

Abstract
Schistosomiasis is a neglected tropical disease that affects more than 200 million people worldwide. The main disease-causing agents, Schistosoma japonicum, S. mansoni and S. haematobium, are blood flukes that have complex life cycles involving a snail intermediate host. In Asia, S. japonicum causes hepatointestinal disease (schistosomiasis japonica) and is challenging to control due to a broad distribution of its snail hosts and range of animal reservoir hosts. In China, extensive efforts have been underway to control this parasite, but genetic variability in S. japonicum populations could represent an obstacle to eliminating schistosomiasis japonica. Although a draft genome sequence is available for S. japonicum, there has been no previous study of molecular variation in this parasite on a genome-wide scale. In this study, we conducted the first deep genomic exploration of seven S. japonicum populations from mainland China, constructed phylogenies using mitochondrial and nuclear genomic data sets, and established considerable variation between some of the populations in genes inferred to be linked to key cellular processes and/or pathogen-host interactions. Based on the findings from this study, we propose that verifying intraspecific conservation in vaccine or drug target candidates is an important first step toward developing effective vaccines and chemotherapies against schistosomiasis.
Schistosomiasis is a neglected tropical disease that still affects more than 200 million people in 70 countries, resulting in a burden of at least 3.31 million disability-adjusted life years1,2. The main disease-causing agents are the blood flukes Schistosoma japonicum, S. mansoni and S. haematobium, which all have complex life cycles involving a snail intermediate host3. Schistosomiasis japonica has affected human populations in many parts of Asia, including the People’s Republic of China, Indonesia and the Philippines1,2. In China, it has been one of the major hepatointestinal diseases in this region for more than 2,100 years4, and is particularly challenging to control due to the wide distribution of its snail hosts (genus Oncomelania) and the range of domestic and wild mammals that act as reservoirs for human infection5,6.
Since the implementation of the National Schistosomiasis Control Program in the mid 1950s, the number of reported cases of this disease in China has decreased significantly5,7. This reduction has been due to changes in China’s health policy, leading to the implementation of snail control (1950s-early 1980s), mass drug administration programmes (mid 1980s to 2003) and subsequent, integrated control regimens (2004 onward) to break the transmission cycle5,8,9. No vaccine is available for use in humans, and the reliance on praziquantel alone to treat/control schistosomiasis over a long period of time carries a risk of the emergence of drug resistance10,11. In spite of the availability of extensive genomic resources for schistosomes12,13,14, little is known about genome-wide changes that take place over time and space in S. japonicum populations, or molecular variation within S. japonicum from humans and different animal hosts. Clearly, more genomic information is required for S. japonicum populations from distinct geographical regions, particularly those that display distinct biological and ecological characteristics15,16. Furthermore, genetic diversity and changes in schistosome populations which are under pressure from repeated treatment with drugs, such as praziquantel, and also environmental biophysical effects (i.e. anthropogenic and environmental change) could represent an obstacle to the sustained control or elimination of schistosomiasis17,18,19,20. Advanced tools are needed to detect and quantify genetic differences and changes in schistosome populations, and to monitor the spread of genetic variants that might affect control strategies. With extensive and prolonged use of only one main drug to combat schistosomiasis, resistance against praziquantel is a distinct possibility11, and new genetic variants could represent a crucial point of vulnerability for any future intervention strategy.
Previous population studies of S. japonicum have been limited to relatively small numbers of genetic loci, largely due to a lack of comprehensive genomic sequence data sets for this blood fluke. For instance, microsatellite, mitochondrial (mt) and enzymatic markers were used to reveal genetic variation among isolates of S. japonicum from various regions in China and surrounding, coastal islands21,22,23,24,25,26, or to identify genetic bottlenecks in laboratory strains of this parasite27. Although these observational studies have been informative, none of them tightly linked genotype to biological traits of the parasite, such as infectivity, pathogenicity and/or immunogenicity.
The advent of high throughput sequencing technologies28,29 and the availability of a draft genome for S. japonicum13 have paved the way toward genome-wide studies of natural and laboratory-adapted populations of S. japonicum from humans and reservoir hosts. To this end, we undertook here the first deep genomic exploration of various S. japonicum populations from China to reveal their systematic relationships as well as considerable genetic variation between some populations, with an impact on numerous genes associated with key metabolic and signalling pathways, cellular processes and/or pathogen-host interactions.
Results
We used an Illumina-based sequencing approach to produce mitochondrial (mt) and nuclear genomic data sets from genomic DNA samples from S. japonicum (Supplementary Fig. 1). This effort yielded 15.5 to 19.8 Gb of high quality genomic sequence data (NCBI BioProject accession no. PRJNA286685) for each of the seven study populations, corresponding to 39- to 50-fold coverage of a reference nuclear genome for S. japonicum (Supplementary Table 1). These data were utilised to estimate genetic diversity among S. japonicum populations from seven provinces in China (Fig. 1A).
Figure 1. Phylogenetic relationships of seven Schistosoma japonicum populations from different parts of China.
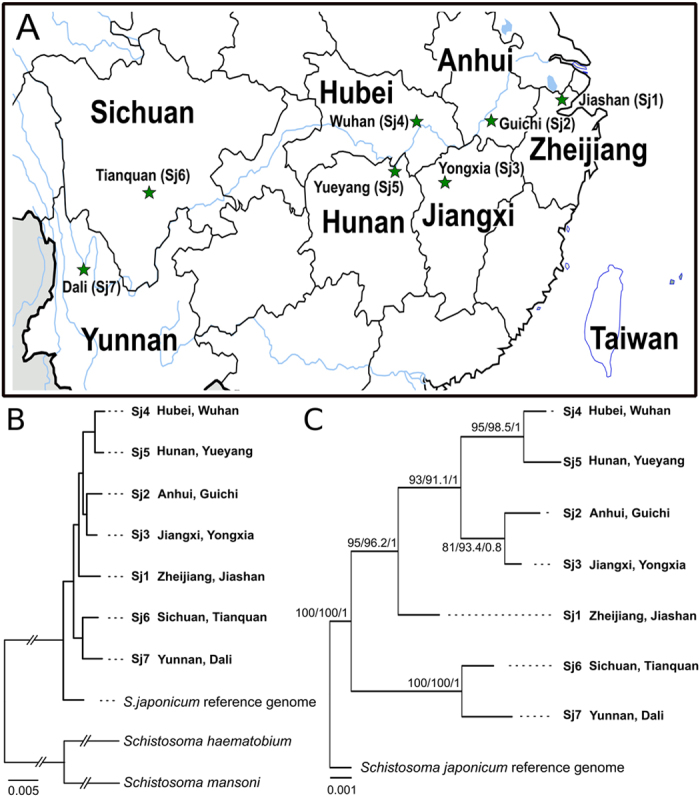
(A) Map indicating the provenance of populations, and their relationships based on Bayesian inference (BI) analysis of (B) nucleotide sequence data representing 4,333 protein-encoding single copy orthologs (SCOs) or (C) four exonic regions within SCOs (designated Sjp_0006080, Sjp_0009700, Sjp_0068320 and Sjp_0102280). The topology of these BI trees (B,C) are the same as those obtained for independent analyses using the maximum parsimony (MP) and maximum likelihood (ML) methods. Absolute nodal support was achieved using each tree building method (B). Nodal bootstrap or posterior probability values are indicated in the following order: ML/MP/BI (C). Map was modified from https://commons.m.wikimedia.org/wiki/File:China_Heilongjiang_Shuangyashan.svg, and was originally created by Joowwww under the creative commons licence [ http://creativecommons.org/licenses/by-sa/3.0/legalcode] and distributed via Wikimedia Commons.
Assessing S. japonicum phylogeny using mitochondrial genomes
First, we de novo-assembled the mt genomes of individual S. japonicum populations (Supplementary Table 2; NCBI GenBank accession nos. KR855668-KR855674) from 1.3–3.3 million paired-end reads, annotated each genome and compared each set of 12 mt protein-encoding genes to those of published mt genomes (Supplementary Table 2) to assess the phylogenetic informativeness of aligned, concatenated nucleotide and amino acid sequence data sets. At the nucleotide level, 7,841 of 10,341 alignment positions were invariable, and 148 (1.43%) were phylogenetically informative (Table 1). At the amino acid level, 2,375 of 3,438 positions were invariable, but only 39 (1.13%) were informative (Table 1). Phylogenetic trees constructed using Bayesian inference (BI; nucleotide and amino acid) and maximum parsimony (MP; nucleotide only) methods revealed two well-supported clades (Supplementary Fig. 2): one including populations Sj6 (Tianquan, Sichuan) and Sj7 (Dali, Yunnan) from provinces in Western China, and the second with Sj1 (Jiashan, Zhejiang), Sj4 (Wuhan, Hubei) and Sj5 (Yueyang, Hunan) from provinces in Eastern China. These results, however, were inconsistent with those obtained by maximum likelihood (ML; nucleotide and protein) and maximum parsimony (MP; protein) analyses, particularly using the aligned mt protein sequence data set. The clustering of other populations, including Sj2 (Guichi, Anhui) and Sj3 (Yongxia, Jiangxi) from Central China, were not well supported (nodal support: <0.8 or <80%) in analyses using any of the three tree-building methods, precluding further interpretation (Supplementary Fig. 2). This lack of resolution led us to explore genetic variation in nuclear genomic data sets among the seven S. japonicum populations.
Table 1. Summary of concatenated mitochondrial and nuclear coding domain alignments and results of phylogenetic analyses.
| Number of genes | Aligned character positions | Constant characters | Informative characters (%)b | Un-informative characters | Bayesian inference likelihood estimates:PSRFc | |
|---|---|---|---|---|---|---|
| Nucleotide – coding only | ||||||
| Mitochondrial | 12 | 10,341 | 7841 | 148 (1.43) | 1024 | −23,185:1 |
| Nucleara | 4946 | 9,947,586 | 8,149,863 | 925,719 (9.31) | 872,004 | −21,562,691:1 |
| PCR primer set (4 exons) | 4d | 3,378 | 3,314 | 43 (1.27) | 21 | −5109:1 |
| Inferred protein translations | ||||||
| Mitochondrial | 12 | 3438 | 2375 | 39 (1.13) | 1024 | −15,788:1 |
| Nucleara | 4946 | 3,315,862 | 2,613,069 | 335,882 (10.13) | 366,911 | −13,453,841:1.6 |
| PCR primer set (4 exons) | 4d | 1,126 | 1,102 | 15 (1.33) | 9 | −3432:1 |
aOrthoMCL single copy orthologues among S. japonicum, S. haematobium and S. mansoni.
bPositions with polymorphic characters supported in two or more species.
cAverage potential scale reduction factor (PSRF).
dFour genes; each represented by single protein-coding exon that can be PCR-amplified (see Supplementary Table 8).
Assessing nuclear genomic variation
We mapped the sequence reads derived from individual populations to a reference nuclear genomic sequence of S. japonicum (designated here as SjRef; Bioproject accession no. PRJEA34885)13. Overall, 82.6 to 88.0% of all reads mapped to this draft reference genome, with ~95% of these mapping as pairs (Supplementary Table 4). Excluding ambiguous positions (i.e. Ns in SjRef), 6,879,937 to 7,509,073 single nucleotide polymorphisms (SNPs) were recorded in individual populations (Table 2), of which 69% (n = 4,767,718 to 5,196,811), 26% (n = 1,784,457 to 1,948,205) and 1.6% (n = 107,446 to 121,775) were within intergenic, intronic and protein-encoding regions, respectively. For individual S. japonicum populations, 55,316 to 64,473 non-synonymous and 51,389 to 57,725 synonymous SNPs were identified in coding domains (Table 2). Employing published genomes, we identified 4,413 single-copy orthologs (SCOs) that were common to S. japonicum13, S. haematobium14 and S. mansoni30. Of the 4,413 single-copy orthologs (SCOs), 697,639 to 768,044 intronic, and 37,273 to 42,333 exonic SNPs were identified in protein-encoding gene regions, with 17,035 to 19,931 non-synonymous and 19,723 to 22,402 synonymous SNPs in individual S. japonicum populations (Table 3). For all SCOs, on average, 6.4 SNPs were detected in S. japonicum per kb of coding sequence (Table 3). The effect of nucleotide polymorphisms in SCOs on variation in the inferred proteins varied considerably; and, variation was reduced by an accumulation of synonymous mutations (recorded as amino acid identity) or mutations substituting amino acid residues with conserved chemical properties (recorded as amino acid similarity) (Fig. 2B and Supplementary Table 5).
Table 2. Single nucleotide polymorphisms (SNPs) recorded following the mapping of genomic sequence read data to the reference genome for Schistosoma japonicum (SjRef)13.
| Isolate code | Origin(County, Province) | Number of SNPs called | Intergenic SNPs | Exonic SNPs | Intronic SNPs | Non-synonymous SNPs | Synonymous SNPs | Sequencing depth within coding domainsa |
|---|---|---|---|---|---|---|---|---|
| Sj1 | Jiashan, Zhejiang | 7,156,718 | 4,924,071 (68.80%) | 114,887 (1.61%) | 1,888,984 (26.39%) | 60,072 | 55,206 | 35.98 ± 42.42 |
| Sj2 | Guichi, Anhui | 7,336,399 | 5,063,834 (69.02%) | 114,136 (1.56%) | 1,920,936 (26.18%) | 59,157 | 55,378 | 42.94 ± 37.71 |
| Sj3 | Yongxia, Jiangxi | 6,936,669 | 4,784,895 (68.98%) | 107,446 (1.55%) | 1,820,674 (26.25%) | 55,316 | 52,507 | 39.80 ± 33.21 |
| Sj4 | Wuhan, Hubei | 7,382,882 | 5,090,987 (68.96%) | 116,471 (1.58%) | 1,938,106 (26.25%) | 60,981 | 55,886 | 43.73 ± 40.64 |
| Sj5 | Yueyang, Hunan | 7,319,896 | 5,044,716 (68.92%) | 115,692 (1.58%) | 1,924,551 (26.29%) | 60,477 | 55,612 | 40.19 ± 40.69 |
| Sj6 | Tianquan, Sichuan | 6,879,937 | 4,767,718 (69.30%) | 108,994 (1.58%) | 1,784,457 (25.94%) | 57,977 | 51,389 | 38.28 ± 52.02 |
| Sj7 | Dali, Yunnan | 7,509,073 | 5,196,811 (69.21%) | 121,775 (1.62%) | 1,948,205 (25.94%) | 64,473 | 57,725 | 39.35 ± 38.35 |
aAverage ± standard deviation.
Table 3. Single nucleotide polymorphisms (SNPs) recorded in the coding domains of 4,413 single-copy orthologs (SCOs) among Schistosoma japonicum, S. haematobium and S. mansoni.
| Isolate code | Origin(County, Province) | Total intronic SNPs | Intronic SNPss per 1 kba | Total exonic SNPs | Exon SNPs per 1 kba | Non-synonymous SNPs | Synonymous SNPs |
|---|---|---|---|---|---|---|---|
| Sj1 | Jiashan, Zhejiang | 747,394 | 14.5 ± 12.0 | 40,677 | 6.5 ± 6.1 | 18,999 | 21,678 |
| Sj2 | Guichi, Anhui | 761,202 | 14.6 ± 12.5 | 40,033 | 6.4 ± 6.4 | 18,344 | 21,689 |
| Sj3 | Yongxia, Jiangxi | 718,704 | 13.9 ± 12.2 | 37,438 | 6.0 ± 6.1 | 17,035 | 20,403 |
| Sj4 | Wuhan, Hubei | 768,044 | 14.9 ± 12.4 | 41,210 | 6.6 ± 6.3 | 19,171 | 22,039 |
| Sj5 | Yueyang, Hunan | 762,258 | 14.8 ± 12.4 | 40,521 | 6.5 ± 6.2 | 18,658 | 21,863 |
| Sj6 | Tianquan, Sichuan | 697,639 | 13.5 ± 12.1 | 37,273 | 5.9 ± 6.1 | 17,550 | 19,723 |
| Sj7 | Dali, Yunnan | 764,339 | 14.9 ± 13.0 | 42,333 | 6.8 ± 6.8 | 19,931 | 22,402 |
aAverage ± standard deviation.
Figure 2. Sequence conservation and variation in single copy orthologs (SCOs) among seven distinct populations of Schistosoma japonicum.

(A) Ranked SCOs, according to pairwise nucleotide sequence identities across coding domains. (B) Locally weighted linear regression (LOWESS) analysis of pairwise nucleotide identity, and amino acid identity and similarity among SCOs, ranked according to pairwise nucleotide sequence identities. Invariable SCOs, with >99.8% pairwise nucleotide identity among all populations, are indicated/boxed in black (left). Variable SCOs with one or more pairwise nucleotide identities ≤98% are indicated/boxed in red (right). Significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) biological pathways and protein families as well as proteins involved in the pathogen-host interplay and other biological processes among (C) invariable and (D) variable SCOs are listed.
Of all 4,413 SCOs, 382 exhibited >2% nucleotide variation between or among populations (Fig. 2). These variable SCOs, 70.2% of which were functionally annotated (Supplementary Table 5), were significantly enriched for functions relating to genetic information processing (ribosomal translation) or metabolic pathways (i.e. glycan biosynthesis and metabolism; amino acid and carbohydrate metabolism) (Fig. 2C; Supplementary Table 6). These SCOs were also significantly enriched for genetic information processing, such as the large subunit of ribosomal protein (see Supplementary Table 7) and the peptidyl prolyl isomerase (PPI) protein folding catalysts, metabolism (protein phosphatases and glycosyltransferases) and cellular signal processing (G protein-coupled receptors [GPCRs] as well as representatives of the cadherin cell adhesion molecule family) (Fig. 2C; Supplementary Tables 6 and 7). By contrast, 574 SCOs each shared >99.8% nucleotide sequence identity between or among all S. japonicum populations, and were thus designated invariable (Fig. 2D). These invariable SCOs, 87.8% of which were functionally annotated (Supplementary Table 5), were significantly enriched for genes associated with genetic information processing (ubiquitin-proteasome complex, ribosomal translation and spliceosome) and environmental information processing [transforming growth factor-β (TGF-β), mitogen-activated protein kinase (MAPK) and hypoxia-inducible factor-1 (HIF-1) signalling and cytokine-cytokine receptor interaction] pathways (Fig. 2D). These invariable SCOs were also significantly enriched for protein families associated predominantly with genetic information processing (ubiquitin-proteosome systems, transcription and translation factors, spliceosome, small subunit ribosomal proteins, DNA repair and remodelling proteins, PPI protein folding catalysts and heat shock proteins), metabolism (protein kinases) or cellular signal processing (cytokine receptors and cell adhesion molecules) (Fig. 2D; Supplementary Table 6).
Genetic variability linked to host response and disease intervention
Pairwise comparisons revealed nucleotide sequence variation of >2% in SCOs encoding structural proteins, molecules recognised to play important roles in regulating or modulating definitive host responses, and known immunogens (Fig. 2C and Supplementary Table 5). Variable structural proteins of cells included five cadherin/protocadherin-like molecules, dynein and actophorin and annexins (Supplementary Table 5). Variable proteins inferred to be involved in the pathogen-host interplay included a disulphide isomerase31, a thioredoxin32, a venom allergen-like (VAL20) protein33, heme-binding protein 134 and an extracellular superoxide dismutase35. Known immunogens included Sm14-like (fatty acid binding protein), Sm29-like36 and four tetraspanins (Fig. 2 and Supplementary Table 8). Sequence variability among some members of the tetraspanin protein family of S. japonicum was similar to a previous observation37 and was detected principally within Sj25 and the surface-exposed extracellular domain 2 (EC2) of the TSP2 ortholog (i.e. Sj-TSP2-EC2; Fig. 3, Supplementary Fig. 3 and Supplementary Table 8)37. Sj-TSP2-EC2 is encoded by a single SCO, and displays considerable nucleotide sequence variation (93.8–98.4%, respectively) within S. japonicum (Fig. 3A and Supplementary Table 8). A comparison of Sj-TSP2-EC2 domains, modelled using the resolved tertiary structure template of Sm-TSP2-EC238 (coverage: 96%; root-mean-square deviations between backbone atomic positions: ~2.4 Å), revealed “stem” regions that mediate contact with the plasma membrane38 and are structurally conserved between S. mansoni and all seven S. japonicum isolates (Fig. 3B). The “head region” of Sj-TSP2 is stabilised by two strictly conserved disulphide bridges38. However, mostly the surface-exposed amino acid residues in the head region are variable (Fig. 3Bb) in this TSP2 moiety between S. japonicum populations and between schistosome species. Compared with Sm-TSP2, the Sj-TSP2 EC2 domain lacks four residues, leading to a loss of the exposed hydrophobic patch in the head region38 (Fig. 3Bb). Other notable differences between S. japonicum populations include variation in features that likely affect protein-protein interactions, such as the change of surface electrostatics (K28T, D35S and K57N) and alterations that increase flexibility and thus allow for structural changes (P52R). In addition to variation in Sj-TSP2 was nucleotide sequence variability in tetraspanin-enriched-microdomain (TEM)38-associated proteins, including calpain (98.0–98.9%), annexin (97.4–100%) and an Sm29-like molecule (91.8–93.6%) (Supplementary Table 8). Importantly, variation in the Sm29-like protein was observed downstream of the N-terminal signal peptide and upstream of the C-terminal hydrophobic transmembrane domain (Supplementary Fig. 3), which has been used to assess immunoprotection in animals against S. mansoni infection39. Interestingly, there was a positive correlation (0.794) in sequence similarity between the Sj-TSP2 and the Sm29-like proteins within individual S. japonicum populations, suggesting that the evolution of these TEM-associated proteins might be linked.
Figure 3. Sequence variability in the extracellular 2 domain (EC2) of the Schistosoma japonicum tetraspanin 2 ortholog (Sj-TSP2) among seven distinct populations.

(A) Nucleotide logos represent the frequency of base calls for each population in sites containing single nucleotide polymorphisms (SNPs). Amino acid logos representing the consensus sequence for all seven populations. SNPs leading to a similar (yellow star) or distinct (white star) change of the translated amino acid are indicated. Each amino acid logo is coloured according to its chemical characteristics; polar residues (G, S, T, Y & C) are green, neutral (Q & N) are purple, basic (K, R & H) are blue, acidic (D & E) are red and hydrophobic (A, V, L, I, P, W, F & M) are black. The extracellular 2 (EC2) domain is highlighted in grey. (B) Comparison of consensus Sj-TSP2-EC2 structures, modelled using the resolved protein structure of Sm-TSP2-EC2 (labelled green; RCSB accession number: 2M7Z) and highlighting structural changes (a & b) in the head region associated with the consensus amino acid sequence composition of each S. japonicum isolate. Proteins structures (S. japonicum) are coloured by percentage amino acid conservation among consensus protein translations.
Robust phylogenomic reconstruction using nuclear data sets
We explored the phylogenomic relationships of the seven S. japonicum study populations, to address current limitations of using small mt or microsatellite DNAs. To do this, we used sequence data sets representing thousands of SCOs, employing the genomes for S. japonicum13, S. haematobium14 and S. mansoni30 as references (Table 1 and Fig. 1B). At the nucleotide level, an alignment of 4,333 of all 4,413 SCO sequences identified 9,947,586 homologous characters, 8,149,863 of which were invariant, and 925,719 (9.3%) of which were variable and phylogenetically informative (Table 2). At the amino acid level, 2,613,069 of 3,315,862 positions were invariant, and 335,882 (10.1%) informative (Table 1). These numbers of informative characters were ~10-fold greater than for mt data sets (cf. Table 1). The trees built from the nuclear data sets (representing 4,333 SCOs) using BI, ML and MP methods produced consensus trees with consistent topology and strongly supported (1.0 or 100%) clades, and unequivocally resolved the relationships among populations from Western (Sj6 and Sj7), Central (Sj4 and Sj5) and Eastern (Sj2 and Sj3) China. The population Sj1 was the farthest east and was basal to those from Eastern China (Fig. 1B). Finally, we sought to define a subset of SCOs containing variable coding regions with conserved flanking sequences within exons, in which primers could be designed and used in future PCR-coupled mutation scanning40 and/or sequencing analyses for large-scale population investigations. In total, we identified 662 SCO regions in which such primers could be designed across all seven S. japonicum populations. By conducting an exhaustive search, four of these SCO regions (Supplementary Table 9) had an adequate signal to reproduce (using BI, ML and MP; see Fig. 1C) trees, whose topology and nodal support values were consistent with those of the final consensus tree constructed using the complete SCO set (cf. Fig. 1B).
Discussion
Although previous mitochondrial and microsatellite DNA studies22,23,27,41,42 had provided some insight into population variation in S. japonicum, nucleotide variation was limited24,25,26,43, often resulting in relationships with limited statistical support. In this study, our initial aim was to assess the utility of large mitochondrial and nuclear genomic sequence data sets to explore molecular variation within and among populations of S. japonicum. The ability to explore such variation in schistosome populations on a genome-wide scale provides a unique opportunity to link genetic variation to genes or gene products associated with important biological and/or disease traits, which has important implications for understanding schistosomiasis, its epidemiology and possibly for its control.
Through the sequencing and analyses of seven new and all publicly available mt genome sequences of S. japonicum44, we confirmed that mitochondrial data sets lacked sufficient signal to reliably establish relationships among populations, consistent with previous findings24,25,43,44. In contrast, we showed that a vast array of single-copy protein-encoding genes (SCOs) in the genome provides a rich source of neutral and adaptive genetic markers45.
The phylogenetic analyses of nuclear SCO data suggest that S. japonicum initially colonised and “stabilised” in the western valleys of the Sichuan and Yunnan provinces, in accord with their snail intermediate host(s)43. They also indicate that S. japonicum radiated eastwards, and established as distinct groups in central and eastern provinces, along the Yangtze River. Taken together, we contend that the biogeographic framework constructed here provides a first, robust foundation for large-scale investigations of molecular variation in S. japonicum in China and/or other parts of Asia, such as the Philippines26,41 and Japan26, and a basis for future population genetic or biogeographic studies. For laboratories without the budget and facilities to undertake genomic sequencing, the four informative SCO loci identified here (and able to be used to reproduce the consensus tree; Fig. 1B) are expected to be useful for systematic and/or population genetic studies. However, these markers need to be sequenced from large numbers of individual female and male worms representing different populations of S. japonicum, in order to assess whether they are neutral or adaptive, and to establish their suitability for particular applications45.
We elected to investigate single copy orthologs (SCOs) shared by at least three schistosome species, to be able draw comparisons among protein homologs of known biological relevance (Fig. 2). While the majority of SCOs could be annotated, interestingly, almost 30% of proteins encoded by variable SCOs lacked a functional annotation. This finding is not surprising and consistent with previous studies12,13,14,46,47,48, demonstrating that flatworms differ substantially genetically, and are evolutionarily very distant, from organisms whose genomes are almost fully characterised, and whose gene sets are functionally annotated. In spite of technical challenges, there is major merit in finding a reliable method(s) to annotate presently uncharacterised genes of S. japonicum and other schistosomes, as they are believed to play important organism- or species-specific roles.
By focusing on the use of coding regions, we aimed to assess variation in any exon of any SCO of S. japonicum with a recognised link to a phenotypic trait, such as host affiliation16,41, infectivity49, pathogenicity, praziquantel susceptibility or resistance50, antigenicity or immunogenicity51,52, and/or to predict how a particular selection pressure might impact on the genotype and/or phenotype of a worm. The power of such an approach is not only in its ability to explore molecular variation within and among worm populations, but, more importantly, among individuals (irrespective of developmental stage) within and among worm populations.
Interestingly, we showed that sequence polymorphism in selected proteins (Sj-TSP2 and Sm29-like, which are predicted to be essential for tegument integrity and are highly antigenic39,53) varies considerably among populations, suggesting that it might affect protein structure and thus influence levels of immunogenicity36,54,55,56. Based on information for S. mansoni38,57, Sj-TSP-2 likely mediates dynamic processes occurring at the tegumental surface and maintains tegumental integrity, and is also a promising vaccine candidate53. The finding that Sj-TSP2-EC2 (encoded by an SCO) varies considerably in S. japonicum agrees with a previous study55 indicating that allelic variation results in protein isoforms, but contrasts the hypothesis that S. japonicum encodes multiple tsp2 genes54. The variation detected here is suggested to relate to an ability of S. japonicum to infect a substantially broader host range than either S. mansoni or S. haematobium55,58. Although it is not known how TSP2 interacts with host molecules or immune system, it is understood that calpain, actin, annexin and Sm29 all associate closely with Sm-TSP2 in tetraspanin-enriched-microdomains (TEMs)38. In S. mansoni, this association has been proposed to underpin the success of Sm-TSP2, annexin and Sm29 as vaccine candidate molecules36,39,53,59, and might be explained by an induction of an immunogenic host response that disrupts the integrity of TEMs, leading to the destruction of the tegument and subsequent death of the parasite38. Interestingly, here, we detected sequence variation in TEM-associated proteins of S. japonicum, including a positive correlation between genetic variation in Sm29-like and Sj-TSP2 molecules. We propose that sequence variation in these molecules might explain inconsistent results (i.e. variable levels of protection, if any) in some vaccination experiments, particularly if the vaccine molecules (i.e. variants of Sj-TSP2-EC2) differ in sequence (particularly in the protective epitope/s) from the homolog present in the parasite used for the challenge infection54,55,60. To date, a limited survey of six S. mansoni individuals from Kenya suggested that sequence polymorphism within the Sm-TSP2-EC2 domain might be less than for Sj-TSP2-EC261; however, future studies using large-scale genomic data sets of individuals across a broader geographic range are needed to test this proposal.
Therefore, based on the present findings, we support recommendations62,63 that, in addition to assessing variation between species64,65, comprehensive assessments of intraspecific conservation in immunogens or their protective epitopes should precede the research and development of any schistosome vaccine, in order to provide an informed position and some confidence that a vaccine would be efficacious in the field. Although our focus here was principally on immunogenic molecules, similar considerations would apply to targets for the development of new anti-schistosomal drugs. Importantly, the genome-wide approach established here should be applicable to a wide range of eukaryotic pathogens for the analysis of genetic variability. Clearly, neutral SCO markers would have advantages for estimating haplotype diversity and population size, and could provide unbiased estimates of random processes, such as genetic drift. On the other hand, adaptive (non-neutral) markers should have practical applications, for example, to the identification of disease-causing genes or other genes that link phenotype to genotype across different environmental conditions.
Methods
Schistosoma japonicum samples
Adults of S. japonicum were available from a previous multilocus enzyme electrophoretic (MEE) study21 and had been stored at −70 °C until 1999, and then at −20 °C from 2000 to mid 2015. In brief, seven isolates of S. japonicum (designated Sj1-Sj7) originated from distinct endemic areas in China (Table 1). Cercariae were obtained from at least 10 infected snails (Oncomelania hupensis) collected from each province. S. japonicum adults were raised (45 days) in rabbits (n = 2 per province), each infected with 1,000 cercariae21. They were perfused from the mesenteric veins and washed extensively in physiological saline.
Genomic DNA library construction, sequencing and pre-processing of reads
High molecular weight genomic DNA (10 μg) was isolated from pooled adult S. japonicum (i.e. male and female en copula; n = 10 pairs) representing each of the isolates Sj1 to Sj7 using a Chemagic DNA Tissue Extraction Kit (Chemagen). Total DNA amounts were determined using a Qubit fluorometer dsDNA HS Kit (Life Technologies), and DNA integrity was verified by agarose gel electrophoresis. High quality genomic DNA was used to construct short-insert (480 bp) genomic DNA libraries, which were then paired-end sequenced (2 × 100 base reads) utilising the TruSeq sequencing chemistry (Illumina) and the HiSeq 2500 sequencing platform (Illumina). High quality sequence data sets were produced by removing low quality bases (<25 Phred quality), adapters and reads of <50 nucleotides (nt) in length using the program Trimmomatic66.
Assembly and curation of mt genomes
High quality paired-end reads were mapped to a reference mt genome sequence for S. japonicum (GenBank accession no. AF215860)67 using Bowtie268. For each data set, paired-reads with at least one read aligned to the reference mt genome were extracted and de novo-assembled using the program SPADES v.3.1069 using a published mt genome sequence (accession no. AF215860)67 for S. japonicum as a “trusted” reference contig. Following assembly, paired-end reads were aligned to their corresponding mt genome using Bowtie2, and nucleotides and genome arrangement were verified using the program PILON70. Subsequently, each mt genome was annotated using an established pipeline71,72, and using the reference S. japonicum mt genome annotation (accession no. AF21586067) and echinoderm/flatworm mt code (NCBI genetic code - Table 9; http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi#SG9).
Functional annotation of predicted protein sequences
S. japonicum proteins were compared by sequence homology (BLASTp; E-value ≤10−5) to proteins in databases representing S. haematobium14, S. japonicum13 and S. mansoni30; Clonorchis sinensis46 and Opisthorchis viverrini47; Swiss-Prot and TrEMBL within UniProtKB73 and Kyoto Encyclopedia of Genes and Genomes (KEGG)74. Conserved protein domains and gene ontology (GO) terms were identified for each inferred amino acid sequence using the program InterProScan75 employing the “–goterms” option. Each protein-encoding gene was assigned to a KEGG orthologous gene (KO) group using established methods76. Individual genes linked to one or more KO terms were assigned to known protein families and biological pathways using the KEGG BRITE and KEGG PATHWAY hierarchies using custom python scripts (available from the corresponding authors upon request). Putative signal peptide and transmembrane domains were predicted using the program Phobius77. In the final annotation, proteins inferred from genes were classified based on their homology (BLASTp; E-value <10−5) to sequences in (a) Swiss-Prot database, (b) the KEGG database, and (c) a recognised, conserved protein domain based on InterProScan analysis. Any predicted proteins without a match (E-value <10−5) in at least one of these databases were designated as hypothetical (or orphans).
Identification and curation of nuclear genome polymorphisms
High quality reads were mapped to scaffolds (≥1000 bases) of the published nuclear (reference) genome of S. japonicum (SjRef; Bioproject PRJEA34885)13 using Bowtie268. Duplicates were removed and insertion-deletion events were assessed and quality of read alignments were established using the sorted BAM files and PICARD tools (http://broadinstitute.github.io/picard) according to best practice (GATK guidelines)78. An MPILEUP format file was created from each BAM file using SAMtools79, and the frequencies of SNPs and insertion-deletion events (indels) were estimated using the program VarScan80. All SNPs and indels were contextualised within the current genome annotation (including exons, introns and intergenic elements) using snpEFF81 and a GFF annotation file available for the reference genome of SjRef13.
Prediction of coding regions and identification of single copy orthologs
To predict the coding domains for each S. japonicum population, variant calls (SNPs only) inferred by VarScan80 were transferred to the genome reference using VCFtools (vcf-consensus)82, ignoring ambiguous positions (N) in the reference sequence of SjRef. Coding domains and amino acid sequences were extracted from each genome using GAG (http://genomeannotation.github.io/GAG) and the genome annotation for SjRef (Bioproject PRJEA34885)13. Single-copy orthologs (SCOs) between or among the genomes representing S. japonicum populations (Sj1-Sj7 and SjRef) and/or the outgroups S. haematobium14 and S. mansoni30 were defined using the program OrthoMCL83. Only SCOs of S. japonicum isolates (Sj1-Sj7) with an average, aligned read depth of >10 across all exonic regions and encoding the same start and stop codon positions as the reference genome (SjRef) were considered for further analysis. If the coding regions of S. japonicum genes homologous to biologically important genes in other schistosomes were only partial in the reference sequence (SjRef), the program PRICE84 and Illumina paired-end genomic reads sequenced in this study were used to extend the genomic region to obtain the full length gene for subsequent analyses.
Phylogenetic analyses of nucleotide and amino acid data sets
To assess the genetic relationships among S. japonicum populations (Sj1-Sj7 and SjRef), mt genes or SCOs were aligned as individual nucleotide or inferred amino acid sequences using MAFFT85, selecting a minimum gap-free alignment length of 20 amino acids, and SCOs with at least one nucleotide or amino acid residue distinct from all others in the alignment. Nucleotide or amino acid sequence alignments were verified by eye, concatenated and then subjected to phylogenetic analyses using the methods Bayesian inference (BI) in MrBayes v.3.2.286, maximum likelihood (ML) in the program RAxML v.8.0.2487 and maximum parsimony (MP) in PAUP* v.4.0 beta88, and including available reference (mt or nuclear) genomes for S. japonicum, and outgroups S. haematobium (nuclear), S. mansoni (nuclear) and Schistosoma mekongi (mt) (Table 2). For mt genome analysis, each protein-encoding gene region was partitioned and the HKY89 nucleotide substitution model was selected for individual genes using the Bayesian Information Criteria (BIC) test in jModeltest v. 2.1.690. Amino acid substitution models were inferred for each gene using MrBayes by creating a consensus from available amino acid models (aamodelpr = mixed). For the analysis of nuclear genomic data, due to computation limitations the general time reversible model of evolution with gamma distribution91 was applied to concatenated SCO protein-encoding domains, and MrBayes created a consensus from available amino acid models (aamodelpr = mixed) for amino sequence alignments. For nucleotide and amino acid data sets, rates of reversible rate matrix, stationary state frequencies, shape of scaled gamma-distribution of site rates, partition-specific rate multiplier, topologies apropriori and branch lengths were unchanged from the default MrBayes v.3.2.286 recommendations. Trees were constructed using sequence data for coding domains or proteins, employing the Monte Carlo Markov chain method (nchains = 4) over 100,000 (nuclear genome) or 2,000,000 (mt genome) generations, with every 100th (nuclear genome) or 200th (mt genome or primer set) tree being saved; 25% (mt) or 50% (nuclear) of the first saved trees were discarded to ensure a stabilisation of the nodal split frequencies. Consensus (50% majority rule) trees were constructed from all remaining trees, with nodal support expressed as a posterior probability (pp).
For ML, the same concatenated mt nucleotide and amino acid alignments were subjected to phylogenetic analysis using the general time reversible (GTR)91 and mtREV92 evolution models, respectively; concatenated alignment blocks were bootstrapped 200,000 times in RAxML to infer nodal support values. Concatenated SCO nucleotide and amino acid alignments were subjected to ML analysis using the general time reversible (GTR)91 and JTT93 evolution models, respectively and setting four discrete rate categories; concatenated alignment blocks were bootstrapped 100 times in RAxML87 to infer nodal support values. For mt and nuclear genome data sets, MP analyses were performed using heuristic searches, utilising tree bisection and reconnection (TBR), for each concatenated sequence alignment. By preserving branch lengths, the concatenated sequence blocks were bootstrapped 1,000 times using PAUP*88. The resultant, bootstrapped trees were then subjected to analysis in the program SumTrees in the DendroPy v.3.12.094 python library to produce a consensus tree and to infer the nodal support values. The consensus trees were drawn and labeled using the program Figtree v.1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Identification of the minimum, phylogenetically informative SCO set
Well-aligned sequence blocks were employed to identify corresponding exonic regions (600–1200 bp). Then, short (20–22 nt), conserved sequences were identified as PCR primers (5′ and 3′) in these regions using the program Primer395. To establish which sets of exonic regions would produce a tree with the same topology as the final consensus tree for all SCOs among all populations, all possible combinations of two regions (designated pairs) were subjected to phylogenetic analysis using the ML-based software RAxML87. Trees matching the exact topology of the final consensus tree using all SCOs (cf. Phylogenetic analyses of nucleotide and amino acid data sets) were studied using Robinson-Foulds metric96 implemented in DendroPy94 python library, and examined further for robustness using 100-fold bootstrapping in the program RAxML87, employing a minimum nodal support value of 70%. Exonic regions fulfilling this stringent criteria were then exhaustively run in triplets and quartets resulting using 100-fold bootstrapping in RAxML. Finally, the best combination of four concatenated amplicons, with >92% nodal support, was selected as the minimum, informative sequence set required for phylogenetic reconstruction of the intraspecific S. japonicum tree. Phylogenetic analysis was the same as for mt data sets, except that a nucleotide substitution model was used.
Selection of variable and invariable gene sets
Individual nucleotide and amino acid sequences representing all SCOs of individual S. japonicum populations were compared in a pairwise manner with their one-to-one orthologs in the S. japonicum reference genome of SjRef13. The nucleotide and amino acid identities as well as similarities were estimated for each pairwise comparison. SCOs were first ranked from least to most variable according to their mean nucleotide identity and standard deviation of mean nucleotide identity. Variable SCOs were defined as those SCO groups with one more SCO with ≤98% nucleotide identity to the SjRef-coding domain. Invariable SCOs were defined as those SCO groups with all pairwise alignments sharing ≥99.8%. nucleotide identity to the SjRef-coding domain. Enriched protein families and biological pathways were defined for selected genes sets using the Fisher’s exact test employing a custom script and linking data to KEGG biological pathway, KEGG BRITE hierarchy and gene ontology (GO) databases.
In silico modeling of protein structure
Amino acid sequences of individual S. japonicum populations homologous to S. mansoni tetraspanin 2 (Sm-TSP2) were aligned using MAFFT85. The protein structure of S. japonicum TSP2 (Sj-TSP2) protein regions that aligned with the Sm-TSP2 extracellular 2 domain (EC2)38 were then modeled using I-TASSER97, and the resolved protein structure of Sm-TSP2-EC2 domain as a template (RCSB accession no. 2M7Z)38,97. Protein structure models were aligned and visualised using Chimera98,99,100,101,102.
Additional Information
How to cite this article: Young, N. D. et al. Exploring molecular variation in Schistosoma japonicum in China. Sci. Rep. 5, 17345; doi: 10.1038/srep17345 (2015).
Supplementary Material
Acknowledgments
Funding from the National Health and Medical Research Council (NHMRC) of Australia and the Australian Research Council and Melbourne Water Corporation is gratefully acknowledged (R.B.G. et al.). This study was funded in part by the Malaysian government through the High Impact Research (HIR) initiative at the University of Malaya (grant numbers H-50001-A000027 and A000001-50001). This project was also supported by a Victorian Life Sciences Computation Initiative (grant number VR0007) on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government (R.B.G.). N.D.Y. holds an NHMRC Early Career Research Fellowship. P.K.K. is the recipient of a scholarship (STRAPA) from The University of Melbourne.
Footnotes
Author Contributions N.D.Y., N.B.C. and B.Q. isolated D.N.A., K.G.C., T.M., R.E.H.J. and Y.L.L. conducted the sequencing and handled the data. N.D.Y., A.H. and P.K.K. conducted the analyses, and Y.L.L., N.M., B.W., D.R. and A.V.K. assisted with selected bioinformatic or phylogenetic components. N.D.Y. and R.B.G. wrote the paper with inputs from A.R.J., N.B.C., G.N.G., D.P.M., P.T. and D.R. and other co-authors. R.B.G. and K.G.C. led and funded the project.
References
- Steinmann P., Keiser J., Bos R., Tanner M. & Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis 6, 411–425 (2006). [DOI] [PubMed] [Google Scholar]
- Hotez P. J., et al. The global burden of disease study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl Trop Dis 8, e2865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despommier D. D., Gwadz R. W., Hotez P. J. & Knirsch C. A. Parasitic Diseases. 5th Edition. Apple Trees Productions, LLC (2005). [Google Scholar]
- Zhou D., Li Y. & Yang X. Schistosomiasis control in China. World Health Forum 15, 387–389 (1994). [PubMed] [Google Scholar]
- Mao S. P. & Shao B. R. Schistosomiasis control in the people’s Republic of China. Am J Trop Med Hyg 31, 92–99 (1982). [PubMed] [Google Scholar]
- Gray D. J., et al. The role of bovines in human Schistosoma japonicum infection in the Peoples’ Republic of China. Am J Trop Med Hyg 81, 301–301 (2009). [Google Scholar]
- Gray D. J., et al. Schistosomiasis elimination: lessons from the past guide the future. Lancet Infect Dis 10, 733–736 (2010). [DOI] [PubMed] [Google Scholar]
- Utzinger J., Zhou X. N., Chen M. G. & Bergquist R. Conquering schistosomiasis in China: the long march. Acta Trop 96, 69–96 (2005). [DOI] [PubMed] [Google Scholar]
- Collins C., Xu J. & Tang S. Schistosomiasis control and the health system in P.R. China. Infect Dis Poverty 1, 8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doenhoff M. J., Kusel J. R., Coles G. C. & Cioli D. Resistance of Schistosoma mansoni to praziquantel: is there a problem? Trans R Soc Trop Med Hyg 96, 465–469 (2002). [DOI] [PubMed] [Google Scholar]
- Wang W., Wang L. & Liang Y. S. Susceptibility or resistance of praziquantel in human schistosomiasis: a review. Parasitol Res 111, 1871–1877 (2012). [DOI] [PubMed] [Google Scholar]
- Berriman M., et al. The genome of the blood fluke Schistosoma mansoni. Nature 460, 352–358 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., et al. The Schistosoma japonicum genome reveals features of host-parasite interplay. Nature 460, 345–351 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. D., et al. Whole-genome sequence of Schistosoma haematobium. Nat Genet 44, 221–225 (2012). [DOI] [PubMed] [Google Scholar]
- Lu D. B., et al. Evolution in a multi-host parasite: chronobiological circadian rhythm and population genetics of Schistosoma japonicum cercariae indicates contrasting definitive host reservoirs by habitat. Int J Parasitol 39, 1581–1588 (2009). [DOI] [PubMed] [Google Scholar]
- Lu D. B., et al. Contrasting reservoirs for Schistosoma japonicum between marshland and hilly regions in Anhui, China–a two-year longitudinal parasitological survey. Parasitology 137, 99–110 (2010). [DOI] [PubMed] [Google Scholar]
- Rogers S. H. & Bueding E. Hycanthone resistance: development in Schistosoma mansoni. Science 172, 1057–1058 (1971). [DOI] [PubMed] [Google Scholar]
- Valentim C. L., et al. Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science 342, 1385–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doenhoff M. J., et al. Praziquantel: its use in control of schistosomiasis in sub-Saharan Africa and current research needs. Parasitology 136, 1825–1835 (2009). [DOI] [PubMed] [Google Scholar]
- Greenberg R. M. New approaches for understanding mechanisms of drug resistance in schistosomes. Parasitology 140, 1534–1546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilton N. B., Bao-Zhen Q., Bogh H. O. & Nansen P. An electrophoretic comparison of Schistosoma japonicum (Trematoda) from different provinces in the People’s Republic of China suggests the existence of cryptic species. Parasitology 119 (Pt 4), 375–383 (1999). [DOI] [PubMed] [Google Scholar]
- Shrivastava J., Qian B. Z., McVean G. & Webster J. P. An insight into the genetic variation of Schistosoma japonicum in mainland China using DNA microsatellite markers. Mol Ecol 14, 839–849 (2005). [DOI] [PubMed] [Google Scholar]
- Rudge J. W., et al. Parasite genetic differentiation by habitat type and host species: molecular epidemiology of Schistosoma japonicum in hilly and marshland areas of Anhui Province, China. Mol Ecol 18, 2134–2147 (2009). [DOI] [PubMed] [Google Scholar]
- Zhao Q. P., Jiang M. S., Dong H. F. & Nie P. Diversification of Schistosoma japonicum in mainland China revealed by mitochondrial DNA. PLoS Negl Trop Dis 6, e1503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M., et al. Geographical genetic structure of Schistosoma japonicum revealed by analysis of mitochondrial DNA and microsatellite markers. Parasit Vectors 8, 150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., et al. Genetic variability among Schistosoma japonicum isolates from the Philippines, Japan and China revealed by sequence analysis of three mitochondrial genes. Mitochondrial DNA 26, 35–40 (2013). [DOI] [PubMed] [Google Scholar]
- Bian C. R., Gao Y. M., Lamberton P. H. & Lu D. B. Comparison of genetic diversity and population structure between two Schistosoma japonicum isolates-the field and the laboratory. Parasitol Res 114, 2357–2362 (2015). [DOI] [PubMed] [Google Scholar]
- Metzker M. L. Sequencing technologies - the next generation. Nat Rev Genet 11, 31–46 (2010). [DOI] [PubMed] [Google Scholar]
- Koboldt D. C., Steinberg K. M., Larson D. E., Wilson R. K. & Mardis E. R. The next-generation sequencing revolution and its impact on genomics. Cell 155, 27–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasio A. V., et al. A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Negl Trop Dis 6, e1455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutapi F., et al. Praziquantel treatment of individuals exposed to Schistosoma haematobium enhances serological recognition of defined parasite antigens. J Infect Dis 192, 1108–1118 (2005). [DOI] [PubMed] [Google Scholar]
- Maggioli G., et al. A recombinant thioredoxin-glutathione reductase from Fasciola hepatica induces a protective response in rabbits. Exp Parasitol 129, 323–330 (2011). [DOI] [PubMed] [Google Scholar]
- Chalmers I. W. & Hoffmann K. F. Platyhelminth Venom Allergen-Like (VAL) proteins: revealing structural diversity, class-specific features and biological associations across the phylum. Parasitology 139, 1231–1245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh S. Q., Glanfield A., Gobert G. N. & Jones M. K. Heme and blood-feeding parasites: friends or foes? Parasit Vectors 3, 108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourao Mde M., Dinguirard N., Franco G. R. & Yoshino T. P. Role of the endogenous antioxidant system in the protection of Schistosoma mansoni primary sporocysts against exogenous oxidative stress. PLoS Negl Trop Dis 3, e550 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso F. C., et al. Schistosoma mansoni tegument protein Sm29 is able to induce a Th1-type of immune response and protection against parasite infection. PLoS Negl Trop Dis 2, e308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W., Cai P., Chen Q. & Wang H. Identification of novel antigens within the Schistosoma japonicum tetraspanin family based on molecular characterization. Acta Trop 117, 216–224 (2011). [DOI] [PubMed] [Google Scholar]
- Jia X., et al. Solution structure, membrane interactions, and protein binding partners of the tetraspanin Sm-TSP-2, a vaccine antigen from the human blood fluke Schistosoma mansoni. J Biol Chem 289, 7151–7163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso F. C., Pacifico R. N., Mortara R. A. & Oliveira S. C. Human antibody responses of patients living in endemic areas for schistosomiasis to the tegumental protein Sm29 identified through genomic studies. Clin Exp Immunol 144, 382–391 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser R. B., et al. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc 1, 3121–3128 (2006). [DOI] [PubMed] [Google Scholar]
- Rudge J. W., et al. Population genetics of Schistosoma japonicum within the Philippines suggest high levels of transmission between humans and dogs. PLoS Negl Trop Dis 2, e340 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D. B., et al. Genetic diversity of Schistosoma japonicum miracidia from individual rodent hosts. Int J Parasitol 41, 1371–1376 (2011). [DOI] [PubMed] [Google Scholar]
- Attwood S. W., Ibaraki M., Saitoh Y., Nihei N. & Janies D. A. Comparative phylogenetic studies on Schistosoma japonicum and Its snail intermediate host Oncomelania hupensis: Origins, dispersal and coevolution. PLoS Negl Trop Dis 9, e0003935 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G. H., et al. A specific PCR assay for the identification and differentiation of Schistosoma japonicum geographical isolates in mainland China based on analysis of mitochondrial genome sequences. Infect Genet Evol 12, 1027–1036 (2012). [DOI] [PubMed] [Google Scholar]
- Kirk H. & Freeland J. R. Applications and implications of neutral versus non-neutral markers in molecular ecology. Int J Mol Sci 12, 3966–3988 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., et al. The draft genome of the carcinogenic human liver fluke Clonorchis sinensis. Genome Biol 12, R107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. D., et al. The Opisthorchis viverrini genome provides insights into life in the bile duct. Nat Commun 5, 4378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cwiklinski K., et al. The Fasciola hepatica genome: gene duplication and polymorphism reveals adaptation to the host environment and the capacity for rapid evolution. Genome Biol 16, 71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley S., et al. Multi-host transmission dynamics of Schistosoma japonicum in Samar province, the Philippines. PLoS Med 5, e18 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H., et al. Transcriptional responses of in vivo praziquantel exposure in schistosomes identifies a functional role for calcium signalling pathway member CamKII. PLoS Pathog 9, e1003254 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., et al. Having a pair: the key to immune evasion for the diploid pathogen Schistosoma japonicum. Sci Rep 2, 346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boamah D., et al. Immunoproteomics identification of major IgE and IgG4 reactive Schistosoma japonicum adult worm antigens using chronically infected human plasma. Trop Med Health 40, 89–102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran M. H., et al. Tetraspanins on the surface of Schistosoma mansoni are protective antigens against schistosomiasis. Nat Med 12, 835–840 (2006). [DOI] [PubMed] [Google Scholar]
- Cai P., et al. Molecular characterization of Schistosoma japonicum tegument protein tetraspanin-2: sequence variation and possible implications for immune evasion. Biochem Biophys Res Commun 372, 197–202 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang W., et al. Inconsistent protective efficacy and marked polymorphism limits the value of Schistosoma japonicum tetraspanin-2 as a vaccine target. PLoS Negl Trop Dis 5, e1166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro C. S., et al. A multivalent chimeric vaccine composed of Schistosoma mansoni SmTSP-2 and Sm29 was able to induce protection against infection in mice. Parasite Immunol 36, 303–312 (2014). [DOI] [PubMed] [Google Scholar]
- Schulte L., et al. Tetraspanin-2 localisation in high pressure frozen and freeze-substituted Schistosoma mansoni adult males reveals its distribution in membranes of tegumentary vesicles. Int J Parasitol 43, 785–793 (2013). [DOI] [PubMed] [Google Scholar]
- He Y. X., Salafsky B. & Ramaswamy K. Host–parasite relationships of Schistosoma japonicum in mammalian hosts. Trends Parasitol 17, 320–324 (2001). [DOI] [PubMed] [Google Scholar]
- Leow C. Y., et al. Crystal structure and immunological properties of the first annexin from Schistosoma mansoni: insights into the structural integrity of the schistosomal tegument. FEBS J 281, 1209–1225 (2013). [DOI] [PubMed] [Google Scholar]
- Yuan C., et al. Schistosoma japonicum: efficient and rapid purification of the tetraspanin extracellular loop 2, a potential protective antigen against schistosomiasis in mammalian. Exp Parasitol 126, 456–461 (2010). [DOI] [PubMed] [Google Scholar]
- Cupit P. M., et al. Polymorphism associated with the Schistosoma mansoni tetraspanin-2 gene. Int J Parasitol 41, 1249–1252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. J. Insights into the functional biology of schistosomes. Parasit Vectors 4, 203 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupalo D. N., Bradic M. & Carlton J. M. The impact of genomics on population genetics of parasitic diseases. Curr Opin Microbiol 23, 49–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippsen G. S., Wilson R. A. & DeMarco R. Accelerated evolution of schistosome genes coding for proteins located at the host-parasite interface. Genome Biol Evol 7, 431–443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealey K. L., Kirk R. S., Walker A. J., Rollinson D. & Lawton S. P. Adaptive radiation within the vaccine target tetraspanin-23 across nine Schistosoma species from Africa. Int J Parasitol 43, 95–103 (2013). [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M. & Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T. H., et al. Mitochondrial gene content, arrangement and composition compared in African and Asian schistosomes. Mol Biochem Parasitol 117, 61–71 (2001). [DOI] [PubMed] [Google Scholar]
- Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., et al. SPAdes: A new genome assembly algorithm and Its applications to single-cell sequencing. J Comput Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B. J., et al. PILON: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jex A. R., Hall R. S., Littlewood D. T. & Gasser R. B. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res 38, 522–533 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas N., et al. Mitochondrial genomes of Trichinella species and genotypes - a basis for diagnosis, and systematic and epidemiological explorations. Int J Parasitol 44, 1073–1080 (2014). [DOI] [PubMed] [Google Scholar]
- Magrane M. Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database 2011, bar009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Sato Y., Furumichi M. & Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40, D109–114 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov E. M. & Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001). [DOI] [PubMed] [Google Scholar]
- Xie C., et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39, W316–322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L., Krogh A. & Sonnhammer E. L. Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res 35, W429–432 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D. C., et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P., et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly (Austin) 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P., et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. & Stoeckert C. J. Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby J. G., Bellare P. & Derisi J. L. PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3 (Bethesda) 3, 865–880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. & Standley D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J. P. & Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001). [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgenbusch J. C. & Swofford D. Inferring evolutionary trees with PAUP*. Curr Protoc Bioinformatics 6, 4–6 (2003). [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Kishino H. & Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22, 160–174 (1985). [DOI] [PubMed] [Google Scholar]
- Santorum J. M., Darriba D., Taboada G. L. & Posada D. jmodeltest.org: selection of nucleotide substitution models on the cloud. Bioinformatics 30, 1310–1311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanave C., Preparata G., Saccone C. & Serio G. A new method for calculating evolutionary substitution rates. J Mol Evol 20, 86–93 (1984). [DOI] [PubMed] [Google Scholar]
- Adachi J. & Hasegawa M. Model of amino acid substitution in proteins encoded by mitochondrial DNA. J Mol Evol 42, 459–468 (1996). [DOI] [PubMed] [Google Scholar]
- Jones D. T., Taylor W. R. & Thornton J. M. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8, 275–282 (1992). [DOI] [PubMed] [Google Scholar]
- Sukumaran J. & Holder M. T. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571 (2010). [DOI] [PubMed] [Google Scholar]
- Untergasser A., et al. Primer3–new capabilities and interfaces. Nucleic Acids Res 40, e115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. F. & Foulds L. R. Comparison of phylogenetic trees. Math Biosci 53, 131–147 (1981). [Google Scholar]
- Roy A., Kucukural A. & Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P. J., Fields C. J., Goto N., Heuer M. L. & Rice P. M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res 38, 1767–1771 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson S. A. EMBOSS opens up sequence analysis. European Molecular Biology Open Software Suite. Brief Bioinform 3, 87–91 (2002). [DOI] [PubMed] [Google Scholar]
- Gaulton A., et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40, D1100–1107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum T., Briesemeister S. & Kohlbacher O. MultiLoc2: integrating phylogeny and Gene Ontology terms improves subcellular protein localization prediction. BMC Bioinformatics 10, 274 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F., et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.