

Abstract
Inositolphosphorylceramide (IPC) and its mannosylated derivatives are the only complex sphingolipids of yeast. Their synthesis can be reduced by aureobasidin A (AbA), which specifically inhibits the IPC synthase Aur1. AbA reportedly, by diminishing IPC levels, causes endoplasmic reticulum (ER) stress, an increase in cytosolic calcium, reactive oxygen production, and mitochondrial damage leading to apoptosis. We found that when Aur1 is gradually depleted by transcriptional downregulation, the accumulation of ceramides becomes a major hindrance to cell survival. Overexpression of the alkaline ceramidase YPC1 rescues cells under this condition. We established hydroxylated C26 fatty acids as a reliable hallmark of ceramide hydrolysis. Such hydrolysis occurs only when YPC1 is overexpressed. In contrast, overexpression of YPC1 has no beneficial effect when Aur1 is acutely repressed by AbA. A high-throughput genetic screen revealed that vesicle-mediated transport between Golgi apparatus, endosomes, and vacuole becomes crucial for survival when Aur1 is repressed, irrespective of the mode of repression. In addition, vacuolar acidification becomes essential when cells are acutely stressed by AbA, and quinacrine uptake into vacuoles shows that AbA activates vacuolar acidification. The antioxidant N-acetylcysteine does not improve cell growth on AbA, indicating that reactive oxygen radicals induced by AbA play a minor role in its toxicity. AbA strongly induces the cell wall integrity pathway, but osmotic support does not improve the viability of wild-type cells on AbA. Altogether, the data support and refine current models of AbA-mediated cell death and add vacuolar protein transport and acidification as novel critical elements of stress resistance.
INTRODUCTION
Yeast sphingolipids have been recognized to be essential for cell growth and survival ever since the initial demonstration by Bob Dickson and colleagues that LCB1 and LCB2, coding for two components of serine palmitoyltransferase, are essential genes (1) (Fig. 1). In 1997 the same group also demonstrated that the essential AUR1 gene encodes the enzyme making inositolphosphorylceramides (IPCs), implying that IPCs are essential for cell growth (2); in contrast, the synthesis of mannosyl-IPCs (MIPCs) and inositol-phospho-MIPCs [M(IP)2Cs] was found to be dispensable (3–5). The essentiality of IPCs, however, has been questioned, since cells lacking all ceramide synthases are highly resistant to the inhibitor aureobasidin A (AbA), a very specific and potent inhibitor of Aur1, which rapidly induces cell death in wild-type (WT) yeast cells (6–9). Nevertheless, several lines of evidence suggest that IPCs are essential for growth. This had been already suggested by early studies showing that the lethality of the lcb1Δ mutation (Fig. 1), eliminating all sphingolipid biosynthesis, is suppressed in cells harboring the SLC1-1 gain-of-function mutation, which allows making phosphatidylinositol (PI) with the very-long-chain fatty acid (VLCFA) C26:0 in the sn-2 position. Structurally, this abnormal PI closely resembles IPC and allows growth of sphingolipid-free and ceramide-free yeast strains (10–13). Furthermore, a recent report showed that AbA causes the endoplasmic reticulum (ER) retention of glycosylphosphatidylinositol (GPI)-anchored proteins, an unfolded protein response, reactive oxygen species (ROS) production, mitochondrial cytochrome c release, and a metacaspase-mediated form of apoptosis that additionally is dependent on the concomitant increase of cytosolic Ca2+ concentrations, all through the reduction of IPCs rather than the accumulation of toxic ceramides, both of which are inevitable consequences of Aur1 depletion/inhibition (14).
FIG 1.

Major pathways of sphingolipid biosynthesis in yeast. Gene names are in italic, essential genes are in green, activities dependent on protein complexes are in gray, and enzyme inhibitors are in bold italic. A detailed description of the biosynthetic reactions can be found in recent reviews (58, 67). Deletions of red and blue genes are predicted or known to reduce ceramide levels; those that scored as suppressors of the growth defect of strains with repressed AUR1 transcription are in red, and those which did not score, although present in the deletion library, are in blue. Deletions in the 5 underlined genes also were identified as suppressors in preliminary screens and were confirmed in serial dilution plating assays (Table 2). Long-chain bases (LCBs) of ceramides are dihydrosphingosine (DHS) or phytosphingosine (PHS); fatty acids of ceramides are indicated by the number of C atoms (e.g., C26) and their hydroxyl (OH) groups.
AbA has fungicidal activity against many pathogenic fungi, including Candida albicans, Cryptococcus neoformans, and some Aspergillus species. Given orally to mice, AbA was effective at curing candidiasis and well tolerated (15). Moreover, AbA-mediated cell death represents a tractable model of sphingolipid-mediated apoptosis.
Here, in an attempt to further understand the mechanisms of AbA-induced apoptosis and to discriminate between effects linked to IPC depletion versus ceramide accumulation, we did chemical/genetic high-throughput screens testing the growth of deletion mutants under Aur1 repression, either when overexpressing YPC1 or when not. YPC1 is an ER ceramidase that has the potential to lower ceramide levels and concomitantly aggravate the reduction of complex sphingolipids. Overexpression of YPC1 has been reported to have various biological effects, e.g., in making cells resistant to exogenous long-chain bases (LCBs) (16) by working in the reverse direction (converting LCBs to ceramides) or to make cells hypersensitive to hydroxyurea (17) and to rescue high-temperature growth of ypk1ts ypk2Δ cells (18) by working as a ceramidase.
MATERIALS AND METHODS
Strains, growth conditions, and materials.
The Saccharomyces cerevisiae strains used are listed in Table S1 in the supplemental material, plasmids in Table S2 in the supplemental material, and the origin of chemicals in the materials section in the supplemental material. Mutant strains and plasmids were generated using standard methods for crossing of single mutants, plasmid transfection, or gene disruption using deletion cassettes generated by PCR. Unless indicated otherwise, cells were grown on rich medium (yeast extract-peptone-dextrose [YPD] or YPGal) supplemented with uracil and adenine or on synthetic complete (SC) medium (yeast nitrogen base [YNB]) (United States Biological) containing 2% glucose (D) or galactose (Gal) as a carbon source plus the amino acids and ingredients contained in the dropout mix. For serial dilution growth assays, agar plates were incubated for 3 days at 30°C unless indicated otherwise.
Construction of plasmids.
To construct plasmid pYPC1-URA3, the coding sequence of YPC1 was amplified by PCR using oligonucleotides 5′-CCTGGGATCCATGGGAATATTTCGTTGGAAC-TATCC-3′ and 5′-CGCCGCGGCCGCTTACTTCTCCTTTTTAACTTC-3′ from genomic DNA of BY4742 cells. The PCR products were doubly digested with NotI and BamHI and ligated into the similarly digested centromeric pNP302 vector carrying the ADH1 promoter to generate pYPC1-URA3. To make the tetAUR1 strain FBY5318, AUR1 was placed under control of the tetO2 promoter by replacing 110 bp preceding the ATG initiator codon by the tetracycline operator cassette containing a repressor-binding site, a tetR-VP16 tTA transactivator, and the kanMX4 marker, amplified from plasmid pCM224 (19). Subsequently, the kanMX4 cassette was replaced by the natMX cassette using a fragment from the marker swap plasmid pAG25 (20).
SGA.
Synthetic genetic array (SGA) analysis was performed as follows. Briefly, the FBY5313 WT, tetAUR1, tetAUR1.YPC1, or tetAUR1.URA3 strain was robotically crossed against an array of 5,044 individual knockouts of nonessential genes, with each cross being done in quadruplicate. After sporulation, colonies were pinned onto medium without histidine, arginine, lysine, and uracil and supplemented with canavanine and thialysine and were left for 2 days at 30°C to select for MATa haploid meiotic progeny (selection 1). When crossing tetAUR1 strains, plates were then replicated on the same medium additionally containing Geneticin (G418) and nourseothricin (clonNAT) (selection 2). After 1 day at 30°C, colonies were replicated onto the same medium (selection 3), the same medium plus 0.03 μg/ml of AbA, or the same medium plus 0.1 μg/ml of doxycycline (Doxy). Pictures were taken after 36 h of growth. The measurement of growth and the analysis and visualization of the data were conducted with the help of the “ScreenMill” software (21), giving positive Z scores for growth defects and negative ones for growth enhancement. Plating of serially 10-fold-diluted cells on nutrient agar was used in the preliminary screens to see whether the synthetic genetic interactions identified by SGA analysis could be reproduced in a simple growth assay.
Fluorescence microscopy.
Cells were imaged using an Olympus BX54 microscope equipped with a piezo-positioner (Olympus). Z sections (0.5 μm thick, 7 to 10/cell) were projected to two-dimensional images and analyzed with the CellM software (Olympus).
Analysis of lipids by MS.
Lipids were extracted and analyzed in negative- and positive-ion modes by direct infusion mass spectrometry (MS) using an LTQ Orbitrap XL mass spectrometer equipped with the automated nanoflow ion source Triversa NanoMate (Advion Biosciences) as described previously (22, 23). Data were expressed either as intensity profiling (lipid analyte intensity normalized to the sum of intensities of all monitored lipid analytes) or as mole percent (based on internal lipid standards) (22, 23).
Detection of IPC/MIPC by metabolic labeling with [3H]inositol.
Cells were grown at 30°C overnight in inositol-free SC medium buffered to pH 5 with 50 mM sodium phosphate and 50 mM sodium succinate and were resuspended in the same medium at an optical density at 600 nm (OD600) of 10 for labeling. To aliquots of 2.5 OD600 units of cells we added AbA (from a 200-fold-concentrated stock in ethanol); after 10 min, [3H]inositol (10 μCi) was added. Every 40 min the labeling media were replenished by adding 1 volume of 5-fold-concentrated SC medium (buffered as described above) to 4 volumes of culture. Labeling was terminated after 2 h, and lipids were extracted as recently described (24) and desalted using butanol-water partitioning or Folch partitioning; the latter does not completely retain M(IP)2C in the organic phase. Lipids were deacylated using NaOH as described previously (25). The final lipid extracts contained 20 to 40% of added radioactivity, depending on the experiment. Lipids were separated by thin-layer chromatography (TLC) using CHCl3–methanol–0.25% KCl (55:45:10) as a solvent and were quantified by two-dimensional Berthold radioscanning. After mild base treatment, only the various forms of IPC, MIPC, and M(IP)2C were left.
Detection of ceramides and IPC/MIPC by metabolic labeling with [14C]serine.
Aliquots of 5 OD600 units of cells were preincubated for 10 min with AbA as required and labeled in 1 ml of serine-free SC medium with 10 μCi of [14C]serine for 5 h at 30°C with shaking and addition of 5-fold-concentrated serine-free SC medium every 40 min. At the end, the optical density of cultures was determined, and trichloroacetic acid (TCA) was added to a final concentration of 5% (wt/vol). Cells were washed in water, and lipids were extracted, deacylated or not by NaOH, desalted by Folch partitioning, and analyzed by TLC in the solvent chloroform–methanol–4.2 N ammonia (9:7:2) followed by radioscanning.
Quinacrine staining.
Quinacrine staining was performed as detailed in the supplemental material.
RESULTS
Overexpression of YPC1 induces futile ceramide hydrolysis and reduces levels of newly synthesized ceramides.
As the double deletion ypc1Δ ydc1Δ strain has normal ceramide levels even in the presence of AbA (24), we investigated whether there was increased ceramide turnover in cells overexpressing YPC1. In view of later experiments, we tested this also in strains in which the natural promoter of AUR1 was replaced by the doxycycline-repressible tetoff promoter (tetAUR1 strains) and which constitutively overexpress either YPC1 (tetAUR1.YPC1) or cytosolic green fluorescent protein (GFP) as a negative control (tetAUR1.URA3). As shown in Fig. 2A, YPC1-overexpressing cells had significantly higher levels of free LCBs than several WT, scs7Δ, or ypc1Δ ydc1Δ (yyΔΔ) cells, and LCB levels could further be increased by AbA treatment, although such treatment did not increase LCB levels in normal cells. As seen in Fig. 2B, free VLCFAs were very low in cells not overexpressing YPC1, and AbA treatment of such cells led to the appearance mainly of nonhydroxylated C26:0;0. YPC1-overexpressing cells generally had higher levels of VLCFAs and, remarkably, had significant amounts of monohydroxylated VLCFAs (C24:0;1 and C26:0;1). The small amounts of C26:0;1 seen in AbA-treated WT cells (asterisk in Fig. 2B) were not detectable in AbA-treated scs7Δ cells lacking the α-hydroxylase for fatty acids (not shown). Lipid profiles generated by MS showed that hydroxylated VLCFAs were not detectable in triacylglycerols (TAGs) or glycerophospholipids of tetAUR1.YPC1 (not shown), suggesting that C26:0-OH-coenzyme A (CoA) either is not made or is not utilized for glycerolipid biosynthesis. Ceramides and IPCs with hydroxylated VLCFAs were reported to be reduced in sur2Δ cells not making phytosphingosine (PHS) (Fig. 1), and this led to the proposal that the VLCFA hydroxylase Scs7 utilizes as substrates only fatty acids that are part of ceramides or IPCs but neither acyl-CoA nor free fatty acids (26). The fact that free hydroxylated VLCFAs become detectable only upon overexpression of YPC1 strongly supports this view. This fact also suggests that in at least some compartment of WT cells, probably the ER, the concentrations of ceramide, LCBs, and fatty acids favor ceramide hydrolysis, i.e., that the ΔG′ for ceramide hydrolysis is negative but that hydrolysis does not occur because ceramidases are rate limiting for this process unless they are overexpressed.
FIG 2.

Overexpression of YPC1 induces ceramide hydrolysis. The indicated strains were grown to an OD600 of 0.7 and extracted either directly or only after further growth in the presence of AbA (0.25 μg/ml) during 4 h. Lipid extracts from equal cell numbers were analyzed by MS. LCBs and VLCFAs are specified by 3 numbers indicating carbon atoms:double bonds;OH groups. (A) All intensities of LCB18:0;3 and LCB20:0;3 detected in the 6 strains, after subtraction of background, were summed and set as 100% and each species in each strain expressed as a fraction thereof. Summed intensities in LCB20:0;3 were only 6% of the total and therefore were multiplied by 10 for better visibility. Results for strains with an asterisk represent averages from two independent experiments and those for others represent a single one; two technical repeats were done in all cases. (B) Plot of the relative abundances (mole percent) of free VLCFA species that were present. Results represent averages from two independent experiments and two technical repeats for each.
AbA treatment caused a normal increase in concentrations of nonhydroxylated VLCFAs (C26:0;0) in various ypc1Δ ydc1Δ cells (see Fig. S1 in the supplemental material). These free VLCFAs therefore are not generated by ceramide hydrolysis, suggesting that AbA increases local ceramide levels to the point that the acyl-CoA-dependent ceramide synthase slows down so that C26:0;0-CoA accumulates and either can be hydrolyzed directly or is used for the synthesis of TAGs from where C26:0;0 can be released by TAG-lipases (27). Metabolic labeling with [14C]serine demonstrated that overexpression of YPC1 indeed can decrease the levels of newly made ceramides, and this was observed even in WT cells in the absence of AbA treatment (see Fig. S2B), although in such cells little C26:0;1 is detectable by MS (Fig. 2B). Different types of ceramides were affected differently by YPC1 overexpression, with dihydrosphingosine-C26 (DHS-C26) apparently not being affected (see Fig. S2B).
Effects of YPC1 overexpression in cells having a reduced Aur1 activity.
The data above suggested that YPC1, by hydrolyzing ceramides, would potentially alter the ceramide levels or ceramide-to-IPC ratio at least at certain subcellular locations, and we therefore compared the growth of serially diluted tetAUR1.YPC1 and tetAUR1.URA3 cells on agar in the presence of AbA or doxycycline (Doxy). As seen in Fig. 3A and B, concentrations of 0.01 μg/ml of AbA or 0.3 μg/ml of Doxy moderately reduced growth of cells on glucose by factors of 10 to 100, i.e., 10- to 100-fold more cells had to be plated on AbA or Doxy to see growth similar to that seen in the absence of drugs. As seen in Fig. 3A, overexpression of YPC1 from the ADH1 or GAL1 promoter enhanced the toxicity of AbA; this effect was observed in most but not all of experiments. In contrast, when AUR1 was downregulated transcriptionally using Doxy, we consistently observed that the overexpression of YPC1 rescued the growth defect (Fig. 3B). This was the case both when YPC1 was overexpressed constitutively and when it was induced by galactose. It seems safe to assume that downregulation of IPC synthase activity in the presence of Doxy sets in more slowly than when cells are simply plated on AbA, and we therefore hypothesized that this kinetic difference may cause different stress situations, which are differently affected by YPC1 overexpression. Indeed, treatment of cells with 10 μg/ml of Doxy required 8 h to reduce incorporation of [3H]inositol into IPCs by >90% (not shown), whereas many previous experiments in our lab show that the effect of AbA on Aur1 is rather instantaneous (28). Also, treatment for 3 h with AbA (0.25 μg/ml) induced strong phosphorylation of the terminal mitogen-activated protein kinase (MAPK) Slt2 of the wall integrity (CWI) pathway and TORC2-dependent phosphorylation of Ypk1 at Thr662 (29, 30), whereas Doxy treatment (10 μg/ml) for 3 h did not increase or only slightly increased phosphorylation of these kinases (Fig. 3C; see Fig. S3 in the supplemental material). Phosphorylation of Slt2 under AbA was reduced when YPC1 was overexpressed (Fig. 3C; see Fig. S3). The chitin-binding Calcofluor white (CFW) can induce strong phosphorylation of Slt2 (31) but failed to do so in our strain background (Fig. 3C; see Fig. S3).
FIG 3.
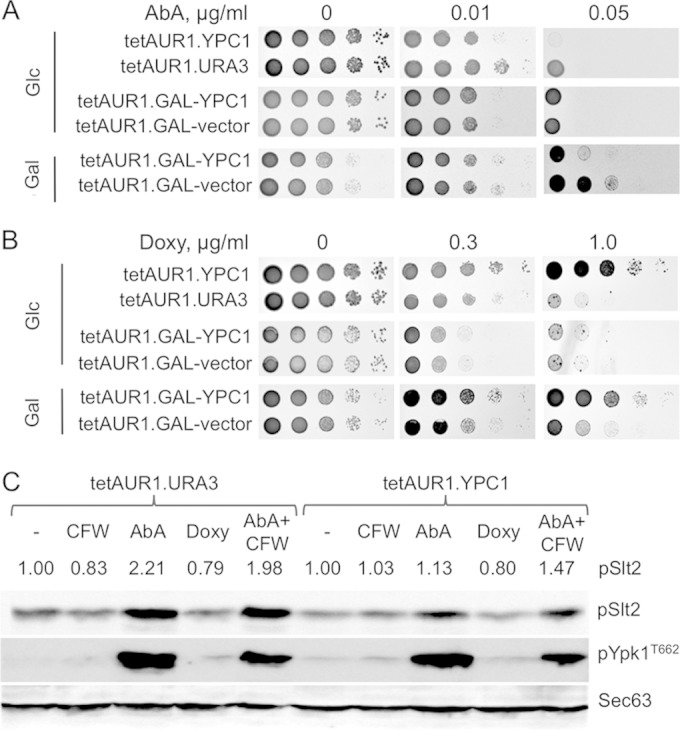
Overexpression of YPC1 influences the growth of tetAUR1 cells depending on the mode of AUR1 repression. (A and B) Tenfold dilutions of cells carrying vectors for overexpression of YPC1 or the control vector were spotted on glucose- or galactose-containing medium. Two centromeric vectors having YPC1 either behind the strong constitutive promoter of ADH1 or behind the galactose-inducible GAL1 promoter were utilized. Growth media were supplemented with AbA or Doxy as indicated. In this experiment, cells plated on glucose or galactose had been precultured on that same carbon source before plating, but the same result was also obtained when all cells had been precultured on glucose (not shown). (C) The indicated strains were grown in SC medium to exponential phase (OD600 = 0.5 to 0.7) and further treated for 3 h with either Calcofluor white (CFW) (5.0 μg/ml), AbA (3.0 μg/ml), or Doxy (10.0 μg/ml). Proteins were extracted and resolved by SDS-PAGE. Phosphorylated Slt2 (pSlt2) and Ypk1 (pYpk1, phosphorylated on Thr662) were detected as described previously (30). Sec63 was used as a loading control. For the quantifications of pSlt2 shown on the tops of lanes, data were normalized with regard to the amount of Sec63 and expressed as a fraction of results for nontreated cells (1.00).
Overexpression of membrane proteins is known to induce an unfolded-protein response (UPR), and the UPR has been shown to increase the synthesis of sphingolipids in yeast (32). To see if overexpression of YPC1 causes the observed effects by inducing or enhancing a UPR, we also overexpressed enzymatically “dead” versions of YPC1 that do not display any Ypc1 activity in vivo or in vitro. As shown in Fig. 4A, overexpression of catalytically dead versions of YPC1 had no effect on the sensitivity of cells to AbA or Doxy, although they were expressed almost as strongly as WT Ypc1 (Fig. 4B).
FIG 4.
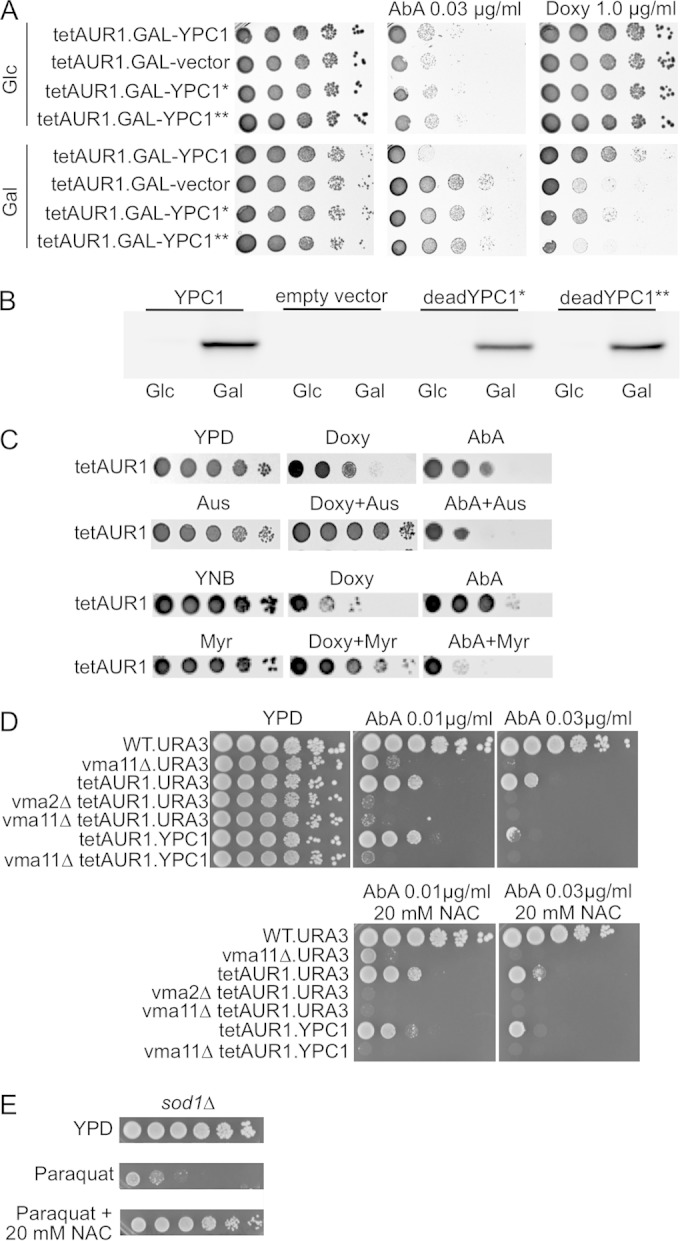
Effects of dead YPC1 alleles, australifungin, and myriocin on cells having reduced IPC synthase activity. (A) Serial dilutions of cells overexpressing either WT or two functionally dead forms of YPC1 (denoted * and **) were plated on plates containing AbA or Doxy. (B) tetAUR1 containing hemagglutinin (HA)-tagged YPC1, empty vector, or the two dead alleles of YPC1 behind the GAL1 promoter were grown in galactose for 20 h, and extracts corresponding to 1 OD600 unit of cells were Western blotted using anti-HA. (C) Upper two rows, serial dilutions of tetAUR1 were spotted on YPD and YPD supplemented with 0.25 μg/ml Doxy, 0.03 μg/ml AbA, 1.50 μg/ml australifungin, or combinations thereof at the same concentrations. Lower two rows, cells were also spotted on media supplemented with 0.50 μg/ml Doxy, 0.01 μg/ml AbA, or 0.05 μg/ml myriocin or combinations thereof at same concentrations. Plates were incubated for 2 days at 30°C. (D) Tenfold dilutions of vma mutants in the BY4742 (WT) or tetAUR1 background were spotted onto YPD plates with the indicated concentrations of AbA and NAC. (E) To demonstrate the efficacy of NAC in antagonizing the deleterious effects of ROS, the ROS-hypersensitive sod1Δ cells, lacking cytosolic superoxide dismutase, were included to show that the growth inhibition caused by paraquat, an oxidant, can be antagonized by NAC.
In view of the different kinetics of AbA- and Doxy-mediated repression of Aur1, we tested if prolonged treatment with Doxy is able to kill cells, as is well described for AbA. Indeed, Doxy treatment for 7 h killed a substantial fraction of WT as well as hypersensitive mutant cells (see Fig. S4 in the supplemental material).
Overexpression of YPC1 modulates AbA and Doxy sensitivity of the tetAUR1 strain by decreasing IPC and ceramide levels, respectively.
We surmised that overexpression of YPC1 rescues the Doxy-induced growth defect either by reducing toxic ceramide levels or by increasing LCB levels but not by further decreasing IPCs. To investigate this positive effect further, we decided to explore alternative ways to reduce ceramides. For this we utilized australifungin, a specific inhibitor of ceramide synthase (33) (Fig. 1). Australifungin, similar to YPC1 overexpression, reduces IPCs and ceramides and increases LCBs. In parallel, we also used myriocin, a specific inhibitor of Lcb1/Lcb2 (34) (Fig. 1), which reduces both ceramides and LCBs. As shown in Fig. 4C, the compromised growth of tetAUR1 cells on Doxy was improved when australifungin or myriocin was present at concentrations which did not have any effect on cell growth on their own. Thus, the data suggest that the growth-promoting effect of YPC1 overexpression on Doxy-treated tetAUR1 cells comes about through reduction of toxic ceramides, not elevation of LCBs.
As to AbA-treated cells, we surmised that the usually observed YPC1-mediated growth reduction could be caused either by reduction of complex sphingolipids or by a rise of LCBs to toxic levels, whereas it seemed a priori excluded that YPC1 would compromise growth on AbA by reducing the elevated ceramide levels. As australifungin and myriocin increased the toxicity of AbA (Fig. 4C), we conclude that AbA is toxic through reduction of complex sphingolipids. This interpretation is in agreement with a recent study of the effect of the conditional lcb1-100 mutation on AbA sensitivity which concluded that the reduction of complex sphingolipid levels but not the increase of ceramide levels is critical for AbA-induced cell death (14).
Thus, we propose that the immediate stress for cells upon acute Aur1 inhibition on AbA may be the lack of IPCs, whereas the growth-limiting factor during a more protracted Aur1 repression under Doxy may be the toxicity of ceramides.
The above-mentioned study and a second one recently reported that AbA (0.05 μg/ml for 4 h or 0.5 μg/ml for 1 h) as well as myriocin induces strong increases of ROS in WT cells (14, 35) and that the membrane-permeating antioxidant N-acetylcysteine (NAC) could rescue growth of cells on 0.4 μg/ml of myriocin. In our hands, NAC did not improve the growth of tetAUR1 cells on AbA (Fig. 4D) or on Doxy (not shown), although it efficiently antagonized the toxic effect of the ROS-generating oxidant paraquat (Fig. 4E). NAC also did not decrease toxicity of AbA in vacuolar acidification (vma) mutants, which are known to be hypersensitive to reactive oxygen radicals (36) and were found to be AbA hypersensitive (see below) (Fig. 4D). Thus, the ROS generated by AbA treatment may not be relevant for its proapoptotic action.
Genetic interaction profile of cells with depressed AUR1 activity as revealed by SGA analysis.
Previous low-throughput studies found sac1Δ, scs7Δ, lcb1-100, stt4ts, aur1ts, gpi1Δ, gpi2-7, gpi3-10, arv1Δ, gaa1-1, lem3Δ, and sch9Δ cells to be hypersensitive to AbA or genetic repression of AUR1 but lac1Δ lag1Δ, lip1, elo3Δ (sur4Δ), sur2Δ, yca1Δ, and pdr5Δ yor1Δ cells to be hyperresistant (14, 29, 37–43). To get a more comprehensive picture, we performed a genetic screen in which a large collection of mutants, each having a nonessential gene replaced by the kanMX resistance marker, were crossed with tetAUR1.YPC1 and tetAUR1.URA3 query strains. After sporulation, haploids that were uracil prototrophs and bore the Doxy-repressible AUR1 and a kanMX4 allele were selected and placed in the last selection round on either 0.03 μg/ml AbA, 0.1 μg/ml Doxy, or control medium, so that we finally observed growth of each nonlethal deletion under 6 different conditions (Fig. 5A).
FIG 5.

Screens identifying gene deletions making cells sensitive or resistant to Aur1 repression. (A) Each of the 4,849 deletion strains was crossed with either tetAUR1.URA3 or tetAUR1.YPC1, and haploid progeny harboring a gene deletion, the tetAUR1 allele, and an URA3 plasmid were selected on plates supplemented with no drug, AbA, or Doxy. The relative growth of each mutant was determined by 5 binary comparisons between conditions linked by a double-pointing arrow. (B) Venn diagram classifying deletion mutants growing worse than average under AUR1 repression (positive Z scores) according to the screen(s) (comparisons 1 to 4 in panel A) that identified them. Different fields denoted by letters are referred to in the text and in Table S5 in the supplemental material and are followed by the number of negative interactors identified in this category. (C) For each deletion in the tetAUR1.URA3 background giving a significant interaction on at least one of the drugs (P < 0.05, |Z score| > 1.95), the Z scores on Doxy were plotted against the Z scores on AbA. (D) Same as for panel C but for deletions in the tetAUR1.YPC1 background. (E) Z scores in comparison 1 (panel A) plotted against their Z scores in comparison 3 for 174 mutants giving significant scores in either one of these screens. Twenty-nine strains giving a significant score in comparison 5 of panel A were excluded. (F) Same as for panel E but plotting scores in comparison 2 versus comparison 4 for 343 mutants after removal of 43 strains giving a significant score in comparison 5. (G) Venn diagram indicating overlaps between gene deletions causing significant growth retardation of hypomorphic aur1 alleles according to BIOGRID and the “high-confidence set” (panel B, fields a to k). (H) Z scores of tetAUR1.URA3 versus those of tetAUR1.YPC1 on AbA (top) and on Doxy (bottom) for deletions in mitochondrial genes.
Five binary comparisons were made, as shown by arrows in Fig. 5A. The growth defect of a given mutant under drug treatment compared to its growth without drug was compared with the average difference of all mutants in a given binary comparison, and only differences with P values of <0.05 (|Z score| > 1.95) were considered to be significant. (The full set of interactions is given in Table S3A in the supplemental material, and deletions giving very strong interactions in most comparisons shown in Fig. 5A are listed in Table S3B in the supplemental material.) Strains highly sensitive to Aur1 repression (positive Z score) in binary comparisons 1 to 4 overlapped only partially (Fig. 5B). This most likely is due not only to the different modes of Aur1 repression and different Ypc1 levels but also to the significant number of false-positive hits generated by the SGA (44, 45). However, if any biological process becomes essential under a particular stress condition tested, one expects an increased frequency of hits among genes involved in that process, i.e., annotated by the corresponding gene ontology (GO) terms describing “biological process” in the Saccharomyces Genome Database (SGD) (http://www.yeastgenome.org/). We find that that negative interactors in each one of the 4 screens shown in Fig. 5B were strongly enriched for genes attributed to GO terms concerning vesicle-mediated transport between the Golgi apparatus, endosomes, and vacuole (Table 1; see Table S4 in the supplemental material). Depending on the localization or their products, genes are also annotated by GO terms describing the “cellular component,” and the GO term enrichments in negative interactors (GENIs) concerning the “cellular component” in our screens similarly are concentrated on the Golgi apparatus and endosomes (Table 1; see Table S4 in the supplemental material). Among the 371 deletions giving significant negative interactions in comparisons 1 to 4 (Fig. 5A), 42 belong to the GO term “vacuolar transport.” Of those, 22 had previously been found to be vacuolar protein sorting (vps) mutants (see Fig. S5A in the supplemental material). vps mutants also stood out qualitatively, as they were overrepresented among the deletions giving the strongest negative interactions on AbA and Doxy (see Table S3B in the supplemental material). The GO terms of Table 1 were of course also strongly enriched in mutants found in more than one screen (fields a to k in Fig. 5B) (see Tables S3B and S5 in the supplemental material), but they were completely absent in mutants found in only a single screen (fields l, m, n, and o in Fig. 5B). Indeed, practically no GO terms were enriched in negative interactors found only in a single screen (see Table S5 in the supplemental material), indicating that many of them may be false-positive hits. In the following, negative interactions found in two or more screens, i.e., comprised in fields a to k of Fig. 5B, are therefore referred to as the “high-confidence set.”
TABLE 1.
GO terms enriched in negative interactors identified by at least one AbA and one Doxy screen from Fig. 5Aa
| GO term | Log10 exponent of P value for the enrichment of the indicated GO term |
|||||||
|---|---|---|---|---|---|---|---|---|
| Interactions from this reportb |
Interactions from BIOGRIDc |
|||||||
| tetAUR1.URA3 |
tetAUR1.YPC1 |
csg2Δ (278) | orm2Δ (267) | gas1Δ (332) | pmr1Δ(249) | |||
| AbA (1) | Doxy (2) | AbA (3) | Doxy (4) | |||||
| Biological processes | ||||||||
| Vesicle-mediated transport | −13 | −4 | −8 | −6 | −13 | −9 | −20 | −22 |
| Endosomal transport | −13 | −4 | −8 | −7 | −2 | −4 | −5 | −5 |
| Vacuolar transport | −10 | −6 | −6 | −11 | −5 | −5 | −5 | −11 |
| Retrograde transport, endosome-Golgi apparatus | −5 | — | −3 | — | — | — | −2 | — |
| Golgi vesicle transport | −5 | — | −2 | −2 | −7 | −5 | −14 | −14 |
| Protein targeting to vacuole | −4 | — | — | −5 | −6 | −3 | −4 | — |
| Post-Golgi vesicle-mediated transport | −3 | — | −3 | −3 | — | — | −7 | — |
| Late endosome-to-vacuole transport | −3 | −3 | −2 | −4 | — | −3 | — | −5 |
| Vacuolar acidification | −12 | — | −19 | — | — | — | — | — |
| Cellular components | ||||||||
| Endosome | −12 | −5 | −13 | −11 | −2 | −4 | −5 | −11 |
| Endosome membrane | −8 | −2 | −9 | −3 | — | −4 | — | −9 |
| Golgi apparatus | −4 | −2 | −3 | — | −15 | −8 | −18 | −22 |
| Vacuolar part | −2 | — | −9 | −3 | — | — | — | — |
| Vacuolar membrane | −2 | — | −8 | −2 | — | — | — | — |
| CORVET complex | — | — | −3 | −6 | — | — | — | — |
Enrichments of GO terms relating to the “biological process” were obtained at http://yeastmine.yeastgenome.org/ with Holm-Bonferroni correction in place. —, P > 0.05.
Numbers in parentheses indicate the arrow number in Fig. 5A.
Numbers in parentheses indicate the number of negative interactors.
Figure 5C and D show that the Z scores on AbA versus Doxy were positively correlated but not very strongly. This again suggests that the stresses on AbA versus Doxy are not quite the same and that cells depend on different genes to overcome them. Figure 5B also clearly shows that the genes identified in the tetAUR1.URA3 background only partially overlap those found in the tetAUR1.YPC1 background, but scores obtained with the two strain backgrounds showed considerable positive correlation (Fig. 5E and F).
Mere overexpression of YPC1 in the absence of drugs (arrow 5 in Fig. 5A) resulted in 81 and 88 significant interactions with positive and negative Z scores, respectively, but these sets showed almost no GO term enrichments (see Table S4 in the supplemental material).
Among the 371 deletions giving significant interactions in comparisons 1 to 4 (Fig. 5A), there also are 63 genes annotated with GO terms containing the term “mitochondrial.” However, they did not score as an enriched GO term, given the high number of such genes in the genome. As shown in Fig. 5H, most of these genes were identified because they gave significant positive or negative interactions on Doxy, not on AbA, suggesting that Aur1 repression by Doxy results in a kind of stress that is more easily modulated by altered mitochondrial functions than the stress caused by AbA.
Screens using AbA identify vacuolar acidification mutants.
Interestingly, mutants of set g (Fig. 5B), i.e., mutants found to give negative interactions only in the two screens done with AbA and not in those done with Doxy, did not show the GENIs mentioned above (Table 1), but on the other hand, they were very strongly enriched in several GO terms describing vacuolar acidification (Table 1; see Tables S4 and S5 in the supplemental material). Indeed, of the 18 genes required to constitute the vacuolar H+-ATPase (14 genes for ATPase subunits and 4 genes required for their assembly), 15 were found in our screen as interacting negatively with AbA (Fig. 5D; see Fig. S6A in the supplemental material). As shown in Fig. S6B in the supplemental material, vacuolar acidification (vma) mutants were found to be AbA hypersensitive on serial dilution plates not only at high pH, at which vma mutants are notoriously unable to grow (46), but also when placed in acidic medium (pH 4.2), in which part of their vacuolar acidification problem may be compensated by endocytosis (47). This suggests that the real problem of cells under AbA stress may be their inability to maintain an alkaline pH in the cytosol. Indeed, in vma2Δ cells grown at pH 5, the cytosolic pH was found to be 0.4 pH unit lower than in the WT (48). Also, it was recently shown that vma mutants endocytose Pma1, thereby reducing their ability to alkalinize the cytosol (49).
Hypersensitivity of vma mutants to AbA is not explained by inositol auxotrophy.
Based on the literature, we wondered whether vma mutants were exhibiting AbA hypersensitivity because of excessive inositol auxotrophy. For one, previous studies by Susan Henry's group demonstrated that SEY6210 WT cells in the presence of 0.05 μg/ml AbA or aur1ts cells are inositol auxotroph (Ino−) (29). Moreover, we realized that there was a 4- to 6-fold-higher-than-expected overlap between our “high-confidence set” of negative interactions and Ino− strains identified through two recent high-throughput screens (48, 50). Strikingly, one of these screens for Ino− mutants had identified all 18 genes coding for the building blocks or essential chaperones required for the construction of the vacuolar proton ATPase (48). Thus, it seemed important to understand whether the hypersensitivity to AbA of our high-confidence set and, in particular, of the 15 vma mutants came about because of an increased inositol requirement under AbA stress. The following results strongly argued against this. (i) All our screens were done in SC medium containing 430 μM inositol, more than the 75 μM used by Susan Henry's group (29). Confirming their results (29), we found that inositol enabled WT cells to grow on AbA and Doxy (see Fig. S7A in the supplemental material) and that 10 μM inositol was sufficient for maximal growth of WT on these drugs (see Fig. S7B in the supplemental material). However, inositol did not support growth of vma2Δ cells on AbA (Fig. S7A). (ii) Of the numerous significantly enriched specific GO terms in the Ino− mutant cells (48, 50), only vacuolar acidification (GO0007035) is also present in our high confidence set (Fig. 5B, fields a to k) (see Table S6 in the supplemental material), and the Ino− mutants identified in these studies are not enriched in vacuolar protein sorting (vps) mutants or other terms listed in Table 1.
AbA causes stronger inhibition of Aur1 in vma cells than in WT cells.
Although AbA is a very hydrophobic cyclic peptide lacking nitrogen atoms that could be protonated, i.e., lacking lysosomotropic properties, we wondered if vacuolar H+-ATPase mutants showed reduced growth on AbA because they take up larger amounts of AbA or degrade it less rapidly, so that the Golgi apparatus-based Aur1 is exposed to higher concentrations of AbA leading to stronger Aur1 repression. We therefore labeled vma and WT cells with [3H]inositol in graded concentrations of AbA in order to test the efficacy of AbA to repress their sphingolipid biosynthesis (Fig. 6). To avoid second-site suppressors, we verified that the vma mutants to be analyzed were unable to grow at high pH in the presence of calcium (Fig. 6A) (46). When cells were labeled with [3H]inositol, it appeared that AbA inhibited IPC and MIPC biosynthesis more strongly in vma and vps6Δ mutants than in the WT, whereas the vps52Δ mutant behaved like the WT (Fig. 6B). Interestingly, the vps6Δ mutant belongs to the small subset of vps mutants that exhibit a vacuolar acidification defect (51). Labeling with [3H]inositol requires preculture of cells in inositol-free medium, and this could compromise the viability of vma mutants since they are inositol auxotrophic (48). Yet, in the experiment of Fig. 6B, the total incorporation of [3H]inositol into lipids in vma mutants was quite normal and was not influenced by the presence of AbA (see Fig. S8A in the supplemental material). To formally exclude any negative effect of inositol deprivation, we chose to also label the cells with [14C]serine, this time in the presence of high concentrations of inositol (430 μM). For this, a 5-h labeling period was used. Here, the presence of AbA reduced total incorporation of [14C]serine into lipids in a concentration-dependent manner (see Fig. S8B in the supplemental material), and incorporation correlated with the number of cell divisions occurring during labeling (see Fig. S8C in the supplemental material). As shown in Fig. 6C, [14C]serine labeling revealed (i) that 0.25 μg/ml of AbA strongly reduced the IPC/MIPC synthesis in the vma2Δ mutant but not in the WT and (ii) that, as judged from lanes treated or not with NaOH, AbA caused a stronger accumulation of ceramides in the vma2Δ mutant than in the WT. These same observations were made in all vma mutants subjected to [14C]serine labeling (Fig. 6D). Thus, both labeling with [3H]inositol and labeling with [14C]serine suggest that the AbA hypersensitivity of vma mutants can be explained by a stronger inhibition of Aur1 by AbA in vma mutants; the reason for this remains to be elucidated. Other interesting observations in the experiment of Fig. 6 are as follows. (i) It appears that [3H]inositol labeling, as we had suspected, overestimates the efficiency of AbA to curtail the synthesis of IPC and MIPCs in vma mutants, since 0.025 μg/ml AbA reduces the synthesis of IPCs and MIPCs in vma mutants by about 50% as judged by [3H]inositol labeling but by only 10% as judged by [14C]serine labeling (Fig. 6B versus D). (ii) Here as well as in many previous experiments, we observed a quantitative increase of ceramides and the appearance of a new band with slightly lower TLC mobility after mild base treatment which could be a ceramide or a free fatty acid (Fig. 6C). It is possible that a significant fraction of ceramides is esterified with the carboxyl group of some polar substance(s), e.g., an amino acid(s), but we have not been able to fully characterize the material comigrating with ceramides after mild base treatment.
FIG 6.

AbA causes stronger inhibition of Aur1 in vma cells than in WT cells. (A) Tenfold dilutions of the indicated strains were plated on YPD or YPD with 50 mM HEPES (pH 7.5) and 50 mM CaCl2. (B) Cells growing for 14 h in inositol-free SC medium were labeled with [3H]inositol for 2 h in the presence of various concentrations of AbA. Lipid extracts were deacylated by mild base treatment and run on TLC. The amounts of mild-base-resistant sphingolipids made in the absence of AbA were set as 100% for each strain, and the amounts of sphingolipids made in the presence of AbA were calculated as a percentage thereof. (NaOH-resistant lipids accounted for 50.6% ± 3.3% and 35.5% ± 0.8% of total lipids in the WT and vma mutants, respectively.) (C) Autoradiograms of TLC plates resolving lipid extracts from WT or vma2Δ cells having been labeled with [14C]serine with or without AbA and deacylated or not with NaOH. In all TLC lanes, material from equivalent numbers of WT and mutant cells was separated. (D) Quantifications of the data of panel C (vma2Δ) plus those from vma7Δ and vma8Δ mutants labeled in the same experiment. For each of the 12 labelings (4 strains labeled under three conditions [0, 0.025, and 0.25 μg/ml of AbA]), the total amounts of label in the nondeacylated lipid extract were set as 100%, and the counts in IPCs plus MIPCs or in the stronger of the two bands in the ceramide region present after NaOH treatment were expressed as a percentage thereof. Strain symbols are as in panel B.
Treatment of cells with aureobasidin A increases vacuolar quinacrine accumulation.
It seemed possible that the heightened AbA sensitivity of IPC biosynthesis in vma mutants derives from the fact that in AbA-treated cells the vacuolar acidification has to compensate for cytosolic acidification or the imbalance of some electrolytes. Quinacrine, a fluorescent compound that accumulates in acidic compartments, was used to estimate the vacuolar acidification under AbA (51, 52). For this, cells were preincubated with AbA for 1 h, stained for 8 min at pH 7.6 with quinacrine, washed, and viewed under the microscope. As shown in Fig. 7 and more extensively in Fig. S9 in the supplemental material, treatment of WT cells with AbA (0.25 μg/ml) for 1 h led to a distinct increase of vacuolar quinacrine accumulation, whereas treatment with Doxy (10 μg/ml) for 7 h did not (not shown). As expected, no quinacrine accumulation was seen in vma2Δ cells with or without AbA treatment (Fig. 7; see Fig. S9 in the supplemental material). The precise reason for the increased vacuolar acidification upon AbA treatment is presently unknown.
FIG 7.
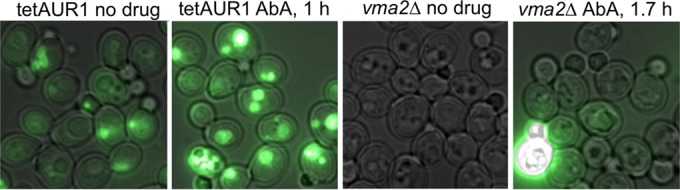
AbA treatment increases vacuolar quinacrine accumulation. Exponentially growing tetAUR1 and vma2Δ cells were harvested at an OD600 of 1.0 and incubated with or without 0.25 μg/ml AbA at 30°C at pH 6.5 for 1 h. Cells were then washed, stained with quinacrine, placed on ice, and viewed within 30 min, during which period early and late viewings did not show any difference. All pictures were taken under the same conditions and processed with ImageJ (http://imagej.nih.gov/ij/) using the same window settings (see Materials and Methods). This is a representative sample of the larger set of pictures shown in Fig. S9 in the supplemental material. The transillumination and fluorescence pictures were merged. Occasional brightly fluorescent cells increased with time and were assumed to be dead cells.
Deletions decreasing ceramide levels enhance growth on doxycycline.
For growth-enhancing interactions (negative Z scores), most of them were found with tetAUR1.YPC1 as a query and by selecting on Doxy (Fig. 5D versus C, lower left quadrants; see Fig. S10 in the supplemental material). The deletions that enhanced growth of tetAUR1.YPC1 on Doxy were enriched for rather generic GO terms such as “regulation of RNA biosynthetic processes,” but, interestingly, the term “sphingolipid biosynthetic processes” (GO 0030148) also was enriched (P = 0.009), with 7 of the 14 genes in this GO term giving significant negative Z scores in our screen (see Table S4 in the supplemental material). Interestingly, gene deletions in all of the 7 hits (TSC3, LCB3, ELO2, ELO3, SUR2, LAC1, or ISC1) (red in Fig. 1) ought to reduce ceramide levels. This fits the notion elaborated based on Fig. 4C that YPC1 overexpression sustains growth of tetAUR1 cells on Doxy by reducing ceramide levels. These data also confirm the reported hyperresistance of elo3Δ and sur2Δ strains under AUR1-repressing conditions mentioned above. Moreover, 5 mutants reducing ceramide levels had already been found in preliminary screens, and all of them showed improved growth in serial dilution growth assays (red underlined in Fig. 1; see Fig. S11 and S12 in the supplemental material).
Twenty-three deletions also enhanced growth of tetAUR1.YPC1 on AbA, and 18 among those also enhanced growth on Doxy (see Fig. S10, fields a, b, d, and j, in the supplemental material), but these 23 were not enriched for any GO term.
Comparison with published high-throughput screens.
While these studies were in progress, a number of genetic interactions with AUR1 damp clones gathered from high-throughput studies were reported in BIOGRID (53, 54) (see Table S7 in the supplemental material). The 287 negative interactions in this set are strongly enriched in the very same GO terms as our 134 high-confidence set (fields a to k in Fig. 5B), but only 23 genes were found to be common between BIOGRID and our high-confidence set (Fig. 5G). However, all 3 subsets in Fig. 5G showed GENIs in vacuolar and endosomal transport, indicating that neither our screen nor the BIOGRID screen was exhaustive (see Table S7 in the supplemental material).
Twenty-eight strains were common between our high-confidence set and the hits identified in the diploid homozygous knockout collection through cocultivation of all nonessential deletion strains in liquid cultures containing 0.02 μg/ml AbA (55) (see Table S8 in the supplemental material). This represents a 4-fold-higher overlap than expected by chance. The 296 most sensitive strains from reference 55 were enriched for “vacuolar acidification” and similar GO terms (0.005 < P < 0.05) but for no other terms in Table 1 (see Table S8 in the supplemental material). It appears that vacuolar acidification also is required when cells are stressed in liquid culture. No significant overlap of AbA-hyperresistant hits in our study or the aur1-damp interactions in BIOGRID with hyperresistant homozygous deletions from reference 55 was found.
AbA-treated cells have a morphologically normal Golgi apparatus but enlarged lipid droplets.
Based on the high sensitivity to Aur1 repression in mutants with affected vesicular traffic between the Golgi apparatus and the vacuole, we tried to see whether AbA or Doxy would cause morphological changes of the Golgi apparatus in tetAUR1 cells expressing the Golgi protein Sec7-GFP or Sed5-GFP. However, no convincing changes were observed (not shown). We also monitored the distribution of GFP-labeled plasma membrane proteins such as Can1, Fur4, and Gas1 under AbA (0.05 or 0.25 μg/ml, 30°C, 1 to 7 h). In this case we also failed to see accumulation of GFP in the ER or Golgi apparatus and only occasionally found cells with increased vacuolar staining (data not shown). Thus, at this level of sensitivity, it did not seem that AbA caused a secretion block or destabilization of plasma membrane proteins. We noted only that the volume of lipid droplets increased upon AbA treatment, as judged from staining with the hydrophobic BODIPY fluorescent dye (see Fig. S13A in the supplemental material), suggesting that AbA may cause alterations of the lipid metabolism beyond the sphingolipid metabolic pathways.
In support of this, mass spectrometric lipid profiling of cells treated for 4 h with AbA indeed revealed major increases in TAGs and sterol esters (SEs) along with strong reductions of phosphatidylserine (PS) and phosphatidylethanolamine (PE) and a small reduction in PI (Fig. 8A), in keeping with an increased volume of lipid droplets in such cells. It also appeared that AbA treatment increased the desaturation of fatty acids in TAGs (not shown). In a pathological process leading to cell death, it is obviously not easy to distinguish between events that cause cell death and events that are just consequences of imminent cell death. However, the same increase in neutral lipids and strong decrease in PS is also found in lag1Δ lac1Δ ypc1Δ ydc1Δ SLC1-1 cells which are not undergoing cell death but suffer from a constitutive lack of complex sphingolipids, as appears under Aur1 repression (S.K. Mallela et al., unpublished data).
FIG 8.

AbA causes the accumulation of neutral lipids. (A) Exponentially growing WT and ypc1Δ ydc1Δ (yyΔΔ) cells were extracted directly or further incubated for 4 h with 0.25 μg of AbA. Lipid extracts were analyzed by MS. Results are expressed as the percentage of lipids identified in positive (SE, TAG, and PE) or negative (PS and PI) ion mode. Error bars for two technical replicates are shown. Only lipids that change with AbA treatment are shown. Results are from one of two experiments giving the same result. (B and C) Tenfold dilutions of exponential-phase WT and pah1Δ lpp1Δ dpp1Δ cells (B) or tenfold dilutions of exponential-phase cells carrying deletions in 3 of the 4 enzymes making sterol esters (SEs) and TAGs (ARE1, ARE2, DGA1, and LRO1) and having the promoter of the genomic copy of the fourth gene, either DGA1 or LRO1, replaced by the GAL1 promoter (C) were plated onto YPD or YPGal with the indicated concentrations of AbA and incubated for 2 (B) or 3 (C) days at 30°C.
To see if the accumulation of sterol esters and TAGs helped cells overcome the AbA-induced stress, we tested pah1Δ lpp1Δ dpp1Δ mutants retaining only 35% of the activity transforming phosphatidic acid into diacylglycerol, the obligatory precursor for TAG biosynthesis (56). As shown in Fig. 8B, this triple mutant showed AbA hypersensitivity. Similarly, nem1Δ cells, lacking the phosphatase required to activate Pah1, were found to be Doxy hypersensitive in our screen. When viewed after BODIPY staining, pah1Δ lpp1Δ dpp1Δ mutants had fewer lipid droplets than the WT and made 3.5-fold less TAG than the corresponding WT, as described before (56, 57) (see Fig. S13B and C in the supplemental material). However, when treated with AbA they nevertheless accumulated dotty hydrophobic material (see Fig. S13B) and still increased their TAG levels (see Fig. S13C), probably using the remaining phosphatidate (PA) phosphatase App1. They also reduced PS and PE, like the WT (see Fig. S13C).
Surprisingly, blocking SE and TAG synthesis by simultaneously deleting/repressing ARE1, ARE2, LRO1, and DGA1 made cells rather AbA resistant (Fig. 8C). It is possible that AbA-treated cells are hypersensitive to PA, which accumulates in pah1Δ cells but not in are1Δ are2Δ lro1Δ dga1Δ cells. PA is a central regulator which sequesters the Opi1 transcriptional repressor and thereby derepresses lipid biosynthesis (48).
DISCUSSION
Our data establish hydroxylated VLCFAs (C24:0;1 and C26:0;1) as an indicator of futile cycling caused by simultaneous presence of alkaline ceramidases and acyl-CoA-dependent ceramide synthases, but we cannot presently assert that they are a quantitative indicator, since we have not excluded that these hydroxylated fatty acids would again be utilized by Ypc1 or Ydc1 for reverse ceramide synthesis, which may occur concomitantly with ceramide hydrolysis. We also have not excluded that they would be activated and used by acyl-CoA-dependent ceramide synthases or be degraded by peroxisomal β-oxidation. Nevertheless, free C24:0;1 and C26:0;1 fatty acids seem to be much better indicators for such futile cycling than LCBs, which can be elevated not only through increased breakdown of ceramides (Fig. 2A) but also by increases in the highly regulated de novo biosynthesis of LCBs or by downregulation of the well-known LCB degradation pathway through Lcb4/Lcb5 and Dpl1 (58).
As mentioned, a recent report showed that the reduction of complex sphingolipids caused by AbA results in ER retention of GPI-anchored proteins, ER stress, an unfolded-protein response, ROS production, mitochondrial cytochrome c release, and a metacaspase-mediated form of apoptosis that additionally is dependent on the concomitant increase of cytosolic Ca2+ concentrations (14). Our report confirms and adds new features to these observations. As said above, we do not find evidence that ROS would play a major role in AbA-induced apoptosis (Fig. 4D). Our data, however, confirm that it is the reduction of IPCs rather than accumulation of toxic ceramides that reduces cell viability under AbA stress, but they also suggest that during the slow depletion of Aur1 function under Doxy, it is rather the accumulation of ceramides which produces the major proapoptotic signal. We also found that the deletion of the unfolded-protein response gene IRE1 or HAC1 aggravates AbA and Doxy toxicity (Fig. 5D; see Table S3 in the supplemental material), in keeping with the fact that AbA induces an ER stress and arguing that the ensuing UPR improves cell growth/survival. One mechanism would be that the UPR relieves the reported accumulation of GPI proteins (14). On the other hand, it is unlikely that a UPR-induced increase in LCB biosynthesis, as observed in the conditional lcb1-100 mutant (32), would help cells to overcome the AbA stress, since (i) such an increase is not seen in WT cells, (ii) a reduction rather than an increase of LCB and ceramide synthesis reduced AbA toxicity (Fig. 4C), and (iii) deletion of ORM1 or ORM2, repressors of LCB1/LCB2, did not help cells under Aur1 repression (see Table S3A in the supplemental material). Our data further confirm that pmr1Δ cells are AbA hypersensitive (Table 2; see Table S3 and Fig. S11C and E in the supplemental material), as reported before (14). Pmr1 acts to reduce cytosolic calcium and prevents calcineurin activation (59), and this probably explains why it helps to avoid the calcium-dependent apoptosis induced by AbA (14).
TABLE 2.
Summary of serial dilution growth tests done with deletion strains identified as sensitive or resistant to Aur1 repression in preliminary screensa
| Phenotype | Identified genesb |
|---|---|
| AbA hypersensitive | BST1, BUD14, ECM33, ERD1, ERG3, ERV14, FKS1, FYV6, GUP1, IML2, OPI9, LEO1, OSH3, PER1, PMR1, RPN4, RVS161, SAC6, SHE2, SLG1, SNT1, SUR1, TEF4, YMR102C, YPK1, YSP2. |
| Doxy hypersensitive | BTS1, COG5, COG6, DOA1, GOS1, GYP1, ITR1, MON2, PEP12, SCS7 |
| Doxy resistant | EGO1, ELO2, ELO3, FAT1, FOX2, GSG1, GUP1, ISC1, KEX2, LCB3, LEO1, MRPL36, SLG1, SUR2, YPT6 |
In preliminary high-throughput SGA screens, the library containing 4,849 strains, each one deleted in a single nonessential gene, had been robotically crossed with either tetAUR1, BY4742 (FBY5313), or BY4742 harboring pYPC1-URA3 (FBY5333) and plated on AbA or Doxy. Some hypersensitive or hyperresistant isolates of interest to us were picked and further tested in the subsequent serial dilution growth assays shown in Fig. S11 and S12 in the supplemental material. Those deletion strains that reproduced the SGA result in the growth test are listed in this table.
Genes involved in sphingolipid metabolism are in bold, and those which, when deleted, cause at the same time AbA hypersensitivity and Doxy resistance are underlined.
We made the new observation that the protein transport to vacuole, late endosome-to-vacuole transport, retrograde endosome-to-Golgi transport, Golgi vesicle transport, and similar processes become critical when Aur1 activity is reduced (Table 1). This was found in each one of the screens 1 to 4 (Fig. 5A), whether Aur1 was repressed pharmacologically or transcriptionally and whether or not YPC1 was overexpressed (see Table S4 in the supplemental material), and this is confirmed by BIOGRID data. Quite in the same line, elo3Δ (sur4Δ) cells, having reduced levels of sphingolipids, were found to be synthetically sick with vps21Δ and vps3Δ cells (60), and our data seem to generalize this finding.
We have no clue to why such vacuolar transport becomes essential when Aur1 is repressed, but it cannot possibly be the aggravation of a defect in endocytosis or Golgi-to-vacuole transport, since both of these processes are nonessential; intra-Golgi transport, however, is part of the essential secretion process.
Lack of IPCs leads to lack of MIPC and M(IP)2C and thereby may affect numerous sphingolipid-dependent processes. We used negative genetic interactions reported in BIOGRID to try to identify such downstream elements, failure of which could render the biological processes indicated by Table 1 more essential. Interestingly, deletion of CSG2 abolishes MIPC biosynthesis without increasing ceramide levels (61), and csg2Δ shows negative interactions with genes involved in the same processes as repressing Aur1, although only 23 mutants were common between negative csg2Δ interactors and our high-confidence set (subsets a to k in Fig. 5B) (Table 1; see Table S9A in the supplemental material); still, this represents 3 times more than expected by chance. Thus, the comparison of GENIs of Aur1-repressed and csg2Δ cells suggests that the essentiality of Golgi-vacuole transport in Aur1-repressed cells may partially be mediated through the lack of MIPC. GO terms connected to Golgi-vacuole traffic are also seen with Doxy-treated cells, indicating that Doxy-treated cells also suffer from a lack of MIPC, although they are greatly relieved by reduction of ceramide levels (Fig. 4C). Deletion of the M(IP)2C synthase gene IPT1 (Fig. 1) did not cause negative interactions enriched in any of the GO terms in Table 1 (see Table S9A in the supplemental material). Lack of MIPC in turn has been shown (i) to cause Slm1/2- and TORC2-dependent activation of the Ypk1 protein kinase leading to inactivation of Orm proteins and activation of LCB and ceramide biosynthesis (30, 62, 63), (ii) to inactivate the aminophospholipid flippase at the plasma membrane (18), and (iii) to activate the calcium-regulated phosphatase calcineurin (61). Deletion of ORM2 mimics the functional Orm2 inactivation through Ypk1-mediated Orm phosphorylation that occurs upon AbA treatment (64, 65), and negative interactors of orm2Δ are enriched in GO terms in Table 1 (see Table S9A and C in the supplemental material). Thus, AbA treatment may render Golgi-vacuole transport essential by activating Ypk1, but at the same time this activation must have a very positive effect, since ypk1Δ mutants show a strong negative interaction with AbA (Fig. 5E; see Fig. S11C and E in the supplemental material). This positive effect may well be the Orm-mediated activation of LCB synthesis and activation of ceramide synthase that may help to increase IPC production under AbA-mediated Aur1-repression (18, 30, 63, 64). On the other hand, deletion of LEM3, an essential component of the DNF1/DNF2/DNF3 aminophospholipid flippase, does not generate the negative genetic interactions found after AbA treatment (Table 1; see Table S9A in the supplemental material). Finally, cnb1Δ cells lacking calcineurin did not genetically interact with Aur1 repression in our screen, nor is such an interaction reported in BIOGRID. Still, genetic but not pharmacological repression of calcineurin was found to enhance resistance to AbA (14). We did not try to reproduce this, but it is possible that part of the toxicity of AbA is mediated by calcium-mediated calcineurin activation.
As shown in Table 1 and Table S9C in the supplemental material, the GENIs of some mutants with mutations affecting the cell wall integrity (gas1Δ, gup1Δ, and pmr1Δ) are somewhat similar to the ones of Aur1-repressed cells. Indeed, acute repression of Aur1 by AbA activates the MAPK cell wall integrity (CWI) pathway (29, 30) (Fig. 3C). Yet, GO terms concerning the fungal cell wall biosynthesis and organization are not enriched in our high-confidence set or the aur1 interactors reported in BIOGRID. Also, deletions in 6 nonessential genes of the MAPK pathway were present in our library (ROM1, ROM2, BCK1, MKK1, MKK2, and SLT2), but essentially none of them showed a negative interaction in our screens or with a hypomorphic aur1 allele according to BIOGRID. This also agrees with the finding that sorbitol could not improve viability of cells on AbA (see Fig. S14 in the supplemental material), as had been mentioned in an earlier report (8). However, vma mutants showed better AbA resistance on sorbitol (see Fig. S14). Thus, in WT cells, AbA treatment readily triggers strong activation of the CWI pathway, but this activation seems to be irrelevant in terms of cell survival, whereas in vma mutants the cell wall defect may contribute to the negative effects of AbA.
The fact that vma mutants are hypersensitive to AbA may be due to higher concentrations of AbA reaching Aur1 and/or a lack of the physiological stimulation of the vacuolar H+-ATPase in reaction to AbA (Fig. 7; see Fig. S9 in the supplemental material). Other effects may also contribute to the AbA hypersensitivity of vma mutants, e.g., the accumulation of cytosolic calcium observed in some vma mutants (59, 66), which can be explained by the fact that the vacuolar pH gradient is necessary to drive Ca2+ import into vacuoles using the vacuolar Ca2+/H+ antiporter Vcx1 (46). The increase in cytosolic calcium may enhance the toxicity of AbA (14) and itself cause the increased acidification of vacuoles in WT cells upon AbA treatment (Fig. 7; see Fig. S9 in the supplemental material).
In summary, our report shows that the mode of repression of AUR1 can influence the spectrum of pathogenic events, that vesicular traffic between Golgi apparatus, endosomes, and vacuole becomes critical for cell survival in Aur1-repressed cells, and that vacuolar acidification is an important defense mechanism when Aur1 is acutely repressed by AbA.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants CRSI33_125232 and 31003AB_131078 from the Swiss National Science Foundation and from the Novartis Foundation for Biological Research to A.C. and by grants from the Lundbeckfonden (R44-A4342) and the Danish Council for Independent Research/Natural Sciences (09-072484) to C.S.E.
We thank Martha Cyert, George Carman, Robbie Loewith, Roger Schneiter, and Akihiko Nakano for plasmids, strains, or antibodies and Hans Kristian Hannibal-Bach for help with mass spectrometric lipid analysis.
ADDENDUM IN PROOF
A similar paper showing the importance of the Golgi complex in cells with reduced sphingolipid biosynthesis has been published since this paper was typeset (F. Fröhlich, C. Petit, N. Kory, R. Christiano, H.-K. Hannibal-Bach, M. Graham, X. Liu, C. S. Ejsing, R. V. Farese, Jr., T. C. Walther, eLife 4:e08712, 2015, http://dx.doi.org/10.7554/eLife.08712)
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00117-15.
REFERENCES
- 1.Buede R, Rinker-Schaffer C, Pinto WJ, Lester RL, Dickson RC. 1991. Cloning and characterization of LCB1, a Saccharomyces gene required for biosynthesis of the long-chain base component of sphingolipids. J Bacteriol 173:4325–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagiec MM, Nagiec EE, Baltisberger JA, Wells GB, Lester RL, Dickson RC. 1997. Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. J Biol Chem 272:9809–9817. [DOI] [PubMed] [Google Scholar]
- 3.Beeler TJ, Fu D, Rivera J, Monaghan E, Gable K, Dunn TM. 1997. SUR1 (CSG1/BCL21), a gene necessary for growth of Saccharomyces cerevisiae in the presence of high Ca2+ concentrations at 37 degrees C, is required for mannosylation of inositolphosphorylceramide. Mol Gen Genet 255:570–579. doi: 10.1007/s004380050530. [DOI] [PubMed] [Google Scholar]
- 4.Uemura S, Kihara A, Inokuchi J, Igarashi Y. 2003. Csg1p and newly identified Csh1p function in mannosylinositol phosphorylceramide synthesis by interacting with Csg2p. J Biol Chem 278:45049–45055. doi: 10.1074/jbc.M305498200. [DOI] [PubMed] [Google Scholar]
- 5.Lisman Q, Pomorski T, Vogelzangs C, Urli-Stam D, de Cocq van Delwijnen W, Holthuis JC. 2004. Protein sorting in the late Golgi of Saccharomyces cerevisiae does not require mannosylated sphingolipids. J Biol Chem 279:1020–1029. doi: 10.1074/jbc.M306119200. [DOI] [PubMed] [Google Scholar]
- 6.Heidler SA, Radding JA. 1995. The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob Agents Chemother 39:2765–2769. doi: 10.1128/AAC.39.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hashida-Okado T, Ogawa A, Endo M, Yasumoto R, Takesako K, Kato I. 1996. AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: a study of defective morphologies in Aur1p-depleted cells. Mol Gen Genet 251:236–244. [DOI] [PubMed] [Google Scholar]
- 8.Endo M, Takesako K, Kato I, Yamaguchi H. 1997. Fungicidal action of aureobasidin A, a cyclic depsipeptide antifungal antibiotic, against Saccharomyces cerevisiae. Antimicrob Agents Chemother 41:672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schorling S, Vallee B, Barz WP, Riezman H, Oesterhelt D. 2001. Lag1p and Lac1p are essential for the Acyl-CoA-dependent ceramide synthase reaction in Saccharomyces cerevisae. Mol Biol Cell 12:3417–3427. doi: 10.1091/mbc.12.11.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagiec MM, Wells GB, Lester RL, Dickson RC. 1993. A suppressor gene that enables Saccharomyces cerevisiae to grow without making sphingolipids encodes a protein that resembles an Escherichia coli fatty acyltransferase. J Biol Chem 268:22156–22163. [PubMed] [Google Scholar]
- 11.Lester RL, Wells GB, Oxford G, Dickson RC. 1993. Mutant strains of Saccharomyces cerevisiae lacking sphingolipids synthesize novel inositol glycerophospholipids that mimic sphingolipid structures. J Biol Chem 268:845–856. [PubMed] [Google Scholar]
- 12.Gaigg B, Toulmay A, Schneiter R. 2006. Very long-chain fatty acid-containing lipids rather than sphingolipids per se are required for raft association and stable surface transport of newly synthesized plasma membrane ATPase in yeast. J Biol Chem 281:34135–34145. doi: 10.1074/jbc.M603791200. [DOI] [PubMed] [Google Scholar]
- 13.Vionnet C, Roubaty C, Ejsing CS, Knudsen J, Conzelmann A. 2011. Yeast cells lacking all known ceramide synthases continue to make complex sphingolipids and to incorporate ceramides into glycosylphosphatidylinositol (GPI) anchors. J Biol Chem 286:6769–6779. doi: 10.1074/jbc.M110.176875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kajiwara K, Muneoka T, Watanabe Y, Karashima T, Kitagaki H, Funato K. 2012. Perturbation of sphingolipid metabolism induces endoplasmic reticulum stress-mediated mitochondrial apoptosis in budding yeast. Mol Microbiol 86:1246–1261. doi: 10.1111/mmi.12056. [DOI] [PubMed] [Google Scholar]
- 15.Takesako K, Kuroda H, Inoue T, Haruna F, Yoshikawa Y, Kato I, Uchida K, Hiratani T, Yamaguchi H. 1993. Biological properties of aureobasidin A, a cyclic depsipeptide antifungal antibiotic. J Antibiot (Tokyo) 46:1414–1420. doi: 10.7164/antibiotics.46.1414. [DOI] [PubMed] [Google Scholar]
- 16.Mao C, Xu R, Bielawska A, Szulc ZM, Obeid LM. 2000. Cloning and characterization of a Saccharomyces cerevisiae alkaline ceramidase with specificity for dihydroceramide. J Biol Chem 275:31369–31378. doi: 10.1074/jbc.M003683200. [DOI] [PubMed] [Google Scholar]
- 17.Matmati N, Metelli A, Tripathi K, Yan S, Mohanty BK, Hannun YA. 2013. Identification of C18:1-phytoceramide as the candidate lipid mediator for hydroxyurea resistance in yeast. J Biol Chem 288:17272–17284. doi: 10.1074/jbc.M112.444802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roelants FM, Baltz AG, Trott AE, Fereres S, Thorner J. 2010. A protein kinase network regulates the function of aminophospholipid flippases. Proc Natl Acad Sci U S A 107:34–39. doi: 10.1073/pnas.0912497106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belli G, Gari E, Aldea M, Herrero E. 1998. Functional analysis of yeast essential genes using a promoter-substitution cassette and the tetracycline-regulatable dual expression system. Yeast 14:1127–1138. [DOI] [PubMed] [Google Scholar]
- 20.Voth WP, Jiang YW, Stillman DJ. 2003. New ‘marker swap’ plasmids for converting selectable markers on budding yeast gene disruptions and plasmids. Yeast 20:985–993. doi: 10.1002/yea.1018. [DOI] [PubMed] [Google Scholar]
- 21.Dittmar JC, Reid RJ, Rothstein R. 2010. ScreenMill: a freely available software suite for growth measurement, analysis and visualization of high-throughput screen data. BMC Bioinformatics 11:353. doi: 10.1186/1471-2105-11-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ejsing CS, Moehring T, Bahr U, Duchoslav E, Karas M, Simons K, Shevchenko A. 2006. Collision-induced dissociation pathways of yeast sphingolipids and their molecular profiling in total lipid extracts: a study by quadrupole TOF and linear ion trap-orbitrap mass spectrometry. J Mass Spectrom 41:372–389. doi: 10.1002/jms.997. [DOI] [PubMed] [Google Scholar]
- 23.Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, Simons K, Shevchenko A. 2009. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci U S A 106:2136–2141. doi: 10.1073/pnas.0811700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voynova NS, Mallela SK, Vazquez HM, Cerantola V, Sonderegger M, Knudsen J, Ejsing CS, Conzelmann A. 2014. Characterization of yeast mutants lacking alkaline ceramidases YPC1 and YDC1. FEMS Yeast Res 14:776–788. doi: 10.1111/1567-1364.12169. [DOI] [PubMed] [Google Scholar]
- 25.Vazquez HM, Vionnet C, Roubaty C, Conzelmann A. 2014. Cdc1 removes the ethanolaminephosphate of the first mannose of GPI anchors and thereby facilitates the integration of GPI proteins into the yeast cell wall. Mol Biol Cell 25:3375–3388. doi: 10.1091/mbc.E14-06-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haak D, Gable K, Beeler T, Dunn T. 1997. Hydroxylation of Saccharomyces cerevisiae ceramides requires Sur2p and Scs7p. J Biol Chem 272:29704–29710. doi: 10.1074/jbc.272.47.29704. [DOI] [PubMed] [Google Scholar]
- 27.Rajakumari S, Rajasekharan R, Daum G. 2010. Triacylglycerol lipolysis is linked to sphingolipid and phospholipid metabolism of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1801:1314–1322. doi: 10.1016/j.bbalip.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Cerantola V, Guillas I, Roubaty C, Vionnet C, Uldry D, Knudsen J, Conzelmann A. 2009. Aureobasidin A arrests growth of yeast cells through both ceramide intoxication and deprivation of essential inositolphosphorylceramides. Mol Microbiol 71:1523–1537. doi: 10.1111/j.1365-2958.2009.06628.x. [DOI] [PubMed] [Google Scholar]
- 29.Jesch SA, Gaspar ML, Stefan CJ, Aregullin MA, Henry SA. 2010. Interruption of inositol sphingolipid synthesis triggers Stt4p-dependent protein kinase C signaling. J Biol Chem 285:41947–41960. doi: 10.1074/jbc.M110.188607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, Walther TC, Loewith R. 2012. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol 14:542–547. doi: 10.1038/ncb2480. [DOI] [PubMed] [Google Scholar]
- 31.de Nobel H, Ruiz C, Martin H, Morris W, Brul S, Molina M, Klis FM. 2000. Cell wall perturbation in yeast results in dual phosphorylation of the Slt2/Mpk1 MAP kinase and in an Slt2-mediated increase in FKS2-lacZ expression, glucanase resistance and thermotolerance. Microbiology 146:2121–2132. doi: 10.1099/00221287-146-9-2121. [DOI] [PubMed] [Google Scholar]
- 32.Epstein S, Kirkpatrick CL, Castillon GA, Muniz M, Riezman I, David FP, Wollheim CB, Riezman H. 2012. Activation of the unfolded protein response pathway causes ceramide accumulation in yeast and INS-1E insulinoma cells. J Lipid Res 53:412–420. doi: 10.1194/jlr.M022186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandala SM, Thornton RA, Frommer BR, Curotto JE, Rozdilsky W, Kurtz MB, Giacobbe RA, Bills GF, Cabello MA, Martin I, et al. 1995. The discovery of australifungin, a novel inhibitor of sphinganine N-acyltransferase from Sporormiella australis. Producing organism, fermentation, isolation, and biological activity. J Antibiot (Tokyo) 48:349–356. [DOI] [PubMed] [Google Scholar]
- 34.Horvath A, Sutterlin C, Manning-Krieg U, Movva NR, Riezman H. 1994. Ceramide synthesis enhances transport of GPI-anchored proteins to the Golgi apparatus in yeast. EMBO J 13:3687–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niles BJ, Joslin AC, Fresques T, Powers T. 2014. TOR complex 2-Ypk1 signaling maintains sphingolipid homeostasis by sensing and regulating ROS accumulation. Cell Rep 6:541–552. doi: 10.1016/j.celrep.2013.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milgrom E, Diab H, Middleton F, Kane PM. 2007. Loss of vacuolar proton-translocating ATPase activity in yeast results in chronic oxidative stress. J Biol Chem 282:7125–7136. [DOI] [PubMed] [Google Scholar]
- 37.Vallee B, Riezman H. 2005. Lip1p: a novel subunit of acyl-CoA ceramide synthase. EMBO J 24:730–741. doi: 10.1038/sj.emboj.7600562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tani M, Kuge O. 2010. Requirement of a specific group of sphingolipid-metabolizing enzyme for growth of yeast Saccharomyces cerevisiae under impaired metabolism of glycerophospholipids. Mol Microbiol 78:395–413. doi: 10.1111/j.1365-2958.2010.07340.x. [DOI] [PubMed] [Google Scholar]
- 39.Tani M, Kuge O. 2010. Defect of synthesis of very long-chain fatty acids confers resistance to growth inhibition by inositol phosphorylceramide synthase repression in yeast Saccharomyces cerevisiae. J Biochem 148:565–571. doi: 10.1093/jb/mvq090. [DOI] [PubMed] [Google Scholar]
- 40.Tani M, Kuge O. 2012. Hydroxylation state of fatty acid and long-chain base moieties of sphingolipid determine the sensitivity to growth inhibition due to AUR1 repression in Saccharomyces cerevisiae. Biochem Biophys Res Commun 417:673–678. doi: 10.1016/j.bbrc.2011.11.138. [DOI] [PubMed] [Google Scholar]
- 41.Kajiwara K, Watanabe R, Pichler H, Ihara K, Murakami S, Riezman H, Funato K. 2008. Yeast ARV1 is required for efficient delivery of an early GPI intermediate to the first mannosyltransferase during GPI assembly and controls lipid flow from the endoplasmic reticulum. Mol Biol Cell 19:2069–2082. doi: 10.1091/mbc.E07-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swinnen E, Wilms T, Idkowiak-Baldys J, Smets B, De Snijder P, Accardo S, Ghillebert R, Thevissen K, Cammue B, De Vos D, Bielawski J, Hannun YA, Winderickx J. 2014. The protein kinase Sch9 is a key regulator of sphingolipid metabolism in Saccharomyces cerevisiae. Mol Biol Cell 25:196–211. doi: 10.1091/mbc.E13-06-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khakhina S, Johnson SS, Manoharlal R, Russo SB, Blugeon C, Lemoine S, Sunshine AB, Dunham MJ, Cowart LA, Devaux F, Moye-Rowley WS. 2015. Control of plasma membrane permeability by ABC transporters. Eukaryot Cell 14:442–453. doi: 10.1128/EC.00021-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 45.Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Menard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C. 2004. Global mapping of the yeast genetic interaction network. Science 303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 46.Kane PM. 2006. The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPase. Microbiol Mol Biol Rev 70:177–191. doi: 10.1128/MMBR.70.1.177-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Munn AL, Riezman H. 1994. Endocytosis is required for the growth of vacuolar H(+)-ATPase-defective yeast: identification of six new END genes. J Cell Biol 127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young BP, Shin JJ, Orij R, Chao JT, Li SC, Guan XL, Khong A, Jan E, Wenk MR, Prinz WA, Smits GJ, Loewen CJ. 2010. Phosphatidic acid is a pH biosensor that links membrane biogenesis to metabolism. Science 329:1085–1088. doi: 10.1126/science.1191026. [DOI] [PubMed] [Google Scholar]
- 49.Smardon AM, Kane PM. 2014. Loss of vacuolar H+-ATPase activity in organelles signals ubiquitination and endocytosis of the yeast plasma membrane proton pump Pma1p. J Biol Chem 289:32316–32326. doi: 10.1074/jbc.M114.574442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Villa-Garcia MJ, Choi MS, Hinz FI, Gaspar ML, Jesch SA, Henry SA. 2011. Genome-wide screen for inositol auxotrophy in Saccharomyces cerevisiae implicates lipid metabolism in stress response signaling. Mol Genet Genomics 285:125–149. doi: 10.1007/s00438-010-0592-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothman JH, Yamashiro CT, Raymond CK, Kane PM, Stevens TH. 1989. Acidification of the lysosome-like vacuole and the vacuolar H+-ATPase are deficient in two yeast mutants that fail to sort vacuolar proteins. J Cell Biol 109:93–100. doi: 10.1083/jcb.109.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisman LS, Bacallao R, Wickner W. 1987. Multiple methods of visualizing the yeast vacuole permit evaluation of its morphology and inheritance during the cell cycle. J Cell Biol 105:1539–1547. doi: 10.1083/jcb.105.4.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M, Deshpande R, Li Z, Lin ZY, Liang W, Marback M, Paw J, San Luis BJ, Shuteriqi E, Tong AH, van Dyk N, Wallace IM, Whitney JA, Weirauch MT, Zhong G, Zhu H, Houry WA, Brudno M, Ragibizadeh S, Papp B, Pal C, Roth FP, Giaever G, Nislow C, Troyanskaya OG, Bussey H, Bader GD, Gingras AC, Morris QD, Kim PM, Kaiser CA, Myers CL, Andrews BJ, Boone C. 2010. The genetic landscape of a cell. Science 327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J. 2011. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol 195:323–340. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, Altman RB, Davis RW, Nislow C, Giaever G. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han G, Gable K, Yan L, Allen MJ, Wilson WH, Moitra P, Harmon JM, Dunn TM. 2006. Expression of a novel marine viral single-chain serine palmitoyltransferase and construction of yeast and mammalian single-chain chimera. J Biol Chem 281:39935–39942. doi: 10.1074/jbc.M609365200. [DOI] [PubMed] [Google Scholar]
- 57.Adeyo O, Horn PJ, Lee S, Binns DD, Chandrahas A, Chapman KD, Goodman JM. 2011. The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J Cell Biol 192:1043–1055. doi: 10.1083/jcb.201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dickson RC, Sumanasekera C, Lester RL. 2006. Functions and metabolism of sphingolipids in Saccharomyces cerevisiae. Prog Lipid Res 45:447–465. doi: 10.1016/j.plipres.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 59.Cunningham KW, Fink GR. 1994. Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J Cell Biol 124:351–363. doi: 10.1083/jcb.124.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Obara K, Kojima R, Kihara A. 2013. Effects on vesicular transport pathways at the late endosome in cells with limited very long-chain fatty acids. J Lipid Res 54:831–842. doi: 10.1194/jlr.M034678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tabuchi M, Audhya A, Parsons AB, Boone C, Emr SD. 2006. The phosphatidylinositol 4,5-biphosphate and TORC2 binding proteins Slm1 and Slm2 function in sphingolipid regulation. Mol Cell Biol 26:5861–5875. doi: 10.1128/MCB.02403-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roelants FM, Breslow DK, Muir A, Weissman JS, Thorner J. 2011. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 108:19222–19227. doi: 10.1073/pnas.1116948108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muir A, Ramachandran S, Roelants FM, Timmons G, Thorner J. 2014. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. eLife 3. doi: 10.7554/eLife.03779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS. 2010. Orm family proteins mediate sphingolipid homeostasis. Nature 463:1048–1053. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gururaj C, Federman R, Chang A. 2013. Orm proteins integrate multiple signals to maintain sphingolipid homeostasis. J Biol Chem 288:20453–20463. doi: 10.1074/jbc.M113.472860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanida I, Hasegawa A, Iida H, Ohya Y, Anraku Y. 1995. Cooperation of calcineurin and vacuolar H(+)-ATPase in intracellular Ca2+ homeostasis of yeast cells. J Biol Chem 270:10113–10119. doi: 10.1074/jbc.270.17.10113. [DOI] [PubMed] [Google Scholar]
- 67.Dickson RC. 2010. Roles for sphingolipids in Saccharomyces cerevisiae. Adv Exp Med Biol 688:217–231. doi: 10.1007/978-1-4419-6741-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.