Importantly, and supporting our hypothesis, the altered fatty acid profiles observed in mutant cells indicate that the changes in lipid composition move these cells in the right direction but that they incorporate into their membranes excessive levels of these shorter-chain and monounsaturated fatty acids, thereby rendering their membranes too fluid.
brl1 mutants display defects in homeoviscous adaptation after treatment with and withdrawal of benzyl alcohol.
The studies presented above indicate that membranes in mutant cells acquire a fatty acid composition that would be expected to render them more fluid than optimal following a shift to 16°C. We wondered how the mutants would modify their membrane lipid composition under conditions where BA rather than temperature was used to induce a shift in composition. Following the addition of BA, membranes would become too fluid until they were able to restore appropriate membrane lipid composition. In this case, restoration of normal fluidity requires a shift to longer fatty acid chain lengths and reductions in the levels of monounsaturated fatty acids. Following the withdrawal of BA, membrane lipid composition would be expected to return to approximately the levels that cells showed before BA treatment.
Interestingly, there have been no reports on the effects of BA on the fatty acid or sterol composition of cells. Because brl1 and brr6 mutant cells are sensitive to BA, possibly due to a failure to control membrane fluidization, we carried out fatty acid and sterol analyses of wild-type cells after treatment with BA at 24°C. Following dilution to restore exponential growth, the cells were exposed to BA and were maintained at 24°C. As expected, following the addition of BA, wild-type cells showed substantial decreases in the levels of C14:0 and C14:1 fatty acids and increased levels of membrane-rigidifying stearic acid (Fig. 5A to C). The levels of C16:0 and C18:1 fatty acids did not differ from those in untreated cells (not shown). When sterols were analyzed, an increase in ergosterol levels following BA treatment was also observed, in accordance with an adjustment to make membranes more rigid (Fig. 5D).
FIG 5.

Adaptive changes in lipid profiles of wild-type and brl1 mutant cells upon treatment with benzyl alcohol. Wild-type cultures grown at 24°C in SC medium were split, diluted to an OD600 of 0.25, and either left untreated or treated with a sublethal dose of BA (0.2%). Cultures with equal ODs were harvested at the indicated time points; lipids were extracted; and fatty acids and sterols were analyzed. Wild-type cells treated with BA show lower levels of the short-chain C14 fatty acids myristic acid (A) and myristoleic acid (B), with concomitant increases in the levels of the long-chain C18:0 saturated fatty acid stearic acid (C) and ergosterol (D). These changes are probably induced to counteract the fluidizing effect of BA. (E and F) Recovery of wild-type and brl1-1 mutant cells from treatment with BA. Cells were cultivated at 24°C and were treated with BA (0.2%) for 5 h. Cells were washed with fresh medium and were recultivated for another 5 h or 10 h. Lipids were extracted, and fatty acid profiles were recorded by GC and were plotted relative to the levels present at the zero time point. Values are means ± standard deviations for three independent experiments.
To test the hypothesis that the brl1 mutants overcompensate in their adaptive changes, we analyzed fatty acid changes in BA-treated brl1-1 cells. Data were normalized to the levels each strain showed prior to treatment with BA. Following BA treatment for 5 h, mutant cells showed smaller reductions in the levels of C14:0 and C14:1 fatty acids than wild-type cells, suggesting that they were unable to adapt fully to the addition of BA. During their recovery after BA was removed, wild-type cells increased their short-chain C14 fatty acid levels to 55% to 72% of their initial levels within 10 h after BA withdrawal (72% for C14:0 and 55% for C14:1). In contrast, brl1-1 cells reached 127% of the initial level of C14:0 and 119% of the initial level of C14:1 (Fig. 5E and F). This indicates that this mutant overcompensates and, within 10 h, reaches C14 levels almost twice as high as those present in similarly treated wild-type cells. Similar, albeit less pronounced, overcompensation was also observed for the C16 and C18 fatty acids (see Fig. S4 in the supplemental material) and in the brl1-2 mutant (data not shown). We conclude that brl1 mutant cells not only sense and respond to BA removal but incorrectly adjust their fatty acid composition to levels expected to cause excess rigidity. The data support the hypothesis that this mutant is defective in homeostatic adaptation of membrane fatty acid composition.
Brl1 localizes to punctate structures within the nuclear envelope and is present in a common complex with Brr6 and Apq12.
Brr6 and Brl1 are low-abundance proteins, and in previous studies, overexpression was used to permit easy detection of these proteins fused to GFP (18, 21, 28). Overexpressed tagged proteins were found primarily in the NE/ER. In an effort to localize them more accurately, we used a highly sensitive microscope setup to localize GFP-tagged versions of the proteins expressed from their normal genomic locations (Fig. 6A). Brl1-GFP localized to a few puncta at the NE, whereas Brr6-GFP displayed a more uniform distribution of puncta over the NE. Apq12, on the other hand, appeared to be localized both to the NE and throughout the ER. This pattern of localization, particularly that of Brr6, resembles the distribution of NPCs, as displayed by Nup84-GFP (Fig. 6A). However, Apq12, Brl1, and Brr6 did not display clustering when analyzed in a nup120Δ mutant background, a strain where NPCs are localized to one or two clusters within the NE (29) (Fig. 6A). Furthermore, puncta containing Brl1 or Brr6 did not display stringent colocalization with the SPB marker Spc42 (Fig. 6A). In cells synchronized by treatment with α-factor, however, partial colocalization of Brl1 and Brr6 with the SPB could be observed (see Fig. S5 in the supplemental material). Interestingly, Brl1 and Brr6 partially colocalized in punctate structures, suggesting that they may associate with each other (Fig. 6A). The punctate localization of Brr6 and Brl1 in the NE was not affected in cells treated with BA, nor did BA affect the colocalization of these two proteins (see Fig. S6 in the supplemental material).
Given that BBA mutant cells display many common phenotypes, we wondered whether the three proteins might interact physically with each other to form a complex. To test this, tagged versions of these proteins were immunoprecipitated, and the presence of the other proteins was detected by Western blotting. These experiments revealed that both Brr6-HA and Brl1-HA, but not Yet3, another transmembrane protein of the NE/ER (30), could be detected when Apq12-GFP was immunoprecipitated (Fig. 6B). Similarly, a pulldown of Brr6–13Myc coprecipitated Brl1-HA in wild-type cells (Fig. 6C). Interestingly, if the co-IP was performed in an apq12Δ mutant strain, significantly less Brl1-HA was coprecipitated with Brr6–13Myc. These data suggest that there may be complexes containing all three BBA proteins as well as some that contain two BBA proteins. These findings are consistent with those of Saitoh et al., who reported earlier that a Brl1–Brr6 interaction could be detected using a two-hybrid system (21).
DISCUSSION
The lipid composition of cellular membranes is a major determinant of their biophysical properties. In response to temperature changes, cells modify the composition of their membranes so as to maintain membrane function, a process known as homeoviscous adaptation (31, 32). The processes by which the cell senses the need to modify lipid composition and assesses when the appropriate composition has been attained are complex and are best characterized in model prokaryotes (33, 34). For example, shifting cells to a lower temperature causes membranes to become more rigid, and this increased rigidity triggers changes in membrane lipid composition leading to the restoration of normal fluidity and flexibility. Changes in lipid composition also occur following heat shock (35). Recently, it was shown that heat shock induces activation of the gene encoding acyl coenzyme A (acyl-CoA) dehydrogenase, which then controls lipid saturation levels and hence membrane fluidity in Caenorhabditis elegans (36). It is not known how the cell senses the need to induce this enzyme. Although medium-chain dehydrogenases are essential enzymes for the degradation of fatty acids through mitochondrial beta-oxidation and are present in humans, it is not known if these enzymes are induced by the same mechanism in humans as they are in C. elegans. In yeast, on the other hand, there is no mitochondrial homolog of the C. elegans enzyme, possibly because beta-oxidation of medium-chain fatty acids is confined to peroxisomes.
Fusion of the inner and outer nuclear membranes is a key event in NPC biogenesis.
NPCs are essential for the proper functioning of eukaryotic cells. During each cell cycle, the number of NPCs is doubled as new NPCs are constructed within the double membrane of the NE. Although highly symmetrical and built from only 30 different proteins, the NPC has a mass of approximately 60 MDa and contains several hundred nucleoporins (3, 4). Construction is likely to be complex, since it requires fusion of the inner and outer nuclear membranes, creating a membrane domain called the pore membrane, the part of the NE that is in close contact with the NPC. The pore membrane contains transmembrane nucleoporins that play an essential role in anchoring the NPC within the nuclear envelope.
In eukaryotic cells, most membrane fusion events occur through the action of SNARE complexes that mediate the apposition of two membranes and then accomplish the task of fusing the inner and outer leaflets of the membranes (37). Membrane fusion during NPC biogenesis does not involve the action of SNARE complexes. Neither the mechanics of this membrane fusion event nor the stage during NPC biogenesis at which fusion occurs has been determined. However, NPC biogenesis requires that the pore membrane domain become highly curved (38). Successful fusion results in the formation of a protein-membrane tunnel that connects the nucleoplasm and the cytoplasm and provides a gated aqueous channel through which nucleocytoplasmic trafficking takes place.
NPCs contain several proteins that have transmembrane domains. Among these, only Ndc1 is essential, and it is the only nucleoporin also found in the SPB (39). All other integral membrane nucleoporins are not essential, but generally only a single one can be absent. It has also been shown that targeting of Pom33, an integral membrane nucleoporin, to the NPC requires its amphipathic helices, which preferentially bind to highly curved membranes (12). Also implicated in NPC biogenesis are the reticulons, which are able to induce membrane curvature and have been shown to be important for the formation of tubule structures of the ER (40). Deletion of both RTN1 and YOP1, encoding two of the reticulons of S. cerevisiae, causes defects in nucleoporin localization and NPC distribution within the NE (10). In metazoan cells, reticulons are required for proper reassembly of the NE following mitosis (41, 42). Using a Xenopus laevis in vitro system where de novo assembly of new NPCs takes place, Dawson et al. (2009) showed that addition of an anti-reticulon antibody to the system led to a block in the assembly of new NPCs (10). Previously, it had been shown that this antibody did not affect the normal expansion of the NE that occurs during the reassembly of nuclei in Xenopus egg extracts, suggesting a specific role for this reticulon in NPC biogenesis.
While some nucleoporins and reticulons are able to bind to and stabilize curved membranes, successful membrane bending and fusion likely require modifications to the biophysical properties of the nuclear membranes at sites of NPC formation, providing locally increased flexibility and fluidity in the pore membrane domain during the actual membrane fusion events. It is not known how these alterations in membrane properties are regulated, but they could be induced as a response to the initial curvature that would result from the interaction of reticulons and some nucleoporins with the nuclear membranes during NPC formation.
Nups comprising the cytoplasmic filaments (CFs) of the yeast NPC were mislocalized to bright cytoplasmic foci in cells carrying mutant alleles of the genes encoding BBA proteins (14, 15) (Fig. 1). The mislocalized nups were sometimes associated with the cytoplasmic face of the NE. In contrast, we saw normal localization for all nuclear basket nups and limited mislocalization of a subset of central framework nups. Experiments to distinguish between defects in NPC assembly and NPC stability in apq12Δ and brr6-1 cells indicated that NPC assembly was affected by the mutations to a much greater degree than the stability of pores formed prior to the shift to a nonpermissive temperature (14, 15). One model that fits with these observations is that NPC assembly might be initiated within the inner nuclear membrane, while the CF nups might be incorporated late during NPC biogenesis. We speculate that membrane fusion events that are part of NPC biogenesis also occur late during NPC assembly and might be required for the incorporation of the CFs as a final step in NPC biogenesis.
Mutations affecting any of the three BBA proteins lead to modest defects in mRNA export that likely result indirectly from defects in NPC biogenesis (14, 15) (Fig. 1). These proteins, particularly Brl1, appear to be involved in SPB biogenesis also (19). In S. cerevisiae and S. pombe, SPB biogenesis involves the construction of a new SPB adjacent to the NE, on its cytoplasmic side, followed by insertion of this structure into the NE.
We identified physical interactions among the three BBA proteins, with both Brl1 and Brr6 coprecipitating with Apq12 (Fig. 6B). This is consistent with the presence of binary Apq12–Brl1 and Apq12–Brr6 complexes, as well as with a ternary complex containing all three proteins. Because the levels of Brr6 and Brl1 that can be coprecipitated are reduced when Apq12 is missing, we think it likely that at least some fraction of these proteins forms a ternary complex. Our data do not indicate how direct the interactions among Brl1, Brr6, and Apq12 are. However, these interactions likely underlie some of the similar phenotypes seen with brl1 and brr6 mutants.
Possible roles for Brl1, Brr6, and Apq12.
After either a shift of mutant cells to a low temperature or recovery from BA treatment, high levels of sterols could be needed to compensate for the increased levels of shorter-chain and monounsaturated fatty acids (Fig. 4 and 5). While BBA mutant cells are able to sense the need to modify membrane properties following a temperature downshift or in response to the addition or removal of BA, their sensitivity to oleic acid and the membrane-fluidizing agent BA suggests that they are unable to detect when proper membrane homeostasis has been restored. This defect results in hyperfluidization of membranes in mutant cells shifted to lower temperatures.
We found that cells respond to the addition of BA by adjusting their membrane composition in an effort to restore membrane homeostasis. In wild-type cells, levels of C14:0 and C14:1 fatty acids decrease, and levels of stearic acid (C18:0) and ergosterol increase (Fig. 5A to D). Wild-type cells thus respond to membrane fluidization by BA with compensatory changes in their membrane lipid composition. When BA is removed, wild-type cells respond with appropriate adjustments in membrane lipid composition.
In contrast, brl1, brr6, and apq12Δ cells are defective in controlling these alterations. In response to low temperatures, their membranes acquire a composition that would be expected to cause excessive fluidity. In response to BA, their membranes appear to become excessively fluid. During recovery from BA treatment, C14:0 and C14:1 levels in brl1-1 mutant cells increased, and the increase was much greater than that in wild-type cells (Fig. 5E and F). This altered adaptive response supports the hypothesis that the BBA mutants are able to sense reduced temperatures or the addition/removal of BA and that they induce changes in their lipid composition. However, they appear unable to assess when proper membrane fluidity has been restored and thus continue to produce lipids that enhance fluidization. One possibility is that mutant cells are defective in the function of some “fluidity regulator” such that the cells become hypersensitive to BA, oleic acid, or a temperature downshift.
This aberrant response to BA, as well as the increased levels of unsaturated fatty acids at all temperatures tested, might suggest that the activity of the fatty acid desaturase Ole1 is affected by the mutation of BRL1 and BRR6 (Fig. 4 and 5). OLE1 expression is regulated by two transcription factors, Mga2 and Spt23, in response to environmental conditions, such as temperature, oxygen levels, and the presence of unsaturated fatty acids in the medium (43). Spt23 and Mga2 are not essential but redundant, since deletion of either gene has little effect on the transcription of OLE1, but deletion of both results in synthetic lethality due to the inability of cells to produce Ole1 (44). Interestingly, mutants of Brl1 and Brr6 are synthetically sick with the deletion of either Mga2 or Spt23, in accordance with enhanced dependence on Ole1 function in these mutants (Table 1).
The biophysical properties of cellular membranes reflect both the degree of fatty acid desaturation and the distribution of fatty acid chain lengths. In principle, short-chain fatty acids should act synergistically with monounsaturated fatty acids in enhancing membrane fluidity. The regulation of both is defective in the BBA mutants. Our findings indicate that Brl1, Brr6, and Apq12 impact multiple key points in lipid metabolism that normally allow cells to maintain lipid homeostasis at the NE/ER membrane.
Link between lipid metabolism and NPC biogenesis.
Several observations link lipid synthesis specifically with the morphology of the NE as well as with the biogenesis of NPCs and the SPB. AccI (acetyl-CoA carboxylase) catalyzes the first step of fatty acid biosynthesis. A mutant allele of ACC1 was identified in a screen for mutants with mutations in genes whose products were required for efficient mRNA export. Mutations affecting AccI also lead to changes in NE morphology and defects in NPC biogenesis (45–47). In budding yeast, inactivation of the Nem1–Spo7 phosphatase complex and its effector protein, Pah1 (phosphatidic acid phosphohydrolase 1), leads to abnormal proliferation of the NE (48). A similar phenotype is observed upon overexpression of the Pah1 antagonist Dgk1 (diacyglycerol kinase 1) (49). Additionally, defective insertion of the SPB into the NE in cold-sensitive cells either overexpressing the SPB component Mps3 or expressing the dominant negative allele Mps3G186K can be rescued by treatment with oleic acid, BA, or the sterol biosynthesis inhibitor terbinafine (50). Lastly, deletion and mutation of genes whose products are involved in the biogenesis and organization of NPCs and the SPB have been shown, by using high-throughput screens, to render cells sensitive to agents that inhibit lipid biosynthesis (see the Yeast Fitness Database [fitdb.stanford.edu/]). Although the mechanism of the regulation of lipid composition by such proteins is unknown, it has been proposed that there is a requirement for modulation of membrane biophysical properties during the construction of NPCs and also for insertion of the new SPB into the NE (2, 51, 52).
Considerable additional work will be required to understand how and when membrane fusion occurs during the complex process of NPC assembly. The nature of the sensors involved in recognizing the need for alteration of membrane biophysical properties and the mechanism by which the cells control the extent of these modifications are not known. Our data suggest that Brl1, Brr6, and Apq12 could play a key role in the local modulation of nuclear membrane composition that is likely needed for nuclear membrane fusion and the completion of NPC biogenesis. Some of the NE abnormalities seen in mutant cells may reflect failure to successfully fuse the two membranes during NPC construction, and this could result from defective control of the biophysical properties of the nuclear membrane.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Westermann for the spindle pole body marker Spc42-mCherry.
The studies reported here were supported by the National Institute of General Medical Sciences, National Institutes of Health, Bethesda, MD, USA; the Canton of Fribourg, Switzerland; and the Swiss National Science Foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00101-15.
REFERENCES
- 1.Baumann O, Walz B. 2001. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol 205:149–214. doi: 10.1016/S0074-7696(01)05004-5. [DOI] [PubMed] [Google Scholar]
- 2.Jaspersen SL, Ghosh S. 2012. Nuclear envelope insertion of spindle pole bodies and nuclear pore complexes. Nucleus 3:226–236. doi: 10.4161/nucl.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aitchison JD, Rout MP. 2012. The yeast nuclear pore complex and transport through it. Genetics 190:855–883. doi: 10.1534/genetics.111.127803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grossman E, Medalia O, Zwerger M. 2012. Functional architecture of the nuclear pore complex. Annu Rev Biophys 41:557–584. doi: 10.1146/annurev-biophys-050511-102328. [DOI] [PubMed] [Google Scholar]
- 5.Raices M, D'Angelo MA. 2012. Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat Rev Mol Cell Biol 13:687–699. doi: 10.1038/nrm3461. [DOI] [PubMed] [Google Scholar]
- 6.Adams RL, Wente SR. 2013. Uncovering nuclear pore complexity with innovation. Cell 152:1218–1221. doi: 10.1016/j.cell.2013.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeshima K, Iino H, Hihara S, Imamoto N. 2011. Nuclear size, nuclear pore number and cell cycle. Nucleus 2:113–118. doi: 10.4161/nucl.2.2.15446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lusk CP, Blobel G, King MC. 2007. Highway to the inner nuclear membrane: rules for the road. Nat Rev Mol Cell Biol 8:414–420. doi: 10.1038/nrm2165. [DOI] [PubMed] [Google Scholar]
- 9.Fichtman B, Ramos C, Rasala B, Harel A, Forbes DJ. 2010. Inner/outer nuclear membrane fusion in nuclear pore assembly: biochemical demonstration and molecular analysis. Mol Biol Cell 21:4197–4211. doi: 10.1091/mbc.E10-04-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson TR, Lazarus MD, Hetzer MW, Wente SR. 2009. ER membrane-bending proteins are necessary for de novo nuclear pore formation. J Cell Biol 184:659–675. doi: 10.1083/jcb.200806174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B. 2007. A general amphipathic alpha-helical motif for sensing membrane curvature. Nat Struct Mol Biol 14:138–146. doi: 10.1038/nsmb1194. [DOI] [PubMed] [Google Scholar]
- 12.Floch AG, Tareste D, Fuchs PF, Chadrin A, Naciri I, Leger T, Schlenstedt G, Palancade B, Doye V. 2015. Nuclear pore targeting of the yeast Pom33 nucleoporin depends on karyopherin and lipid binding. J Cell Sci 128:305–316. doi: 10.1242/jcs.158915. [DOI] [PubMed] [Google Scholar]
- 13.Cook A, Bono F, Jinek M, Conti E. 2007. Structural biology of nucleocytoplasmic transport. Annu Rev Biochem 76:647–671. doi: 10.1146/annurev.biochem.76.052705.161529. [DOI] [PubMed] [Google Scholar]
- 14.Scarcelli JJ, Hodge CA, Cole CN. 2007. The yeast integral membrane protein Apq12 potentially links membrane dynamics to assembly of nuclear pore complexes. J Cell Biol 178:799–812. doi: 10.1083/jcb.200702120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodge CA, Choudhary V, Wolyniak MJ, Scarcelli JJ, Schneiter R, Cole CN. 2010. Integral membrane proteins Brr6 and Apq12 link assembly of the nuclear pore complex to lipid homeostasis in the endoplasmic reticulum. J Cell Sci 123:141–151. doi: 10.1242/jcs.055046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker KE, Coller J, Parker R. 2004. The yeast Apq12 protein affects nucleocytoplasmic mRNA transport. RNA 10:1352–1358. doi: 10.1261/rna.7420504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hieronymus H, Yu MC, Silver PA. 2004. Genome-wide mRNA surveillance is coupled to mRNA export. Genes Dev 18:2652–2662. doi: 10.1101/gad.1241204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Bruyn Kops A, Guthrie C. 2001. An essential nuclear envelope integral membrane protein, Brr6p, required for nuclear transport. EMBO J 20:4183–4193. doi: 10.1093/emboj/20.15.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamm T, Grallert A, Grossman EP, Alvarez-Tabares I, Stevens FE, Hagan IM. 2011. Brr6 drives the Schizosaccharomyces pombe spindle pole body nuclear envelope insertion/extrusion cycle. J Cell Biol 195:467–484. doi: 10.1083/jcb.201106076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneiter R, Cole CN. 2010. Integrating complex functions: coordination of nuclear pore complex assembly and membrane expansion of the nuclear envelope requires a family of integral membrane proteins. Nucleus 1:387–392. doi: 10.4161/nucl.1.5.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saitoh YH, Ogawa K, Nishimoto T. 2005. Brl1p—a novel nuclear envelope protein required for nuclear transport. Traffic 6:502–517. doi: 10.1111/j.1600-0854.2005.00295.x. [DOI] [PubMed] [Google Scholar]
- 22.Lo Presti L, Cockell M, Cerutti L, Simanis V, Hauser PM. 2007. Functional characterization of Pneumocystis carinii brl1 by transspecies complementation analysis. Eukaryot Cell 6:2448–2452. doi: 10.1128/EC.00321-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. 2002. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30:e23. doi: 10.1093/nar/30.6.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cole CN, Heath CV, Hodge CA, Hammell CM, Amberg DC. 2002. Analysis of RNA export. Methods Enzymol 351:568–587. doi: 10.1016/S0076-6879(02)51869-3. [DOI] [PubMed] [Google Scholar]
- 25.Jacquier N, Choudhary V, Mari M, Toulmay A, Reggiori F, Schneiter R. 2011. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Sci 124:2424–2437. doi: 10.1242/jcs.076836. [DOI] [PubMed] [Google Scholar]
- 26.Czabany T, Athenstaedt K, Daum G. 2007. Synthesis, storage and degradation of neutral lipids in yeast. Biochim Biophys Acta 1771:299–309. doi: 10.1016/j.bbalip.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Sandager L, Gustavsson MH, Stahl U, Dahlqvist A, Wiberg E, Banas A, Lenman M, Ronne H, Stymne S. 2002. Storage lipid synthesis is non-essential in yeast. J Biol Chem 277:6478–6482. doi: 10.1074/jbc.M109109200. [DOI] [PubMed] [Google Scholar]
- 28.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. 2003. Global analysis of protein expression in yeast. Nature 425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 29.Heath CV, Copeland CS, Amberg DC, Del Priore V, Snyder M, Cole CN. 1995. Nuclear pore complex clustering and nuclear accumulation of poly(A)+ RNA associated with mutation of the Saccharomyces cerevisiae RAT2/NUP120 gene. J Cell Biol 131:1677–1697. doi: 10.1083/jcb.131.6.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson JD, Barlowe C. 2010. Yet1p and Yet3p, the yeast homologs of BAP29 and BAP31, interact with the endoplasmic reticulum translocation apparatus and are required for inositol prototrophy. J Biol Chem 285:18252–18261. doi: 10.1074/jbc.M109.080382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinensky M. 1974. Homeoviscous adaptation—a homeostatic process that regulates the viscosity of membrane lipids in Escherichia coli. Proc Natl Acad Sci U S A 71:522–525. doi: 10.1073/pnas.71.2.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aguilar PS, de Mendoza D. 2006. Control of fatty acid desaturation: a mechanism conserved from bacteria to humans. Mol Microbiol 62:1507–1514. doi: 10.1111/j.1365-2958.2006.05484.x. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YM, Rock CO. 2008. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol 6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 34.de Mendoza D. 2014. Temperature sensing by membranes. Annu Rev Microbiol 68:101–116. doi: 10.1146/annurev-micro-091313-103612. [DOI] [PubMed] [Google Scholar]
- 35.Balogh G, Péter M, Glatz A, Gombos I, Török Z, Horváth I, Harwood JL, Vígh L. 2013. Key role of lipids in heat stress management. FEBS Lett 587:1970–1980. doi: 10.1016/j.febslet.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 36.Ma DK, Li Z, Lu AY, Sun F, Chen S, Rothe M, Menzel R, Sun F, Horvitz HR. 2015. Acyl-CoA dehydrogenase drives heat adaptation by sequestering fatty acids. Cell 161:1152–1163. doi: 10.1016/j.cell.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jahn R, Scheller RH. 2006. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol 7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 38.Antonin W. 2009. Nuclear envelope: membrane bending for pore formation. Curr Biol 19:R410–R412. doi: 10.1016/j.cub.2009.03.053. [DOI] [PubMed] [Google Scholar]
- 39.Lau CK, Giddings TH Jr, Winey M. 2004. A novel allele of Saccharomyces cerevisiae NDC1 reveals a potential role for the spindle pole body component Ndc1p in nuclear pore assembly. Eukaryot Cell 3:447–458. doi: 10.1128/EC.3.2.447-458.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibata Y, Hu J, Kozlov MM, Rapoport TA. 2009. Mechanisms shaping the membranes of cellular organelles. Annu Rev Cell Dev Biol 25:329–354. doi: 10.1146/annurev.cellbio.042308.113324. [DOI] [PubMed] [Google Scholar]
- 41.Kiseleva E, Morozova KN, Voeltz GK, Allen TD, Goldberg MW. 2007. Reticulon 4a/NogoA locates to regions of high membrane curvature and may have a role in nuclear envelope growth. J Struct Biol 160:224–235. doi: 10.1016/j.jsb.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson DJ, Hetzer MW. 2008. Reshaping of the endoplasmic reticulum limits the rate for nuclear envelope formation. J Cell Biol 182:911–924. doi: 10.1083/jcb.200805140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin CE, Oh CS, Jiang Y. 2007. Regulation of long chain unsaturated fatty acid synthesis in yeast. Biochim Biophys Acta 1771:271–285. doi: 10.1016/j.bbalip.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 44.Zhang S, Skalsky Y, Garfinkel DJ. 1999. MGA2 or SPT23 is required for transcription of the Δ9 fatty acid desaturase gene, OLE1, and nuclear membrane integrity in Saccharomyces cerevisiae. Genetics 151:473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadowaki T, Chen S, Hitomi M, Jacobs E, Kumagai C, Liang S, Schneiter R, Singleton D, Wisniewska J, Tartakoff AM. 1994. Isolation and characterization of Saccharomyces cerevisiae mRNA transport-defective (mtr) mutants. J Cell Biol 126:649–659. doi: 10.1083/jcb.126.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneiter R, Hitomi M, Ivessa AS, Fasch EV, Kohlwein SD, Tartakoff AM. 1996. A yeast acetyl coenzyme A carboxylase mutant links very-long-chain fatty acid synthesis to the structure and function of the nuclear membrane-pore complex. Mol Cell Biol 16:7161–7172. doi: 10.1128/MCB.16.12.7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneiter R, Brugger B, Amann CM, Prestwich GD, Epand RF, Zellnig G, Wieland FT, Epand RM. 2004. Identification and biophysical characterization of a very-long-chain-fatty-acid-substituted phosphatidylinositol in yeast subcellular membranes. Biochem J 381:941–949. doi: 10.1042/BJ20040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santos-Rosa H, Leung J, Grimsey N, Peak-Chew S, Siniossoglou S. 2005. The yeast lipin Smp2 couples phospholipid biosynthesis to nuclear membrane growth. EMBO J 24:1931–1941. doi: 10.1038/sj.emboj.7600672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han GS, O'Hara L, Carman GM, Siniossoglou S. 2008. An unconventional diacylglycerol kinase that regulates phospholipid synthesis and nuclear membrane growth. J Biol Chem 283:20433–20442. doi: 10.1074/jbc.M802903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friederichs JM, Ghosh S, Smoyer CJ, McCroskey S, Miller BD, Weaver KJ, Delventhal KM, Unruh J, Slaughter BD, Jaspersen SL. 2011. The SUN protein Mps3 is required for spindle pole body insertion into the nuclear membrane and nuclear envelope homeostasis. PLoS Genet 7:e1002365. doi: 10.1371/journal.pgen.1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothballer A, Kutay U. 2013. Poring over pores: nuclear pore complex insertion into the nuclear envelope. Trends Biochem Sci 38:292–301. doi: 10.1016/j.tibs.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 52.Zhang D, Oliferenko S. 2013. Remodeling the nuclear membrane during closed mitosis. Curr Opin Cell Biol 25:142–148. doi: 10.1016/j.ceb.2012.09.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.