

Abstract
Transcription factors (TFs) bind to thousands of DNA sequences in mammalian genomes, but most of these binding events appear to have no direct effect on gene expression. It is unclear why only a subset of TF bound sites are actively involved in transcriptional regulation. Moreover, the key genomic features that accurately discriminate between active and inactive TF binding events remain ambiguous. Recent studies have identified promoter-distal RNA polymerase II (RNAP2) binding at enhancer elements, suggesting that these interactions may serve as a marker for active regulatory sequences. Despite these correlative analyses, a thorough functional validation of these genomic co-occupancies is still lacking. To characterize the gene regulatory activity of DNA sequences underlying promoter-distal TF binding events that co-occur with RNAP2 and TF sites devoid of RNAP2 occupancy using a functional reporter assay, we performed cis-regulatory element sequencing (CRE-seq). We tested more than 1000 promoter-distal CCAAT/enhancer-binding protein beta (CEBPB)-bound sites in HepG2 and K562 cells, and found that CEBPB-bound sites co-occurring with RNAP2 were more likely to exhibit enhancer activity. CEBPB-bound sites further maintained substantial cell-type specificity, indicating that local DNA sequence can accurately convey cell-type–specific regulatory information. By comparing our CRE-seq results to a comprehensive set of genome annotations, we identified a variety of genomic features that are strong predictors of regulatory element activity and cell-type–specific activity. Collectively, our functional assay results indicate that RNAP2 occupancy can be used as a key genomic marker that can distinguish active from inactive TF bound sites.
The spatial and temporal control of gene expression is critical for proper cellular development, differentiation, and maintenance of distinct cell types, all of which are key hallmarks of complex metazoan systems (Tjian and Maniatis 1994; Levine 2010; Bulger and Groudine 2011; Sakabe et al. 2012). This concerted transcriptional control is driven by DNA-binding transcription factor (TF) proteins that exert their regulatory effects by binding to thousands of discrete, largely nonprotein coding, sites throughout mammalian genomes (Tjian and Maniatis 1994; Levine 2010; Bulger and Groudine 2011; Sakabe et al. 2012). The association of TFs with DNA sequences leads to the recruitment of transcriptional cofactors, including chromatin modifying enzymes and the basal transcriptional machinery, through both DNA–protein and protein–protein interactions (Bulger and Groudine 2011; Sakabe et al. 2012). The formation of these macromolecular complexes leads to the initiation or augmentation of transcription at target gene promoters, some of which are more than a million base pairs away from their corresponding regulatory elements (Lettice et al. 2003). The faithful transfer of this molecular signal is believed to occur through direct enhancer–promoter looping interactions in three-dimensional space and/or via RNAP2 tracking along the DNA sequence from enhancer to target gene promoter (Bulger and Groudine 2011).
Although chromatin immunoprecipitation followed by next-generation DNA sequencing (ChIP-seq) studies support the association of TFs with thousands of sites throughout the genome (The ENCODE Project Consortium 2007, 2012; Mouse ENCODE Consortium et al. 2012), many of these interactions are thought to have a minimal impact on gene regulation. For instance, inducible transcription factors have been shown to bind to at least ten times more genomic loci than the number of affected target genes (Cheng et al. 2009; Reddy et al. 2009; Gertz et al. 2012). Technical artifacts, regulatory element redundancy, transcriptional epistasis, and/or RNA stability may explain part of this discrepancy, but collectively these data suggest that a considerable portion of sites bound by TFs do not affect transcription. Indeed, these seemingly passive interactions may represent stochastic associations of TF proteins with DNA segments, or they may play a functional role to control local TF concentrations and/or TF localization in the nuclear milieu. As a result, the predictive regulatory activity of a discrete TF binding event is currently limited without secondary information.
Several genomic features have been proposed as potential predictors of active enhancer elements, including the presence of histone acetyltransferase EP300 (Visel et al. 2009), local chromatin modifications (Heintzman et al. 2009; Ernst and Kellis 2010), promoter-distal RNAP2 occupancy (De Santa et al. 2010), enhancer RNA (eRNA) production (Hah et al. 2013; Li et al. 2013), and long-range looping interactions (Fullwood et al. 2009). Although these correlative genomic features allude to transcriptional roles, large-scale functional assays that evaluate the gene regulatory activities of DNA sequences enriched for these distinct genomic features are clearly warranted to determine and to validate their predictive value for the demarcation of enhancer function. Several recent analyses that use next-generation DNA sequencing combined with massively parallel reporter assays have addressed some of these outstanding challenges (Patwardhan et al. 2012; Kheradpour et al. 2013; White et al. 2013; Kwasnieski et al. 2014). These methods allow for the functional assessment of thousands of DNA sequences in parallel through distinct RNA transcript barcoding strategies. Massively parallel reporter analyses have found that chromatin state predictions, based on the integration of many ChIP-seq data sets, can predict functional regulatory activity (Kheradpour et al. 2013; Kwasnieski et al. 2014). However, functional discrepancies in regulatory activity from distinct chromatin states have been documented (Kwasnieski et al. 2014), suggesting that our understanding of histone modifications is not exhaustive and further stressing the need to identify more reliable predictors of active regulatory sequences. Rather than relying on chromatin state predictions, which require many distinct ChIP-seq experiments to explore a cell type, here we determine the predictive ability of RNAP2 promoter-distal binding, assayed simply by ChIP-seq with an antibody against RNAP2, to discriminate active from inactive TF-bound sites. To perform this analysis in a controlled manner, we took a different experimental approach by comparing loci bound by CCAAT/enhancer-binding protein beta (CEBPB) that overlap or do not overlap with promoter-distal RNAP2. CEBPB serves as an ideal model because high-quality ChIP-seq data is publicly available (The ENCODE Project Consortium 2007, 2012), and CEBPB exhibits extensive cell-type–specific genome binding, allowing for the additional characterization of cell-type specificity on regulatory activities.
Although chromatin states and accessibility can vary dramatically between cell types (Ernst et al. 2011; Kasowski et al. 2013; McVicker et al. 2013), it is unclear if an enhancer's cell-type–specific activity is intrinsic to the underlying local DNA sequence, or if regulatory element activity is primarily governed by more global sequence-defined chromatin dynamics. Massively parallel reporter assays have also assessed this to varying degrees. Mutation of TF binding motifs at tested DNA sequences overlapping chromatin state predictions ablated regulatory activity (Kheradpour et al. 2013), while a second analysis documented minimal activity of H1-ESC-specific chromatin states in K562 cells (Kwasnieski et al. 2014). These data collectively support a local DNA-driven mechanism in the coordination of cell-type–specific enhancer activity, wherein local sequence information near TF binding events largely contributes to proper regulatory activity, as opposed to global sequence features that influence chromatin structure. To expand on these observations, we address this question more thoroughly through direct cross comparisons of the regulatory activities of HepG2- and K562-specific CEBPB-bound sites in opposing cell types, including an assessment of CEBPB sites shared across both cell types. In line with previous observations (Kasowski et al. 2013; Kilpinen et al. 2013; McVicker et al. 2013), our results support the notion that global sequence features that designate chromatin state events play a secondary role to determining enhancer activity, with local DNA sequence alone able to accurately recapitulate cell-type–specific regulation. Our data illustrate that CEBPB binding sites coincident with promoter-distal RNAP2 binding display substantially stronger enhancer activity than CEBPB sites devoid of RNAP2. We also perform quantitative analyses that show a significant enrichment for the presence of eRNAs at active regulatory elements. This study is the first large-scale functional test of promoter-distal RNAP2 binding events, underscoring this genomic feature as an accurate discriminator of active regulatory sequences. We further demonstrate the importance of local DNA sequence for directing proper cell-type specificity and identify additional genomic features enriched at active CEBPB binding sites to better understand cis-regulatory architecture.
Results
Identification of RNAP2 occupancy at promoter-distal CEBPB-bound sites in HepG2 and K562 cells
To evaluate the function of TF binding sites coincident with RNAP2, we focused on CEBPB TF sites identified through ChIP-seq in HepG2 cells, a hepatocellular carcinoma cell line, and K562 cells, a chronic myelogenous leukemia cell line (The ENCODE Project Consortium 2007, 2012). CEBPB is a bZIP domain TF that can form homodimers or heterodimers with other members of the CCAAT/enhancer-binding protein family (Ramji and Foka 2002). Publicly available ChIP-seq data (The ENCODE Project Consortium 2007, 2012) indicate that CEBPB reproducibly binds (across two biological replicates) to 18,125 genomic loci in HepG2 and 22,240 genomic loci in K562 cells (Supplemental Fig. 1A). Most bound sites contain a CEBPB binding motif: 78% of CEBPB-bound sites in HepG2 and 68% of CEBPB-bound sites in K562 (Supplemental Fig. 1B). Of 34,810 CEBPB-bound sites present in either HepG2 or K562, only 5555 (15.9%) loci are bound by CEBPB in both cell types (Supplemental Fig. 1A). As a result of the limited fraction of shared sites, CEBPB binding also serves as a reasonable model for evaluating cell-type–specific enhancer activity.
We further assessed the co-occurrence of RNAP2 at extragenic CEBPB sites in both HepG2 and K562 cells. Only reproducible RNAP2 binding events identified across two biological replicate ChIP-seq experiments were used. To ensure RNAP2 binding was promoter-distal, we assessed histone modifications at proximal and distal RNAP2 bound sites to determine the distance from transcription start sites (TSSs) at which RNAP2 sites switch from promoter-like to enhancer-like elements. Acetylation of lysine 27 on histone H3 (H3K27ac) was high at all RNAP2-bound sites (data not shown); however, when using a distance cutoff of 5 kb from TSSs (Fig. 1A,B), TSS proximal RNAP2 bound sites exhibit a high ratio of histone H3 lysine 4 trimethylation to histone H3 lysine 4 monomethylation (H3K4me3:H3K4me1), indicative of promoters, whereas TSS distal sites had a low ratio of H3K4me3:H3K4me1, an established hallmark of enhancer sequences (Heintzman et al. 2007, 2009). Given this transition, we used RNAP2 sites at least 5 kb away from the nearest TSS for identifying co-occupancy with CEBPB binding at candidate enhancers. In HepG2 cells, 646 (3.6% of total sites) CEBPB-bound sites overlapped with distal RNAP2 binding events, whereas in K562 cells, 1479 (6.7% of total sites) of CEBPB-bound loci overlap with promoter-distal RNAP2. CEBPB binding events common to both HepG2 and K562 cells exhibit comparable co-occurrence with RNAP2 (241 sites; 4.3% of shared sites).
Figure 1.

CRE-seq of CEBPB binding sites. Analysis of H3K4me1 (in purple) and H3K4me3 (in red) enrichment at CEBPB binding sites <5 kb from promoters (A) and >5 kb from promoters (B). Average ChIP-seq signal enrichment is displayed on the y-axis, whereas the distance from transcription start site (TSS) is displayed on the x-axis. At >5 kb from promoters, a substantial enrichment in H3K4me1 is observed. (C) Schematic of CRE-seq experimental platform. Oligonucleotides are generated harboring 120 bp of sequence centered on CEBPB binding site summits, unique 9-bp barcodes, and restriction enzyme sites for cloning. Oligonucleotides are cloned into a plasmid backbone, upstream of the backbone poly-A sequence, while a minimal promoter and luciferase coding sequence is subsequently cloned in between binding sites and barcodes within oligonucleotides, generating a pool of approximately 12,000 unique plasmids. HepG2 and K562 cells are transfected, in replicates, into HepG2 and K562 cells. DNA and RNA (cDNA) is extracted, barcodes from plasmids (DNA) and transcribed RNA molecules are amplified and prepared for next-generation sequencing. Regulatory activity for each barcode is determined by normalizing the RNA-derived barcode counts with counts from DNA (plasmids). The median activity across all barcodes for an individual element was used to determine activity in each replicate experiment. The mean activity across replicate experiments for each element was used to calculate the final activity. (D,E) Luciferase assay results in HepG2 (D) and K562 (E) cells are also displayed. The log2-transformed activity using luciferase assay (y-axis) and CRE-seq assay (x-axis) is shown (where fc denotes fold change). The Rank correlation for each data set is shown at the bottom right of each graph.
CRE-seq of CEBPB-bound sites in HepG2 and K562 cells
We used CRE-seq, a method that measures the gene regulatory effects of thousands of DNA sequences in a parallel manner, to evaluate the functional activity of CEBPB binding events (Kwasnieski et al. 2012). We designed array-synthesized oligos spanning 120 bp of promoter-distal CEBPB-bound sites centered on the ChIP-seq signal summit. These CEBPB sites were randomly selected to equally represent HepG2-specific, K562-specific, and common sites that were concurrent with or devoid of RNAP2 binding. We also designed control DNA sequences by scrambling the selected 120-bp genomic regions while still preserving dinucleotide frequencies. The scrambled sequences control for random effects on regulatory activity due to differences in dinucleotide frequencies and provide a background distribution for the analysis of significant activity (White et al. 2013; Kwasnieski et al. 2014). Both genomic and scrambled DNA sequences were tailed with 9-bp DNA barcodes and restriction enzyme sites necessary for CRE-seq plasmid construction, resulting in 186-mer oligos (Methods). Each DNA element was engineered with four unique barcodes, resulting in four distinct oligonucleotides per DNA sequence tested, providing a metric for reproducibility and robust measurement of activity. Collectively, more than 12,000 unique 186-mer oligos were synthesized (see Supplemental Table 1 for barcode and binding site sequence information). Our cloning strategy situated CEBPB binding sites upstream of a minP minimal promoter driving a luciferase gene that contained barcodes in the 3′ untranslated region, specific for each binding site tested (Fig. 1C).
A schematic outline of our experimental design is illustrated in Figure 1C. CRE-seq plasmids were transfected into HepG2 and K562 cells in replicate experiments and harvested 24 h after transfection. Extracted cellular RNA and DNA underwent a multistep library construction before next-generation DNA sequencing of barcodes from RNA molecules and plasmids respectively (see Methods and Supplemental Data for RNA and DNA barcode counts). The plasmid DNA barcodes provide an internal control for plasmid abundance that can confound expression levels. DNA sequences represented by at least two independently sequenced plasmid barcodes in each replicate experiment and DNA barcodes that reached a conservative cutoff of 200 read counts were utilized. This read cutoff controlled for technical artifacts arising from low read counts that negatively impact assay reproducibility (Methods; Supplemental Fig. 2). Regulatory activity for each barcode was calculated by normalizing the RNA barcode sequence count with the plasmid DNA barcode abundance. The composite regulatory activity of each element was calculated by taking the median regulatory activity across all barcodes for the element, as has been previously described (Kwasnieski et al. 2014). We assessed regulatory activity of CEBPB-bound sites and compared this transcriptional potential with associated scrambled control sequences. We identified active binding sites as the subset of sequences that activated gene expression higher than the 95th percentile of matched scrambled sequences (Kwasnieski et al. 2014). A complete list of all site categories and the number of sites above this cutoff is shown in Supplemental Table 2.
To evaluate assay reproducibility, we quantified the concordance of read tag counts for DNA and RNA measurements between replicates (Supplemental Figs. 2, 3). Our assay retained high reproducibility (R2 > 0.93) between replicates for all DNA and RNA barcode counts in both HepG2 and K562 cells (Supplemental Fig. 3). As a secondary validation of our regulatory measurements, we tested a subset of enhancer sequences spanning a wide range of activities in HepG2 and K562 cells through traditional, individual clone-based luciferase reporter assays (Fig. 1D,E). Despite distinct assay platforms, these results are in good agreement with our CRE-seq data (HepG2 r = 0.66 and K562 r = 0.6). We also divided our test binding sites into groups based on their CRE-seq activity into low, middle, and high activity sequences (six per group). For this analysis, we ranked our binding sites based on CRE-seq regulatory activity (independently for HepG2 and K562 cells) and split these ranked sites evenly into each activity category. Using this binning approach, we identified significant differences in luciferase activity between distinct groups of sites (HepG2 high versus low activity P < 3.5 × 10−4; HepG2 middle versus low activity P < 0.02; K562 high versus low activity P < 0.03) (Supplemental Fig. 4). Along with our correlation data, these additional analyses further support our CRE-seq activity observations (Supplemental Fig. 4).
RNAP2 co-occupied sites display stronger enhancer activity
We examined the regulatory activity of groups of CEBPB-bound sites relative to a set of associated scrambled control sequences within each cell line (Fig. 2A,B). For this analysis, we utilized sites found exclusively in HepG2 or K562 cells. Our data supported an enrichment of CEBPB TF binding site enhancer activities above scrambled control sequences. At the 95th percentile of scrambled sequence activity, 21.2% (178 of 841 sites) of HepG2-specific CEBPB binding events and 11.1% (61 of 549 sites) of K562-specific CEBPB sites displayed stronger regulatory activity in HepG2 and K562 cells, respectively.
Figure 2.
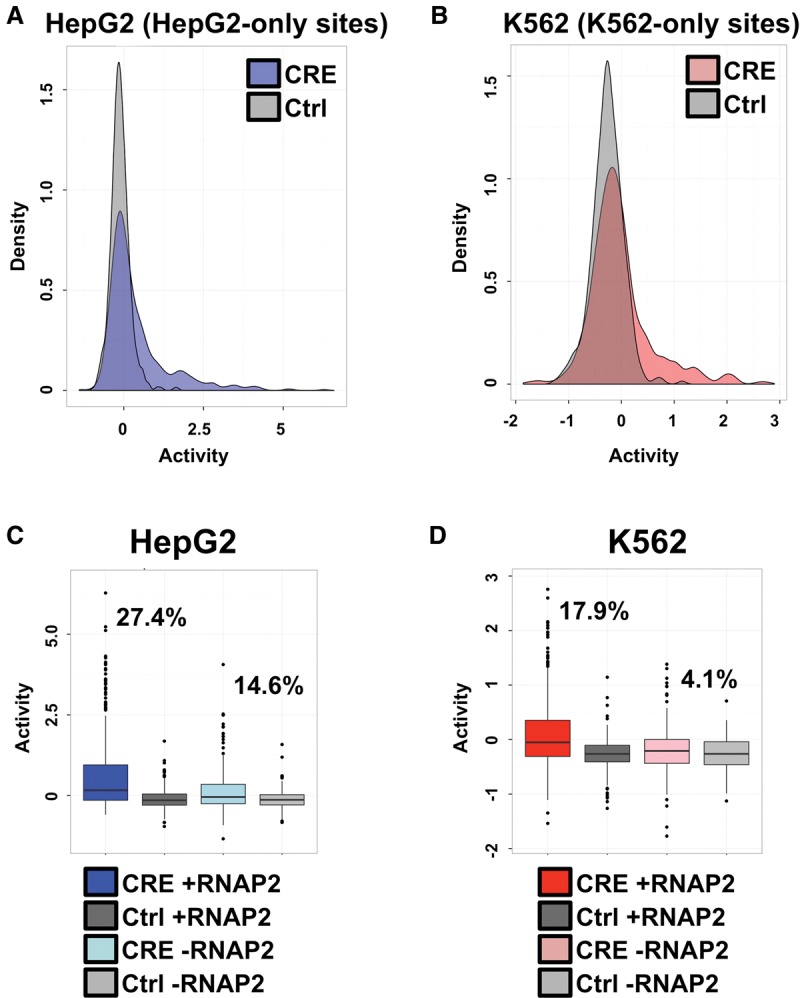
RNAP2-associated sites exhibit stronger activity. (A) Density plots of HepG2-specific CEBPB binding sequence activity in HepG2 cells. The x-axis displays the distribution of log2-transformed regulatory activity (RNA/DNA barcode counts), while the y-axis displays the density across distinct regulatory activities. HepG2 binding site CRE-seq activity distributions are shown in blue and compared with associated scrambled control sequence activities (shown in gray). (B) Density plots of K562-specific CEBPB binding sequence activity in K562 cells. The x-axis displays the distribution of log2-transformed regulatory activity (RNA/DNA barcode counts), while the y-axis displays the density across distinct regulatory activities. K562 binding site CRE-seq activity distributions are shown in red and compared with associated scrambled control sequence activities (shown in gray). (C) HepG2 CRE-seq activity for distinct classes of binding sites is shown as box plots. The log2-transformed CRE-seq activity (RNA/DNA barcode counts) is displayed on the y-axis. Binding sites that are coincident with promoter-distal RNAP2 are shown in dark blue, and the scrambled control sequences for these RNAP2-associated binding sites are displayed in dark gray. HepG2 binding sites devoid of promoter-distal RNAP2 binding are displayed in light blue, and the scrambled control sequences for these RNAP2-devoid sites are shown in light gray. The percentage of RNAP2-associated and RNAP2-devoid HepG2 sites above the 95th percentile of associated scrambled control sites are displayed on the plot. (D) K562 CRE-seq activity for distinct classes of binding sites is shown as box plots. The log2-transformed CRE-seq activity (RNA/DNA barcode counts) is displayed on the y-axis. Binding sites that are coincident with promoter-distal RNAP2 are shown in dark red, and the scrambled control sequences for these RNAP2-associated binding sites are displayed in dark gray. K562 binding sites devoid of promoter-distal RNAP2 binding are displayed in light red, and the scrambled control sequences for these RNAP2-devoid sites are shown in light gray. The percentage of RNAP2-associated and RNAP2-devoid K562 sites above the 95th percentile of associated scrambled control sites are displayed on the plot.
We further examined the relative contribution of RNAP2-association in regulatory activity at cell-type–specific CEBPB binding sites. For this analysis, we split HepG2- and K562-specific sites into groups of binding events that co-occurred or that were depleted for RNAP2 binding within each cell type and compared regulatory activities of each set of sites with their associated set of scrambled control sequences. We observed substantially stronger expression from CEBPB sites coincident with RNAP2 across both cell types, supporting a role for RNAP2 as a key discriminator of active versus inactive CEBPB binding events (Fig. 2C,D; Supplemental Table 2). In HepG2 cells, 27.4% (118 of 431 sites) of RNAP2 binding events maintained expression above the 95th percentile of matched control sequences, compared to only 14.6% (60 of 410 sites) for non-RNAP2-associated CEBPB sites (Supplemental Table 2). In K562 cells, 17.9% (50 of 280 sites) of RNAP2-enriched CEBPB binding sites held activity above the 95th percentile of scrambled sequences compared with only 4.1% (11 of 269 sites) of RNAP2-devoid sites (Supplemental Table 2).
We also assessed a role for RNAP2 at shared CEBPB binding sites (CEBPB binding sites identified in both HepG2 and K562 cells) by subdividing these sites shared based on RNAP2 occupancy (Supplemental Table 2; Supplemental Fig. 5). Shared sites that were coincident with RNAP2 also displayed stronger activity compared with non-RNAP2 sites; in HepG2, 34.3% (60 of 175 sites) of shared RNAP2 sites were active compared to 19.4% (32 of 165 sites) of shared non-RNAP2 sites, whereas in K562 cells, 26.3% (46 of 175 sites) of shared RNAP2 sites had activity compared to 10.9% (18 of 165 sites) of shared non-RNAP2 sites (Supplemental Fig. 5).
Consistent with the functional role of RNAP2, we also found that RNAP2 ChIP-seq signal was significantly higher (P = 3.211 × 10−10 in HepG2; P = 4.059 × 10−8 in K562) at sites above the 95th percentile of scrambled sequences in both cell lines (Supplemental Fig. 6). Because promoter-distal RNAP2 binding has the potential to produce eRNAs, we compared our data with GRO-seq data generated in K562 cells (Core et al. 2014) to determine if the expression of eRNAs was predictive of enhancer activity in our data. We calculated GRO-seq read counts near active and inactive CEBPB-bound sites based on the CRE-seq data and observed a highly significant enrichment in GRO-seq signal at active sites compared to inactive binding events (P = 5.65 × 10−5) (Supplemental Fig. 7). The strong enrichment of eRNAs and RNAP2 binding observed at active sites confirms a link between RNAP2 binding and eRNA production. Importantly, these data provide a large-scale functional analysis of endogenous eRNA activity as a predictor of regulatory activity within a well-controlled experimental system and indicate that both eRNA levels and RNAP2 binding are accurate predictors of the regulatory activity of local DNA sequence.
DNA-encoded enhancer activity is cell-type–specific
The use of a common plasmid pool containing HepG2-specific, K562-specific, and shared CEBPB binding events allowed for the direct assessment of cell-type–specific regulatory information. For this analysis, we determined enhancer activities of cell-type–specific CEBPB-bound sites (HepG2-specific and K562-specific sites only) co-occurring with RNAP2 in the opposing cell line (HepG2-specific overlapping RNAP2 sites in K562 cells and K562-specific overlapping RNAP2 sites in HepG2 cells) and compared those results with the fraction of active RNAP2-associated CEBPB sites from sites identified within the same cell type (HepG2-specific overlapping RNAP2 sites in HepG2 cells and K562-specific overlapping RNAP2 sites in K562 cells). The use of stringent criteria for identifying CEBPB binding events (sites that were reproducibly identified in two biological replicates) may lead to the inclusion of CEBPB binding events that are inappropriately categorized as cell-type–specific. To control for this, we therefore also compared the activities of cell-type–specific sites with the activity of RNAP2-associated CEBPB sites shared between HepG2 and K562 cells.
We observed a pronounced cell-type–specific effect for CEBPB binding events (Fig. 3A,B). In HepG2 cells, 34.3% (60 of 175 sites) of shared CEBPB binding events (shared sites co-occurring with RNAP2) and 27.4% (118 of 431 sites) of HepG2-specific sites (HepG2-specific site co-occurring with RNAP2) maintained expression above the 95th percentile of matched control sequences in HepG2 cells, compared to only 10% (28 of 280 sites) for K562-specific sites (K562-specific site co-occurring with RNAP2). K562 displayed less extensive cell-type specificity; 26.3% (46 of 175 sites) of shared CEBPB binding and 17.9% (50 of 280 sites) of K562-specific binding events maintained expression above the 95th percentile of matched control sequences compared to only 10.2% (44 of 431 sites) for HepG2-specific sites in K562 cells. To characterize these observations more thoroughly, we measured Spearman rank correlations in activity between cell types for shared and cell-type–specific sites (Fig. 3C). In line with our previous observations of cell-type specificity, shared sites exhibit correlated activity, whereas cell-type–specific sites were not as strongly correlated or negatively correlated between cell types. Collectively, these data support a strong role for local DNA sequence information in dictating enhancer activity.
Figure 3.
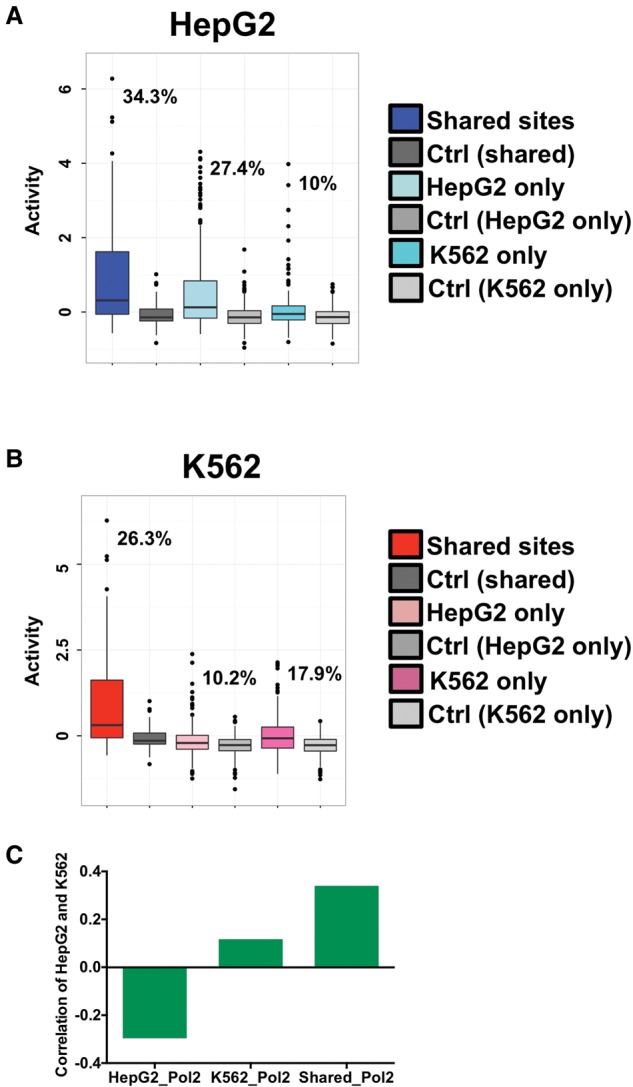
Cell-type specificity analysis of RNAP2-associated sites. (A) HepG2 CRE-seq activity for distinct classes of RNAP2-associated elements is shown as box plots. The log2-transformed CRE-seq activity (RNA/DNA barcode counts) is displayed on the y-axis. HepG2 CRE-seq activities are given for binding sites that are shared between HepG2 and K562 cells (Shared sites), as well as cell-type–specific sites that are found in only HepG2 cells (HepG2 only) or only in K562 cells (K562 only). Associated HepG2 CRE-seq activities of scrambled control sequences for each of the three classes of sites are also plotted. A key is given on the right of the graph. The percentage of elements from each of the three classes of sites above the 95th percentile of associated scrambled control sites are displayed on the plot. (B) K562 CRE-seq activity for distinct classes of RNAP2-associated elements is shown as box plots. The log2-transformed CRE-seq activity (RNA/DNA barcode counts) is displayed on the y-axis. K562 CRE-seq activities are given for binding sites that are shared between HepG2 and K562 cells (Shared sites), as well as cell-type–specific sites that are found in only HepG2 cells (HepG2 only) or only in K562 cells (K562 only). Associated K562 CRE-seq activities of scrambled control sequences for each of the three classes of sites are also plotted. A key is given on the right of the graph. The percentage of elements from each of the three classes of sites above the 95th percentile of associated scrambled control sites are displayed on the plot. (C) Spearman rank correlations between HepG2 and K562 CRE-seq activities for RNAP2-associated shared sites (Shared_Pol2), HepG2-specific sites (HepG2_Pol2), and K562-specific sites (K562_Pol2) are given. Shared sites exhibit positive correlation between CRE-seq activities in HepG2 and K562 cells, whereas cell-type–specific sites are not as strongly correlated (K562-specific sites) or negatively correlated (HepG2-specific sites).
DNA sequence and functional genomic features distinguish active from inactive binding events
We interrogated surrounding DNA sequence information at our tested CEBPB binding sites to determine DNA sequence features that may predict enhancer activity. For this analysis, we compared active versus inactive CEBPB sites (using the 95th percentile cutoff), regardless of CEBPB binding site category. We performed discriminative motif finding (Bailey 2011) to identify motifs that are more common in active sites compared with nonactive sites. We identified the HNF4A TF motif (P < 1 × 10−10, 2.5-fold enriched in active versus inactive sites) and the AP1 family motif (P < 2 × 10−16, 3.4-fold enriched in active versus inactive sites), which includes JUND and FOSL2 TFs, as enriched at active CEBPB binding sites in HepG2 cells (Fig. 4A,B). In K562 cells, an ETS motif (P < 1 × 10−6, 3.4-fold enriched in active versus inactive sites) exhibited significant enrichment for activity (Fig. 4C). Active sites were depleted for Alu repetitive elements (P = 0.00321), consistent with a recent finding that most human Alus lack epigenetic features of active enhancers (Su et al. 2014). We also detected significant differences in sequence conservation between active and inactive sequences (GERP scores for active and inactive elements P = 4.069 × 10−5).
Figure 4.
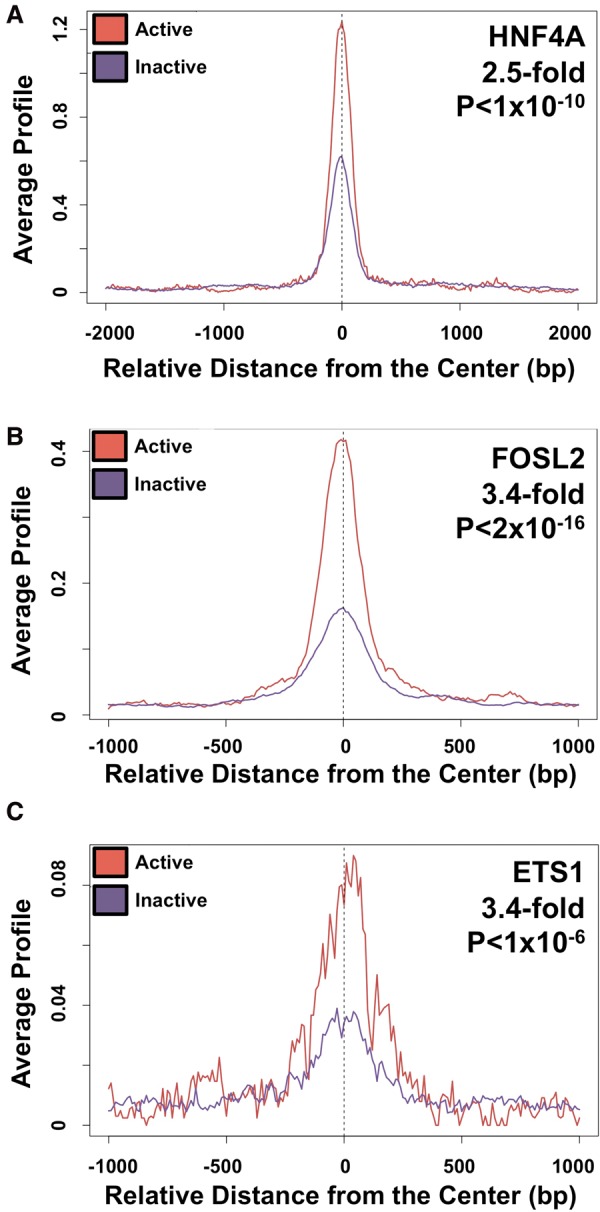
Motif and functional genomic analyses of CEBPB binding sites. (A) Motif analysis depicts enrichment for the HNF4A motif at active (in red) and inactive (in purple) CEBPB-bound sites in HepG2 cells. Motif fold enrichments and P-values are given in the top right. The location and orientation from the center of each element is shown on the x-axis. (B) Motif analysis depicts enrichment for the FOSL2 motif at active (in red) and inactive (in purple) CEBPB-bound sites in HepG2 cells. (C) Motif analysis depicts enrichment for the ETS1 motif at active (in red) and inactive (in purple) CEBPB-bound sites in K562 cells.
We next capitalized on publicly available genome-wide annotations from the ENCODE Project Consortium (2007, 2012), including ChIP-seq, DNase I hypersensitivity, and chromatin state data sets, to identify additional functional genomic features enriched at active CEBPB binding sites. This data set includes more than 1000 independent annotations, allowing for a thorough analysis of the predictive value of a wide array of genomic markers. This large data set serves as a complement to our analyses of RNAP2 binding and cell-type specificity (see cell-type–specific annotations data below), and was further agnostic to RNAP2 status and the 95th percentile activity cutoff, as we solely utilized raw activity counts (RNA/DNA barcode counts) for these correlative evaluations. We performed Wilcoxon tests to determine whether particular annotations were associated with increased (or decreased) expression activity in HepG2 or K562 cells. DNase I hypersensitivity exhibited the strongest association with increased expression in each cell line (HepG2 P = 3.07 × 10−13; K562 P = 7.93 × 10−23). We also identified various ENCODE TF ChIP-seq data sets that were highly associated with increased CEBPB-bound site expression (Supplemental Table 3). Supporting the sequence motif analyses above, JUND (P = 3.87 × 10−11), FOSL2 (P = 4.64 × 10−10), and HNF4A (P = 1.69 × 10−6) HepG2 binding events were identified in HepG2 cells, whereas ELF1 (P = 2.79 × 10−15) and ETS1 (P = 3.17 × 10−5) K562 sites were identified in K562 cells, further indicating that particular TF binding motifs may explain activity for a subset of sites (Supplemental Table 3). In line with a role for promoter-distal RNAP2 as a marker of active enhancers, RNAP2 binding was highly significant in HepG2 (RNAP2 HepG2 binding P = 8.16 × 10−11) and K562 (RNAP2 K562 binding P = 1.74 × 10−18) cells. A complete list of all functional genomic annotations and associated P-values for both cell lines is given in Supplemental Table 3. Additionally, we found subtly stronger activity for more distal regulatory elements (linear regression of log expression activity versus log of absolute distance to nearest TSS; P = 0.012, R2 = 0.011). These data show there is no detectable influence of promoters, suggesting that our experimental design which focused on distal regulatory elements worked effectively. Overall, these data identify distinct sequence and functional hallmarks, in addition to the presence of RNAP2, at active CEBPB binding sites.
We finally utilized this large annotation repository to ascertain the predictive value of distinct genomic features on cell-type–specific activity of CEBPB-bound sites. For this detailed analysis, we calculated the significance of enrichment of each annotation separately for HepG2 and K562 activity and ranked the resulting ratios for sites that preferentially predicted HepG2 activity (HepG2 P-value divided by K562 P-value) or K562 activity (K562 P-value divided by HepG2 P-value). We identified HepG2 DNase I hypersensitivity (P-value ratio = 3.62 × 10−13) as well as HepG2 ATF3 (P-value ratio = 4.23 × 10−11), JUND (P-value ratio = 1.2 × 10−10), RNAP2 (P-value ratio = 1.62 × 10−9), and FOSL2 (P-value ratio = 3.1 × 10−9) binding as the strongest predictors of HepG2-specific activity, whereas K562 DNase I hypersensitivity (P-value ratio = 3.35 × 10−21) as well as K562 RNAP2 (P-value ratio = 4.24 × 10−17), JUND (P-value ratio = 1.85 × 10−14), TAF1 (P-value ratio = 7.07 × 10−12), and EGR1 (P-value ratio = 7.56 × 10−12) binding as some of the best indicators of K562-specific activity (Supplemental Table 4). A complete list of our analysis is given in Supplemental Table 4. These results suggest that regulatory activity is encoded by the presence of other transcription factors and functional genomic features.
Discussion
Accurate transcriptional regulation is a cornerstone of complex metazoan systems, but our understanding of the cis-regulatory logic that governs the spatial and temporal regulation of genes is still rudimentary. Here, we systematically assessed the regulatory activity of CEBPB binding sites through CRE-seq functional assays to (1) characterize extragenic RNAP2 occupancy at TF binding sites as a viable marker of active enhancers; (2) evaluate the role of DNA sequence in defining cell-type specificity; and (3) leverage publicly available genome data to identify both DNA sequence and functional features that predict cis-regulatory activity. By choosing a particular TF's binding sites to pursue, we were able to perform these studies in a well-controlled manner. CEBPB represented an ideal model because of the high quality data available and the cell-type–specific nature of CEBPB genomic binding.
Our data illustrate that promoter-distal TF binding events that co-occur with RNAP2 are more likely to exhibit enhancer-like functional properties. Importantly, these results highlight the utility of promoter-distal RNAP2 as an important marker for active TF binding events, supporting a direct functional role for RNAP2 at enhancer elements that have been suggested by other groups (Hah et al. 2013). This is a promising finding because it is more challenging to define chromatin states than to identify promoter-distal RNAP2 bound sites, which can even be analyzed in a small amount (∼25 mg) of frozen tissue (Savic et al. 2013). Moreover, functional results from chromatin state predictions have previously led to conflicting results (Kwasnieski et al. 2014), supporting the utility of promoter-distal RNAP2 as a more straightforward marker of active TF binding events. However, we do note that additional features are likely to be important for regulatory activity as a subset of CEBPB sites devoid of RNAP2 exhibit activity, albeit not at the same level as sites co-occupied by RNAP2.
Our results provide the first large-scale functional validation of RNAP2-associated regulatory sequences. An interesting avenue for follow-up of these results should center on the mechanism(s) leading to gene regulatory activity. Specifically, enhancers may be recruiting RNAP2 and/or depositing this machinery at distal promoter sites. Although not mutually exclusive, another possibility is that distal sites loop to promoters to activate the transcriptional machinery that is already present at the promoter site. However, current ChIP-based approaches cannot distinguish between these two possibilities. For instance, RNAP2 ChIP-seq signal at cis-regulatory sequences may indirectly reflect increased promoter–enhancer interactions via chromatin crosslinking, indicative of highly active regulatory sequences. Our data further supports RNAP2 co-occupancy as being associated with eRNA production, a feature that has been observed at candidate enhancer elements and is also correlated with stronger three-dimensional enhancer–promoter looping interactions (Hah et al. 2013; Lam et al. 2013; Li et al. 2013). This association with eRNA expression further suggests that RNAP2 does bind to the enhancer at some point, since transcription occurs with the enhancer as the template. When coupled, the statistically significant enrichment of RNAP2 (P = 3.211 × 10−10 in HepG2; P = 4.059 × 10−8 in K562) and concomitant enrichment for local eRNA transcripts at active CEBPB sites (P = 5.65 × 10−5) supports RNAP2 recruitment and subsequent eRNA production as accurate markers and important steps in enhancer activation as has been previously proposed (Hah et al. 2013; Lam et al. 2013; Li et al. 2013). Additional functional manipulations and approaches that use a combination of genome editing technologies (Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013) and genomic assays will be necessary for defining the distinct functional roles of RNAP2 and eRNAs at cis-regulatory sequences with gene regulatory activity.
We further assessed the ability of local DNA sequence information alone to recapitulate proper cell-type–specific gene regulation. Chromatin state plays a critical role in demarcating specific regulatory features across mammalian genomes, including active and repressed promoters, enhancers, and gene bodies (Ernst and Kellis 2010; Ernst et al. 2011). However, it has not been definitively established whether chromatin state is a cause or a consequence of genomic activity. For instance, does chromatin state at repressed loci restrict TF interactions or does the lack of TF binding lead to a repressed state? Taken from another perspective, does the local DNA sequence information that directs TF-DNA interactions or more global sequence context that defines the chromatin landscape dictate proper regulatory activities? Recent work highlights the role of DNA sequences in directing TF binding that guide subsequent chromatin modifications (Kasowski et al. 2013; Kilpinen et al. 2013; McVicker et al. 2013). Our data is in line with these observations and further shows that local DNA sequence information, removed from its native chromatin context, often accurately predicts cell-type specificity of enhancer activity. However, we do note differences in the degree of cell-type specificity between cell types, with HepG2 exhibiting stronger specificity, which could reflect either the underlying biology of these cell lines or differences stemming from technical artifacts. Included among the latter possibilities is the use of stringent criteria for identifying true CEBPB binding sites, potentially resulting in CEBPB binding events harboring weak signal in one cell line being inappropriately defined as cell-type–specific, which may explain some of these inconsistencies. Supporting this idea, comparisons of cell-type specificity using shared binding sites led to the strongest effects.
There are several reasons we believe that our results may underestimate regulatory activity. For instance, we cannot exclude potential artifacts in our data due to the use of a non-native promoter, including the possibility that this may explain the inactivity of a subset of CEBPB binding sites (Guilluy et al. 2011). However, previous analyses observed concordant results using distinct minimal promoters (Kwasnieski et al. 2014). It is also possible that the use of a 120-bp segment for enhancer activity limits activity for a subset of our tested sequences. Collectively, these limitations highlight several experimental parameters that may lead to false negative results in our data set, and therefore the percentage of active CEBPB sites in our assay should be viewed as a conservative estimate. Indeed, by analyzing thousands of sites in a controlled platform, we assessed trends that transcend our technical limitations and do not expect a perfect separation of active and inactive sites. Despite this experimental design, we identified a substantial enrichment (approximately twofold on average) in regulatory activity at RNAP2-associated sites compared to sites devoid of RNAP2. The percentage of active sites is likely an underestimate due to limitations discussed above; however, we are confident in the enrichment of active sites that overlap promoter-distal RNAP2. Overall, our results support the observation that a comparatively modest number of genes are altered in response to the activation and binding of TFs to thousands of genomic loci (Reddy et al. 2009; Gertz et al. 2012).
Using more than 1000 diverse genomic data sets, including both DNA sequence information and publicly available genomic assay results, we further identified genomic features that predict regulatory activity as well as cell-type–specific enhancer behavior. Notably, AP1 and ETS motifs were enriched at active CEBPB binding sites in HepG2 and K562 cells, respectively, and these results were validated by enrichments of AP1 (JUND and FOSL2) and ETS (ELF1 and ETS1) family TFs binding events in the same cell line. Intriguingly, the conclusions of an independent investigation also supported a role for AP1 motifs in regulatory activity (Kwasnieski et al. 2014). These secondary co-occurrences suggest that combinations of TFs may be key for distinguishing active from inactive TF-bound loci. Furthermore, the binding motif and associated TF events at active CEBPB binding sites is in line with the biological properties of K562 and HepG2 cells. As a hematopoietic-derived cell line, the identification of the ETS family motif and ETS family TF binding events (such as ELF1 and ETS1) at active CEBPB binding sites is supported by the underlying biology of K562 cells (Yang et al. 2011; Yu et al. 2011; Liu et al. 2015). In HepG2 cells, a hepatic cancer-derived cell line, the identification of AP1 TF family binding events (such as JUND and FOSL2) at active CEBPB sites is supported by their recognized functions in hepatocytes (Stepniak et al. 2006; Marden et al. 2008), whereas HNF4A binding events and motifs at active HepG2 CEBPB sites have a prominent function in liver tissue (DeLaForest et al. 2011). We also determined DNase I hypersensitivity as the most pronounced genomic feature that predicted cell-type–specific enhancer activity in addition to several distinct TFs that are implicated in cell-type–specific enhancer activity (see Supplemental Table 3). Additional analyses of these secondary genomic features may provide a better understanding of cis-regulatory logic and complex spatial and temporal gene regulation.
Our results demonstrate the utility of massively parallel reporter assays for the thorough characterization of cis-regulatory logic. These high-throughput platforms also provide a rational and straightforward methodology to functionally validate diverse genomic data sets such as ChIP-seq (Johnson et al. 2007). Collectively, our data strongly support promoter-distal RNAP2 as a powerful hallmark of active cis-regulatory sequences while further pointing to a key role for DNA-encoded sequence information in dictating cell-type specificity of gene regulation.
Methods
Selection of CEBPB binding sequences
Publicly available CEBPB and RNAP2 (8WG16) ChIP-seq data in HepG2 and K562 cells from the ENCODE Project Consortium was downloaded from the University of California Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway). Binding sites concordant between two biological replicates in each cell line were utilized. Comparisons of binding site coordinates between cell lines identified cell-type–specific binding events and CEBPB binding sites shared across both cell lines. We further identified promoter-distal RNAP2 occupancy at CEBPB sites using a 5-kb distance cutoff from transcription start sites identified by GENCODE (version 14). CEBPB binding sites were randomly selected and encompassed all combinations of CEBPB binding categories (cell-type–specific sites, shared sites, RNAP-associated sites, RNAP2-non-associated sites). In K562, all sites contained a CEBPB motif as identified by Patser (Hertz et al. 1990), while in HepG2, we chose a subset of sites that did not contain a significant CEBPB binding motif. For all binding sites, we utilized 120-bp of sequence for our assays centered on the CEBPB ChIP-seq binding site summits as determined by MACS (Zhang et al. 2008). Control DNA sequences were generated by scrambling CEBPB binding sites while preserving dinucleotide sequences.
CRE-seq plasmid construction
A pool of more than 12,000 array-synthesized 186-mer oligos were ordered from Agilent Technologies. Each unique oligo sequence contained the following structure: 5′ priming sequence (GTAGCG TCTGTCCGT)/EcoR1 restriction enzyme (RE) site (GAATTC)/120-bp CEBPB binding site or scrambled sequence/Spe1 and Sph1 RE site separated by a cytosine (ACTAGTCGCATGC)/9-bp barcode/Not1 RE site (GCGGCCGC)/3′ priming sequence (CAACT ACTACTACAG). Plasmid libraries were generated through a two-step cloning process (Kwasnieski et al. 2012, 2014; White et al. 2013). Briefly, array-generated oligos were amplified (four cycles), gel purified and digested with restriction enzymes (Not1 and EcoR1), PAGE purified, and cloned into a pRho-dsRED vector (Kwasnieski et al. 2012). Subsequently, the minimal promoter and luciferase coding sequence from pGL4.23 (Promega) was amplified with tailed primers containing Sph1 and Xba1 (compatible with Spe1) RE sites (Forward Sph1 tailed = TTTAGCATGCAGA GGGTATATAATGGAAGCTCGACTT; Reverse Xba1 tailed = TTTAT CTAGATTACACGGCGATCTTGCCGC) for cloning between test sequences and the 9-bp barcodes. This cloning strategy engineered plasmids containing a DNA element upstream of a minimal promoter driving the expression of a luciferase gene containing unique 3′ UTR barcodes. To ensure DNA sequence and barcode complexity in the final plasmid pool, cloning was performed on a large scale; approximately 70,000 bacterial colonies were pooled at each step, and we recovered all barcodes in our final plasmid pool. Sanger sequencing was performed to confirm barcode complexity prior to transfection and library preparation.
Cell culture and transfection
HepG2 and K562 cells were grown under standard growth conditions. HepG2 cells were grown in Dulbecco's Modified Eagle's Media (DMEM; Life Technologies) containing 10% fetal bovine serum (FBS; Sigma Aldrich) and 1% penicillin-streptomycin (Pen Strep; Life Technologies), while K562 cells were grown in RPMI Media 1640 (Life Technologies) containing 10% FBS (Sigma Aldrich) and 1% Pen Strep (Life Technologies). Cells were seeded in media devoid of antibiotics 24 h prior to transfection. Cells were transfected at ∼75% confluence using FuGENE reagent (Promega) for HepG2 cells or Lipofectamine LTX (Life Technologies) for K562 cells. Following a 24-h incubation, cells were lysed with RLT buffer (Qiagen) and stored at −80°C until further use.
CRE-seq sequencing library preparation
Next-generation sequencing library preparation was performed as previously described (Kwasnieski et al. 2012, 2014; White et al. 2013). Nucleic acids were purified from cell lysates using Norgen Total RNA Purification Kit (Norgen Biotek Corporation) and DNeasy Blood and Tissue Kit (Qiagen) for RNA and DNA, respectively. RNA was treated with DNase I (Turbo DNase; Life Technologies) and reverse transcription was performed using SuperScript II reverse transcriptase (Life Technologies). The 3′ UTR of the luciferase gene containing barcode sequences was amplified (Phusion high fidelity master mix; New England Biolabs) from cellular RNA (cDNA) or DNA (Forward Primer = GCGGCAAG ATCGCCGTGTAAGCATGC; Reverse Primer = CAGTCGAATTCTA GCCAGAAGTCAGATGCTCAAG) and then ligated to next-generation sequencing adaptors: Barcode 1 Paired End (PE) 1 = (Forward) ACTCTTTCCCTACACGACGCTCTTCCGATCTGCTCGATCATG, (Reverse)/5Phos/ATCGAGCAGATCGGAAGAGCGTCGTGTAGG GAAAGAGT; Barcode 2 (PE1) = (Forward) ACTCTTTCCCTACACG ACGCTCTTCCGATCTTAGACTATCATG, (Reverse)/5Phos/ATAGT CTAAGATCGGAAGAGCGTCGTGTAGGGAAAGAGT; Barcode 3 (PE1) = (Forward) ACTCTTTCCCTACACGACGCTCTTCCGATCTC GCTACCCTCATG (Reverse)/5Phos/AGGGTAGCGAGATCGGAA GAGCGTCGTGTAGGGAAAGAGT; Barcode 4 (PE1) = (Forward) ACTCTTTCCCTACACGACGCTCTTCCGATCTATAGTGGACACA TG, (Reverse)/5Phos/TGTCCACTATAGATCGGAAGAGCGTCGTG TAGGGAAAGAGT; Barcode 5 (PE1) = (Forward) ACTCTTTCCCT ACACGACGCTCTTCCGATCTGTCAGTAGGTACATG, (Reverse)/5Phos/TACCTACTGACAGATCGGAAGAGCGTCGTGTAGGGAA AGAGT; PE 2 barcode = (Forward)/5Phos/A*ATTAACCTCAAGAT CGGAAGAGCGGTTCAGCAGGAATGC, (Reverse) GCATTCCTG CTGAACCGCTCTTCCGATCTTGAGGTT. Barcodes sequences were amplified for eight cycles (Forward Primer = AATGATACGG CGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTT; Reverse Primer = CAAGCAGAAGACGGCATACGAGATCGGTCTC GGCATTCCTGCTGAACCGCTC) prior to next-generation sequencing. Each replicate experiment consisting of five libraries (HepG2 DNA, HepG2 RNA, K562 DNA, K562 RNA, and Plasmid DNA), and each replicate experiment was sequenced as a pool on one lane on an Illumina HiSeq 2000 sequencer.
Luciferase reporter assays
Individual plasmids (18 total) were purified from bacterial colonies. Empty pGL4.23 plasmids were used as a control. Cells were seeded in 96-well format, and each plasmid was transfected (see above) in triplicates (HepG2) or quadruplicates (K562). After a 24-h incubation, activity was determined using the Steady-Glo Luciferase Assay System (Promega) and analyzed on a luminometer. The average luminescence of each construct from replicate transfections was used to determine regulatory activity. The luminescence from each tested CRE-seq construct was further normalized to pGL4.23 empty vector activity.
Data analysis
We evaluated regulatory activity as previously described (Kwasnieski et al. 2012, 2014; White et al. 2013). RNA and DNA read counts for each barcode were tabulated. Only data from barcodes with more than 200 reads for DNA and DNA elements that had at least two independent barcodes measurements that passed the 200-count threshold were evaluated. Regulatory activity for each barcode sequence was calculated by normalizing the RNA read counts with DNA read counts. The activity of each tested sequence was determined by taking the median activity (RNA/DNA) of all barcodes for each element, and the average activity of each element across independent replicate transfection experiments was used to determine the final activity of all tested elements. Comparisons of regulatory activity with scrambled control sequences were performed as previously described (White et al. 2013; Kwasnieski et al. 2014). Annotations studies were performed on all tested elements in each cell line. For annotation analyses, Wilcoxon rank-sum tests were performed on the CRE-seq activities of sites overlapping an annotation with nonoverlapping elements for each annotation and independently for each cell line. Cell-type–specific activity annotation ranking assessments were calculated through P-value ratios by dividing the HepG2 and K562 P-values for each annotation. RNAP2 and GRO-seq signal was determined for the 1000 bp surrounding the summit of each individual CEBPB binding site, and the signal for inactive versus active sites was compared using Wilcoxon rank-sum test.
Genomic data resources
We utilized publicly available genomic data from the ENCODE Project Consortium (2007, 2012) on the University of California Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway) for identifying secondary genomic features correlated with enhancer activity. The DREME program was used for interrogation of sequence motif information (Bailey 2011). GRO-seq data was obtained from Core et al. (2014).
Data access
All CRE-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE73183. All DNA and RNA sequence barcode counts for tested elements and scrambled sequences can be found in Supplemental Data.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants U54 HG006998-0 (to R.M.M.) and R00 HG006922 (to J.G.). We thank Barbara Wold, Ross Hardison, Gregory Barsh, Timothy Reddy, Ali Mortazavi, and Rachel Cosby for helpful suggestions and advice. We also thank the HudsonAlpha Genomics Services Laboratory, especially Dr. Shawn Levy and Ms. Angela Jones, for their contributions to the sequencing and primary analysis for this study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.191593.115.
References
- Bailey TL. 2011. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. 2011. Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wu W, Kumar SA, Yu D, Deng W, Tripic T, King DC, Chen KB, Zhang Y, Drautz D, et al. 2009. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res 19: 2172–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. 2014. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. 2010. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 8: e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaForest A, Nagaoka M, Si-Tayeb K, Noto FK, Konopka G, Battle MA, Duncan SA. 2011. HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development 138: 4143–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2010. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 28: 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. 2009. An oestrogen-receptor-α-bound human chromatin interactome. Nature 462: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Reddy TE, Varley KE, Garabedian MJ, Myers RM. 2012. Genistein and bisphenol A exposure cause estrogen receptor 1 to bind thousands of sites in a cell type-specific manner. Genome Res 22: 2153–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C, Zhang Z, Bhende PM, Sharek L, Wang L, Burridge K, Damania B. 2011. Latent KSHV infection increases the vascular permeability of human endothelial cells. Blood 118: 5344–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. 2013. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23: 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz GZ, Hartzell GW III, Stormo GD. 1990. Identification of consensus patterns in unaligned DNA sequences known to be functionally related. Comput Appl Biosci 6: 81–92. [DOI] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. 2013. RNA-programmed genome editing in human cells. eLife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kasowski M, Kyriazopoulou-Panagiotopoulou S, Grubert F, Zaugg JB, Kundaje A, Liu Y, Boyle AP, Zhang QC, Zakharia F, Spacek DV, et al. 2013. Extensive variation in chromatin states across humans. Science 342: 750–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. 2013. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res 23: 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Waszak SM, Gschwind AR, Raghav SK, Witwicki RM, Orioli A, Migliavacca E, Wiederkehr M, Gutierrez-Arcelus M, Panousis NI, et al. 2013. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 342: 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. 2012. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci 109: 19498–19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA. 2014. High-throughput functional testing of ENCODE segmentation predictions. Genome Res 24: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. 2013. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. 2003. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12: 1725–1735. [DOI] [PubMed] [Google Scholar]
- Levine M. 2010. Transcriptional enhancers in animal development and evolution. Curr Biol 20: R754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. 2013. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498: 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Li D, Yu YY, Kang I, Cha MJ, Kim JY, Park C, Watson DK, Wang T, Choi K. 2015. Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep 16: 654–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marden JJ, Zhang Y, Oakley FD, Zhou W, Luo M, Jia HP, McCray PB Jr, Yaniv M, Weitzman JB, Engelhardt JF. 2008. JunD protects the liver from ischemia/reperfusion injury by dampening AP-1 transcriptional activation. J Biol Chem 283: 6687–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G, van de Geijn B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK. 2013. Identification of genetic variants that affect histone modifications in human cells. Science 342: 747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse ENCODE Consortium, Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, Canfield T, et al. 2012. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol 13: 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. 2012. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30: 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramji DP, Foka P. 2002. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J 365(Pt 3): 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM. 2009. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19: 2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe NJ, Savic D, Nobrega MA. 2012. Transcriptional enhancers in development and disease. Genome Biol 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic D, Gertz J, Jain P, Cooper GM, Myers RM. 2013. Mapping genome-wide transcription factor binding sites in frozen tissues. Epigenetics Chromatin 6: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, Hui L, Wagner EF. 2006. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev 20: 2306–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Han D, Boyd-Kirkup J, Yu X, Han JD. 2014. Evolution of Alu elements toward enhancers. Cell Rep 7: 376–385. [DOI] [PubMed] [Google Scholar]
- Tjian R, Maniatis T. 1994. Transcriptional activation: a complex puzzle with few easy pieces. Cell 77: 5–8. [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Myers CA, Corbo JC, Cohen BA. 2013. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc Natl Acad Sci 110: 11952–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZF, Drumea K, Cormier J, Wang J, Zhu X, Rosmarin AG. 2011. GABP transcription factor is required for myeloid differentiation, in part, through its control of Gfi-1 expression. Blood 118: 2243–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Cui K, Jothi R, Zhao DM, Jing X, Zhao K, Xue HH. 2011. GABP controls a critical transcription regulatory module that is essential for maintenance and differentiation of hematopoietic stem/progenitor cells. Blood 117: 2166–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]