

Abstract
DNA is organized into complex three-dimensional chromatin structures, but how this spatial organization regulates gene expression remains a central question. These DNA/chromatin looping structures can range in size from 10–20 kb (enhancers/repressors) to many megabases during intra- and inter-chromosomal interactions. Recently, the influence of telomere length on chromatin organization prior to senescence has revealed the existence of long-distance chromatin loops that dictate the expression of genes located up to 10 Mb from the telomeres (Telomere Position Effect–Over Long Distances [TPE-OLD]). Here, we demonstrate the existence of a telomere loop at the 4q35 locus involving the sorbin and SH3 domain-containing protein 2 gene, SORBS2, a skeletal muscle protein using a modification of the chromosome conformation capture method. The loop reveals a cis-acting mechanism modifying SORBS2 transcription. The expression of this gene is altered by TPE-OLD in myoblasts from patients affected with the age-associated genetic disease, facioscapulohumeral muscular dystrophy (FSHD1A, MIM 158900). SORBS2 is expressed in FSHD myoblasts with short telomeres, while not detectable in FSHD myoblasts with long telomeres or in healthy myoblasts regardless of telomere length. This indicates that TPE-OLD may modify the regulation of the 4q35 locus in a pathogenic context. Upon differentiation, both FSHD and healthy myotubes express SORBS2, suggesting that SORBS2 is normally up-regulated by maturation/differentiation of skeletal muscle and is misregulated by TPE-OLD-dependent variegation in FSHD myoblasts. These findings provide additional insights for the complexity and age-related symptoms of FSHD.
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common neuromuscular disorders (Padberg et al. 1995; Mostacciuolo et al. 2009; Deenen et al. 2014), yet the molecular mechanisms causing FSHD are unclear. FSHD is an autosomal dominant myopathy associated with a variable age of onset (Padberg et al. 1995; Pastorello et al. 2012). Clinical manifestations are heterogeneous; and in 90%–95% of FSHD cases, the disease onset is linked to contraction of multiple 3.3-kb tandem D4Z4 repeats located in the subtelomeric region of the long arm of Chromosome 4 (FSHD1A; 4q35 locus). A fraction of the 5%–10% remaining cases (FSHD1B) are believed to be caused by mutations in the SMCHD1 gene (structural maintenance of chromosomes flexible hinge domain containing 1) (Lemmers et al. 2012; Sacconi et al. 2013) on Chromosome 18 (18p11), and SMCHD1 might act as a modifier of the clinical severity in FSHD1A patients (Sacconi et al. 2013).
A model explaining the pathology implicates the DUX4 retrogene encoded by D4Z4 and transcribed from the last repeat and through the distal 4qA region (Gabriëls et al. 1999; Lemmers et al. 2010). This gene encodes a double homeobox protein ectopically overexpressed in bursts in patients’ myotubes that might activate a number of target genes and induce toxicity (Geng et al. 2012) and apoptosis (Bosnakovski et al. 2008; Block et al. 2013). In agreement with this model is the higher prevalence of the A-type haplotype among patients with FSHD1A compared to the general population (Lemmers et al. 2010; Scionti et al. 2012). However, in a number of cases, the role of DUX4 remains unclear due to the complexity of its expression pattern (Dixit et al. 2007; Snider et al. 2010; Tassin et al. 2013), low mRNA and protein abundance (Snider et al. 2010; Ferreboeuf et al. 2014), and the presence of DUX4 in unaffected individuals (Jones et al. 2012; Broucqsault et al. 2013). Thus, DUX4 might not be sufficient to explain the wide variability of the pathology, the muscle specificity and asymmetry, or the age-related onset suggesting that other factors are likely involved (Cabianca et al. 2012; Caruso et al. 2013; Stadler et al. 2013; Mariot et al. 2015; Puppo et al. 2015).
FSHD symptoms appear during adolescence, with 95% of the individuals carrying the genetic defect affected by the age of 20 (Pastorello et al. 2012; Deenen et al. 2014). Although the underlying genetic alteration is present at birth, additional factors or epigenetic changes may trigger disease onset and participate in disease variability and penetrance. Among them, we recently investigated the role of progressive telomere shortening in FSHD due to its age-related onset and because the genetic locus linked to the disease is only 25–50 kb from the Chromosome 4q telomere. Since telomeres are known to progressively shorten with increased cell turnover, we hypothesized that telomere proximity might contribute to the disease through telomere position effect (TPE) (Stadler et al. 2013).
Telomeres are complex 5′-(TTAGGG)n repetitive nucleoprotein structures found at the end of each chromosome. Their main functions are to prevent telomeres’ ends from being recognized as double-strand DNA breaks triggering the DNA damage response (DDR) (d'Adda di Fagagna et al. 2004; O'Sullivan and Karlseder 2010) and to complete DNA replication without loss of genetic information with each cell division (Gilson and Geli 2007). With age and progressive cell divisions, telomere shortening occurs in most tissues, including skeletal muscle (Daniali et al. 2013), and is accelerated in certain muscular dystrophies due to increased stem cell turnover (Decary et al. 2000). Shortening of telomeres induces a widespread change in gene expression, and some of those modulations can be attributed to differential expression of genes proximal to telomeres resulting from chromatin modifications, a phenomenon called telomere position effect (TPE) (Sandell et al. 1994; Baur et al. 2001; Koering et al. 2002; Mason et al. 2003; Pedram et al. 2006). Classic TPE is a conserved phenomenon occurring in many organisms that results in a silencing mechanism spreading from the telomeres toward subtelomeric regions (Ottaviani et al. 2008).
The mechanism of TPE has been extensively studied in yeast but not in other organisms, in particular humans (Blackburn 2001; Ottaviani et al. 2008; Stadler et al. 2013; Robin et al. 2014). In both yeast and human, TPE involves chromatin modifications (acetylation, methylation) (Smith et al. 2000; Zhou et al. 2011; Weuts et al. 2012; Buxton et al. 2014) and chromatin remodeling factors such as SIRT6 (Tennen et al. 2011; Anderson et al. 2014). TPE has also been implicated with telomere length changes and telomere-associated proteins (Blackburn 2001; Weuts et al. 2012; Ferreira et al. 2013). In FSHD, telomere length influences expression of DUX4 and FRG2 (FSHD region gene 2) (Stadler et al. 2013), suggesting that this region is sensitive to TPE. This is similar to studies in budding yeast in which telomeres can exert their repressive influence well beyond the immediate subtelomeric region (Maillet et al. 1996; Vega-Palas et al. 1997; Fourel et al. 1999). So far, only a few endogenous genes have been reported to be influenced by TPE in human cells and tissues (Lou et al. 2009; Boussouar et al. 2013), but based on microarray analyses, there are likely to be hundreds of genes near telomeres that are modified by a telomere length-dependent and DNA damage-independent mechanism, termed telomere position effect–over long distance (TPE-OLD) (Robin et al. 2014). At the genome-wide level, this mechanism might influence gene expression well before replicative senescence and explain the increased incidence of certain diseases with increased age without imposing a DNA damage signal from a too-short telomere (Wright and Shay 1992; Campisi 1997; Ye et al. 2014).
One hypothesis to explain how the contraction of D4Z4 leads to deregulation of nearby genes in FSHD (van Geel et al. 1999; Jiang et al. 2003; Celegato et al. 2006; Caruso et al. 2013) on 4q is via chromosomal looping and/or epigenetic modifications (Balog et al. 2012; Gaillard et al. 2014; Jones et al. 2014). The number of D4Z4 repeats might influence the ability of repressor or enhancer binding sequences within the repeats to loop and form contacts with other genes in the 4q region and thereby influence transcription (Pirozhkova et al. 2008; Tsumagari et al. 2008; Bodega et al. 2009; Ottaviani et al. 2009). This implies that looping is disturbed upon D4Z4 array contraction in FSHD1A. In FSHD1B, chromosomal looping might be modified by SMCHD1 mutation, which is implicated in chromosomal structural maintenance (Blewitt et al. 2008) and/or D4Z4 hypomethylation. However, it remains unclear whether or not FSHD cells have a different 4q35 chromatin organization (Pirozhkova et al. 2008; Bodega et al. 2009; Pope et al. 2011), and no reports have integrated the possible influence of telomere length on the chromatin structure of the 4q35 end (e.g., the last 5 Mb of Chromosome 4).
Since we recently demonstrated that long-range chromosome looping involving telomeres influences gene expression over several Mb at chromosome ends by TPE-OLD (Robin et al. 2014), in the present study, we investigated whether this phenomenon occurs in FSHD (myoblasts).
Results
Chromosomal organization of the 4q35 locus revealed by Hi-C
To address the hypothesis that telomere length may impact the expression of genes on the 4q35 region via TPE-OLD, we used a system in which telomere length can be controlled to analyze the impact of telomere shortening in isogenic clones (Stadler et al. 2013; Robin et al. 2014). This cell culture system avoids the inherited diversity of telomere length observed in primary cell cultures isolated from individuals (Londoño-Vallejo et al. 2001) and also eliminates variations in gene expression that may be due to differences in time in culture. Importantly, cells were analyzed prior to telomere length-induced senescence (Supplemental Figs. S1, S2).
We then applied to this cellular model, the unbiased technique, Hi-C chromosome conformation capture (van Berkum et al. 2010) that permits identification of chromosomal interactions over long distances. We modified the technique by introducing a second capture step that retrieved interactions involving the small genomic region around the 4q35 locus (0.15% of the genome) as illustrated (Supplemental Figs. S3, S4). To recover most of the 4q35 region from the Hi-C preparation, we used a total of 23,000 RNA baits (120 mers) covering ∼55% of the distal 5 Mb of the 4q end. The location of the RNA baits is presented in Figure 1A (green lines). To minimize overlap of the RNA baits with the 10q Chromosome end (98% homology with the 40–50 kb most telomeric part of the 4q35) and other loci, baits were designed to uniquely match 120-mer sequences located in the 4q35 region. In most cases, baits surround if not cover genes present in the last 5 Mb subtelomeric region of Chromosome 4 (Supplemental Fig. S5). We subjected FSHD and control myoblasts with long (11 kb) and short (5–6 kb) telomeres (Supplemental Fig. S2) to our modified locus-specific Hi-C technique to obtain subtelomere–chromosome interaction maps (Fig. 1A; Supplemental Figs. S6, S7).
Figure 1.

Long-range interactions revealed by locus-specific Hi-C at the 4q35 locus. (A) Circos plot (Krzywinski et al. 2009) of the 3D organization of the 4q35.2 locus in FSHD myoblasts with long telomeres after Hi-C 4q35-specific experiment. Data were corrected and normalized to background using HOMER software. Only true significant interactions are shown (red: P < 0.05; blue: P < 0.001). The last 8 Mb of Chromosome 4q are represented as a circle, the location of known genes are shown as segments in a purple inner circle, and the location of biotinylated bait sequences is shown in green. The locations of the probes used for chromosome conformation capture (3C) and in situ hybridization are indicated in colored boxes (green for FRG1 and red for SORBS2). (B) Schematic representation of the Hi-C validation by 3C for a 4.6-Mb loop between FRG1 and SORBS2. A forward primer adjacent to a HindIII site in the FRG1 sequence was used with a variety of reverse primers adjacent to HindIII sites in the SORBS2 sequence. Following crosslinking, digestion with HindIII, ligation, and crosslink reversion, DNA was amplified with the different primer pairs. Each assay was done with droplet digital PCR (ddPCR) and normalized to a 3C assay performed in a control region. (C) Graphical representation of 3C quantification of the FRG1-SORBS2 loop in control and FSHD myoblasts with long and short telomeres. Only FSHD myoblasts with short telomeres differ, with a decrease of 48.5% in the signal corresponding to the center of interaction. Assays were done in triplicate and measured in duplicate (Mean ± SD shown for each primer pair).
A majority of the sequences (>80%) recovered from the paired-end sequencing (HiSeq 2500, Illumina) contained at least one end corresponding to the 4q35 region (Supplemental Fig. S5). Circular plots, generated with HOMER (Heinz et al. 2010), revealed many long-range interactions at the 4q35 locus in myoblasts with long telomeres (Fig. 1A; Supplemental Figs. S6, S7). We observed several regions where interactions were highly significant (P < 0.001) with the formation of interactions between the 4q telomeric regions and sites at a distance of 4.8 Mb from the 4q telomeric region. In particular, after correction, the longest and most significant interaction was found between the region proximal to the promoter of FRG1 (FSHD region gene 1) and SORBS2 (P < 0.001) (Fig. 1A; Supplemental Figs. S6, S7). Interestingly, these maps reveal the existence of long-distance interactions across the 4q35 region, conserved between the different conditions (i.e., long, short, FSHD versus control).
Hi-C identifies a 4.8-Mb loop that is lost in FSHD myoblasts with short telomeres
To investigate whether the FRG1-SORBS2 interactions identified with the Hi-C plot vary with telomere length or are altered in FSHD myoblasts, we used chromosome conformation capture (3C). The 3C technique analyzes local interactions with primers designed on each side of the interacting DNA sequences (“one versus one” technique) (Fig. 1B). We confirmed the interaction between the region next to the FRG1 promoter and SORBS2 as revealed by Hi-C and observed a significantly decreased frequency of interaction in FSHD myoblasts with short telomeres (48.5%) compared to control myoblasts (FSHD-Short telomere versus Control-Short telomere [P = 0.02] and versus Control-Long telomere [P = 0.006]) and FSHD myoblast with long telomeres (P = 0.03) (Fig. 1C). Additionally, we observed a similar phenomenon when quantifying another interaction found between FRG1 and FAM149A (family with sequence similarity 149 member A), a gene located 4 Mb from FRG1 (Supplemental Fig. S8).
Using 3D-FISH, a fluorescence in situ hybridization technique, we further confirmed the FRG1-SORBS2 interactions (Fig. 2). We used two probes corresponding either to the FRG1 gene and surrounding sequences (Fig. 2A, green) or SORBS2 (Fig. 2A, red). The two probes were in close proximity (described as adjacent) in control myoblasts regardless of telomere length as observed in FSHD myoblasts with long telomeres. However, the two probes behaved differently in FSHD myoblasts upon telomere shortening. For 47% of the chromosomes analyzed, the distance between the two probes significantly increased in isogenic FSHD myoblasts with short telomeres (Fig. 2B,C).
Figure 2.
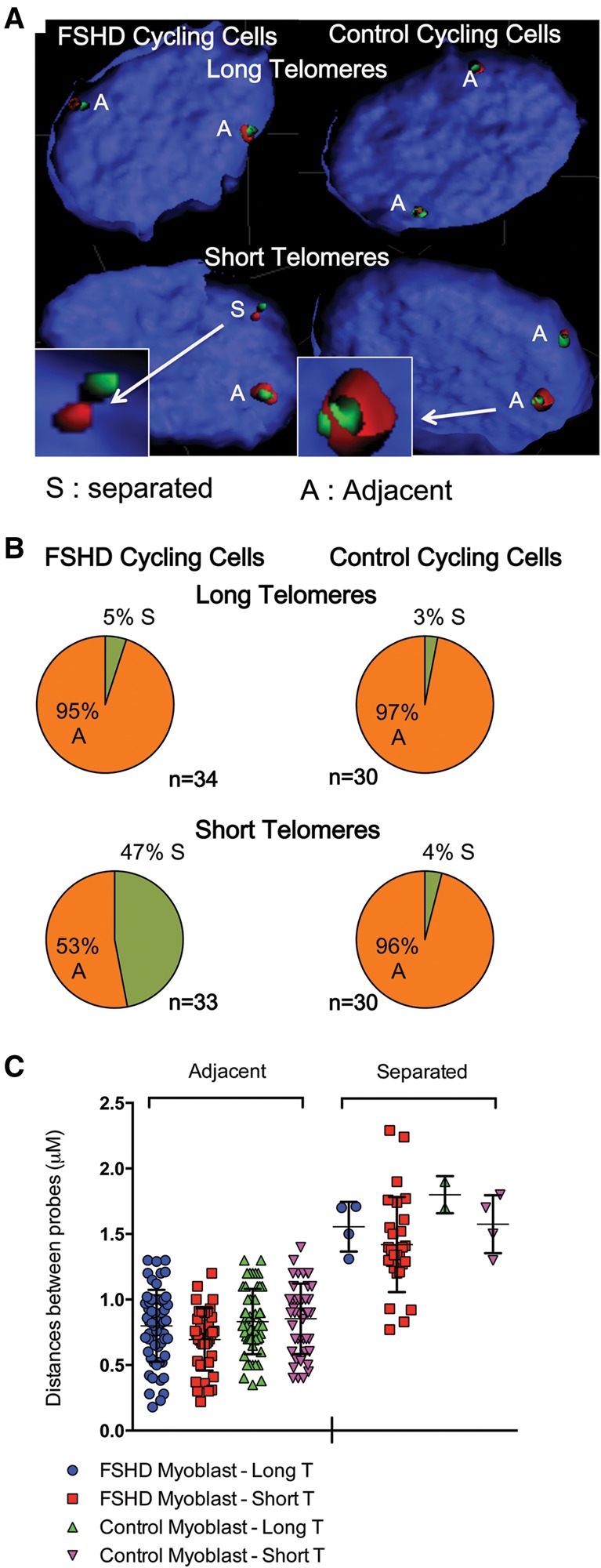
3D DNA-FISH of the SORBS2-FRG1 interaction. (A) Processed images of in situ hybridization using an FRG1 (green) and SORBS2 (red) probe. Images were processed with IMARIS for N∼30 nuclei per condition. (B) Quantification of the number of adjacent (A) and separated (S) signals. (C) Mean of the distances measured between the gravity centers of each signal. Respective mean ± SEM of the adjacent distances are shown. Independent t-test between all conditions for binary choice: P < 10 × 10−7; t-test of the mean distance between centers: P = 0.01.
FSHD is an autosomal dominant disease with one 4q allele containing a contracted number of D4Z4 repeats (between one and 10 units) and one normal 4q allele (>100 units). The loss of interaction between the FRG1 promoter and SORBS2 is only seen in FSHD cells. This suggests that in the context of short telomeres, long-range interactions are likely modulated by TPE-OLD for the allele carrying the short D4Z4 array, thus inducing a modified folding of one of the two 4q35 alleles in the disease context. In agreement with those results, we do not observe any topological difference at the 1p36 locus between the interferon-stimulated gene 15 (ISG15 gene) regulated by TPE-OLD and a more distal subtelomeric probe between FSHD and control cells with short telomeres (Supplemental Fig. S9; Robin et al. 2014). In addition, we showed by 3D-FISH that probes corresponding to ZFP42 zinc finger protein (ZFP42) and FRG1 remain adjacent regardless of telomere or D4Z4 length, suggesting that the modification of the FRG1–SORBS2 loop in the context of a short D4Z4 array is specific for the FSHD disease locus and not resulting from a global DNA relaxation of the 4q35 region (Supplemental Fig. S9). Overall, our results suggest that shortening of the D4Z4 array (affected locus) in the context of short telomeres modifies the chromatin organization of the 4q35 region in cis.
Telomere shortening correlates with TPE-OLD and D4Z4 hypomethylation in FSHD
To test if telomere shortening in FSHD myoblasts mimicked DNA methylation patterns observed at the 4q35 locus, we analyzed individual CpGs for three different regions within D4Z4 (Fig. 3; Gaillard et al. 2014). In control myoblasts, DNA methylation is not affected by telomere length. However, we observed a significant decrease in methylation in FSHD myoblasts with short telomeres (BIS-Mid region percent methylation values: FSHD-Short telomere = 49% versus FSHD-Long telomere = 63% [P = 0.03]; versus Control-Long telomere = 83% [P = 0.001]; versus Control-Short telomere = 73% [P = 0.001]), suggesting a link between the length of the telomeres and the level of D4Z4 methylation, especially in the DUX4 promoter region (Mid), which is activated by TPE (Stadler et al. 2013).
Figure 3.

Telomere shortening is associated with D4Z4 hypomethylation in FSHD cells. The absolute level of DNA methylation at a single nucleotide level was determined by sodium bisulfite sequencing for three different regions within the D4Z4 sequence: BIS-5′ (21 CpGs represented by dots), BIS-mid (31 CpGs), and BIS-3′ (14 CpGs). For each sequence, several clones were analyzed from DNA obtained from control myoblasts with long or short telomeres and FSHD myoblasts with long or short telomeres. The distribution of methylated (black dots) or unmethylated (white dots) is represented for the different clones analyzed in the different conditions. The average percentage of methylation is indicated for each sequence.
TPE-OLD modulates gene expression in FSHD
We next determined if modification in the 4q35 organization modifies SORBS2 expression at the mRNA and protein level. SORBS2 is a 371-kb gene composed of 42 exons with a complex expression pattern that produces 118 alternative splicing variants and nine proteins (NCBI Gene) (Supplemental Fig. S10). SORBS2 protein has previously been reported to be present in cardiac but not skeletal muscle (Wang et al. 1997; Sanger et al. 2010). We used immunohistochemistry (IHC) for protein localization and RT-PCR for RNA expression to examine whether SORBS2 was expressed in skeletal muscle and whether its expression was modified in FSHD biopsies. We analyzed paraffin-embedded tissues from cardiac and skeletal (trapezius) muscle from a non-FSHD individual by IHC. We observed that SORBS2 is enriched at the intercalated disk and Z-line of cardiac cells and at the Z-line of skeletal muscle fibers (Fig. 4A,B).
Figure 4.

SORBS2 gene expression in skeletal muscle biopsies. Immunohistochemistry (IHC) staining of transversal biopsies of cardiac (A) and skeletal (B) muscle for the detection of SORBS2 (Epitomics, RB19714). (C) RT-PCR of a SORBS2 transcript variant (Exon 17–42) in skeletal muscle biopsies from biceps (bic) and deltoid (del) isolated from FSHD patients (FSHD A, B) and their unaffected siblings (control) for a total of 12 cohorts. For each sample, three PCRs were performed using 35, 40, or 45 amplification cycles. In these RT-PCR assays, all transcript variants that include exons 17 and 42 (23 variants, including eight protein-coding forms) are amplified.
Next, we addressed the level of transcription of SORBS2 in skeletal muscle (Fig. 4C) by generating cDNAs from muscle biopsies isolated from 14 FSHD individuals and 11 unaffected first-degree relatives from a total of 11 different families (cohorts) (Rahimov et al. 2012). Primers were designed to detect all transcripts containing exons 17 and 42. The assay includes eight of the nine transcripts encoding proteins. We found a 2.2-kb transcript that codes for 23 different isoforms (including eight protein-coding variants) in 89% of FSHD and 87% of control biopsies (Fig. 4C). Thus, SORBS2 is present in skeletal muscle biopsies (control and FSHD), and the protein is localized at the Z-lines of muscle fibers.
To further investigate whether SORBS2 is associated with FSHD and regulated by TPE-OLD, we determined the presence of two different exon–exon pairs in both control and FSHD myoblasts with long and short telomeres by RT-PCR (Fig. 5; Supplemental Fig. S11). We found that transcripts containing exons 31 through 42 (Ex31–Ex42) were present regardless of the sample status; these transcripts correspond to a total of 56 variants regrouped in two distinct clusters (±exon 40); the variants excluding the exon represent 13 forms, among which nine are protein-coding transcripts (Fig. 5B; Supplemental Figs. S11, S12). However, using primers encompassing Ex17–Ex42, the eight protein-coding transcripts were only detected in FSHD cells with short telomeres (Fig. 5A; Supplemental Fig. S11). These observations can be interpreted to indicate that TPE-OLD not only influences the level of SORBS2 expression, but also directly or indirectly modifies splicing of transcripts, specifically in the context of FSHD.
Figure 5.

TPE-OLD modifies SORBS2 expression in FSHD myoblasts. (A,B) Amplification of SORBS2 transcripts in FSHD and sibling control myoblasts with long and short telomeres. (A) RT-PCR of SORBS2 transcripts using primers amplifying all known transcript variants that include exons 17 and 42 (23 variants). Assay performed in FSHD#12 and sibling control, FSHD#11, and FSHD#15 myoblasts with long and short telomeres. Only FSHD myoblasts with short telomeres express the 2-kb splice forms (23 variants, including eight protein-coding forms). (B) RT-PCR performed using a set of primers amplifying all known transcript variants between exons 31 and 42 (a total of 30 forms). Among the 30 isoforms detected, 13 exclude the totality of exon 40 (smaller form at ∼530 bp), among which are the nine protein-coding variants. (C) Gene expression analysis of SORBS2. For all four assays, we report the predicted transcripts according to the NCBI database. ddPCR results are expressed as the number of molecules detected in a 12.5-ng RT input; values are averages ± SEM.
To determine if SORBS2 transcription fluctuates during the differentiation process, we next quantified expression of different exon–exon pairs in myoblasts and myotubes isolated from three different cohorts (FSHD#11, #12, and #15, respectively) with long and short telomeres. Using droplet digital PCR (ddPCR), we analyzed a set of eight assays (Fig. 5; Supplemental Fig. S10) each detecting a different exon–exon junction across SORBS2. Figure 5C presents the results of four quantitative assays from the 5′ UTR to the 3′ UTR region of SORBS2. Each assay detects the abundance of transcripts containing a particular exon–exon junction, hence, the detection of a combination of different transcripts. In the 3′ UTR assay (50 possible variants), we observed a sixfold significant increase in FSHD myotubes compared to FSHD myoblasts (P = 0.034), whereas in control cells, we observed a nonstatistically significant threefold increase in myotubes (P = 0.18). For the Ex17–Ex18 assay (23 possible variants), we observed a statistically significant 25-fold increase in control myotubes compared to myoblasts (P = 0.018) and a highly significant 75-fold increase in FSHD cells (P < 0.0001), suggesting that variants containing the 3′ UTR and exons 17–18 are induced upon differentiation. Moreover, similar observations were made in the assay on the abundance of transcripts including Ex30–Ex31 (33 variants) with a 30-fold and 155-fold significant increase upon differentiation in control and FSHD samples, respectively (Control, P = 0.0071; FSHD, P < 0.0001).
No significant differences were reported between FSHD and control myoblasts (undifferentiated) in the three assays. However, we observed a significant twofold increase in FSHD myotubes compared to controls in the Ex30–Ex31 assay (P = 0.0006), suggesting that increased level of SORBS2 transcripts are associated with the onset of the disease. Finally, in the 5′ UTR assay, we noticed a significant increase upon differentiation (control, P = 0.0001; FSHD, P < 0.0001) along with a significant twofold increase in FSHD myotubes compared to control myotubes (P < 0.001). In conclusion, seven of the eight assays performed showed a significant increase of SORBS2 transcripts in myotubes (Fig. 5C; Supplemental Fig. S10), suggesting an up-regulation of SORBS2 upon differentiation (myoblasts versus myotubes). In addition, we observed a significant telomere length-dependent increase in FSHD myoblasts with short telomeres compared to other myoblasts (Tukey's multiple comparisons test, FSHD-Short telomere versus FSHD-Long telomere [P = 0.0037]; versus Control myoblast long telomere [P < 0.0001]; versus Control-Short telomeres [P = 0.0054]). Interestingly, this up-regulation in myoblasts with short telomeres correlates with the detection of the transcript encompassing exons 17–42 (Fig. 5A) and the loss of the 4.8-Mb TPE-OLD loop described in Figure 1. We did not detect any significant change in the level of expression of other 4q35 genes present in the 4.8-Mb loop such as FAT1, ZFP42, FAM149A, FRG1, PDLIM3, or TRIML1 (Supplemental Fig. S13). We show that in FSHD1A, TPE-OLD allows for the earlier transcription of SORBS2 in cells with short telomeres, that could contribute to the disease phenotype by prematurely initiating differentiation.
Stimulated by recent studies on muscle maturation (Millay et al. 2013, 2014), we also evaluated the expression of a newly proposed master regulatory gene involved in muscle maturation, TMEM8C (Myomaker). In cells with short telomeres, TMEM8C is up-regulated in myoblasts (Fig. 6), suggesting that this gene located 4.5-Mb from the q end of Chromosome 9 is another potential gene regulated by TPE-OLD. However, TMEM8C is not restricted to FSHD because we did not observe any difference between control and FSHD cells upon telomere shortening (Fig. 6). Altogether, this suggests a mild defect in the differentiation program, with an early expression of a gene involved in the fusion of myoblasts upon telomere shortening. These results suggest that in FSHD myoblasts with short telomeres, the combined up-regulation of the SORBS2 and TMEM8C might result in the premature initiation of the differentiation program.
Figure 6.

Gene expression analysis of MYOD1 and TMEM8C. RNA was isolated from 1 × 106 cells and cDNAs made in triplicate. ddPCR results are expressed as the number of molecules detected in a 12.5-ng RT input; values shown as averages ± standard error of the mean (assay done in duplicate using three cohorts of matched siblings from two biopsies; total of 12 measures per condition). MYOD1 expression does not depend on telomere length (long versus short) nor disease status (control versus FSHD) using the Holm-Sidak's multiple comparisons test (alpha = 0.05). In myoblasts, TMEM8C expression increased in FSHD cells with short telomeres when compared to control myoblast with long telomeres (adjusted P-value = 0.0178; Holm-Sidak's multiple comparisons test) or FSHD myoblast with long telomeres (adjusted P-value = 0.0178; Holm-Sidak's multiple comparisons test).
Discussion
Facioscapulohumeral dystrophy (FSHD) is a genetic disease linked to rearrangements in the 4q35 locus located at a short distance from the telomere. This subtelomeric region can be regulated by telomeric position effects in a telomere length-dependent manner (Ottaviani et al. 2009; Arnoult et al. 2010; Stadler et al. 2013). In the present studies, we analyzed the chromatin organization of the 4q35 locus upon telomere shortening and differentiation by comparing control and FSHD cells. To this end, we have developed a specific Hi-C assay allowing us to analyze long-range interactions within the 4q35 locus.
We found long-range interactions between the telomere proximal FRG1 gene and the more centromere-proximal SORBS2 gene that are lost upon telomere shortening in FSHD but not in normal myoblasts. By quantifying the 3D-chromatin organization of the 4q35 locus by 3C and 3D-FISH (Figs. 1C, 2), we report that the organization is similar between conditions when telomeres are long, a finding that is supported by previous replication timing experiments (Pope et al. 2011). However, this chromatin organization is sensitive to telomere length in FSHD isogenic clones with short telomeres (Figs. 1, 2). Using control conditions (Supplemental Fig. S8), we have characterized an interaction between the proximal region of FRG1 and SORBS2. Consistent with changes occurring on only one of the two 4q35 alleles, these topological modifications were specifically observed in ∼50% of the chromosomes analyzed in FSHD samples (Supplemental Fig. S9). This suggests that telomere length is associated with changes in chromatin folding in the context of a short contracted disease-associated allele (six copies of the D4Z4 macrosatellite repeat in cells used in the present study) and correlates with D4Z4 hypomethylation. Since no changes were observed in control myoblasts carrying two wild-type alleles (more than 11 repeats), we conclude that telomere shortening together with D4Z4 array contraction induces a modification of the 4q35 folding accompanied by a more pronounced chromatin relaxation (Fig. 3). This indicates that the length-dependent heterochromatin nature of telomeres might contribute in cis to the condensation of the 4q35 region.
Interestingly, this chromatin relaxation is restricted to a specific region corresponding to the SORBS2 gene (Supplemental Fig. S9) and occurs at the nuclear periphery as no significant changes were detected in the positioning of the locus within the nuclear volume. Nevertheless, one can hypothesize that the modified loop alleviates accessibility to regulatory elements, subsequently changing gene expression (Fig. 5; Supplemental Figs. S10, S13). A key to the understanding of this mechanism might rely on the presence of lamina-associated domains (LADs) in the vicinity of SORBS2 and FRG1. Anchoring to the nuclear periphery at D4Z4 or at a different position along the 4q35 region together with TPE-OLD-dependent folding might contribute to the higher-order organization and regulation of this subtelomeric region.
SORBS2 has previously been detected at the Z-line of cardiac muscle, and overexpression in cardiac cells causes sarcomere disruption (Sanger et al. 2010). SORBS2 is also localized at the intercalated disk of cardiomyocytes (Sanger et al. 2010) and has been shown to interact with multiple proteins (Roignot and Soubeyran 2009; Lamberti et al. 2011; Murase et al. 2012; Roignot et al. 2014). As a member of the Sorbin family, SORBS2 interacts with actin and can mediate signal-transduction events, from cell adhesion (Murase et al. 2012), migration (Roignot and Soubeyran 2009), proliferation (Haglund et al. 2004), to cell homeostasis (Roignot et al. 2014). SORBS2 participates in the scaffolding of muscle cells (cardiomyocytes) and may act as an adapter protein to assemble signaling complexes in stress fibers; but prior to our present studies, nothing has been reported on its function in skeletal muscles. By immunohistochemistry, we confirmed the presence of SORBS2 in cardiac muscle but also at the Z-line in skeletal muscle (Fig. 4A,B).
The telomere length-dependent alterations of SORBS2 chromatin organization and expression in FSHD myoblasts suggest a role for this protein in the disease, but additional investigations will be required to confirm and expand on our findings. In fact, we detected SORBS2 protein-coding transcripts in both healthy and FSHD patient biopsies (88% of positive samples) (Fig. 4C). We also tested whether the SORBS2 transcripts were also observed in other cohorts (FSHD patients and age-matched relatives) and in different types of muscle (Figs. 4C, 5A; Supplemental Figs. S11, S12). Interestingly, modification in the 3D-chromatin organization correlates with changes in the SORBS2 expression and splicing pattern in myoblasts (Figs. 2, 5), whereas expression of other genes located at the 4q35 locus remains unchanged (Supplemental Fig. S13).
In summary, the early expression of SORBS2 in FSHD myoblasts with short telomere influences the differentiation program and may contribute to the pathology with other FSHD candidate genes such as the DUX4 toxic protein (Winokur et al. 2003; Cheli et al. 2011; Tsumagari et al. 2011; Broucqsault et al. 2013). Due to the complexity of SORBS2 (42 exons, 118 putative transcripts), previous results are often contradictory, with some observing up-regulation (Celegato et al. 2006), and others reporting no change in the expression of this gene (Masny et al. 2010; Rahimov et al. 2012). These results are not surprising if the major changes involve modifications of the splicing pattern rather than a global change in the transcription level. Although we were unable to distinguish between the different types of SORBS2 transcripts, our studies highlight the necessity of further investigations to decipher the function and regulation of this gene. Interestingly, we found that SORBS2 is up-regulated in myotubes, suggesting a critical function of SORBS2 in skeletal muscle differentiation/maturation. We did not analyze other genes involved in differentiation, as no difference in gene expression or myotube formation were previously found in these cells (Supplemental Fig. S13; Stadler et al. 2013).
Replicative aging and telomere shortening are relevant in dystrophies such as DMD (Duchenne muscular dystrophy), due to the increased cycles of degeneration–regeneration observed in skeletal muscle. It is less clear whether this occurs in FSHD, since no overt differences in telomere length have been found in FSHD biopsies compared to controls. However, telomeres shorten at similar rates in all tissues throughout their lifespans (Daniali et al. 2013), suggesting that technical issues and sample limitations may be responsible for the observations made in skeletal muscle about telomere shortening in FSHD. Our data are consistent with the idea that in FSHD, the genetic condition leading to the accumulation of the defects in myoblasts is enhanced by telomere shortening. Based on our observations, we propose a model in which chromatin conformation and altered gene expression dependent on telomere shortening is a hallmark of FSHD muscle.
Methods
Cell culture
CD56-positive myoblasts from primary muscle biopsies were infected with retroviral constructs containing CDK4 and a floxable human TERT. Individual clones were then treated with Cre-recombinase at different times and cultivated for approximately the same number of population doublings to generate isogenic subclones with different telomere lengths (Supplemental Fig. S1). For further details (i.e., cell growth conditions), see Supplemental Material.
4q35 Hi-C; 3C
Samples both for Hi-C and 3C were prepared as described (van Berkum et al. 2010). Hi-C was modified to enrich for the 4q35 region. In brief, proteins within intact cells were crosslinked for 10 min in 1% formaldehyde. Reaction was stopped with glycine. Cells were lysed in the presence of protease inhibitors. DNA was digested with HindIII restriction enzyme leaving an overhang that is filled in by biotinylated dCTP. Blunt ends were ligated under very dilute conditions that favor ligation of ends held in close proximity. Ligation products containing biotin labels were pulled-down with streptavidin. The full Hi-C library was produced by PCR amplification. The library was hybridized to RNA biotinylated probes (targeting the 4q35 locus), reamplified after purification, and then analyzed by paired-end sequencing (Illumina, HiSeq 2500). For further details (i.e., step by step procedure), see Supplemental Material.
3D-FISH
Probes for FISH were produced following the manufacturer's instructions (nick translation kit, Abbott Molecular). Templates were chosen with the UCSC Genome Browser and ordered from the CHORI institute (SORBS2, RP11-1L10; proximal region of FRG1, G248P88732C10). BACS and fosmids were verified by PCR and/or metaphase spreads. For further details (i.e., used solutions), see Supplemental Material.
Methylation assay by sodium bisulfite sequencing
Assay was performed as previously described (Gaillard et al. 2014) with 2 μg of genomic DNA denatured for 30 min at 37°C in NaOH 0.4 N and incubated overnight in a solution of sodium bisulfite 3 M pH5 and 10 mM hydroquinone. For further details, see Supplemental Material.
Data access
4q Hi-C sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP062778.
Acknowledgments
We thank Kathryn Wagner for tissue samples and Fedick Rahimov for the cDNAs produced from the muscle biopsies (Wellstone Foundation). We also thank Min Chen for bioinformatics assistance (UT Dallas) and Jean-Marie Rouillard from Biodiscovery, LLC, for providing reagents. M.C.G. is the recipient of a fellowship from the Association Française contre les Myopathies (AFM). This work was supported by AG01228 from the US National Institute of Aging (W.E.W.) and NIH Postdoctoral Training Fellowship (T32 CA124334, A.T.L.). This work was performed in laboratories constructed with support from National Institutes of Health grant C06 RR30414.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.190660.115.
References
- Anderson MZ, Gerstein AC, Wigen L, Baller JA, Berman J. 2014. Silencing is noisy: Population and cell level noise in telomere-adjacent genes is dependent on telomere position and sir2. PLoS Genet 10: e1004436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult N, Schluth-Bolard C, Letessier A, Drascovic I, Bouarich-Bourimi R, Campisi J, Kim SH, Boussouar A, Ottaviani A, Magdinier F, et al. 2010. Replication timing of human telomeres is chromosome arm–specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet 6: e1000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balog J, Thijssen PE, de Greef JC, Shah B, van Engelen BG, Yokomori K, Tapscott SJ, Tawil R, van der Maarel SM. 2012. Correlation analysis of clinical parameters with epigenetic modifications in the DUX4 promoter in FSHD. Epigenetics 7: 579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Zou Y, Shay JW, Wright WE. 2001. Telomere position effect in human cells. Science 292: 2075–2077. [DOI] [PubMed] [Google Scholar]
- Blackburn EH. 2001. Switching and signaling at the telomere. Cell 106: 661–673. [DOI] [PubMed] [Google Scholar]
- Blewitt ME, Gendrel AV, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N, et al. 2008. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet 40: 663–669. [DOI] [PubMed] [Google Scholar]
- Block GJ, Narayanan D, Amell AM, Petek LM, Davidson KC, Bird TD, Tawil R, Moon RT, Miller DG. 2013. Wnt/β-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum Mol Genet 22: 4661–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodega B, Ramirez GD, Grasser F, Cheli S, Brunelli S, Mora M, Meneveri R, Marozzi A, Mueller S, Battaglioli E, et al. 2009. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol 7: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosnakovski D, Xu Z, Gang EJ, Galindo CL, Liu M, Simsek T, Garner HR, Agha-Mohammadi S, Tassin A, Coppée F, et al. 2008. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J 27: 2766–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussouar A, Barette C, Nadon R, Saint-Léger A, Broucqsault N, Ottaviani A, Firozhoussen A, Lu Y, Lafanechère L, Gilson E, et al. 2013. Acacetin and chrysin, two polyphenolic compounds, alleviate telomeric position effect in human cells. Mol Ther Nucleic Acids 2: e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broucqsault N, Morere J, Gaillard MC, Dumonceaux J, Torrents J, Salort-Campana E, Maues De Paula A, Bartoli M, Fernandez C, Chesnais AL, et al. 2013. Dysregulation of 4q35- and muscle-specific genes in fetuses with a short D4Z4 array linked to facio-scapulo-humeral dystrophy. Hum Mol Genet 22: 4206–4214. [DOI] [PubMed] [Google Scholar]
- Buxton JL, Suderman M, Pappas JJ, Borghol N, McArdle W, Blakemore AI, Hertzman C, Power C, Szyf M, Pembrey M. 2014. Human leukocyte telomere length is associated with DNA methylation levels in multiple subtelomeric and imprinted loci. Sci Rep 4: 4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D. 2012. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149: 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. 1997. The biology of replicative senescence. Eur J Cancer 33: 703–709. [DOI] [PubMed] [Google Scholar]
- Caruso N, Herberth B, Bartoli M, Puppo F, Dumonceaux J, Zimmermann A, Denadai S, Lebossé M, Roche S, Geng L, et al. 2013. Deregulation of the protocadherin gene FAT1 alters muscle shapes: implications for the pathogenesis of facioscapulohumeral dystrophy. PLoS Genet 9: e1003550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celegato B, Capitanio D, Pescatori M, Romualdi C, Pacchioni B, Cagnin S, Viganò A, Colantoni L, Begum S, Ricci E, et al. 2006. Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics 6: 5303–5321. [DOI] [PubMed] [Google Scholar]
- Cheli S, Francois S, Bodega B, Ferrari F, Tenedini E, Roncaglia E, Ferrari S, Ginelli E, Meneveri R. 2011. Expression profiling of FSHD-1 and FSHD-2 cells during myogenic differentiation evidences common and distinctive gene dysregulation patterns. PLoS One 6: e20966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Teo SH, Jackson SP. 2004. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev 18: 1781–1799. [DOI] [PubMed] [Google Scholar]
- Daniali L, Benetos A, Susser E, Kark JD, Labat C, Kimura M, Desai K, Granick M, Aviv A. 2013. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat Commun 4: 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decary S, Hamida CB, Mouly V, Barbet JP, Hentati F, Butler-Browne GS. 2000. Shorter telomeres in dystrophic muscle consistent with extensive regeneration in young children. Neuromuscul Disord 10: 113–120. [DOI] [PubMed] [Google Scholar]
- Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, Weinreich SS, Verbeek AL, van Engelen BG. 2014. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology 83: 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit M, Ansseau E, Tassin A, Winokur S, Shi R, Qian H, Sauvage S, Mattéotti C, van Acker AM, Leo O, et al. 2007. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci 104: 18157–18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreboeuf M, Mariot V, Bessières B, Vasiljevic A, Attié-Bitach T, Collardeau S, Morere J, Roche S, Magdinier F, Robin-Ducellier J, et al. 2014. DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles. Hum Mol Genet 23: 171–181. [DOI] [PubMed] [Google Scholar]
- Ferreira HC, Towbin BD, Jegou T, Gasser SM. 2013. The shelterin protein POT-1 anchors Caenorhabditis elegans telomeres through SUN-1 at the nuclear periphery. J Cell Biol 203: 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourel G, Revardel E, Koering CE, Gilson E. 1999. Cohabitation of insulators and silencing elements in yeast subtelomeric regions. EMBO J 18: 2522–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriëls J, Beckers MC, Ding H, De Vriese A, Plaisance S, van der Maarel SM, Padberg GW, Frants RR, Hewitt JE, Collen D, et al. 1999. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 236: 25–32. [DOI] [PubMed] [Google Scholar]
- Gaillard MC, Roche S, Dion C, Tasmadjian A, Bouget G, Salort-Campana E, Vovan C, Chaix C, Broucqsault N, Morere J, et al. 2014. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 83: 733–742. [DOI] [PubMed] [Google Scholar]
- Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, van der Maarel SM, Ruzzo WL, Gentleman RC, Tawil R, et al. 2012. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell 22: 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson E, Geli V. 2007. How telomeres are replicated. Nat Rev Mol Cell Biol 8: 825–838. [DOI] [PubMed] [Google Scholar]
- Haglund K, Ivankovic-Dikic I, Shimokawa N, Kruh GD, Dikic I. 2004. Recruitment of Pyk2 and Cbl to lipid rafts mediates signals important for actin reorganization in growing neurites. J Cell Sci 117: 2557–2568. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Yang F, van Overveld PG, Vedanarayanan V, van der Maarel S, Ehrlich M. 2003. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum Mol Genet 12: 2909–2921. [DOI] [PubMed] [Google Scholar]
- Jones TI, Chen JC, Rahimov F, Homma S, Arashiro P, Beermann ML, King OD, Miller JB, Kunkel LM, Emerson CP Jr, et al. 2012. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet 21: 4419–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TI, Yan C, Sapp PC, McKenna-Yasek D, Kang PB, Quinn C, Salameh JS, King OD, Jones PL. 2014. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin Epigenetics 6: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koering CE, Pollice A, Zibella MP, Bauwens S, Puisieux A, Brunori M, Brun C, Martins L, Sabatier L, Pulitzer JF, et al. 2002. Human telomeric position effect is determined by chromosomal context and telomeric chromatin integrity. EMBO Rep 3: 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberti A, Sanges C, Chambery A, Migliaccio N, Rosso F, Di Maro A, Papale F, Marra M, Parente A, Caraglia M, et al. 2011. Analysis of interaction partners for eukaryotic translation elongation factor 1A M-domain by functional proteomics. Biochimie 93: 1738–1746. [DOI] [PubMed] [Google Scholar]
- Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, et al. 2010. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329: 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJ, Tawil R, Petek LM, Balog J, Block GJ, Santen GW, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, et al. 2012. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet 44: 1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londoño-Vallejo JA, DerSarkissian H, Cazes L, Thomas G. 2001. Differences in telomere length between homologous chromosomes in humans. Nucleic Acids Res 29: 3164–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Z, Wei J, Riethman H, Baur JA, Voglauer R, Shay JW, Wright WE. 2009. Telomere length regulates ISG15 expression in human cells. Aging 1: 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet L, Boscheron C, Gotta M, Marcand S, Gilson E, Gasser SM. 1996. Evidence for silencing compartments within the yeast nucleus: a role for telomere proximity and Sir protein concentration in silencer-mediated repression. Genes Dev 10: 1796–1811. [DOI] [PubMed] [Google Scholar]
- Mariot V, Roche S, Hourdé C, Portilho D, Sacconi S, Puppo F, Duguez S, Rameau P, Caruso N, Delezoide AL, et al. 2015. Correlation between low FAT1 expression and early affected muscle in FSHD. Ann Neurol 78: 387–400. [DOI] [PubMed] [Google Scholar]
- Masny PS, Chan OY, de Greef JC, Bengtsson U, Ehrlich M, Tawil R, Lock LF, Hewitt JE, Stocksdale J, Martin JH, et al. 2010. Analysis of allele-specific RNA transcription in FSHD by RNA-DNA FISH in single myonuclei. Eur J Hum Genet 18: 448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JM, Konev AY, Biessmann H. 2003. Telomeric position effect in Drosophila melanogaster reflects a telomere length control mechanism. Genetica 117: 319–325. [DOI] [PubMed] [Google Scholar]
- Millay DP, O'Rourke JR, Sutherland LB, Bezprozvannaya S, Shelton JM, Bassel-Duby R, Olson EN. 2013. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 499: 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millay DP, Sutherland LB, Bassel-Duby R, Olson EN. 2014. Myomaker is essential for muscle regeneration. Genes Dev 28: 1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostacciuolo ML, Pastorello E, Vazza G, Miorin M, Angelini C, Tomelleri G, Galluzzi G, Trevisan CP. 2009. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clin Genet 75: 550–555. [DOI] [PubMed] [Google Scholar]
- Murase K, Ito H, Kanoh H, Sudo K, Iwamoto I, Morishita R, Soubeyran P, Seishima M, Nagata K. 2012. Cell biological characterization of a multidomain adaptor protein, ArgBP2, in epithelial NMuMG cells, and identification of a novel short isoform. Med Mol Morphol 45: 22–28. [DOI] [PubMed] [Google Scholar]
- O'Sullivan RJ, Karlseder J. 2010. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 11: 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Gilson E, Magdinier F. 2008. Telomeric position effect: from the yeast paradigm to human pathologies? Biochimie 90: 93–107. [DOI] [PubMed] [Google Scholar]
- Ottaviani A, Rival-Gervier S, Boussouar A, Foerster AM, Rondier D, Sacconi S, Desnuelle C, Gilson E, Magdinier F. 2009. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet 5: e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padberg GW, Frants RR, Brouwer OF, Wijmenga C, Bakker E, Sandkuijl LA. 1995. Facioscapulohumeral muscular dystrophy in the Dutch population. Muscle Nerve Suppl 2: S81–S84. [PubMed] [Google Scholar]
- Pastorello E, Cao M, Trevisan CP. 2012. Atypical onset in a series of 122 cases with FacioScapuloHumeral Muscular Dystrophy. Clin Neurol Neurosurg 114: 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram M, Sprung CN, Gao Q, Lo AW, Reynolds GE, Murnane JP. 2006. Telomere position effect and silencing of transgenes near telomeres in the mouse. Mol Cell Biol 26: 1865–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirozhkova I, Petrov A, Dmitriev P, Laoudj D, Lipinski M, Vassetzky Y. 2008. A functional role for 4qA/B in the structural rearrangement of the 4q35 region and in the regulation of FRG1 and ANT1 in facioscapulohumeral dystrophy. PLoS One 3: e3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Tsumagari K, Battaglia D, Ryba T, Hiratani I, Ehrlich M, Gilbert DM. 2011. DNA replication timing is maintained genome-wide in primary human myoblasts independent of D4Z4 contraction in FSH muscular dystrophy. PLoS One 6: e27413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppo F, Dionnet E, Gaillard MC, Gaildrat P, Castro C, Vovan C, Bertaux K, Bernard R, Attarian S, Goto K, et al. 2015. Identification of variants in the 4q35 gene FAT1 in patients with a facioscapulohumeral dystrophy-like phenotype. Hum Mutat 36: 443–453. [DOI] [PubMed] [Google Scholar]
- Rahimov F, King OD, Leung DG, Bibat GM, Emerson CP Jr, Kunkel LM, Wagner KR. 2012. Transcriptional profiling in facioscapulohumeral muscular dystrophy to identify candidate biomarkers. Proc Natl Acad Sci 109: 16234–16239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin JD, Ludlow AT, Batten K, Magdinier F, Stadler G, Wagner KR, Shay JW, Wright WE. 2014. Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev 28: 2464–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roignot J, Soubeyran P. 2009. ArgBP2 and the SoHo family of adapter proteins in oncogenic diseases. Cell Adh Migr 3: 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roignot J, Bonacci T, Ghigo E, Iovanna JL, Soubeyran P. 2014. Oligomerization and phosphorylation dependent regulation of ArgBP2 adaptive capabilities and associated functions. PLoS One 9: e87130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacconi S, Lemmers RJ, Balog J, van der Vliet PJ, Lahaut P, van Nieuwenhuizen MP, Straasheijm KR, Debipersad RD, Vos-Versteeg M, Salviati L, et al. 2013. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet 93: 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Gottschling DE, Zakian VA. 1994. Transcription of a yeast telomere alleviates telomere position effect without affecting chromosome stability. Proc Natl Acad Sci 91: 12061–12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger JM, Wang J, Gleason LM, Chowrashi P, Dube DK, Mittal B, Zhukareva V, Sanger JW. 2010. Arg/Abl-binding protein, a Z-body and Z-band protein, binds sarcomeric, costameric, and signaling molecules. Cytoskeleton 67: 808–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scionti I, Greco F, Ricci G, Govi M, Arashiro P, Vercelli L, Berardinelli A, Angelini C, Antonini G, Cao M, et al. 2012. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am J Hum Genet 90: 628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer C, Wolberger C, et al. 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci 97: 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, et al. 2010. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 6: e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler G, Rahimov F, King OD, Chen JC, Robin JD, Wagner KR, Shay JW, Emerson CP Jr, Wright WE. 2013. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol 20: 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassin A, Laoudj-Chenivesse D, Vanderplanck C, Barro M, Charron S, Ansseau E, Chen YW, Mercier J, Coppée F, Belayew A. 2013. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J Cell Mol Med 17: 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennen RI, Bua DJ, Wright WE, Chua KF. 2011. SIRT6 is required for maintenance of telomere position effect in human cells. Nat Commun 2: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumagari K, Qi L, Jackson K, Shao C, Lacey M, Sowden J, Tawil R, Vedanarayanan V, Ehrlich M. 2008. Epigenetics of a tandem DNA repeat: chromatin DNaseI sensitivity and opposite methylation changes in cancers. Nucleic Acids Res 36: 2196–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumagari K, Chang SC, Lacey M, Baribault C, Chittur SV, Sowden J, Tawil R, Crawford GE, Ehrlich M. 2011. Gene expression during normal and FSHD myogenesis. BMC Med Genomics 4: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkum NL, Lieberman-Aiden E, Williams L, Imakaev M, Gnirke A, Mirny LA, Dekker J, Lander ES. 2010. Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp 10.3791/1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geel M, Heather LJ, Lyle R, Hewitt JE, Frants RR, de Jong PJ. 1999. The FSHD region on human chromosome 4q35 contains potential coding regions among pseudogenes and a high density of repeat elements. Genomics 61: 55–65. [DOI] [PubMed] [Google Scholar]
- Vega-Palas MA, Venditti S, Di Mauro E. 1997. Telomeric transcriptional silencing in a natural context. Nat Genet 15: 232–233. [DOI] [PubMed] [Google Scholar]
- Wang B, Golemis EA, Kruh GD. 1997. ArgBP2, a multiple Src homology 3 domain-containing, Arg/Abl-interacting protein, is phosphorylated in v-Abl-transformed cells and localized in stress fibers and cardiocyte Z-disks. J Biol Chem 272: 17542–17550. [DOI] [PubMed] [Google Scholar]
- Weuts A, Voet T, Verbeeck J, Lambrechts N, Wirix E, Schoonjans L, Danloy S, Marynen P, Froyen G. 2012. Telomere length homeostasis and telomere position effect on a linear human artificial chromosome are dictated by the genetic background. Nucleic Acids Res 40: 11477–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winokur ST, Chen YW, Masny PS, Martin JH, Ehmsen JT, Tapscott SJ, van der Maarel SM, Hayashi Y, Flanigan KM. 2003. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet 12: 2895–2907. [DOI] [PubMed] [Google Scholar]
- Wright WE, Shay JW. 1992. Telomere positional effects and the regulation of cellular senescence. Trends Genet 8: 193–197. [DOI] [PubMed] [Google Scholar]
- Ye J, Renault VM, Jamet K, Gilson E. 2014. Transcriptional outcome of telomere signalling. Nat Rev Genet 15: 491–503. [DOI] [PubMed] [Google Scholar]
- Zhou BO, Wang SS, Zhang Y, Fu XH, Dang W, Lenzmeier BA, Zhou JQ. 2011. Histone H4 lysine 12 acetylation regulates telomeric heterochromatin plasticity in Saccharomyces cerevisiae. PLoS Genet 7: e1001272. [DOI] [PMC free article] [PubMed] [Google Scholar]