

Abstract
We describe a genome reference of the African green monkey or vervet (Chlorocebus aethiops). This member of the Old World monkey (OWM) superfamily is uniquely valuable for genetic investigations of simian immunodeficiency virus (SIV), for which it is the most abundant natural host species, and of a wide range of health-related phenotypes assessed in Caribbean vervets (C. a. sabaeus), whose numbers have expanded dramatically since Europeans introduced small numbers of their ancestors from West Africa during the colonial era. We use the reference to characterize the genomic relationship between vervets and other primates, the intra-generic phylogeny of vervet subspecies, and genome-wide structural variations of a pedigreed C. a. sabaeus population. Through comparative analyses with human and rhesus macaque, we characterize at high resolution the unique chromosomal fission events that differentiate the vervets and their close relatives from most other catarrhine primates, in whom karyotype is highly conserved. We also provide a summary of transposable elements and contrast these with the rhesus macaque and human. Analysis of sequenced genomes representing each of the main vervet subspecies supports previously hypothesized relationships between these populations, which range across most of sub-Saharan Africa, while uncovering high levels of genetic diversity within each. Sequence-based analyses of major histocompatibility complex (MHC) polymorphisms reveal extremely low diversity in Caribbean C. a. sabaeus vervets, compared to vervets from putatively ancestral West African regions. In the C. a. sabaeus research population, we discover the first structural variations that are, in some cases, predicted to have a deleterious effect; future studies will determine the phenotypic impact of these variations.
Nonhuman primates (NHPs), compared with rodents, display a far greater level of conservation with humans at all levels of biology, providing essential disease models for systems where humans and rodents are particularly divergent, including inflammatory, infectious, and metabolic diseases, and disorders of brain and behavior. However, the lack of tools for large-scale, genome-level investigations has limited the utility of NHPs as genetic models for common, complex disorders. Given that the vervet is among the most widely used NHP in biomedical research, we established the International Vervet Genome Consortium to develop genomic resources, beginning with the reference genome described here.
Caribbean vervets are uniquely valuable for genetic research, as a very small number of West African C. a. sabaeus vervets introduced to the West Indies as early as the 17th century (Long 2003) gave rise to wild populations on the islands of St. Kitts, Nevis, and Barbados that were recently estimated at more than 50,000–100,000 individuals (Jasinska et al. 2012). The rapid expansion from an extreme bottleneck has likely enabled deleterious variants to attain a relatively high frequency in these populations, facilitating detection of their association with phenotypes (Service et al. 2014). These Caribbean vervet populations provided the founding monkeys for several research colonies on St. Kitts and in North America that now contain large numbers of phenotyped monkeys from a homogeneous and restricted genetic background (Jasinska et al. 2013). In particular, the Vervet Research Colony (VRC), which included the male monkey whose DNA we used to generate the reference genome, is managed as a single extended pedigree, now up to nine generations deep.
A second motivation for vervet genomic efforts derived from the opportunity to identify host genomic features that evolved in relation to simian immunodeficiency virus (SIV), and thereby gain insight into the biology of human immunodeficiency virus (HIV), which originated through mutations in SIV (Hirsch et al. 1989; Gao et al. 1999). The main vervet subspecies (C. a. sabaeus, C. a. tantalus, C. a. pygerythrus, C. a. aethiops, and C. a. cynosurus) co-evolved with distinct SIV strains (Fukasawa et al. 1988; Fomsgaard et al. 1990; Allan et al. 1991; Hirsch et al. 1993; Jin et al. 1994) which are endemic in their natural populations across sub-Saharan Africa and which appear to infect their hosts without causing immunodeficiency diseases. Vervet genome sequencing will enable comparative studies to identify variations between vervets and other primates, including humans (which have had only recent exposure to HIV), and rhesus macaques (which have been exposed to SIV only experimentally), both of which, when infected, progress to AIDS. Additionally, host genome-level analyses will permit comparative studies within C. a. sabaeus to evaluate the hypothesis that balancing selection (Cagliani et al. 2010) may have maintained some as yet unknown protective alleles at a higher frequency in Africa than in the Caribbean, where wild vervet populations are SIV-free.
Finally, a high-quality reference assembly is a prerequisite for characterizing the structural genomic features that differentiate Cercopithecini (including vervets) from the other Cercopithecidae and from catarrhines, generally, including humans. This divergence is important for reconstructing primate evolutionary biology as well as for efforts to identify the genomic basis for phenotypic differences between these taxa (Fig. 1). The vervet genome differs from most other primate genomes in its higher chromosome number (2n = 60), which mainly reflects chromosome breakages (Finelli et al. 1999; Jasinska et al. 2007). Seven chromosome fission events resulted in 29 vervet autosomes, compared to 21 or 22 in most other catarrhines (Stanyon et al. 2012). With few exceptions, such as the gibbon (Carbone et al. 2014) and owl monkey (Ruiz-Herrera et al. 2005), primate chromosomes reveal little change from the inferred ancestral karyotype. The chromosomal variation in gibbon likely resulted from a gibbon-specific retrotransposon that moved into regions harboring chromosomal segregation genes (Carbone et al. 2014). The vervet provides a different kind of model for studying chromosome stability since the fission events are likely more recent, having occurred since the split between Cercopithecini and other members of Cercopithecinae ∼11.5–14.1 million years ago (Mya) (Perelman et al. 2011; Pozzi et al. 2014).
Figure 1.

A phylogenetic tree depicting the position of vervet. The ultrametric tree with branch lengths is labeled in millions of years for the 11 mammalian species used in this study. Divergence times obtained from TimeTree (www.timetree.org/).
In summary, we have built a high-quality vervet genome reference to enable genetic investigations of complex phenotypes, to compare the vervet genome to those of other primates, to describe the genetic relatedness among Chlorocebus subspecies, to measure intra-specific diversity at the major histocompatibility (MHC) locus within C. a. sabaeus, and to examine structural variations within this biomedically relevant research population.
Results
Genome reference build
Biomedical research on the vervet largely utilizes C. a. sabaeus monkeys of Caribbean origin. In the final male vervet assembly, referred to as Chlorocebus_sabaeus 1.1, we ordered and oriented a total of 2.78 Gb in 29 autosomes (CAE1-CAE29) (Supplemental Fig. S1) and two sex chromosomes (CAEX and CAEY), with only 17 Mb remaining unplaced. The assembled size of vervet Chromosome Y (6 Mb) was ∼21% of that estimated for the closely related rhesus macaque Chromosome Y (Hughes et al. 2012). As this highly repetitive chromosome was present in half the number of copies as the autosomes, and was assembled de novo, further investigations will be required to fully characterize Chromosome Y (Hughes et al. 2012). Similarly, we expected the male vervet Chromosome X to be assembled in fewer bases when compared to the assembled female Chromosome X of rhesus macaque (Zimin et al. 2014). Assembled sizes for the male vervet and female rhesus macaque X Chromosomes are 144 and 148 Mb with spanned gaps of 5445 and 4281, respectively, suggesting slightly lower overall sequence representation in the vervet (Supplemental Table S1).
The vervet assembly displayed a greater degree of sequence contiguity than other NHP assemblies with a similar total of assembled bases (Supplemental Table S2). About 50% of the assembled genome size is in contigs >90 kb (compared to 13–86 kb reported in other NHPs) and scaffolds >81 Mb (Supplemental Table S2). The vervet assembly was supported by >8.5-fold coverage in spanning BACs (91% have a unique paired-end alignment position in the assembly), and only 1.6% were residual discordant clones, where the vervet assembly requires improvement. The human genome is the only other primate genome curated to this degree of detail for chromosomal accuracy.
To annotate the vervet genome for gene content, we utilized the collective methods deployed at NCBI (http://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/), taking as input RNA sequencing (RNA-seq) data from total RNA and small RNA cDNA libraries derived from a diverse set of tissue types, as well as human RefSeq and GenBank transcripts and primate proteins. The currently identified set of protein coding genes (21,128) and noncoding genes (8520) is comparable in number to that observed for other primate genomes (http://www.ncbi.nlm.nih.gov/genome/annotation_euk/Chlorocebus_sabaeus/100/; Supplemental Table S2). We aligned 97.9% of 46,823 human known RefSeq (Pruitt et al. 2014) transcripts to the vervet genome at an average coverage of 98.8% (Kapustin et al. 2008). To further assess the vervet genome for quality of gene representation, we aligned with BLAT (Kent 2002) de novo assembled transcripts (Grabherr et al. 2011), derived from vervet blood, caudate, pituitary, fibroblast, and adrenal RNA-seq data, against the vervet genome reference at a 90% identity threshold, and found that >95% of vervet transcripts constructed in a reference-free manner were represented in the assembly at 95% of their length. Additionally, we produced an independent set of predicted protein-coding genes (19,165) using the Ensembl annotation process (Flicek et al. 2014) that adds additional gene query options.
Primate genome conservation
The vervet karyotype contains seven additional autosomes compared to the inferred ancestral catarrhine karyotype (Finelli et al. 1999; Stanyon et al. 2012). To discern whether vervets also display more frequent smaller-scale rearrangements, we first conducted a comparative analysis of total interspersed repeats, then performed detailed comparisons of synteny between the vervet genome and those of human and rhesus macaque. Total transposable elements (TEs) occupy ∼48% of the vervet genome and are distributed in a pattern comparable to that observed in rhesus macaque (Zimin et al. 2014) and human (Supplemental Tables S3–S6; International Human Genome Sequencing Consortium 2001). Elements such as L1_RS, AluYR, and ERV(L)-MaLR showed a high proportion of full-length copies, indicating their recent activity and possible specificity for catarrhines. The chromosomal distribution of selected TEs (SINEs, LINEs, LTRs, and DNA transposons) showed the highest total coverage (>50%) on the sex chromosomes and on CAE 6 and 28, and the lowest coverage (44%) on CAE 8. Among specific elements, SINEs were significantly overrepresented on CAE 5, 6, 16, 19, and 28; and LINEs, LTRs, and DNA elements were significantly underrepresented on CAE 6, 19, and 28.
To analyze small-scale rearrangements, we utilized alignment of vervet BAC end and chromosome sequences to the genomes of human (GRCh38) and rhesus macaque (rheMac2) (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007) and fluorescent in situ hybridization (FISH) of human BACs. First, we aligned the vervet (CHORI-252) BAC end sequences to the human reference (GRCh38) to evaluate overall synteny. Of 136,002 BAC clones with a pair of uniquely aligning end sequences, 96.6% aligned in a collinear fashion. Intra-chromosomal rearrangements (2.1%) outnumbered inter-chromosomal rearrangements (1.2%). Genome-wide, the occurrence of inter-chromosomal and intra-chromosomal rearranged BAC clones was 1.03 and 1.07 clones/Mb, respectively. Human Chromosome 14 (corresponding to CAE24 and 29) was the most conserved (0.85 intra- and 0.36 inter-chromosomal rearranged BACs/Mb) and human Chromosome 10 (corresponding to vervet Chromosome 9) the most divergent between the species (2.31 intra- and 2.99 inter-chromosomal rearranged BACs/Mb). Whole genome alignments of human, vervet, and rhesus macaque using LASTZ (Supplemental Fig. S2; Harris 2007) confirmed that seven fission events would best explain the representation of human sequence (from Chromosomes 1, 3–7, and 15) dispersed among the 29 vervet autosomes. Human Chromosome 2 formed through a human lineage-specific fusion of two ancestral chromosomes (Jdo et al. 1991); therefore, its synteny to CAE10 and CAE14 does not reflect a fission event in the vervet lineage. Vervet chromosomes that underwent fission events ranged in size from 23 to 134 Mb. Overall, our observations indicated a high degree of genome collinearity between vervet and human, with confirmation of known chromosome fissions representing the major form of observed chromosomal rearrangement.
A previous report used chromosome painting to compare the human karyotype with those of several catarrhine species, including vervet (Stanyon et al. 2012). We conducted a higher resolution molecular cytogenetic comparison of the vervet, rhesus macaque, and human karyotypes, narrowing each breakpoint region through reiterative FISH experiments. These studies cohybridized on vervet metaphase chromosomes, 570 well-spaced human BAC clones, used previously to characterize rhesus macaque (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007). We found that sixteen vervet chromosomes display breaks in synteny with respect to rhesus macaque (CAE2, CAE4, CAE7, CAE13, CAE15, CAE17, and CAE20-29) (Fig. 2A). Comparison of vervet and human with their inferred common catarrhine ancestor identified 58 gross synteny breaks overall; 27 of these breaks occurred in the human lineage and 31 in the vervet lineage (11 inversions, 8 fissions, and 1 fusion). The Old World monkey (OWM) ancestor had 46 chromosomes; several rearrangements have increased the vervet diploid chromosome count to 60 (Stanyon et al. 2012). A detailed summary of the recent evolutionary history of each vervet chromosome with respect to rhesus macaque and human is included at http://www.biologia.uniba.it/vervet/. An example of a vervet-specific fission (Fig. 2B) shows the results of a cohybridization FISH experiment, with three almost overlapping human BACs from Chromosome 5 mapping within an interval of ∼622 kb. The external BACs (RP11-450M18 and RP11-1115K14) map to CAE Chromosomes 4 and 23, respectively, while the central BAC RP11-435K10 shows a splitting signal. Moreover, a single rhesus macaque BAC that mapped to separate vervet chromosomes (Supplemental Fig. S3) provided a higher resolution example of a fission that seeded a centromere on CAE24 and a telomere on CAE29. Analysis of synteny to a 284-kb region on human Chromosome 14 (Supplemental Fig. S4) revealed a complex array of LINE repeats and an 82-kb segmental duplication (SD).
Figure 2.
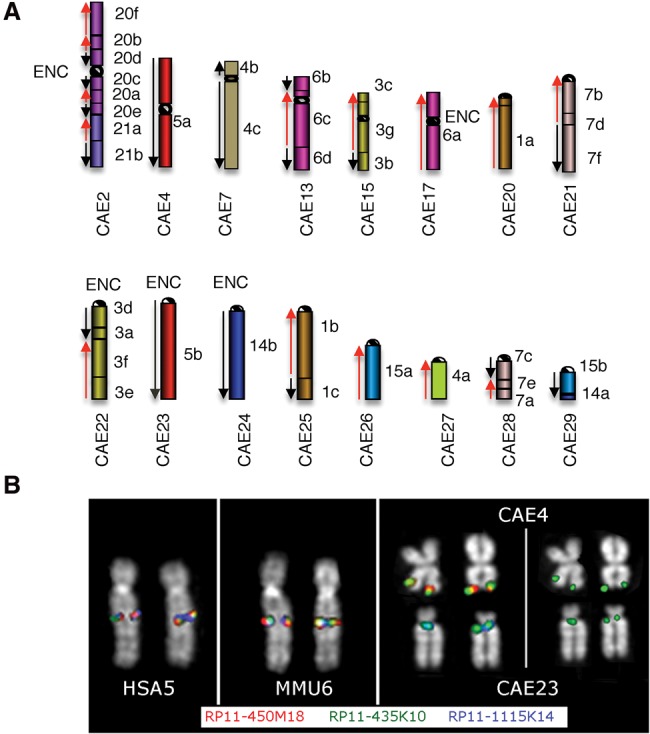
Vervet chromosomes showing synteny breakage with respect to rhesus macaque. (A) On each vervet chromosome, on the right, the corresponding human synteny blocks are reported and also colored for ease of interpretation. Black arrows indicate an identical sequence orientation; red arrows indicate inverted orientation. “ENC” denotes the approximate location of evolutionary neocentromeres. (B) FISH results using RP11-450M18, RP11-435K10, and RP11-1115K14 human BACs (mapping on Chromosome 5) to homologous chromosomes of rhesus macaque and vervet (see Methods).
Evolutionary neocentromeres (ENC) are centromeres that repositioned along the chromosome without any inversion or sequence transposition or that were seeded on an acentric fragment generated by a fission event. ENCs are typically discovered while performing chromosomal marker order comparison, usually by FISH, among closely related species (Stanyon et al. 2012). Nine ENCs were reported as shared by all OWM species and are OWM-specific, based on human and rhesus macaque studies (Ventura et al. 2007). Our FISH experiments (see Fig. 2B example) revealed vervet ENCs on five chromosomes (CAE2, CAE17, CAE22–CAE24) (Fig. 2A). The centromere was repositioned along CAE17, while those on CAE2 and CAE22–CAE24 were seeded at the fission point. The latter events generated acentric fragments that were rescued by the emergence of the ENCs. The neocentromere of CAE23 was near the telomere and expanded without disturbing the sequences recognized by the BAC that split each chromosome (Fig. 2B).
Structural variation
Naturally occurring structural variants (SVs) in NHPs are therefore of great scientific and clinical interest as they may include duplications or deletions of gene regulatory or protein-coding regions. To identify candidate SVs potentially contributing to vervet trait variation, we selected six vervets with 59 direct descendants (collectively) and a range of kinship coefficients (from 0 [essentially unrelated] to 0.0625 [first cousin]). These six monkeys spanned multiple generations (date of birth 1985–1996), and each was sequenced to an average depth of 32× (Supplemental Table S7). We detected numerous deletions, considering a predetermined size range of 500 bp to 2 Mb size (Supplemental Table S8).
To identify a high-confidence set of deletions, we used CNVnator (Abyzov et al. 2011), which relies on sequence depth, and LUMPY (Layer et al. 2014), which relies on split and paired-end sequence discordance. Among the VRC monkeys, we found 556 unique deletions, with 60–116 deletions per monkey (Fig. 3A; Supplemental Table S8), as defined by CNVnator genotypes falling below a read-depth threshold of 1.5 (Abyzov et al. 2011). Of these unique deletions, a majority (64%) were <2 kb in size (Supplemental Table S9) and at least 7% were homozygous (Supplemental Table S8). From 2.3 Mb of total unique deletion space, we found 77 deletions that disrupt coding sequence of one or more genes (Fig. 3A). However, most of these deletions occurred in and around pseudogenes or tandemly arranged gene families of the immune system, e.g., MHC and Ig heavy chain genes. We also measured deletion variants shared by at least three individuals; they accounted for 5.3 Mb and represented loss of at least a portion of exon sequence for 57 genes (Supplemental Table S8), with 61% of these variants under 2 kb in length (Supplemental Table S10). Overall, this set of shared deletions was enriched for genes harboring immunoglobulin domains, but most deleted genes were characterized as having unknown function.
Figure 3.

Structural variation among vervets. (A) The total deletions unique to each vervet for a size range of 500 bp–1 Mb are shown in green bars, and those genes where any exon space is deleted in one chromosome or greater are displayed as black bars. (B) Shared segmental duplications among vervets are blue bars, non-sample-specific duplications are red bars, and sample-specific duplications are green bars. (C) Shared duplications among vervets and other primate species; a minimum of six samples were previously sequenced per species population (Prado-Martinez et al. 2013). In all box plots, the vertical limits of the box represent one standard deviation around the mean, the horizontal line within the box is the median, and the whiskers extend from the box to the 25th and 75th percentiles.
We also identified multiple segmental duplications, highly identical expanding sequences that can serve as templates for recombination and crucibles for gene birth and death (Bailey and Eichler 2006), which we defined as genome sequences >5 kb in size found more than once with >90% identity, and considered them without a specific restriction on size-range. While vervet chromosomes that formed through fissions displayed no difference in average counts of SDs compared to other autosomes (P-value = 0.94) (Supplemental Fig. S5), closer scrutiny of several syntenic breakpoints by BAC mapping revealed numerous SDs, supporting the hypothesis that they have played an important role in chromosome evolution, including fissions (Kehrer-Sawatzki and Cooper 2008; Marques-Bonet et al. 2009; Carbone et al. 2014).
On average, over 44 Mb of SDs were found in each monkey across all autosomes (Fig. 3B). Most of these SDs were shared (36 Mb, or 81%) among all samples, including the reference genome, while only a small fraction (1%–3%) was sample-specific (Fig. 3B; Supplemental Table S11). The total number of duplicated bases shared among VRC monkeys was 6.7-fold greater than the number of deleted bases (both unique and shared). Investigations in great apes have reported, similarly, a 7.7-fold increase in fixed duplications relative to deleted bases (Sudmant et al. 2013). Upon intersection of fixed SD sequence (36 Mb in total) with gene positions, we found a total of 1601 protein-coding genes, of which 321 have ascribed gene function. On CAE6, we observed an elevated number of zinc finger genes relative to other chromosomes (19 of 33 total). Rhesus macaque Chromosome 6 is also particularly enriched for zinc finger genes (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007). Canonical pathway enrichment analysis of unique genes embedded in SDs revealed several classifications potentially relevant to diseases, including immunological pathways related to pathogen response (P < 0.04) (Supplemental Table S12). For example, a member of the leukocyte receptor cluster, LENG1, whose interacting protein partners are involved in viral release from host cells, was found in three copies compared to one in other sequenced primates (Supplemental Fig. S6). Comparatively, the percentage (∼92%–96%) of shared SDs is higher within hominids (human, chimpanzee) than in vervet (81%) (Fig. 3C), most likely representing their natural low variability (Prado-Martinez et al. 2013). We carried out a complementary analysis of duplications in vervet and 11 other mammalian species to detect, for all predicted genes, those rapidly expanding gene families specific to vervet; we identified 38 specific expansions, compared to a high of 288 and low of 8 for human and gibbon, respectively (Supplemental Table S13).
Subspecies relationships
We sought to clarify the relationships among the taxa (variously classified as subspecies or species) that constitute the genus Chlorocebus by comparing the reference assembly from Caribbean-origin C. a. sabaeus to whole-genome sequencing data from one representative of each of the five principal African subspecies: C. a. sabaeus; C. a. pygerythrus; C. a. aethiops; C. a. tantalus; and C. a. cynosurus (Supplemental Table S14). We discovered 34 million sites with single nucleotide variants (SNVs) relative to reference; 19.7 million of these SNVs passed quality control filters and allowed inference of the ancestral allele, using the rhesus macaque reference assembly (Supplemental Fig. S7). The level of sequence divergence across these five vervet subspecies was 0.21%–0.33%, comparable to the level of divergence between subspecies of other nonhuman primates: chimpanzee, 0.17%–0.19%; gorilla, 0.16%–0.20% (Prado-Martinez et al. 2013); and macaque, 0.31% (Yan et al. 2011). Heterozygosity estimates for vervet subspecies ranged from 0.08% (C. a. aethiops) to 0.18% (C.a. pygerythrus). Further, 36% of the SNVs displayed fixed differences between vervet subspecies (22% in a single sample rhesus macaque study), and 28% varied both within and between vervet subspecies (12.5% in rhesus macaque). In addition to the latter variations, 16% of the fixed differences were incompatible with our inferred species tree (Fig. 4A), suggesting that these sites either constituted ancestral polymorphisms that might still be segregating or were subject to gene flow across subspecies boundaries. Thus, despite the small sample size, a large proportion of vervet genetic variation (34%) was shared across subspecies, suggesting recent or ineffective reproductive isolation among the subspecies.
Figure 4.

The phylogenetic tree, geographical distribution, and population history of vervet subspecies. (A) Subspecies relationships were obtained by applying a clustering algorithm to the pairwise distance matrix. The tree is rooted using rhesus macaque as an outgroup, and the estimated geographical distribution of each subspecies based on previous field studies used to characterize endangered species (www.iucnredlist.org) is displayed to the right. (B) The inferred effective population size across time (both on log-scale) for each subspecies sample inferred with the multiple sequentially Markovian coalescent (MSMC) software in two-haplotype mode (Schiffels and Durbin 2014).
To describe the relationship between the subspecies, we calculated the pairwise distance across all SNVs for each pair of chromosomes and inferred average divergence times (Supplemental Table S15). By subtracting the average divergence time within individuals from the pairwise comparisons, we estimated divergence dates for the subspecies under the assumption of no subsequent gene flow (Supplemental Tables S16, S17). A phylogenetic tree (Fig. 4A) supported C. a. cynosurus and C. a. pygerythrus as the most closely related subspecies, with an inferred divergence date around 129 thousand years ago (kya); C. a. tantalus diverged from these two subspecies most recently (265 kya), followed by C. a. aethiops (446 kya), and then C. a. sabaeus (531 kya). For each subspecies, branch lengths were roughly consistent with known range proximities within Africa (Fig. 4A). In particular, applying a clustering algorithm to geographic distances yielded a similar tree, except that C. a. tantalus was sister to C. a. aethiops. This inconsistency might be explained by a considerable desert-like environment separating the range of C. a. aethiops from that of the other subspecies, which increases its effective geographic distance from them. We further note that the estimated times for splitting of the subspecies (Supplemental Table S16) are considerably more recent than the corresponding sequence divergence times (coalescent times) (Supplemental Table S17).
To further reconstruct the evolutionary history of the vervet, we inferred the effective population size of each subspecies using MSMC (multiple sequentially Markovian coalescent) (Schiffels and Durbin 2014). The results (Fig. 4B) suggest that effective population sizes across the five subspecies started to diverge ∼300 kya (the apparent, more ancient divergence of effective population size for C. a. aethiops represents a known artifact of this method). Perhaps the most striking result is the decrease in the effective population size of C. a. aethiops beginning ∼200 kya, followed by a mild recovery of effective population size around 20 kya and an even more extreme decline in more recent times. C. a. sabaeus and C. a. cynosurus show intermediate effective population sizes for most of their evolutionary history, with a much weaker decline than C. a. aethiops 200 kya and an earlier and stronger recovery peaking at around 30 kya. Estimates point to a recent (∼10 kya) relative decline in effective population size in C. a. sabaeus and a slight increase in C. a. cynosurus. The evolutionary history of C. a. tantalus is characterized by a remarkable stability in effective population size until a sharp decline around 10 kya. Finally, C. a. pygerythrus, the subspecies with the largest geographic range, shows a minimum of effective population size before 100 kya, followed by a strong increase, a phase of constantly large effective population size, and an even more extreme increase in the most recent past. We note that the divergence of effective population sizes between C. a. cynosurus and C. a. pygerythrus for times more ancient than the split-time inferred above can be explained by population structure in the ancestral population.
MHC diversity among C. a. sabaeus vervets
We hypothesize that natural resistance to SIV progression among vervet populations could derive, in part, from the action of protective genetic variants in genes that regulate immune responses. We therefore have characterized MHC diversity in vervets sampled from West Africa, St. Kitts, and from several North American primate centers. As a first step towards identifying MHC variants, we sequenced BAC clones spanning the MHC region of the reference genome to obtain unambiguous haploid sequence paths. After sequencing and organizing a tile path of sequences that comprise a total of 4 Mb of the MHC reference haplotype, only five known gaps remained (Supplemental Fig. S8). We then aligned the ungapped regions to a nonreference-derived rhesus macaque MHC (Daza-Vamenta et al. 2004); we used these data because the MHC region of the rheMac7 reference (Zimin et al. 2014) contains several errors due to the complex SDs that characterize the MHC of all rhesus macaques examined to date (Daza-Vamenta et al. 2004; Watanabe et al. 2007). When compared to the rhesus macaque single haploid MHC region (Daza-Vamenta et al. 2004), the vervet displayed highly conserved synteny with only minor structural variation (Fig. 5A). However, gaps that flank the Chsa-A locus and another localized within the Chsa-B region (the analogous region in the rhesus macaque MHC included a tandem array of nineteen closely related Mamu-B loci) prevented us from drawing definitive conclusions for sequence orientation in some apparently syntenic regions.
Figure 5.

The characterization of the vervet MHC region and its diversity. (A) A tile path of sequenced and assembled BACs were aligned to a single haplotype region of the rhesus macaque MHC (Daza-Vamenta et al. 2004); vervet sequence aligned in the correct orientation with macaque is colored blue, and gaps are denoted as missing color. (B) Pie charts show MHC haplotype distribution in (left) captive, (middle) Caribbean, and (right) West African vervets. Six ancestral MHC class I haplotypes (B1–B6) identified by pyrosequencing accounted for all major haplotypes observed in vervets (n = 51) from US primate centers. The remaining haplotypes reflected simple recombination events between these six ancestral haplotypes. The distribution of MHC class I haplotypes of feral vervets (n = 21) from St. Kitts is virtually indistinguishable from that of US primate center monkeys. Eleven feral vervets from Ghana exhibited at least 16 distinct Chsa-B haplotypes. Only the Chsa-B5 haplotype (yellow slice) was identical with the class IB region of the B5 haplotype in St. Kitts-origin vervets. Chsa-A haplotypes in the individuals from Ghana also exhibited much greater diversity compared to the Caribbean-origin population (data not shown).
To assess MHC locus diversity in C. a. sabaeus, we sequenced MHC class I transcripts from 83 vervets. Using RNA isolated from blood samples (whole blood or peripheral blood mononuclear cells [PBMC]) obtained from vervets at five US primate centers (Supplemental Table S18), we performed high resolution pyrosequencing of cDNA-PCR amplicons spanning the second to fourth exons, which encode the highly polymorphic peptide binding domain of class I gene products (Wiseman et al. 2013). We observed extensive MHC class I allele sharing in 51 apparently unrelated individuals from different primate centers (Fig. 5B); all MHC class I diversity could be accounted for by six inferred haplotypes together with simple recombinants (Fig. 5B; Supplemental Table S19). Four BAC clones (CH252-276C1, CH252-124F12, CH252-5C22, and CH252-175F15) contained genomic sequences that were identical to six class I transcripts from the M4 haplotype of Caribbean-origin vervets (Chsa-A*01:01, -B*07:01, -B*09:01, B*21:nov:01, B*23:nov:01, and E*01:nov:02) (Supplemental Table S19). Despite at least one gap of unknown size in the BAC tile path for the Chsa-B region, genomic sequences corresponding to all known Chsa-B transcripts were identified, with the exception of Chsa-B*01:01.
We then obtained MHC sequences, using a similar approach, from vervets sampled at seven distinct locations on St. Kitts (N = 21) and at four locations in Ghana (N = 11), a country considered a potential source of the vervets brought to the Caribbean during the colonial period. All six MHC class 1 haplotypes observed in vervets from US primate centers (n = 51) were shared with 75% of sequenced vervets from St. Kitts, with the remainder of the St. Kitts vervets predominantly displaying recombinant versions of one of these haplotypes (Fig. 5B). The near-identity between the haplotype sets observed in the St. Kitts sample and in the (independently collected) US primate center samples (Fig. 5B) is consistent with the hypothesis that the Caribbean vervet populations have expanded dramatically from a recent bottleneck and demonstrates that samples of captive Caribbean-origin vervets provide a good genetic approximation of the wild populations on St. Kitts. The substantially more diverse array of novel MHC class I transcripts observed in the Ghana samples compared to those from St. Kitts provides additional evidence in favor of the suggestion of a recent founder effect in the St. Kitts population (Fig. 5B; Supplemental Table S19). Specifically, in comparing variation at the Chsa-B haplotypes (the only ones that we could reconstruct in the Ghana samples), we observed much higher allelic variation of Chsa-B sequences in the Ghana vervets than in the St. Kitts vervets, despite the smaller size of the Ghana sample. The B5 Chsa-B haplotype (Fig. 5B), seen in a single Ghana vervet, is the only haplotype showing a complete identity of alleles between the Ghana, St. Kitts, and US vervet samples. More conclusive evaluation of vervet MHC diversity will require sequencing of samples from Nevis, Barbados, and from a wider range of African populations, while fuller characterization of the MHC gene repertoire awaits a more detailed haploid reconstruction.
Discussion
The importance of the vervet as a biomedical model system rests in large part on the utility for genetic studies of the large, richly phenotyped C. a. sabaeus populations, within the Caribbean and in North American colonies of Caribbean descent. To support such studies, we provide a high-quality vervet genome reference generated from a single C. a. sabaeus monkey. In constructing this assembly, we optimized sequence contiguity, but not at the expense of accuracy, producing vervet gene models that are more complete (98.8%) when aligning to human RefSeq transcripts than those for other recently assembled OWMs: 94.6% and 95.8% for olive baboon (Papio anubis) and golden snub-nosed monkey (Rhinopithecus roxellana), respectively (http://www.ncbi.nlm.nih.gov/genome/annotation_euk/status/). Extensive RNA-seq data (22 Gb of sequence from 174 individual samples across five different tissues) provide additional validation of this assembly. A variety of measures support our contention that the vervet genome reference is of sufficient quality to launch large-scale whole-genome and transcriptome sequencing studies. The fact that 99% of whole-genome sequences generated from Chlorocebus subspecies map to the vervet reference assembly further substantiates its use in studies of the African subspecies.
We observed a high level of sequence conservation in comparing vervet to humans and rhesus macaque using both in silico and experimental approaches, implying that investigations in vervets will have good translatability to human studies. However, members of the Cercopithecini, including vervets, have experienced several lineage-specific chromosomal fissions, and therefore their karyotypes diverge substantially from those of most other catarrhines, including humans (Finelli et al. 1999). Broadly, complex Robertsonian and non-Robertsonian fissions are the source of this chromosomal variation, but among the biomedically relevant OWMs, only vervets have experienced significant dysploidy, mostly as a result of noncentromeric sequence exchanges. Three of these fissions generated an acentric fragment whose stability appears to result from the seeding of an ancient neocentromere. Similar events have been reported in OWMs following the fission of the chromosome corresponding to human Chromosome 3 (Ventura et al. 2004). Macaque neocentromeres harbor large blocks of alphoid DNA (except Chromosome Y), whose sequence is shared among all of them, unlike humans, in which many alphoid subsets are chromosome-specific. In vervet, the alphoid blocks of the five neocentromeres in vervet are not distinguishable from those of other chromosomes, suggesting that these noecentromeres probably arose in the Cercopithecini ancestor.
Unlike other NHPs adopted for the study of human disease, vervets used in biomedical research descend mainly from small founder populations that have expanded from extreme bottlenecks, making them particularly suited for studying the phenotypic impact of structural variations. We have so far evaluated gene-containing deletions and duplications observed in the VRC extended pedigree. In this setting, observed sequence gain has considerably outpaced loss, a pattern similar to that observed in great apes (Sudmant et al. 2013). Overall, CAE6, a chromosome that attracts a proportionally higher number of SINEs than other vervet chromosomes, displays the most frequent sequence losses. Intriguingly, human Chromosome 19, which is homologous to CAE6, also has, among human chromosomes, incurred the greatest number of losses, based on examination of numerous SV events in the database of genome variation (Zarrei et al. 2015). A closer examination of the breakpoints shared in the ancestral state of CAE6 and other chromosomes should provide further insight regarding the molecular mechanisms responsible for these occurrences. Among rare autosomal deletions (observed in a single vervet) that perturb exon sequences, we found very few in homozygous states (only six in a total of 77 genes containing such deletions), and most of these are in pseudogenes, presumably not subject to purifying selection. This finding is consistent with the suggestion of Gokcumen et al. (2013) that purifying selection has contributed little to the SV differences observed among primates.
Few prior studies have used sequence data to extensively evaluate copy number variation in NHP populations (Gokcumen et al. 2013; Sudmant et al. 2013). Sudmant et al. (2013) have reported that, in the common chimpanzee/bonobo ancestor, 57 genes likely experienced exonic deletions; similar to our observations in vervets, they found that these genes were enriched for immune and olfactory functions (Sudmant et al. 2013). Ours is the first study to characterize vervet SVs, and those monkeys in which we observed such variants were apparently healthy. We therefore presumed that none of these variants is severely deleterious, although neurodevelopmental phenotypes predominately associated with SVs in humans (Cooper et al. 2011) would likely not be readily detected in most primate colonies. However, detailed assessment of neurocognitive function in vervets is feasible (Elsworth et al. 2015), and we propose that conducting such assessments in vervets carrying SVs identified here (and in future studies) and matched controls could yield novel models for such disorders. In fact, iterative phenotyping of humans identified based on loss-of-function mutations is at an early stage (Alkuraya 2015; Johnston et al. 2015) and to our knowledge has not been initiated in NHPs.
The palindromic nature of SDs has enabled a high degree of genome plasticity in primate evolution (Kehrer-Sawatzki and Cooper 2008) and has led to their association with disease-related instability in various human chromosomes (Antonacci et al. 2014). We describe the first collection of vervet SDs; their localization will facilitate future studies focused on the evolution of OWMs or examining their phenotypic impact in vervet populations. Specifically, evaluation of the occurrence within these regions of protein-coding genes may shed light on broad trajectories of primate evolution. Examples include the observation that some vervet SDs harbor a greater than expected number of zinc finger genes, a feature conserved between vervet and rhesus macaque (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007), or the enrichment in vervet SDs of genes that function in immunological pathways related to pathogen response. Notable examples include LENG1, whose interacting protein partners are involved in viral release from host cells, and JAK2 and NOS2, both of which act in response to opportunistic leishmania infection in HIV+ individuals (Pasquau et al. 2005). Furthermore, we uncovered 38 rapid expansions within hierarchical gene families for oxidative stress, a correlate also relevant to human health. As also seen in previous characterizations of SDs among primates, we observed substantial variation in their span and copy number (Gokcumen et al. 2013; Prado-Martinez et al. 2013; Sudmant et al. 2013); such variation could have broad evolutionary implications, either through its stimulation of regional de-stabilization or through its role in propagating adaptive gene families.
Understanding vervet population genetic history, including the genetic relationships among vervet subspecies, is essential for identifying loci that may contribute to susceptibility or resistance to a vast array of viral, microbial, and parasitic pathogens. We found that the most geographically isolated subspecies, C. a. aethiops, displayed the lowest heterozygosity, while C. a. pygerythrus, the subspecies with the broadest geographic range, displayed the highest. Previous taxonomic studies relied on smaller data sets and reached conclusions that are not directly comparable (Rosenberg and Nordborg 2002; Perelman et al. 2011; Guschanski et al. 2013). In particular, the divergence dates inferred in this first whole-genome analysis of vervet subspecies are considerably more recent than previous estimates (Perelman et al. 2011; Guschanski et al. 2013), largely owing to the fact that we take within-subspecies diversity into account. The phylogenetic relationships presented here are consistent with a proposed geographic origin of the genus in the Congo basin (Kingdon 1984) or the Rift Valley (Osman Hill 1966) and with purported migration routes (Osman Hill 1966). Yet, contrary to previous speculation (Osman Hill 1966), our results do not support a recent common origin of C. a. sabaeus and C. a. tantalus but indicate that C. a. sabaeus, C. a. aethiops, and C. a. tantalus represent independent descendants of the ancestral lineage that subsequently migrated to their respective ranges. Consistent with Osman Hill (1966), our results suggest that C. a. cynosurus split off from C. a. pygerythrus on its way south. Inferences of ancestral population sizes for particular subspecies using MSMC (Schiffels and Durbin 2014) are consistent with information about their geographic ranges. For example, MSMC indicates that C. a. tantalus maintained a relatively stable population size over much of its evolutionary history, supporting the suggestion that, among the subspecies, the current range of C. a. tantalus is closest to its ancestral range. Similarly, our MSMC results suggest that C. a. aethiops had the smallest effective population size for most of its evolutionary past, an observation consistent with its geographical isolation. To obtain a clearer picture of within-subspecies diversity, and to get a more nuanced view of the demographic history of the five subspecies, including historical patterns of gene flow, it will be necessary to sequence additional representatives of each of them.
Despite substantial progress with the experimental use of a variety of species deemed “model organisms,” the biological differences of such species from humans are often a hindrance to the development of human-specific therapies. This problem may be particularly important for infectious and immunological diseases, which are major contributors to the human health burden. Interest in vervets as a biomedical model species derives in part from its status as the most abundant natural host of SIV (Goldstein et al. 2000; Ma et al. 2013). As with other well-adapted natural simian hosts, vervets do not progress to immunodeficiency disease upon infection with species-specific SIV (Ma et al. 2014). A nearly complete high-quality reference MHC haplotype embedded within CAE17 (Fig. 5A; Supplemental Fig. S8) shows a high conservation of MHC structure between vervet and a rhesus macaque single-haplotype MHC region (Daza-Vamenta et al. 2004) but also demonstrates the need for further work to close remaining gaps and infer OWM ancestral origins of the MHC. With PCR typing of class I transcripts, we confirmed that Caribbean-origin vervets in US primate centers provide an accurate representation of the Caribbean populations from which they descend. Corroborating our findings, a recent study using other methods has also reported restricted MHC diversity in St. Kitts and Barbados vervet populations (Aarnink et al. 2014).
Restricted MHC diversity in the Caribbean-origin vervet population is remarkably similar to that observed in Mauritian-origin cynomolgus macaques (Wiseman et al. 2013). The macaque population of this isolated Indian Ocean island was characterized by only seven extended MHC haplotypes, together with recombinants between these ancestral haplotypes. As with the Caribbean-origin vervets, this macaque population was founded in the 17th century by a small number of monkeys introduced by Europeans (Bonhomme et al. 2008).
In summary, we provide the foundation for comparative studies of vervet subspecies and the different populations that comprise them. More importantly, this report provides an extensive but preliminary resource of single base variants, SDs, and deletions for Chlorocebus a. sabaeus. This ultrastructural framework can support trait mapping as well as preliminary studies into functional consequences of segregating protein-coding genes that have damaged structure.
Methods
Ethics statement
This study was carried out in strict accordance with the recommendations described in the Guide for the Care and Use of Laboratory Animals of the National Institute of Health, the Office of Animal Welfare, and the United States Department of Agriculture. All animal work was approved by the NIAID Division of Intramural Research Animal Care and Use Committees (IACUC), in Bethesda, MD (protocol # LMM-12E and LMM-6). The animal facilities are accredited by the American Association for Accreditation of Laboratory Animal Care. All procedures were carried out under ketamine anesthesia by trained personnel under the supervision of veterinary staff, and all efforts were made to ameliorate the welfare and to minimize animal suffering in accordance with the “Weatherall report for the use of nonhuman primates” recommendations. All techniques for trapping, sedation, and sampling were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Milwaukee. Collections were conducted under permits and permissions issued by the following: Ethiopian Wildlife Conservation Authority; Zambia Wildlife Authority; Wildlife Division, Forestry Commission, Republic of Ghana; and the Gambia Department of Parks and Wildlife Management.
Sequencing, assembly, and gene annotation
Genomic DNA from a male individual vervet (Chlorocebus a. sabaeus; animal ID#1994-021) within the pedigreed VRC, previously used to create the BAC library CHORI-252, provided the source material for all sequencing. An ABI3730 instrument was utilized to sequence BAC ends, and Roche 454 Titanium and Illumina HiSeq 2000 instruments to generate two independent assemblies; sequencing libraries of single fragment, overlapping, or paired 3- and 8-kbp reads were made according to requirements of the platform and the intended assembly algorithm. For assembly one, sequence coverage was generated on the Roche 454 Titanium instrument: fragment- 10x, 3kbp- 8x, and 8kbp- 1x based on an estimated 3-Gb genome. For assembly two, sequence coverage was generated on the Illumina HiSeq 2000 instrument, 100-bp read length: 45x overlapping, 45x 3kbp and 10x 8kbp, all on the. All 454 and ABI3730 reads were assembled with the Newbler algorithm (Roche); all Illumina reads were assembled with ALLPATHS (Gnerre et al. 2011). These versions were merged (Yao et al. 2012), misassembled regions were identified with BES mapping and corrected, and then data from finished BAC clones (n = 143) were integrated where gaps remained (Kurtz et al. 2004). This error-corrected assembly was used to construct individual chromosome files (Supplemental Methods). We utilized the fully described NCBI gene annotation pipeline (http://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/) for gene model predictions that were supported by RNA-seq samples not associated with this study: SAMN02356306, SAMN02439943, SAMN02665515, SAMN02665584, SAMN02665615.
Primate genome synteny
The synteny block arrangements of vervet chromosomes were defined in cytogenetic hybridization experiments, BAC end, and comparative whole-genome alignments. For cytogenetics experiments, using 570 human BAC clones, each position defined on the human genome (hg19) was positionally examined throughout the vervet genome. For each putative breakpoint, reiterative FISH experiments were performed to narrow the breakpoint interval. Examination of breakpoints shared with rhesus macaque used FISH previously performed on this species (http://www.biologia.uniba.it/macaque2013) (Ventura et al. 2007). All sequenced BAC ends were then aligned to the assembly to define synteny with human. Finally, human sequence was aligned with the rhesus macaque (rheMac2) and vervet (ChlSab1.1) genomes. Reciprocal best nets were created using the standard UCSC chain/net process; thus for every human base, the best single alignment to rhesus and vervet was found (Supplemental Fig. S2).
Repeats
The transposed element landscape was derived by a local version of RepeatMasker (http://www.repeatmasker.org/RMDownload.html) using the latest rhesus monkey transposon library (http://www.girinst.org/server/RepBase/index.php), as detailed in Supplemental Methods. Unique vervet sequences represent the best source for detecting new lineage-specific genomic elements, including active retrotransposons, and were extracted as described in Supplemental Methods. Coordinates of vervet lineage-specific sequences were then correlated with the two-way alignment and repetitive elements from the RepeatMasker report. Overlapped coordinates were then used to calculate the genome-wide coverage of lineage-specific repetitive elements, which were then used to populate a database containing unique vervet regions occupied by more than 69% of repetitive elements (purified from satellite and low complexity DNA) to compile lineage-specific TE insertions and activities (Supplemental Table S3). Potentially active elements were defined as being (1) those elements in unique regions that occupied more than 1% of their total genomic representation, (2) those that yielded more than 15 hits per element type, and (3), especially for those that were long terminal repeat elements (LTRs), all full-length retroviral elements or their remaining solitary LTRs (Supplemental Table 3). Finally, genome-wide information of the chromosomal percentage occupation of specific element families extracted from the vervet, human, and rhesus macaque RepeatMasker reports (Supplemental Tables S4–S6), were evaluated for their under- or overrepresentation in the different taxa by a two-sided confidential interval test (P < 0.05).
Structural variation detection
Examination of deletions and SDs used previously published methods robust for each SV type (Alkan et al. 2009; Layer et al. 2014). A total of six individual vervets (each belonging to the VRC), sequenced to average sequence coverage of 32×, were separately used to call shared and unique deletions (Supplemental Table S7). All sequences were aligned to the ChlSab1.1 reference with BWA-MEM (Li and Durbin 2009) prior to sequence alignment discordance filtering. To detect deletions, overlapping split-read and paired-end discordance coordinates were generated with LUMPY (Layer et al. 2014). A secondary read-depth filter was used to validate matching concordance (Abyzov et al. 2011). CNVnator (Abyzov et al. 2011) version 0.3 was used in conjunction with a bin size of 100 bp to call CNVs on all autosomes. CNVnator was then used to generate histogram files corresponding to each chromosome for each of the six samples.
For the discovery of SDs, the repetitive regions detected by RepeatMasker (www.repeatmasker.org) and Tandem Repeat Finder (Benson 1999) were first masked to remove most of the repetitive regions present in the ChlSab1.1 assembly, and a k-mer approach was used to further mask potential hidden repeats (see Supplemental Methods; Supplemental Fig. S9). Supplemental Table S20 summarizes the number of reads, mapping percentage, and coverage prior to and after removing PCR duplicates. Assessment of canonical pathway enrichment for genes linked with deleted or duplicated bases utilized a hypergeometric test (P < 0.05) against the human genome enabled in WebGestalt (Wang et al. 2013), while examination of gene family expansion and contraction used the updated CAFE3 (see Supplemental Methods; Han et al. 2013).
Subspecies sequencing and variant calling
Short insert libraries (200–400 bp) were used to generate ∼10× sequence coverage for five subspecies (Supplemental Table S14) (C.a. aethiops, C.a. tantalus, C.a. pygerythrus, C. a. cynosurus, and C. a. sabaeus [West Africa]). All sequences were aligned against the ChlSab1.1 reference using BWA-MEM. The Genome Analysis Toolkit (GATK) was used for SNP calling (McKenna et al. 2010). Following the recommended GATK workflow, a first low-threshold round of variant calling was used to perform local realignment and base quality recalibration. The steps of duplicate marking, local realignment, and quality score recalibration were performed as suggested by the GATK best practices guide. Given the lack of existing polymorphism data, a first round of conservative variant calling provided input to the quality score recalibration procedure. SNPs were then called using the GATK UnifiedGenotyper algorithm on all samples simultaneously. Subsequently, to reduce the amount of false positives, hard filters were applied to the SNP set, and variants were removed that failed any of the filters (Supplemental Table S22). The number of SNPs per chromosome, SNP density, and the percentage of SNPs that did not pass a given filter were calculated (Supplemental Fig. S7). To infer the ancestral state for each SNP, ChlSab1.1 was aligned against rheMac2 (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007) using NUCmer (Kurtz et al. 2004), with filtering for one-to-one mappings of a minimum length of 200 bp. Each SNP was checked for a mapping to the rhesus macaque genome for this region, and the state in rhesus macaque was assumed to be the ancestral state.
Analysis of vervet subspecies
The polymorphic sites were used to calculate the pairwise difference matrix across vervets (Supplemental Table S15). Diagonal entries correspond to the comparison of the two chromosomes within each diploid individual; off-diagonal entries correspond to an average of the four possible pairwise sequence comparisons. For each subspecies, coalescent effective population sizes were calculated using the relation π = 4NeµL, where π is the diagonal entries in Supplemental Table S15, µ = 1.5 × 10−8 is the expected per base-pair mutation rate, and L = 2.5 × 109 is the mutational target size—in our case the genome size minus all masked intervals and positions where the reference base is missing.
In a neutral model, the number of pairwise differences is proportional to the genomic average of the time to the common ancestor of each sequence pair (coalescent time). An UPGMA (unweighted pair group method with arithmetic mean) tree was constructed which reflects these relationships using the R software package “phangorn” (Schliep 2011). To get from individual relationships to species relationships, the average within-species coalescent time was subtracted from the between-species coalescent time, i.e., Tsplit = 1/(2µl) × (πbetween−πwithin) was estimated for each pair of subspecies. This estimate assumes that the effective size of each ancestral population before the split was an average of the current effective population sizes (see Supplemental Table S16, where a generation time of 8.5 yr was assumed). These estimates assume instantaneous speciation and the absence of subsequent gene flow. MSMC software in two-haplotype mode (Schiffels and Durbin 2014) was used to estimate effective population size among vervet subspecies.
MHC diversity
To define synteny differences, the rhesus macaque MHC region (Daza-Vamenta et al. 2004) was aligned to a tile path of finished vervet BAC sequences representing the MHC region (Supplemental Fig. S8) using MegaBLAST at a word size of 100 and an e-value of 0.05. The resulting alignments were filtered with Genome Workbench (http://www.ncbi.nlm.nih.gov/tools/gbench/). Whole blood or PBMC samples obtained from 51 captive and 32 feral vervets (Supplemental Table S18) were used for characterization of MHC class I sequences and MHC haplotypes. Total RNAs were isolated from whole blood or PBMC. First-strand cDNAs were used as templates for PCR with universal primers that bind highly conserved sequences in exons 2 and 4 that flank the highly polymorphic peptide binding domain of all NHP MHC class I gene products studied to date (Wiseman et al. 2013). PCR products were purified with AmPure-XP SPRI beads (Beckman Coulter), normalized, and pooled as MID-tagged amplicons. The resulting amplicon pools were subjected to pyrosequencing with a GS Junior instrument (Roche/454). After index binning of quality-filtered sequence data, and assembly of unidirectional contigs of identical sequence reads, BLAST was run to align contigs against a custom allele database of vervet class I sequences.
Data access
The vervet genome assembly in this study has been submitted to NCBI GenBank (http://www.ncbi.nlm.nih.gov/assembly/) under assembly accession number GCA_000409795.2. Deletion variants have been submitted to the NCBI database of genomic structural variation (dbVar; http://www.ncbi.nlm.nih.gov/dbvar) under accession number nstd114. The raw sequence data from this study have been submitted to the NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA240242.
Acknowledgments
Funding to R.K.W. was provided by NIH-NHGRI grant 5U54HG00307907. Support for the Vervet Research Colony was provided by NIH grant RR019963/OD010965 to J.R.K. Funding to N.B.F. was provided by NIH grants R01RR016300 and R01OD010980. The French National Agency for Research on AIDS and Viral Hepatitis (ANRS) provided funding to M.C.M.-T. Funding to M.R. and R.S. was provided by the Ministero della Universita’ e della Ricerca. Funding to K.D. was provided by Genome Canada and Genome Quebec. B.A. and R.N. acknowledge support from the Wellcome Trust (grant number WT095908) and the European Molecular Biology Laboratory. We thank the following organizations for providing sampling permits and permissions: Ethiopian Wildlife Conservation Authority; Zambia Wildlife Authority; Wildlife Division, Forestry Commission, Republic of Ghana; and Gambia Department of Parks and Wildlife Management. We thank Dr. James L. Blanchard and Dr. Ivona Pandrea for vervet samples used to characterize MHC. We thank Josh McMichael and Josh Peck for figure assistance. We thank Joanne Nelson and Barbara Gillam for data submission. We thank the members of the library production group led by Catrina Fronick and sequencing led by Matt Cordes. We also thank the Medical Research Council Unit (MRC), The Gambia for their assistance, as well as all the veterinarians who worked with us to safely obtain samples.
Author contributions: Project leadership and coordination: W.C.W., N.B.F., R.K.W., G.M.W., A.J.J. Vervet resources: J.R.K., T.R.T., M.J.J., C.A.S., E.R., A.N., O.-W.C., Y.J.J., C.A.S., B.J., M.C.M.-T., J.M.B., M.D., M.A., N.T. Sub-species phylogeny: H.S., M.N. Assembly curation: K.D., L.W.H., P.M., J.W., N.J., J.H., V.R., Y.H. Gene annotation: B.A., D.W., F.T.-N., G.P.S. Repeat analysis: J.S., G.C., A.N. Genome synteny: B.J.R., M.R., N.A., O.C., R.S. Structural variation: C.T., M.J.M., K.K., R.G.-P., T.M.-B. MHC data: R.W.W., J.J.T., J.A.K., J.E.S., D.H.O'C., M.K., T.G., R.Z. Gene family evolution: M.W.H., G.W.C.T.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.192922.115.
References
- Aarnink A, Jacquelin B, Dauba A, Hebrard S, Moureaux E, Muller-Trutwin M, Blancher A. 2014. MHC polymorphism in Caribbean African green monkeys. Immunogenetics 66: 353–360. [DOI] [PubMed] [Google Scholar]
- Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. 2009. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 41: 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkuraya FS. 2015. Natural human knockouts and the era of genotype to phenotype. Genome Med 7: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan JS, Short M, Taylor ME, Su S, Hirsch VM, Johnson PR, Shaw GM, Hahn BH. 1991. Species-specific diversity among simian immunodeficiency viruses from African green monkeys. J Virol 65: 2816–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Dennis MY, Huddleston J, Sudmant PH, Steinberg KM, Rosenfeld JA, Miroballo M, Graves TA, Vives L, Malig M, et al. 2014. Palindromic GOLGA8 core duplicons promote chromosome 15q13.3 microdeletion and evolutionary instability. Nat Genet 46: 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Eichler EE. 2006. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet 7: 552–564. [DOI] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhomme M, Blancher A, Cuartero S, Chikhi L, Crouau-Roy B. 2008. Origin and number of founders in an introduced insular primate: estimation from nuclear genetic data. Mol Ecol 17: 1009–1019. [DOI] [PubMed] [Google Scholar]
- Cagliani R, Riva S, Biasin M, Fumagalli M, Pozzoli U, Lo Caputo S, Mazzotta F, Piacentini L, Bresolin N, Clerici M, et al. 2010. Genetic diversity at endoplasmic reticulum aminopeptidases is maintained by balancing selection and is associated with natural resistance to HIV-1 infection. Hum Mol Genet 19: 4705–4714. [DOI] [PubMed] [Google Scholar]
- Carbone L, Harris RA, Gnerre S, Veeramah KR, Lorente-Galdos B, Huddleston J, Meyer TJ, Herrero J, Roos C, Aken B, et al. 2014. Gibbon genome and the fast karyotype evolution of small apes. Nature 513: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. 2011. A copy number variation morbidity map of developmental delay. Nat Genet 43: 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daza-Vamenta R, Glusman G, Rowen L, Guthrie B, Geraghty DE. 2004. Genetic divergence of the rhesus macaque major histocompatibility complex. Genome Res 14: 1501–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsworth JD, Jentsch JD, Groman SM, Roth RH, Redmond ED Jr, Leranth C. 2015. Low circulating levels of bisphenol-A induce cognitive deficits and loss of asymmetric spine synapses in dorsolateral prefrontal cortex and hippocampus of adult male monkeys. J Comp Neurol 523: 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli P, Stanyon R, Plesker R, Ferguson-Smith MA, O'Brien PC, Wienberg J. 1999. Reciprocal chromosome painting shows that the great difference in diploid number between human and African green monkey is mostly due to non-Robertsonian fissions. Mamm Genome 10: 713–718. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2014. Ensembl 2014. Nucleic Acids Res 42: D749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomsgaard A, Allan J, Gravell M, London WT, Hirsch VM, Johnson PR. 1990. Molecular characterization of simian lentiviruses from east African green monkeys. J Med Primatol 19: 295–303. [PubMed] [Google Scholar]
- Fukasawa M, Miura T, Hasegawa A, Morikawa S, Tsujimoto H, Miki K, Kitamura T, Hayami M. 1988. Sequence of simian immunodeficiency virus from African green monkey, a new member of the HIV/SIV group. Nature 333: 457–461. [DOI] [PubMed] [Google Scholar]
- Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, Cummins LB, Arthur LO, Peeters M, Shaw GM, et al. 1999. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397: 436–441. [DOI] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci 108: 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokcumen O, Tischler V, Tica J, Zhu Q, Iskow RC, Lee E, Fritz MH, Langdon A, Stutz AM, Pavlidis P, et al. 2013. Primate genome architecture influences structural variation mechanisms and functional consequences. Proc Natl Acad Sci 110: 15764–15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S, Ourmanov I, Brown CR, Beer BE, Elkins WR, Plishka R, Buckler-White A, Hirsch VM. 2000. Wide range of viral load in healthy African green monkeys naturally infected with simian immunodeficiency virus. J Virol 74: 11744–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschanski K, Krause J, Sawyer S, Valente LM, Bailey S, Finstermeier K, Sabin R, Gilissen E, Sonet G, Nagy ZT, et al. 2013. Next-generation museomics disentangles one of the largest primate radiations. Syst Biol 62: 539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MV, Thomas GW, Lugo-Martinez J, Hahn MW. 2013. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol 30: 1987–1997. [DOI] [PubMed] [Google Scholar]
- Harris RS. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, Pennsylvania State University, University Park, PA. [Google Scholar]
- Hirsch VM, Olmsted RA, Murphey-Corb M, Purcell RH, Johnson PR. 1989. An African primate lentivirus (SIVsm) closely related to HIV-2. Nature 339: 389–392. [DOI] [PubMed] [Google Scholar]
- Hirsch VM, McGann C, Dapolito G, Goldstein S, Ogen-Odoi A, Biryawaho B, Lakwo T, Johnson PR. 1993. Identification of a new subgroup of SIVagm in tantalus monkeys. Virology 197: 426–430. [DOI] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, Kremitzki C, et al. 2012. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 483: 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Jasinska AJ, Service S, Levinson M, Slaten E, Lee O, Sobel E, Fairbanks LA, Bailey JN, Jorgensen MJ, Breidenthal SE, et al. 2007. A genetic linkage map of the vervet monkey (Chlorocebus aethiops sabaeus). Mamm Genome 18: 347–360. [DOI] [PubMed] [Google Scholar]
- Jasinska AJ, Lin MK, Service S, Choi OW, DeYoung J, Grujic O, Kong SY, Jung Y, Jorgensen MJ, Fairbanks LA, et al. 2012. A non-human primate system for large-scale genetic studies of complex traits. Hum Mol Genet 21: 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasinska AJ, Schmitt CA, Service SK, Cantor RM, Dewar K, Jentsch JD, Kaplan JR, Turner TR, Warren WC, Weinstock GM, et al. 2013. Systems biology of the vervet monkey. ILAR J 54: 122–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jdo JW, Baldini A, Ward DC, Reeders ST, Wells RA. 1991. Origin of human chromosome 2: an ancestral telomere–telomere fusion. Proc Natl Acad Sci 88: 9051–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MJ, Hui H, Robertson DL, Muller MC, Barre-Sinoussi F, Hirsch VM, Allan JS, Shaw GM, Sharp PM, Hahn BH. 1994. Mosaic genome structure of simian immunodeficiency virus from West African green monkeys. EMBO J 13: 2935–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Lewis KL, Ng D, Singh LN, Wynter J, Brewer C, Brooks BP, Brownell I, Candotti F, Gonsalves SG, et al. 2015. Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations. Am J Hum Genet 96: 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustin Y, Souvorov A, Tatusova T, Lipman D. 2008. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Cooper DN. 2008. Molecular mechanisms of chromosomal rearrangement during primate evolution. Chromosome Res 16: 41–56. [DOI] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingdon J. 1984. East African mammals: an atlas of evolution. University of Chicago Press, Chicago. [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, Hall IM. 2014. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. 2003. Introduced mammals of the world. CSIRO Publishing, Clayton South, Victoria, Australia. [Google Scholar]
- Ma D, Jasinska A, Kristoff J, Grobler JP, Turner T, Jung Y, Schmitt C, Raehtz K, Feyertag F, Martinez Sosa N, et al. 2013. SIVagm infection in wild African green monkeys from South Africa: epidemiology, natural history, and evolutionary considerations. PLoS Pathog 9: e1003011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Jasinska AJ, Feyertag F, Wijewardana V, Kristoff J, He T, Raehtz K, Schmitt CA, Jung Y, Cramer JD, et al. 2014. Factors associated with simian immunodeficiency virus transmission in a natural African nonhuman primate host in the wild. J Virol 88: 5687–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Ryder OA, Eichler EE. 2009. Sequencing primate genomes: what have we learned? Annu Rev Genomics Hum Genet 10: 355–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman Hill WC. 1966. Primates: comparative anatomy and taxonomy. Vol. 6, Catarrhini, Cercopithecoidea, Cercopithecinae Edinburgh University Press, Edinburgh, UK. [Google Scholar]
- Pasquau F, Ena J, Sanchez R, Cuadrado JM, Amador C, Flores J, Benito C, Redondo C, Lacruz J, Abril V, et al. 2005. Leishmaniasis as an opportunistic infection in HIV-infected patients: determinants of relapse and mortality in a collaborative study of 228 episodes in a Mediterreanean region. Eur J Clin Microbiol Infect Dis 24: 411–418. [DOI] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y, et al. 2011. A molecular phylogeny of living primates. PLoS Genet 7: e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi L, Hodgson JA, Burrell AS, Sterner KN, Raaum RL, Disotell TR. 2014. Primate phylogenetic relationships and divergence dates inferred from complete mitochondrial genomes. Mol Phylogenet Evol 75: 165–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, Veeramah KR, Woerner AE, O'Connor TD, Santpere G, et al. 2013. Great ape genetic diversity and population history. Nature 499: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, et al. 2014. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 42: D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhesus Macaque Genome Sequencing and Analysis Consortium, et al. 2007. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316: 222–234. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Nordborg M. 2002. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat Rev Genet 3: 380–390. [DOI] [PubMed] [Google Scholar]
- Ruiz-Herrera A, Garcia F, Aguilera M, Garcia M, Ponsa Fontanals M. 2005. Comparative chromosome painting in Aotus reveals a highly derived evolution. Am J Primatol 65: 73–85. [DOI] [PubMed] [Google Scholar]
- Schiffels S, Durbin R. 2014. Inferring human population size and separation history from multiple genome sequences. Nat Genet 46: 919–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep KP. 2011. phangorn: phylogenetic analysis in R. Bioinformatics 27: 592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service SK, Teslovich TM, Fuchsberger C, Ramensky V, Yajnik P, Koboldt DC, Larson DE, Zhang Q, Lin L, Welch R, et al. 2014. Re-sequencing expands our understanding of the phenotypic impact of variants at GWAS loci. PLoS Genet 10: e1004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanyon R, Rocchi M, Bigoni F, Archidiacono N. 2012. Evolutionary molecular cytogenetics of catarrhine primates: past, present and future. Cytogenet Genome Res 137: 273–284. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Huddleston J, Catacchio CR, Malig M, Hillier LW, Baker C, Mohajeri K, Kondova I, Bontrop RE, Persengiev S, et al. 2013. Evolution and diversity of copy number variation in the great ape lineage. Genome Res 23: 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Weigl S, Carbone L, Cardone MF, Misceo D, Teti M, D'Addabbo P, Wandall A, Bjorck E, de Jong PJ, et al. 2004. Recurrent sites for new centromere seeding. Genome Res 14: 1696–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M, Antonacci F, Cardone MF, Stanyon R, D'Addabbo P, Cellamare A, Sprague LJ, Eichler EE, Archidiacono N, Rocchi M. 2007. Evolutionary formation of new centromeres in macaque. Science 316: 243–246. [DOI] [PubMed] [Google Scholar]
- Wang J, Duncan D, Shi Z, Zhang B. 2013. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 41: W77–W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Shiina T, Shimizu S, Hosomichi K, Yanagiya K, Kita YF, Kimura T, Soeda E, Torii R, Ogasawara K, et al. 2007. A BAC-based contig map of the cynomolgus macaque (Macaca fascicularis) major histocompatibility complex genomic region. Genomics 89: 402–412. [DOI] [PubMed] [Google Scholar]
- Wiseman RW, Karl JA, Bohn PS, Nimityongskul FA, Starrett GJ, O'Connor DH. 2013. Haplessly hoping: macaque major histocompatibility complex made easy. ILAR J 54: 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, Cooper DN, Li Q, Li Y, van Gool AJ, et al. 2011. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol 29: 1019–1023. [DOI] [PubMed] [Google Scholar]
- Yao G, Ye L, Gao H, Minx P, Warren WC, Weinstock GM. 2012. Graph accordance of next-generation sequence assemblies. Bioinformatics 28: 13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrei M, MacDonald JR, Merico D, Scherer SW. 2015. A copy number variation map of the human genome. Nature Rev Genet 16: 172–183. [DOI] [PubMed] [Google Scholar]
- Zimin AV, Cornish AS, Maudhoo MD, Gibbs RM, Zhang X, Pandey S, Meehan DT, Wipfler K, Bosinger SE, Johnson ZP, et al. 2014. A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol Direct 9: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]