

Abstract
Physiological and behavioral circadian rhythms are driven by a conserved transcriptional/translational negative feedback loop in mammals. Although most core clock factors are transcription factors, post-transcriptional control introduces delays that are critical for circadian oscillations. Little work has been done on circadian regulation of translation, so to address this deficit we conducted ribosome profiling experiments in a human cell model for an autonomous clock. We found that most rhythmic gene expression occurs with little delay between transcription and translation, suggesting that the lag in the accumulation of some clock proteins relative to their mRNAs does not arise from regulated translation. Nevertheless, we found that translation occurs in a circadian fashion for many genes, sometimes imposing an additional level of control on rhythmically expressed mRNAs and, in other cases, conferring rhythms on noncycling mRNAs. Most cyclically transcribed RNAs are translated at one of two major times in a 24-h day, while rhythmic translation of most noncyclic RNAs is phased to a single time of day. Unexpectedly, we found that the clock also regulates the formation of cytoplasmic processing (P) bodies, which control the fate of mRNAs, suggesting circadian coordination of mRNA metabolism and translation.
Almost all life on Earth exhibits an evolutionary adaptation to the 24-h light-dark cycle in the form of physiological and behavioral circadian rhythms (Edgar et al. 2012). These rhythms are ubiquitous, presumably because the maintenance of these rhythms has been shown experimentally to confer selective advantages (Woelfle et al. 2004; Dodd et al. 2005), and the disruption of these rhythms has been linked to various human pathologies (for reviews, see Barnard and Nolan 2008; Reddy and O'Neill 2010), illustrating their biological importance.
The central driver of these rhythms in mammals consists of auto-regulatory molecular feedback loops (Mohawk et al. 2012). Within the major loop, the trans-activators CLOCK and ARNTL (also known as BMAL1) form a heterodimer and activate the transcription of a large number of target genes, including their own negative regulators, the period (PER1-3) and cryptochrome (CRY1-2) genes. Over time, PER and CRY proteins accumulate, heterodimerize, and translocate to the nucleus to repress the CLOCK-ARNTL complex and inhibit their own transcription, closing the negative feedback loop. Next, the PER-CRY complex degrades in a timely fashion, which allows the CLOCK-ARNTL heterodimer to initiate a new round of transcription. This cycle takes ∼24 h to complete and regulates transcriptional output at the appropriate times of day.
While the transcriptional feedback loop that is thought to drive the circadian clock is well characterized, major questions regarding how a 24-h oscillation is generated and maintained remain unanswered. For instance, a delay in negative feedback is necessary for self-sustained limit cycle oscillations (Novak and Tyson 2008). The delay can come in a variety of different forms, and in the case of the circadian clock, it has been reported that the PER and CRY proteins exhibit delayed accumulation relative to the expression of their respective mRNAs (Field et al. 2000; Lee et al. 2001; Reppert and Weaver 2001). This observation suggests the involvement of some type of post-transcriptional control, but the nature of the regulation is not known.
Historically, most studies of gene expression in the mammalian timekeeper have focused on RNA abundance measurements, partly due to the relative ease of obtaining such data with the advent of microarrays and RNA sequencing technologies. Although these methodologies are informative and have led to major advances in understanding clock gene expression, this has naturally led to a focus on transcriptional regulatory networks. Correspondingly little work has occurred investigating translation or post-translational regulation. In support of translational regulation, past studies have found that a significant number of genes oscillate at the protein level without a corresponding oscillation at the transcript level (Reddy et al. 2006; Lim and Allada 2013). In addition, a growing body of research suggests a role for translational control in regulating circadian gene expression in different model organisms (Mauvoisin et al. 2015). However, the extent of this type of regulation, and its relevance to the delays mentioned above, remains unclear.
To investigate the contribution of translational control to circadian clock mechanisms and output in an autonomous circadian clock model, we conducted around-the-clock ribosome profiling experiments in human U2OS cells and compared these results with RNA-seq. Ribosome profiling is based on the protection of small lengths of mRNAs (∼30 nt) by the ribosome during translation (Steitz 1969). Sequencing libraries can be generated from these ribosome-protected fragments (RPFs) to determine which mRNAs are actively translated at any given time on a genome-wide scale with single codon resolution (Ingolia et al. 2009). Using this method, we report on the effect of circadian rhythms on the global translation of rhythmic genes.
Results
Ribosome profiling over a circadian time course in U2OS cells
To reconfirm that a functional molecular clock was present in our experimental system (Vollmers et al. 2008), we grew U2OS cells in tissue culture and synchronized them with dexamethasone. This cell type was chosen due to the relative ease with which samples can be collected and compared to whole tissues. In addition, U2OS cells are known to oscillate and are commonly used in circadian studies (Hirota et al. 2008; Vollmers et al. 2008; Baggs et al. 2009; Hughes et al. 2009; Maier et al. 2009). The cells showed robust oscillations of luciferase activity derived from a PER2 promoter-luciferase fusion. As expected, these oscillations were abolished with siRNAs targeting ARNTL (henceforth referred to as siARNTL cells) to disrupt circadian rhythms (Supplemental Fig. S1A,B).
For our experiments, we synchronized cells with and without treatment with siRNAs targeting ARNTL (also known as BMAL1) and began the collection 24 h post-synchronization (Supplemental Fig. S1A) for a 24-h time course, with 2-h resolution (in duplicate). The collected cells were used to generate sequencing libraries of ribosome-protected fragments at each time point. We aligned these RPFs to the human reference genome using STAR (Dobin et al. 2013) and quantified their distribution using protein-coding gene models from the UCSC Known Gene annotation (Hsu et al. 2006) (see Supplemental Methods for full details; mapping statistics in Supplemental Table S1).
To confirm that the sequenced RNA fragments were generated through the authentic protection of mRNAs by ribosomes, we examined if RPFs mapped correctly to coding regions of the genome and determined their size and periodicity. First, we found that ∼70% of the putative RPFs mapped to exons and ∼10% mapped to introns (Fig. 1A). The remaining ∼20% mapped to inter-genic regions, which may correspond to lincRNAs, as suggested by others (Ingolia et al. 2011). This pattern was consistent in both wild-type and siARNTL cells. When we further examined the exonic subset of reads in wild-type cells, ∼95% mapped to protein-coding regions, while ∼5.5% mapped to 5′ UTRs, and the remaining ∼0.5% mapped to 3′ UTRs (Fig. 1B). Again, the distribution of reads was similar in the siARNTL cells. Second, we examined the length of the RPFs across the entire data set and found that the distribution of read lengths was centered on ∼28 nt (Supplemental Fig. S2), in agreement with the known ribosome footprint (Ingolia et al. 2009). Third, others have shown that RPFs display a characteristic triplet periodicity along translated transcripts that corresponds to the translocation of the ribosome during elongation (Ingolia et al. 2009). To determine if this was the case in our data set, we took the 1000 genes with the largest mean number of mapped RPFs in the wild-type data set, and for each gene we mapped the putative A-site for each RPF from 80 bp flanking the start and stop of the coding region (i.e., RPFs adjacent to either the start codon or the stop codon). The A site of the ribosome is the site at which an aminoacyl-tRNA enters to extend a growing polypeptide. Previous studies have shown that the first nucleotide of the A-site codon is typically located +15 nt from the 5′ end of a given RPF read (Guo et al. 2010; Michel et al. 2012). We used this definition to map the locations of A-sites along the lengths of protein-coding transcripts. Last, we combined and plotted these A-site data from all 1000 genes (Fig. 1C) and observed a strong aggregate 3-nt periodicity in RPFs for these 1000 genes, as well as a steep drop in RPF signal when transitioning between the coding region and the UTR. Both of these observations are typical of ribosome profiling experiments. Thus, we find that the majority of our RPFs map to protein-coding regions of the genome, correspond to the expected size of a ribosome footprint, and are periodic in nature. These three pieces of evidence strongly suggest that our data are generated from the authentic protection of mRNA by ribosomes.
Figure 1.

Analysis of ribosome profiling data. (A) A bar graph displaying the fraction of total RPF reads mapping to introns, exons, and inter-genic regions of the genome. (B) A bar graph displaying the fraction of total exon reads mapping to coding sequence, 5′, or 3′ untranslated regions of genes. (C) The distribution of ribosome A-sites by nucleotide position within a gene, relative to the start and stop codons. Data are aggregated from the 1000 genes with the highest number of RPF reads in the wild-type data.
To provide a direct, parallel comparison between transcription and translation, we also created total RNA-seq libraries from the same cells used to generate the RPF libraries. These libraries were used as a read-out for steady state mRNA abundance. We saw excellent correlations between replicates across all time points and treatment conditions, for both the RPF and RNA-seq data (Supplemental Fig. S3). To further validate our RNA-seq data, we compared it to published U2OS microarray data (Hughes et al. 2009). We examined several known clock genes (ARNTL, CRY1, PER2, NR1D1, also known as REV-ERBAalpha, and NR1D2) and a known output gene, thyrotrophic embryonic factor (TEF), and found that our data were in good agreement with this previous study (Supplemental Fig. S4). Transcriptional rhythms of these same genes were disrupted in the siARNTL cells. Taken together, this confirms the validity of our mRNA abundance measurements for subsequent use.
Circadian rhythms in translation are bimodal
Most genes only had a few RPF reads mapping to them. To reduce noise due to extremely low expression and RPF occupancy, we filtered out all genes with <10 RPF reads across all wild-type samples and those genes with no total RNA-seq reads in any sample. This left 9942 protein-coding transcripts, which are used for all subsequent analyses. This number of expressed protein-coding transcripts is within the expected range, given previous literature that has studied RNA expression across many human and mouse tissues and cell lines (Su et al. 2004; Ramskold et al. 2009). We identified rhythms in transcript accumulation and translation using the JTK_CYCLE algorithm (Hughes et al. 2010). Using a false-discovery rate (FDR) cutoff of 0.2, we found 590 genes with rhythms in RPF occupancy. However, 35% of these genes maintain oscillations in the data obtained from the siARNTL cells (Supplemental Table S2). This is not unheard of, as a recent study showed that over half of mouse liver genes with rhythmic protein accumulation maintain their rhythms in the absence of a functional molecular clock (Mauvoisin et al. 2014). While these residual rhythms may be driven by feeding cycles in a whole organism, they may arise from interaction with the cell cycle in our cellular model. This hypothesis is supported by DAVID analysis of these genes, which showed significant enrichment for cell-cycle proteins (Supplemental Table S3; Huang et al. 2009). In order to focus on those genes most likely to be regulated by the molecular clock, we removed all genes that maintained RPF or RNA rhythms in the siARNTL data. The only exceptions to this filter were 58 genes that showed phase differences ≥4 h between the two treatment conditions (marked in Supplemental Tables S4, S5). While these genes were also enriched for cell-cycle proteins (Supplemental Table S6), we retained them in our analyses since the detectable phase-shift in the siARNTL condition suggests their rhythms are regulated in part by the molecular clock.
By examining this filtered set of genes and varying the JTK_CYCLE false-discovery rate cutoff, we identified 122 (FDR = 0.1), 224 (FDR = 0.15), and 321 (FDR = 0.2) genes with oscillations in RPF occupancy, including the clock genes ARNTL, PER2/3, CRY1/2, and NR1D1/NR1D2 (Supplemental Table S4). While we display the results for the 321 genes (FDR = 0.2) for simplicity's sake, we demonstrate that our findings are the same for all cutoffs. For the total RNA-seq data, we used a standard FDR of 5% and found 592 genes with rhythmic RNA accumulation (Supplemental Table S5). Our data indicated a bimodal pattern for general cellular RNA accumulation as well as translation. Thus, each of the two processes—RNA accumulation and translation—displayed peaks ∼5 and 17 h after the start of collection, which correspond to 29 and 41 h following dexamethasone treatment, respectively (Supplemental Fig. S5). These two anti-phased peaks in expression/translation are in agreement with recent studies of the mammalian circadian transcriptome and proteome (Hoffmann et al. 2014; Mauvoisin et al. 2014; Zhang et al. 2014).
Next, we focused on three distinct sets of genes: (1) those with RNA oscillations, but no RPF oscillations (RNA-only) (Fig. 2A,D); (2) those with RPF and RNA oscillations (Fig. 2B,E); and (3) those with RPF oscillations, but no RNA oscillations (RPF-only) (Fig. 2C,F). We defined RNA-only cyclers as those genes with RNA oscillations (FDR = 0.05), and P ≥ 0.2 in the RPF data. By using the higher P-value threshold in the RPF data, we sought to reduce the impact of genes which had actual RPF oscillations but were just outside our significance cutoffs. Similarly, we defined RPF-only cyclers as those genes with RPF oscillations (FDR = 0.1, 0.15, and 0.2), and P ≥ 0.2 in the RNA data. Again, using this P-value cutoff should yield a more accurate list of genes oscillating only at the RPF level. Lastly, we defined genes oscillating both at the level of RNA (FDR = 0.05) and RPF accumulation (FDR = 0.1, 0.15, and 0.2).
Figure 2.

Peak RNA and RPF timing in cycling genes. (A–C) Radial diagrams displaying circadian phase information for those genes that (A) oscillate in the RNA data only (RNA FDR < 0.05; RPF P > 0.2), (B) oscillate in the RNA and RPF data (RNA FDR < 0.05; RPF FDR < 0.2), and (C) oscillate in the RPF data only (RPF FDR < 0.2; RNA P > 0.2). Note that B depicts phase data for both RNA and RPF, as both show peaks at the same time of day; A and C depict phases for RNA and RPF, respectively. The number of genes in each set is displayed below the corresponding radial diagrams. The blue and red radial diagrams display phase information for RNA and RPF data, respectively. All phases were calculated using JTK_CYCLE. (D–F) RNA and RPF traces for two representative genes in each of the above groups are plotted as a function of circadian time. (D) MAPK6 and SATB1 oscillate in the RNA data only. (E) CDC20 and PLCE1 oscillate in both the RNA and RPF data. (F) PABPC3 and ERAL1 oscillate in the RPF data only. Blue traces for the RNA data use the axes on the left. Red traces for the RPF data use the axes on the right. Points from both replicates are displayed, and the lines are plotted using a moving average (see Supplemental Methods for further detail). Traces from siARNTL libraries are displayed in Supplemental Figure S8.
Interestingly, peak RNA expression of genes with RNA-only oscillations occurred at one of two major times of day (Fig. 2A). Likewise, translation of genes showing rhythmicity at the level of both RNA and RPF showed the bimodal pattern noted above (Fig. 2B). However, genes with RPF-only oscillations showed a unimodal pattern in peak phase, in that they tended to be translated at a single time of day (Fig. 2C). This pattern was maintained regardless of FDR cutoff (Supplemental Fig. S6). We also found genes that cycled in both the RNA and RPF data had higher amplitude oscillations in both data sets than those that cycled only at the level of RNA or RPF (Supplemental Fig. S7). This difference was largely lost with siARNTL treatment, suggesting this trend is the result of a functional molecular clock. In addition to these distinct patterns in peak oscillation phase and amplitude, these different sets of genes showed enrichment for different pathways and biological processes. Using DAVID, we found the RNA-only cyclers were enriched for transcription factors and genes involved in MAPK, Wnt, and GnRH signaling. For example, MAPK6 and the Wnt signaling mediator SATB1 (Fig. 2D; Supplemental Fig. S8A; Notani et al. 2010) show rhythmic mRNA accumulation without RPF oscillations. Those genes with both RNA and RPF oscillations were enriched largely for circadian rhythms (this group contained the core clock genes), with weaker associations to cell-cycle regulators and GTP signal transduction (Supplemental Table S6). These include CDC20, a regulator of the Anaphase Promoting Complex (APC; an E3-ubiquitin ligase), and the phospholipase PLCE1, which show good concordance between RPF and RNA oscillations (Fig. 2E; Supplemental Fig. S8B). Note that CDC20 was one of the genes phase-shifted in the siARNTL condition, suggesting it may be dually regulated by the circadian clock and the cell cycle. Lastly, the RPF-only cyclers are enriched for ribosome-related proteins, as well as genes involved in translation and RNA splicing. Two such examples are the mitochondrial rRNA chaperone ERAL1 and the poly(A) binding protein regulator PABPC3 (Fig. 2F; Supplemental Fig. S8C). This agrees with a recent study finding that the clock both regulates ribosome biogenesis and consolidates it to a single time of day (Jouffe et al. 2013). Taken together, these results suggest that the clock regulates distinct cellular processes at both the transcriptional and translational level. Furthermore, the majority of this translational control is consolidated into a single circadian phase at approximately CT (circadian time) 05.
The delay in clock gene expression is not generated through translational control
Given that circadian genes were enriched among those with rhythms in both RNA and RPF, we sought to examine the relationship between RNA accumulation and translation. It is thought that one of the requirements for the generation of the 24-h circadian oscillation is a delay in the accumulation of the transcriptional inhibitors, PER and CRY. This delay appears to be present in our cycling U2OS cells when we compare the steady state levels of CRY2 protein with corresponding mRNA over the entire 24-h time course (Fig. 3A). While many mechanisms could create this pattern, one hypothesis is that delayed protein synthesis causes the lag between mRNA and steady state protein accumulation.
Figure 3.

The transcription and translation of the transcriptional repressors of the circadian clock are synchronous. (A, top) The average CRY2 band intensity from three protein immunoblots in wild-type synchronized U2OS cells is plotted as a function of time. All measurements were normalized to beta-actin. Error bars represent standard error of the mean. mRNA abundance measurements for CRY2 from total RNA-seq are plotted alongside for comparison. This is the same line displayed in part C of this figure. (A, bottom) Representative protein immunoblots for CRY2 and beta-actin are shown. (B) The lag in time between peak RPF and peak mRNA level is plotted for all genes that oscillate in both RNA and RPF data. Phases were calculated using JTK_CYCLE. (C) RNA and RPF traces for the negative regulators of the circadian clock are plotted as a function of time. Blue traces for the RNA data use the axes on the left. Red traces for the RPF data use the axes on the right. Points from both replicates are displayed, and the lines are plotted using a moving average (see Supplemental Methods for further detail). Traces from siARNTL libraries are displayed in Supplemental Figure S9.
To test this hypothesis, we focused on those genes that displayed oscillations in RPF and mRNA accumulation (Fig. 2B). We calculated the phase lag for this subset of genes as the absolute value of the time difference between peak RPF and peak mRNA, corrected for edge cases (e.g., RPF peak at CT 02; RNA peak at CT 22 should have a 4-h phase lag, not a 20-h phase lag). The timing of peak phases was determined by the JTK_CYCLE algorithm (see Supplemental Methods for further detail). The majority of these genes (92%) showed phase lags ≤2 h (Fig. 3B). This means that most of these genes have either no difference in phase, or the phases are too close together to distinguish with a 2-h sampling resolution. In either case, these phase lags are much smaller than the 6-h delay between mRNA and protein accumulation that others have observed using mass spectrometry (Mauvoisin et al. 2014; Robles et al. 2014). Interestingly, this subset contained all of the major clock genes. The negative regulators in particular, PER1/2 and CRY1/2, exhibited phase differences of 0–1 h. Given the limitations of our 2-h temporal resolution, we also directly compared RPF and mRNA abundance measures for the transcriptional repressors of the circadian clock (PER1/2, CRY1/2, NR1D1/NR1D2) and found transcription and translation to be synchronous (Fig. 3C; Supplemental Fig. S9). Taken together with our previous results, these observations imply that the translation of these circadian transcriptional repressors occurs concurrently with RNA accumulation and that the delay in clock protein accumulation required for a self-sustaining circadian clock is generated through a post-translational control mechanism.
The circadian clock regulates a subset of genes solely at the level of translation
Oscillations in RPF counts can arise from multiple sources of regulation. In the case of the core clock genes, rhythms in RPF appear to arise due to the oscillations in available RNA and reflect regulation of translation through transcriptional control. RPF rhythms can also arise from pure translational control, like those of PABPC3, which has no rhythms in RNA accumulation. However, additional control at the level of translation may be imposed on genes that also show rhythmic RNA accumulation. In order to remove the effect of RNA abundance on RPF counts, previous studies have calculated the translational efficiency (Ingolia et al. 2011; Brar et al. 2012). Briefly, translational efficiency is calculated by dividing RPF counts for a given gene by the RNA read counts for the same gene at the same time (see Supplemental Methods for further details). We found 40 genes (Supplemental Table S7) with oscillations in translational efficiency, many of which were among the list of RPF cyclers. Only two of these genes (CDC25B and KIF3C) with rhythmic translational efficiency cycle in both the RNA and RPF data. However, we also found 18 genes with rhythmic translational efficiency missed by our earlier analyses. While there are too few genes for DAVID analysis, we noticed genes associated with small GTPase activity/signaling (RABL3, GNB2L1), cellular transport and the cytoskeleton (TUBA4A, VIM, BIN1), a GPI anchoring protein (PGAP1), and splicing factors (SNRNP70, SNRPA). The patterns of translational efficiency across many of these genes diverged from their accumulated RNA levels (Fig. 4A). We confirmed the rhythmic accumulation of SNRNP70 protein levels by Western blot and found that the pattern showed a strong similarity to our RPF data (Fig. 4B), while SNRNP70 mRNA levels were found to be nonoscillatory by quantitative PCR (Fig. 4C), consistent with previous findings in U2OS cells (Hughes et al. 2009). Taken together, these oscillations in translational efficiency provide further evidence that the circadian clock regulates a diverse set of biological processes solely at the level of translation.
Figure 4.

Analysis of genes that are translationally regulated by the circadian clock. (A) Traces for RNA and translational efficiency (TE) from PGAP1, VIM, SNRNP70, and RABL3 are plotted as a function of time. Blue traces for the RNA data use the axes on the left. Yellow traces for the TE data use the axes on the right. Points from both replicates are displayed, and the lines are plotted using a moving average (see Supplemental Methods for further detail). Traces from siARNTL libraries are displayed in Supplemental Figure S10. (B) The average SNRNP70 band intensity from eight protein immunoblots in wild-type synchronized U2OS cells is plotted as a function of time. A representative Western blot is shown at the bottom. (C) Average quantitative PCR measurements of SNRNP70 mRNA. All measurements were normalized to beta-actin. Error bars represent standard error of the mean.
Mechanisms underlying rhythmic transcription and translation in U2OS cells are unclear
In an effort to understand the different mechanisms underlying the regulation of the three groups of genes we observe in our study (RNA-only cyclers, RNA and RPF cyclers, RPF-only cyclers), we searched gene promoters (+5 kb: −1 kb of the transcriptional start site) for transcription factor binding sites, and UTRs for RNA-binding protein (RBP) interaction sites. In order to assess if direct activation by the core molecular clock affects the transcription of our cycling genes, we used an existing ChIP-seq data set (Hoffmann et al. 2014). This data set identifies the locations of CLOCK, ARNTL, and CRY1 binding sites in unsynchronized U2OS cells. By assigning these binding sites to gene promoters (see Supplemental Methods for full details), we found only 42 genes that cycle at the level of RNA or RPF are directly bound by these clock factors (marked in Supplemental Tables S4, S5). Of these, only five (CRY1, CRY2, PER2, TEF, and EIF5A2) were directly bound by all three clock factors, and all of these genes cycled in both the RNA and RPF data. Additionally, the major expression peaks in our data (CT 17 and CT 05) are ∼3–4 h out of phase with the components of the core oscillator (ARNTL, CRY1, CRY2, PER1, PER2, PER3). These results suggest that the majority of transcriptional cycling observed in our data is likely to arise from circadian outputs downstream from the core oscillator and not due to direct transcriptional regulation by the CLOCK/ARNTL complex. Next, we used oPOSSUM (Kwon et al. 2012) to search gene promoters for overrepresented transcription factor binding motifs. Notably, we found enrichment of ROR/RRE binding sites and D-box binding sites (well-characterized circadian regulatory/output motifs) in the RNA/RPF cyclers and RNA-only cyclers, respectively. In both of these cases, only those genes with peak expression clustered around CT 17 showed enrichment for these binding sites, while there were no binding sites significantly enriched over background in the genes peaking around CT 05. Those genes that cycle only in the RPF data showed significant enrichment for the joint MYC::MAX binding site, as well as the ELK4 binding site. None of these transcription factors have rhythmic RNA or RPF expression in our data. While these data suggest that some of these transcriptional rhythms could be due, in part, to regulation by RRE and D-box elements, there are still substantial numbers of genes that lack these motifs in their promoters, particularly the group of genes with peak transcription occurring around CT 05.
In order to identify potential avenues of translational regulation, we searched for enriched RBP binding sites and regulatory motifs in the 5′ and 3′ UTRs of genes. The AURA database (Dassi et al. 2014) contains a curated set of 754 RNA-RBP interactions and miRNA binding sites derived from the literature and various experimental data sets. We used this resource to search for regulatory motifs enriched in each of our three gene groups of interest (see Supplemental Methods for full details). Separating genes by peak phase in transcription/translation, we found enrichment of RBM10 interaction sites in the RPF-only cycling genes and miR-335 binding sites among the RNA/RPF and RNA-only cycling genes. However, RBM10 does not cycle in our data, and when we manually inspected the primary transcript of miR-335, we found no evidence of expression. While this analysis is limited to the ∼1.2 million RNA-RBP interaction events described by the AURA database, there does not appear to be a singular RBP responsible for the translational rhythms we observe in the RPF-only cycling genes or the lack of translational rhythms we observe in the RNA-only cycling genes. We also compared the lengths of the UTR and coding regions between these groups of genes, as there has been some evidence that UTR length is associated with transcriptional/translational efficiency (Tanguay and Gallie 1996; Cenik et al. 2010). The only difference we found was that RPF-only cycling genes tended to have shorter 3′ UTRs than genes in the other two groups (Wilcoxon rank-sum test P-value <9 × 10−4). Upon closer inspection, this difference appears to be driven largely by the rRNA genes included in the set of RPF-only cyclers, as they tend to have shorter 3′ UTR sequences than other genes (Ledda et al. 2005). The mechanisms underlying differential circadian regulation of transcription and translation in the U2OS system remain unclear.
The translation of uORFs in genes with rhythmic transcription and translation
One regulatory control mechanism that could account for differences in mRNA accumulation and RPF occupancy is the translation of open reading frames located upstream of the coding sequence (upstream ORFs or uORFs). Translation of these uORFs can affect the translation of the main coding sequence (CDS) of their associated gene, leading to disrupted translation, translation from an alternative start codon, or targeting the mRNA for nonsense-mediated decay (for review, see Barbosa et al. 2013). In order to search for uORFs in our data, we first identified complete ORFs contained in the 5′ UTRs of protein-coding genes, beginning with an AUG. In order to accurately differentiate between the uORF signal and that of the main CDS, we only considered those uORFs located at least 30 bp upstream of the main start codon. After quantifying RPF reads mapping to these putative uORFs and filtering out those with low quantities of mapped reads, we found that 101 genes with oscillations in either mRNA or RPF accumulation also had translated uORFs (Supplemental Table S8). While JTK_CYCLE identified 13 genes with rhythmic uORF translation, the uORF signals appeared to be noisier than the RNA and RPF data from whole transcripts/CDSs. Through manual inspection of the data, we found additional genes that appear to have cycling uORFs, including NR1D1 and ARNTL. Looking at the distribution of all uORFs among the three groups of cycling genes (RNA-only cyclers, RNA and RPF, RPF-only), we observed 22% of RNA and RPF cyclers, 12% of RNA-only cyclers, and only 7% of RPF-only cyclers contained uORFs detectable in our data. The RNA and RPF cyclers showed significant enrichment of uORFs over the background (Wilcoxon rank-sum test P-value = 5.448 × 10−4). Among these genes, we found cases where the uORF was translated with a temporal pattern similar to the main CDS (NR1D1), the uORF was translated in the opposite temporal pattern as the main CDS (C2CD5), and cases where the uORF and CDS followed no common pattern in translation (SUPT7L) (Fig. 5A,B). As a rough measure of the similarity between the uORF and CDS data, we first normalized the RPF read counts by the lengths (kb) of these features, and then calculated the Spearman correlation between the normalized uORF and CDS signals. Interestingly, when we compared these correlations between the three groups of cycling genes, we noticed RNA and RPF cyclers tended to have the best correlations between uORF and CDS signals, while RNA-only cyclers tended to have very low correlations, and RPF-only cyclers tended toward anti-correlation between uORF and CDS signals (Fig. 5C). Furthermore, this trend largely disappeared when comparing uORF and CDS signals in the siARNTL data. This mirrored our earlier finding that genes cycling in both the RNA and RPF data tended to have higher amplitudes. We also searched these uORFs for enriched sequence motifs using the MEME suite (Bailey et al. 2009) and for enriched RBP interaction sites using the AURA analysis described above but found no significant results. While there does not appear to be one overarching mechanism by which the clock uses uORFs to regulate downstream translation, the enrichment of translated uORFs among RNA and RPF cyclers suggests that translation of these uORFs in U2OS cells may either reinforce, or is a consequence of, robust transcriptional and translational cycles.
Figure 5.

Translation of upstream open reading frames. (A) Coverage plots for RPF data mapped to the NR1D1, C2CD5, and SUPT7L loci are plotted. The locations of the uORFs are identified by the black bar above the coverage plot. The gene models for these genes are displayed in blue below the coverage plots. Blocks correspond to exons, and lines indicate introns. The thick blocks indicate the annotated coding region, while the thin blocks correspond to the annotated untranslated region. The chevrons within the intronic lines indicate the direction of transcription. CDS regions are displayed in black below the gene models. The regions displayed are truncated for visualization purposes. (B) RPF traces from the uORF and coding sequences are plotted as a function of time. The black traces are from the wild-type RPF libraries, while the gray traces are from the siARNTL RPF libraries. (C) Box plots for the distribution of Spearman correlation coefficients between uORF and CDS data are displayed for each of the three gene groups (RNA-only cyclers, RNA and RPF cyclers, RPF-only cyclers). These distributions are plotted for both the wild-type and siARNTL data.
Circadian regulation of LSM1 expression leads to oscillating processing body formation
The enrichment of genes associated with mRNA processing, splicing, and metabolism among those displaying oscillations in RPF accumulation (Supplemental Table S6) suggests that the clock influences other post-transcriptional regulatory processes. There are a number of previous reports that link the regulation of multiple post-transcriptional events to the mammalian clock (Gatfield et al. 2009; Kojima et al. 2010, 2012; McGlincy et al. 2012; Morf et al. 2012). The LSM1 gene showed rhythmic RPF accumulation and an expressed, albeit nonoscillatory, uORF. A recent study has implicated other LSM genes as regulators of the plant and mammalian clocks (Perez-Santangelo et al. 2014). Interestingly, LSM1 encodes an RNA binding protein that is involved in mRNA metabolism and is a necessary component for the formation of processing bodies, more commonly known as P bodies, in human cells (Andrei et al. 2005; Chu and Rana 2006). These P bodies are cytoplasmic foci where translationally silent mRNAs are stored and where their ultimate fate—either continued translation or degradation—is decided (Eulalio et al. 2007; Kulkarni et al. 2010; Decker and Parker 2012).
Since the ribosome occupancy of LSM1 appeared to be circadian, we were interested in determining if the circadian clock had an effect on P body formation. To do this, we conducted immunofluorescence microscopy for the P body components, DDX6 (also known as P54/RCK) and EDC4 (also known as GE-1/HEDLS), in synchronized U2OS cells (Fig. 6A; Kedersha and Anderson 2007). In particular, we looked at two representative time points roughly corresponding to the peak and nadir of LSM1 ribosome occupancy (Fig. 6B). We found that ∼20% of cells possessed P bodies when LSM1 ribosome occupancy was high (at 4 h), while we saw a ∼50% drop in P body formation when LSM1 ribosome occupancy was lower, at 16 h (Fig. 6C). To confirm that this was clock-dependent, we repeated this experiment in siARNTL cells and found decreased P body formation and no significant difference between the two time points. Interestingly, the frequency of P bodies was also found to be lower at both time points in siARNTL cells when compared to wild type (Fig. 6D), although this difference did not pass our cutoff for statistical significance. Regardless, this trend implies that clock integrity may be necessary for proper control of P body dynamics. Taken together, this suggests that the circadian clock plays a role in regulating P body dynamics and perhaps in regulating global mRNA decay under periods of cellular stress.
Figure 6.
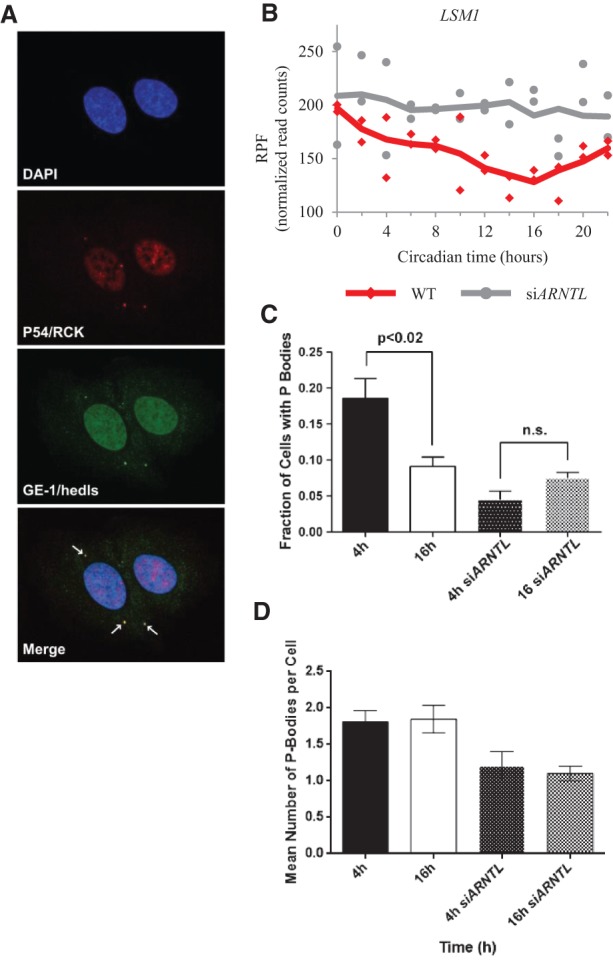
The circadian clock regulates cytoplasmic processing body formation. (A) Confocal immunofluorescence images of the P body markers, P54/RCK and GE-1/HEDLS, are shown at a representative time point when P bodies were found to be present in U2OS cells. DAPI was used as a nuclear counterstain. P body foci are indicated with arrows in the merged image. (B) Normalized RPF reads for the LSM1 gene as a function of time for both the wild-type (red) and siARNTL (gray) data. Points from both replicates are displayed, and the lines are plotted using a moving average (see Supplemental Methods for further detail). (C) A graph representing the number of cells with P bodies at the 4- and 16-h time points for both synchronized wild-type and siARNTL U2OS cells. Error bars represent standard deviations. (D) A graph representing the average number of P bodies per cell at the 4- and 16-h time points for both synchronized wild-type and siARNTL U2OS cells. Error bars represent standard deviations.
Discussion
Here, we used ribosome profiling in a cell autonomous human clock model to examine the global circadian control of translation in U2OS cells. To our knowledge, this is the first study to use ribosome profiling to conduct a survey of global circadian translational control in mammalian cells. We found that rhythmic RNA accumulation and translation is consolidated into two distinct peaks per day, which parallels recent findings using other methods in clock cells obtained from whole head extracts of Drosophila melanogaster (Huang et al. 2013) and in the mammalian circadian system (Zhang et al. 2014). These peaks occur at ∼5 and 17 h after the start of collection (Supplemental Fig. S5). Those genes regulated through translational, but not transcriptional, control mechanisms tend to show peak translation 5 h after the start of collection (Fig. 2C), while genes with rhythms in RNA and RPF accumulation show a bimodal pattern in translation, peaking at 5 and 17 h after the start of collection (Fig. 2B). This implies that translation-specific rhythms are somehow further restricted to a particular period of time during the day, through other pathways/mechanisms aside from mRNA availability.
Previous studies have shown that the accumulation of protein for the negative regulators PER and CRY is delayed, relative to their peak mRNA levels (Field et al. 2000; Lee et al. 2001; Reppert and Weaver 2001). We have shown similar data here in U2OS cells for CRY2 (Fig. 3A). It is thought that this delay is required for a self-sustaining circadian oscillation, but the mechanism by which this delay is created is not well understood. While we initially hypothesized that changes in translational efficiency are responsible for this delay, we were surprised to find that the core clock genes, and the majority of genes with rhythms in both RNA and RPF, showed almost no delay between RNA accumulation and translation. This observed discrepancy may be due to differences between our model systems, or an indirect effect of genetic manipulation. Regardless, it remains possible that the delay is cell-type–specific. Our evidence suggests the delay is primarily caused by post-translational mechanisms, such as regulated protein stability.
If this delay is not made through changes in translational efficiency, this naturally leads to the question: Why do these translational control mechanisms impinge on clock genes in the first place? The control of translation is unique in that it provides the cell an immediate and selective way to control protein expression, when compared to control imparted at the transcriptional level. As a result, translational control mechanisms are often used under stress conditions where immediate changes in protein levels are required, such as ultraviolet irradiation (Deng et al. 2002; Jiang and Wek 2005), NO signaling (Lee et al. 2003), and temperature changes (Lu et al. 2001). We speculate that translational control mechanisms converging on the clock are used in response to acute environmental changes to ensure accurate timekeeping.
While the translation of uORFs provides an attractive mechanism for the clock's regulation of translation, the exact role they play in establishing translation rhythms in U2OS cells remains unclear. We noted a clock-dependent correlation between uORF and CDS RPF signals, but relatively few uORFs showed detectable translational rhythms in our data. It is possible many of these uORFs show rhythmic translation in full organisms, where they may be driven by stronger entrainment queues and more robust peripheral oscillators. Furthermore, those genes that are only rhythmic at the transcriptional level in U2OS cells may gain translational rhythms in full organisms if they are accompanied by rhythmic uORF translation. Future work is needed to define the role circadian translation of uORFs plays in systems beyond U2OS cells and to examine uORFs beginning with noncanonical start codons.
To assess translational efficiency independent of mRNA expression, we used the calculation introduced by previous ribosome profiling papers (Ingolia et al. 2011; Brar et al. 2012). For the most part, this measurement has been used to analyze differential expression style experiments, with only a few conditions. Circadian expression analyses require specialized algorithms to identify complex patterns spread over multiple samples, making them more subject to biological noise, particularly when attempting to detect low amplitude rhythms (Hughes et al. 2010). Given that the translational efficiency calculation involves the normalization of one signal by another, this interference may be masking circadian signals in the translational efficiency measurement. This may explain why we observed fewer genes with rhythms in translational efficiency, rather than by looking at the RPF or RNA-seq data alone. Future work may overcome this deficit by using higher sampling resolution and multiple days of data.
One caveat to our findings is the observation that there are a number of genes that have RPF oscillations in the siARNTL cells. This observation is consistent with findings from other groups (Mauvoisin et al. 2014), and it remains a possibility that the cell cycle is responsible for the persistence of these rhythms. However, there are two pieces of evidence that suggest that these oscillations in RPF are cell-cycle–independent. Firstly, it has been shown that treatment of mammalian cells with dexamethasone arrests the cell cycle in a concentration-dependent manner (Reil et al. 1999). Secondly, treatment of mammalian cells with dexamethasone at lower concentrations will synchronize the cell cycle with a non-24-h period (Yeom et al. 2010). Since we have filtered our hits for oscillating genes with 24-h periods in the siARNTL cells, it is unlikely that their oscillations are cell-cycle–generated. However, we cannot completely rule out this possibility, so we removed these genes from our analyses.
The question remains as to how the translation of these genes is mechanistically controlled by the clock. As stated previously, oscillations in ribosome biogenesis may play a role in regulating global circadian translation. Another possibility includes the temporally controlled expression of trans-acting factors that may regulate translational control elements on a given mRNA, such as iron regulatory proteins in the translational control of ferritin (Gray and Hentze 1994). This mechanism is a likely candidate, given that we see enrichment of translation factors in the set of genes with translational rhythms. While we did see evidence for transcriptional control by ROR/RRE and D-box elements in a large number of our rhythmic genes, we found no clear RNA interactions with cycling RBPs. This may mean many of these translational rhythms are conferred by an as-yet unannotated RBP-RNA interaction, or by regulation of subcellular localization of these RNAs. One speculative possibility is the circadian regulation of a “ribosome filter” (Mauro and Edelman 2007), whereby circadian control of the ribosome itself through temporal modification of rRNAs (Yoon et al. 2006) and ribosomal proteins (Otto et al. 2002) could selectively regulate its binding and translation of mRNAs. These hypotheses remain to be tested.
Since we found overrepresentation of RNA processing and metabolism genes, including LSM1, in our data, we were interested in determining if the clock affects the role of LSM1 in cytoplasmic P body formation. These foci play an important role in various types of mRNA decay and miRNA-induced silencing and are, unsurprisingly, comprised of numerous proteins involved in those pathways. Although some evidence suggests that P body formation is influenced by the phase of the cell cycle in U2OS cells (Aizer et al. 2013), a mechanistic link with the cell cycle is not known. To our surprise, we found that P body formation is regulated by the circadian clock (Fig. 6), with high LSM1 RPF levels at the 4-h time point corresponding to greater levels of P body formation. Lower RPF counts at the 16-h time point corresponded to lower levels of P body formation. This trend disappears in siARNTL cells. In addition, the frequency of P bodies in cells drops in siARNTL cells when compared to wild type, suggesting that clock integrity is required for proper control of P body formation.
Interestingly, P body formation distinguishes the two phases of circadian gene translation reported here. In other words, when P bodies are high, genes that are synthesized through circadian changes in translational efficiency (independent of mRNA abundance) are produced. Conversely, when P bodies are low, circadian gene translation appears to be driven through changes in mRNA abundance.
In our view, these findings are consistent with the idea that the two processes of general translation and mRNA decay are tightly coupled and often sit in direct opposition to one another. For example, the initial steps of general mRNA decay (removal of the 5′ cap and the shortening of the poly(A) tail) correspond to the removal of mRNA features that stimulate canonical translation initiation. In addition, other factors that stimulate general translation, such as EIF4E, are known to inhibit the components of the mRNA de-capping machinery (Schwartz and Parker 1999, 2000). Thus, we believe it is likely that proteins translated independently of mRNA abundance are preferentially made during periods of time when general mRNA decay is high. This idea is also consistent with our observations that genes whose oscillations are translationally, but not transcriptionally, controlled peak with the same phase. Thus, from our results, it appears that circadian clock integrity is required to regulate P body dynamics, and it may play a role in regulating the mRNA cycle and general metabolism of cytoplasmic mRNAs during periods of cellular stress, when P bodies are present in the majority of cells.
Methods
Reagents
Oligos used for ribosome-protected fragment library generation and quantitative PCR were obtained from IDT. Custom siRNAs to target ARNTL were obtained from Qiagen. All oligo sequences are given in Supplemental Table S9. Primary antibodies for GE-1/HEDLS (sc-8418) and SNRNP70 (sc-9571) were obtained from Santa Cruz Biotechnology. Antibodies for P54/RCK (A300-461A) and CRY2 (A302-615A) were obtained from Bethyl Laboratories. The antibody for beta-actin (ab8224) was obtained from Abcam.
Cell culture
A U2OS human osteosarcoma cell line expressing luciferase under the control of the PER2 promoter was used. Cells were maintained in a humidified 37°C incubator at 5% CO2 in DMEM (Life Technologies), supplemented with 10% FBS, 4 mM L-glutamine, 50 units/mL penicillin, and 50 μg/mL streptomycin.
Cell synchronization
To monitor luciferase cycling, 0.122 × 106 cells were seeded per 35-mm plate. For the purposes of collection, 2.25 × 106 cells were seeded per 15-cm plate. These cells were grown overnight, and the media was then replaced with phenol red-free DMEM containing 0.1 mM luciferin and 0.1 μM dexamethasone to synchronize the cells. Plates were then sealed with sterile vacuum grease (Corning) and incubated in a 36°C, nonhumidified incubator at 5% CO2. Luciferase cycling in 35-mm plates was monitored by a LumiCycle luminometer (Actimetrics).
ARNTL siRNA targeting
In order to target ARNTL expression using siRNAs, 3.0 × 104 cells were seeded per 35-mm plate for the purposes of monitoring. For collection, 5.6 × 105 cells were seeded per 15-cm plate. These cells were grown overnight in complete DMEM without antibiotics, and transfected using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's protocols. A mixture of siRNAs that target ARNTL (siARNTL-1 through siARNTL-5) was utilized, and 12 pmol of this mixture were added to each 35-mm plate, and 120 pmol of siRNA mixture were added to each 15-cm plate. As a negative control, an equal amount of AllStars Negative Control siRNA (Qiagen) was used for transfection in parallel. Cells were then synchronized with dexamethasone following transfection as described above.
Quantitative PCR
cDNAs were produced from isolated RNAs from U2OS cells by using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). These cDNAs were used for quantitative PCR, using SYBR Green PCR Master Mix (Life Technologies). Thermal cycling and analysis were conducted on an ABI Prism 7000 Sequence Detection System (Applied Biosystems). Primetime qPCR primers (Hs.PT.58.2126556, Integrated DNA Technologies) were used to assay for SNRNP70 mRNA levels. All other qPCR primers used are listed in Supplemental Table S9.
Cell collection
Cells in 15-cm plates were treated with 100 μg/mL cycloheximide for 3 min at 36°C, 24.5 h post-synchronization when luciferase levels were at their nadir. Cells were 80% confluent at the time of collection. All subsequent steps were done on ice. Spent media was aspirated from the plates, and the cells were washed with 10 mL of cold PBS before treatment with 790 μL of cold lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 100 μg/mL cycloheximide, 1% Triton X-100, and 0.02 units/μL Turbo DNase). Cells were recovered from the plate using a cell scraper, triturated with a 1.0-mL pipette, and incubated on ice for 10 min with occasional mixing. The lysate was then triturated 10 times through a 26-gauge needle and centrifuged at 20,000g for 10 min at 4°C. One hundred fifty microliters of the cleared lysate was added to 700 μL of QIAzol lysis reagent and stored at −80°C for total RNA preparation. The remaining cleared lysate was used for RPF preparation and flash-frozen in a dry ice/ethanol bath before storage at −80°C. Collection of cells was repeated every 2 h for an entire 24-h time course.
Generation of ribosome-protected fragments, library preparation, and sequencing
Frozen cleared lysate was thawed on ice, and RNase I (Life Technologies) was added to 400 μL of cleared lysate to a final activity of 0.75 units/μL. This mixture was incubated for 45 min at room temperature with gentle mixing on a tube rotator, and the nuclease was subsequently inactivated by the addition of 20 μL of SUPERase•In (Life Technologies). The digestion was carefully layered onto a 0.9-mL sucrose cushion (20 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 100 μg/mL cycloheximide, 20 units/mL SUPERase•In, 34% sucrose, [w/v]) in an 11 × 34-mm polycarbonate ultracentrifuge tube (Beckman Coulter), and centrifuged for 4 h at 78,000 rpm in a prechilled TLA-100.2 rotor. The ribosome pellet was resuspended in 700 μL of QIAzol lysis reagent and processed according to the Qiagen miRNeasy Quick-Start protocol. RNA was then isopropanol-precipitated from the elution, and resuspended in 5 μL of 10 mM Tris, pH 8.0. RNA samples were run on a 15% TBE-urea gel (Life Technologies), stained with SYBR Gold, and visualized with a Dark Reader blue light transilluminator (Clare Chemical Research). The 26- to 34-nt region demarcated by the size selection RNA oligos NI-NI-19 and NI-NI-20 was excised from the gel, and the RPFs were extracted and isopropanol precipitated from the gel fragment. Library generation from RPFs was performed as described previously (Ingolia et al. 2012), with some modifications. The full details are provided in the Supplemental Methods. PCR-amplified libraries were pooled and sequenced on a HiSeq 2000 using a V3 flowcell at the University of Pennsylvania Next Generation Sequencing Core. Pooled libraries were sequenced as single 50-bp reads.
Library preparation from total RNA and sequencing
Cleared lysate frozen with QIAzol lysis reagent was processed according to the Qiagen miRNeasy Quick-Start protocol. RNA integrity was verified by Bioanalyzer (Agilent Technologies) before library preparation as described according to the TruSeq Stranded Total RNA Sample Prep kit (Illumina). PCR-amplified libraries were pooled and sequenced on a HiSeq 2000 using a V3 flow cell at the University of Pennsylvania Next Generation Sequencing Core. Pooled libraries were sequenced as single 100-bp reads.
Data alignment, quantification, and analysis for cycling transcripts
Raw FASTQ files from RPF library sequencing were preprocessed as previously described (Ingolia et al. 2012). Following preprocessing, the remaining reads were aligned to the reference human genome (GRCh37/hg19) using STAR (Dobin et al. 2013), quantified using HTSeq (Anders et al. 2015), and normalized with DESeq2 (Anders and Huber 2010; Love et al. 2014), all using default parameters. While hg38 represents a newer human genome build, we have chosen to keep our data aligned to the hg19 version because there are several important resources that have not been mapped to the hg38 version. These include the ChIP-seq data from Hoffmann et al. (2014), the ENCODE Project data sets, and the Genotype-Tissue Expression (GTEx) data set. Furthermore, given our focus on well-studied, protein-coding regions, we do not expect that alignment to hg38 would significantly change the conclusions of this work. Total RNA-seq data were aligned using the same methods as the RPF data, without any of the preprocessing steps. Oscillations in quantification data for these genes were detected using the JTK_CYCLE algorithm (Hughes et al. 2010), with parameters set to handle replicates and to look for oscillating transcripts with period lengths of 24 h. For more detailed information about data analysis, please see the Supplemental Methods.
Processing body immunostaining
U2OS cells were grown on chamber slides and synchronized as described above. P body immunostaining was done as described by others (Kedersha and Anderson 2007). In brief, cells were rinsed with PBS and fixed with 4% paraformaldehyde at the indicated time point. Blocking was done by incubation in 5% normal horse serum (Life Technologies) in PBS for 45 min at room temperature. Primary antibodies were prepared in blocking buffer and used to incubate cells for 1 h at room temperature before washing in PBS and treatment with secondary conjugates. Slides were then washed with PBS and mounted in Vectashield with DAPI (Vector Labs) as a nuclear counterstain.
Confocal microscopy
Image stacks were obtained using a Leica SP5 confocal microscope, using a 40× oil immersion objective lens. Confocal settings were roughly identical for all images captured but were adjusted in some cases to obtain a high-quality image for analysis. A two-dimensional image was generated for analysis by merging Z-planes through each set of cells, and the number of cells with punctate cytoplasmic colocalization of the P body markers GE-1/HEDLS and P54/RCK were counted by eye. All counting was done in a blinded fashion.
Data access
Raw and processed RPF-seq and RNA-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE56924.
Acknowledgments
We thank Dr. Nicholas Ingolia for technical assistance with ribosome profiling and Dr. Nancy Kedersha for guidance on P body immunostaining. This work is supported by the National Institutes of Health (R01-NS-048471 to A.S.), the National Institute of Neurological Disorders and Stroke (1R01NS054794-06 to J.B.H.), the Defense Advanced Research Projects Agency (DARPA-D12AP00025, to John Harer, Duke University), and by the Penn Genome Frontiers Institute under an HRFF grant with the Pennsylvania Department of Health. A.S. is an Investigator of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.191296.115.
References
- Aizer A, Kafri P, Kalo A, Shav-Tal Y. 2013. The P body protein Dcp1a is hyper-phosphorylated during mitosis. PLoS One 8: e49783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei MA, Ingelfinger D, Heintzmann R, Achsel T, Rivera-Pomar R, Luhrmann R. 2005. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 11: 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggs JE, Price TS, DiTacchio L, Panda S, Fitzgerald GA, Hogenesch JB. 2009. Network features of the mammalian circadian clock. PLoS Biol 7: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa C, Peixeiro I, Romao L. 2013. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet 9: e1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard AR, Nolan PM. 2008. When clocks go bad: neurobehavioural consequences of disrupted circadian timing. PLoS Genet 4: e1000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. 2012. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik C, Derti A, Mellor JC, Berriz GF, Roth FP. 2010. Genome-wide functional analysis of human 5′ untranslated region introns. Genome Biol 11: R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CY, Rana TM. 2006. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol 4: e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassi E, Re A, Leo S, Tebaldi T, Pasini L, Peroni D, Quattrone A. 2014. Aura 2. Translation 2: e27738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Parker R. 2012. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol 4: a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg N. 2002. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol 12: 1279–1286. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd AN, Salathia N, Hall A, Kevei E, Toth R, Nagy F, Hibberd JM, Millar AJ, Webb AA. 2005. Plant circadian clocks increase photosynthesis, growth, survival, and competitive advantage. Science 309: 630–633. [DOI] [PubMed] [Google Scholar]
- Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, Xu Y, Pan M, Valekunja UK, Feeney KA, et al. 2012. Peroxiredoxins are conserved markers of circadian rhythms. Nature 485: 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8: 9–22. [DOI] [PubMed] [Google Scholar]
- Field MD, Maywood ES, O'Brien JA, Weaver DR, Reppert SM, Hastings MH. 2000. Analysis of clock proteins in mouse SCN demonstrates phylogenetic divergence of the circadian clockwork and resetting mechanisms. Neuron 25: 437–447. [DOI] [PubMed] [Google Scholar]
- Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F, Ruskeepaa AL, Oresic M, Esau CC, Zdobnov EM, et al. 2009. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 23: 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NK, Hentze MW. 1994. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J 13: 3882–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Lewis WG, Liu AC, Lee JW, Schultz PG, Kay SA. 2008. A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3β. Proc Natl Acad Sci 105: 20746–20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann J, Symul L, Shostak A, Fischer T, Naef F, Brunner M. 2014. Non-circadian expression masking clock-driven weak transcription rhythms in U2OS cells. PLoS One 9: e102238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. 2006. The UCSC Known Genes. Bioinformatics 22: 1036–1046. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Huang Y, Ainsley JA, Reijmers LG, Jackson FR. 2013. Translational profiling of clock cells reveals circadianly synchronized protein synthesis. PLoS Biol 11: e1001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, Hogenesch JB. 2009. Harmonics of circadian gene transcription in mammals. PLoS Genet 5: e1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, Kornacker K. 2010. JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms 25: 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7: 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Wek RC. 2005. GCN2 phosphorylation of eIF2αa activates NF-κB in response to UV irradiation. Biochem J 385: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouffe C, Cretenet G, Symul L, Martin E, Atger F, Naef F, Gachon F. 2013. The circadian clock coordinates ribosome biogenesis. PLoS Biol 11: e1001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. 2007. Mammalian stress granules and processing bodies. Methods Enzymol 431: 61–81. [DOI] [PubMed] [Google Scholar]
- Kojima S, Gatfield D, Esau CC, Green CB. 2010. MicroRNA-122 modulates the rhythmic expression profile of the circadian deadenylase Nocturnin in mouse liver. PLoS One 5: e11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Sher-Chen EL, Green CB. 2012. Circadian control of mRNA polyadenylation dynamics regulates rhythmic protein expression. Genes Dev 26: 2724–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni M, Ozgur S, Stoecklin G. 2010. On track with P-bodies. Biochem Soc Trans 38: 242–251. [DOI] [PubMed] [Google Scholar]
- Kwon AT, Arenillas DJ, Worsley Hunt R, Wasserman WW. 2012. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda) 2: 987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledda M, Di Croce M, Bedini B, Wannenes F, Corvaro M, Boyl PP, Caldarola S, Loreni F, Amaldi F. 2005. Effect of 3′UTR length on the translational regulation of 5′-terminal oligopyrimidine mRNAs. Gene 344: 213–220. [DOI] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. 2001. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107: 855–867. [DOI] [PubMed] [Google Scholar]
- Lee J, Ryu H, Ferrante RJ, Morris SM Jr, Ratan RR. 2003. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc Natl Acad Sci 100: 4843–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C, Allada R. 2013. Emerging roles for post-transcriptional regulation in circadian clocks. Nat Neurosci 16: 1544–1550. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Han AP, Chen JJ. 2001. Translation initiation control by heme-regulated eukaryotic initiation factor 2α kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21: 7971–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Wendt S, Vanselow JT, Wallach T, Reischl S, Oehmke S, Schlosser A, Kramer A. 2009. A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev 23: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro VP, Edelman GM. 2007. The ribosome filter redux. Cell Cycle 6: 2246–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvoisin D, Wang J, Jouffe C, Martin E, Atger F, Waridel P, Quadroni M, Gachon F, Naef F. 2014. Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Proc Natl Acad Sci 111: 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvoisin D, Dayon L, Gachon F, Kussmann M. 2015. Proteomics and circadian rhythms: It's all about signaling! Proteomics 15: 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, Valomon A, Chesham JE, Maywood ES, Hastings MH, Ule J. 2012. Regulation of alternative splicing by the circadian clock and food related cues. Genome Biol 13: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AM, Choudhury KR, Firth AE, Ingolia NT, Atkins JF, Baranov PV. 2012. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res 22: 2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. 2012. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci 35: 445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morf J, Rey G, Schneider K, Stratmann M, Fujita J, Naef F, Schibler U. 2012. Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science 338: 379–383. [DOI] [PubMed] [Google Scholar]
- Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, Mehta S, Purbey PK, Joseph J, Galande S. 2010. Global regulator SATB1 recruits β-catenin and regulates TH2 differentiation in Wnt-dependent manner. PLoS Biol 8: e1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak B, Tyson JJ. 2008. Design principles of biochemical oscillators. Nat Rev Mol Cell Biol 9: 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto GA, Lukavsky PJ, Lancaster AM, Sarnow P, Puglisi JD. 2002. Ribosomal proteins mediate the hepatitis C virus IRES–HeLa 40S interaction. RNA 8: 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Santangelo S, Mancini E, Francey LJ, Schlaen RG, Chernomoretz A, Hogenesch JB, Yanovsky MJ. 2014. Role for LSM genes in the regulation of circadian rhythms. Proc Natl Acad Sci 111: 15166–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramskold D, Wang ET, Burge CB, Sandberg R. 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol 5: e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AB, O'Neill JS. 2010. Healthy clocks, healthy body, healthy mind. Trends Cell Biol 20: 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AB, Karp NA, Maywood ES, Sage EA, Deery M, O'Neill JS, Wong GK, Chesham J, Odell M, Lilley KS, et al. 2006. Circadian orchestration of the hepatic proteome. Curr Biol 16: 1107–1115. [DOI] [PubMed] [Google Scholar]
- Reil TD, Sarkar R, Kashyap VS, Sarkar M, Gelabert HA. 1999. Dexamethasone suppresses vascular smooth muscle cell proliferation. J Surg Res 85: 109–114. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. 2001. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol 63: 647–676. [DOI] [PubMed] [Google Scholar]
- Robles MS, Cox J, Mann M. 2014. In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS Genet 10: e1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. 1999. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol 19: 5247–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. 2000. mRNA decapping in yeast requires dissociation of the cap binding protein, eukaryotic translation initiation factor 4E. Mol Cell Biol 20: 7933–7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz JA. 1969. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature 224: 957–964. [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci 101: 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanguay RL, Gallie DR. 1996. Translational efficiency is regulated by the length of the 3′ untranslated region. Mol Cell Biol 16: 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmers C, Panda S, DiTacchio L. 2008. A high-throughput assay for siRNA-based circadian screens in human U2OS cells. PLoS One 3: e3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelfle MA, Ouyang Y, Phanvijhitsiri K, Johnson CH. 2004. The adaptive value of circadian clocks: an experimental assessment in cyanobacteria. Curr Biol 14: 1481–1486. [DOI] [PubMed] [Google Scholar]
- Yeom M, Pendergast JS, Ohmiya Y, Yamazaki S. 2010. Circadian-independent cell mitosis in immortalized fibroblasts. Proc Natl Acad Sci 107: 9665–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, Ruggero D. 2006. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312: 902–906. [DOI] [PubMed] [Google Scholar]
- Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. 2014. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci 111: 16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]