

Abstract
Epigenetic processes in the brain involve the transfer of information arising from short-lived cellular signals and changes in neuronal activity into lasting effects on gene expression. Key molecular mediators of epigenetics include methylation of DNA, histone modifications, and noncoding RNAs. Emerging findings in animal models and human brain tissue reveal that epilepsy and epileptogenesis are associated with changes to each of these contributors to the epigenome. Understanding and influencing the molecular mechanisms controlling epigenetic change could open new avenues for treatment. DNA methylation, particularly hypermethylation, has been found to increase within gene body regions and interference with DNA methylation in epilepsy can change gene expression profiles and influence epileptogenesis. Posttranscriptional modification of histones, including transient as well as sustained changes to phosphorylation and acetylation, have been reported, which appear to influence gene expression. Finally, roles have emerged for noncoding RNAs in brain excitability and seizure thresholds, including microRNA and long noncoding RNA. Together, research supports strong effects of epigenetics influencing gene expression in epilepsy, suggesting future therapeutic approaches to manipulate epigenetic processes to treat or prevent epilepsy.
Complex changes in epigenetic modifications and gene expression patterns are associated with epileptogenesis. Some changes confer beneficial adaptive changes that reduce brain excitability; others are pathogenic.
The process of epileptogenesis is characterized by widespread changes to gene expression—both turn-on and turn-off—many of which are thought to underlie causal pathogenic processes, such as neuronal death, gliosis, neuroinflammation, changes to ion channel and neurotransmitter receptors, axonal and dendritic plasticity, and network-level remodeling (Pitkanen and Lukasiuk 2011; Sharma 2012). Maintenance of the epileptic state also features lasting changes to the expression of genes involved in many of the same processes. Transcription is driven by transcription factors and their associated protein complexes, but the ability to access and transcribe is strongly influenced by the state of the nucleosome, the histone–DNA assembly that is the basic unit of chromatin. Epigenetics is the umbrella term for processes that influence gene transcription by altering the chromatin state and that persist long after the initial stimulus has ceased. This includes chemical modifications to DNA bases and surrounding protein structures, thereby influencing chromatin compaction and the readability of the DNA code.
Epigenetic processes are required for the gene silencing that is essential for organism development, for example, X-chromosome inactivation in females and tissue-specific gene expression. Epigenetic control of gene expression is of major importance in normal brain development and function where transient stimuli, such as changes in patterns of neuronal activity, may need to be converted to long-lasting changes in gene expression (Graff et al. 2011). Epigenetics is increasingly recognized to play major roles in neurological diseases, including epilepsy. Indeed, a number of neurologic disorders (e.g., Rett syndrome) are caused by errors in genes that encode components of the epigenetic machinery, serving to highlight the relationship between epigenetics and epilepsy (Jakovcevski and Akbarian 2012).
The processes that contribute to establishing the epigenome of the epileptic brain include covalent modifications of DNA, such as cytosine methylation, posttranslational histone modifications, and noncoding RNA (ncRNA) expression (Graff et al. 2011; Sweatt 2013). This article attempts to provide an overview of the main epigenetic changes in experimental and human epilepsy and reflect on what functional interventions targeting these processes tell us about the role of epigenetics in disease pathogenesis, maintenance of the chronic epileptic state, and as targets for therapeutics. The reader is referred to excellent reviews for more in-depth analysis of specific epigenetic processes (Jaenisch and Bird 2003; Allis et al. 2008; Graff et al. 2011; Sweatt et al. 2013).
EPIGENETICS: MECHANISMS AND FUNCTIONS
Epigenetics (“epi” derived from the Greek for above or over) refers to changes to the physical structure that supports genes (Graff et al. 2011). Originally used to describe heritable changes independent of changes to the DNA code, epigenetics is now broadly applied to describe the regulation and structure of chromatin. Because epigenetic changes in nondividing neurons cannot be inherited, the term “neuroepigenetics” has been proposed to encompass those epigenetic processes in the brain (Sweatt 2013). The epigenome refers to the various structural modifications to DNA and histones that influence the three-dimensional shape of DNA and thereby support or oppose transcription. Epigenetic processes are important determinants of gene expression. This is perhaps most clear from the discordance between monozygotic twins, which increases with aging and environmental factors (Urdinguio et al. 2009). In the adult brain, the epigenome remains plastic where it plays a key role in memory formation and other processes. Most, and perhaps all, epigenetic marks appear to be reversible (Jakovcevski and Akbarian 2012).
Among the key epigenetic processes for which there are data in the field of epilepsy are (1) covalent modification of DNA, (2) posttranslational modification (PTM) of histones, and (3) ncRNA (Fig. 1).
Figure 1.

Basic epigenetics processes. Cartoon shows the basic structure of the nucleosome, comprising DNA (black line) wrapped around a core of histone proteins (green) and three of the major epigenetic processes including (1) methylation (Me) of DNA, (2) histone posttranslational modifications (PTMs) including Me, phosphorylation (P) and acetylation (Ac), and (3) noncoding RNAs (depicted as long hairpins and short double-stranded microRNAs) (based on data in Dulac 2010).
Data on other epigenetic processes, for example, nucleosome remodeling (Becker and Horz 2002), will not be discussed as data in epilepsy remain limited or absent. Control of the epigenome is vastly complex and still poorly understood. The epigenetic markings of DNA methylation and histone modifications are written and erased by large families of enzymes, whereas the reading of the epigenome further involves large numbers of proteins. Different cell types appear to express varying amounts of specific epigenetic factors. For example, MeCP2 is much more abundant in neurons than glia (Akbarian et al. 2001). Here follows a short overview of these three epigenetic processes.
DNA Methylation
Methylation of DNA, which typically has a silencing effect on transcriptional activity, is considered the most long lasting of the epigenetic changes (Dulac 2010). The process involves covalent addition of a methyl group from S-adenosylmethionine (SAM), the cell’s primary methyl donor, to a cytosine base to form 5-methylcytosine (5-mC). In somatic cells, 5-mC is primarily restricted to palindromic CpG dinucleotides (cytosines that are followed by a guanine), which are typically methylated in a symmetric manner. Methylation of cytosine in a non-CpG context (CpH, H = A, T, C) is prevalent in plants, but has also been reported in mammals for embryonic stem cells, induced pluripotent stem cells, oocytes, and mature neurons (Clark et al. 1995; Grandjean et al. 2007; Barres et al. 2009; Ziller et al. 2011; Ichiyanagi et al. 2013; Lister et al. 2013; Varley et al. 2013). Although CpG methylation is thought to lock and protect the genome providing a basal repressive mark, the biological impact of non-CpG methylation remains unsolved. Given that this mark is highly present in the mammalian brain, but rare or absent in other differentiated cell types, a unique role in brain development and function can be assumed (Lister et al. 2013).
Maintenance of DNA methylation patterns during cell division is crucial to animal development. DNA methyltransferase (DNMT)1 primarily recognizes and methylates hemimethylated CpG sites, and is, therefore, the major player in maintaining DNA methylation patterns during replication. Genomic methylation patterns in somatic differentiated cells are generally stable and heritable but, during gametogenesis and early embryonic development, methylation patterns are extensively reprogrammed (Reik et al. 2001). Thereby, a substantial part of the genome is actively demethylated, and subsequently remethylated, in a cell- and/or tissue-specific manner. De novo methylation is mediated by DNMT3A and B, but it remains unknown how specific patterns of methylation are established in the genome (Jaenisch and Bird 2003). DNMTs show dynamic expression during all stages of central nervous system (CNS) development. Homozygous deletion of DNMT genes in mice has been associated with embryonic lethality supporting the overall physiological importance of DNA methylation (Li et al. 1992; Okano et al. 1999). Conditional knockout of DNMTs in neural precursor cells of mice affected viability, whereas ablation of DNMT1, 3A, or both in postmitotic neurons altered cell survival, morphology, synaptic plasticity, learning, and memory (Fan et al. 2001; Feng et al. 2010). DNA methylation is, therefore, a requirement for normal CNS function, and altered DNMT expression and enzymatic activity have been identified in many neurological disorders including autism spectrum disorders (James et al. 2004; Feinberg 2007), schizophrenia (Grayson et al. 2005), Alzheimer’s disease (Scarpa et al. 2003; Graff et al. 2012), brain tumors (Hegi et al. 2005), spinal muscular atrophy (Hauke et al. 2009), and epilepsy.
The process of active DNA demethylation involves a set of less-well-understood processes and proteins that include TET1 and GADD45β (Bhutani et al. 2011). TET proteins are Fe2+- and 2-oxoglutarate-dependent dioxygenases that successively oxidize 5-mC to 5-hydroxymethylcytosine (5-hmC). The function of 5-hmC is still matter of debate, but there is some evidence for 5-hmC-mediated epigenetic dynamics during postnatal development and aging in the rodent and human brain (Szulwach et al. 2011; Wang et al. 2012; Hahn et al. 2013). It could be shown that transcriptional activity is associated with intragenic enrichment of hydroxymethylation. Although cell-type-specific genes show conserved 5-hmC patterns in the fetal and adult brain, loss of 5-hmC is associated with transcriptional down-regulation during development (Lister et al. 2013). 5-hmC is involved in higher-order brain function, given that Tet1 knockout animals show significant reduction in 5-hmC levels in the cortex and hippocampus accompanied by abnormal hippocampal long-term depression and impaired memory extinction (Rudenko et al. 2013). Consistent with this, postmortem brains of Alzheimer patients displayed broad alterations in 5-hmC patterns (Chouliaras et al. 2013).
Methyl-CpG-binding domain proteins, such as MBD1-4, MeCP2, or KAISO, are the established “readers” of DNA methylation. Hence, the current consensus for their function is 5-mC-dependent binding and repression. MeCP2 is preferentially expressed in the brain promoting a specific role in brain development and function. Despite its role in 5-mC binding and associated gene repression, MeCP2 has also been identified as the major 5-hmC-binding protein in the brain. Both marks, 5-hmC and MeCP2, were highly enriched in active genes in differentiated CNS cell types in vivo supporting a new role in neuronal gene activation (Mellen et al. 2012). There is also evidence for a yet-unappreciated interaction between long noncoding (lnc)RNA and MeCP2 facilitating MeCP2 target recognition and mediated gene regulation (Maxwell et al. 2013). Mutations of MeCP2 are the primary cause of classic Rett syndrome (Amir et al. 1999). Clinical manifestations of Rett syndrome, such as mental retardation, seizures, muscular hypotonia, and acquired microcephaly, may result from aberrant expression of target genes that escape proper regulation on the loss-of-function of MeCP2.
Although most DNA methylation is stable in the brain, a fraction is dynamically regulated by neuronal activity (Martinowich et al. 2003; Guo et al. 2011), which may well have implications in the pathogenesis epilepsy. Although hypermethylation of DNA in promoter regions is invariably associated with transcriptional silencing, methylation of DNA within the gene body can promote transcription, for example, from alternative promoters, or affect splicing (Ball et al. 2009; Laurent et al. 2010). The persistence of DNA methylation marks is context specific and can be either transient or lifelong.
Histone Modification
Histones are a family of conserved basic proteins, which are core components of the nucleosome. DNA is wrapped around an octamer of histones, normally comprising two subunits of core histones H2A, H2B, H3, and H4 (Graff et al. 2011). The amino terminal of these histones contains sites that are amenable to PTMs that affect the affinity for DNA and other binding proteins. Histone PTMs include acetylation, methylation, phosphorylation, and ubiquitination. The number of possible different combinations of histone PTMs is estimated to be >100 (Jakovcevski and Akbarian 2012). PTMs affect the compaction of chromatin and, therefore, change their accessibility to the transcriptional machinery. Combinations of these histone marks form a code that influences the transcriptional activation state of a particular gene locus (Graff et al. 2011). Expression of noncore (so-called variant) histones is also associated with transcriptional effects (Jakovcevski and Akbarian 2012).
The influence of histone PTMs on gene transcription is not as well understood as that of DNA methylation. Single or combined histone PTMs are not uniquely predictive of transcriptional silencing or activation and are thought to influence the capacity for changes in transcription to ensue, rather than directly contributing to silencing per se (Dulac 2010). Moreover, histone PTMs are not thought to be as persistent as DNA methylation, ranging from minutes to hours.
Acetylation of histones on lysine residues is invariably associated with transcriptional activity at the associated gene locus. This is thought to be because the addition of the acetyl group reduces the affinity of the protein for DNA (Graff et al. 2011). The process is catalyzed by histone acetyltransferases (HATs). Removal of an acetyl group is performed by histone deacetylases (HDACs), and the degree of acetylation is dependent on the balance of HAT and HDAC activity (Hwang et al. 2013). Methylation of histones also influences transcription by attracting chromatin-modifying enzymes but, unlike acetylation, can either promote or repress transcription depending on the site and number of histone methylations. For example, H3K27/9me3 is a repressive mark, whereas H3K4me and H4K12ac promote a transcriptionally active state (Jakovcevski and Akbarian 2012). Histones can also be phosphorylated on a number of residues, and this is usually associated with transcriptional activity (Graff et al. 2011).
ncRNA
ncRNA is usually included as a subgroup of the epigenetic process (Hwang et al. 2013; Qureshi and Mehler 2013; Sweatt 2013). ncRNA refers to any transcribed RNA, which does not code or cannot be translated into protein and is usually divided according to size into short (<200 nt) and long (>200 nt) forms. Molecules in both classes of ncRNA have been implicated in regulating epigenetic effects and direct roles in relation to epilepsy have been suggested in recent studies (Qureshi and Mehler 2012; Henshall 2014).
MicroRNAs (miRNAs) are the best understood class of short ncRNAs. Their mature sequence is 19 to 24 nt in length and their principal mechanism of action is posttranscriptional gene silencing in the cytoplasm. miRNAs achieve this by sequence-specific binding to complementary bases in a region of the target messenger RNA (mRNA), usually the 3′ untranslated region. To function, miRNAs are uploaded to the RNA-induced silencing complex (RISC) that contains argonaute-2 (AGO2), which brings together the miRNA and target mRNA resulting in either degradation of the mRNA or translational repression. Although only ∼2500 miRNAs have been identified in the human genome, targeting requires just a 7–8 nt “seed” match. As a result, an individual miRNA can influence expression of perhaps hundreds of mRNAs. Evidence for this multitargeting mechanism of action in the brain was recently provided for a single miRNA (miR-128). The mouse brain RISC contained ∼1000 predicted mRNA targets, whereas deletion of miR-128 produced changes to the expression levels of a similar number of mRNAs (Tan et al. 2013). This supports miRNA as metacontrollers of gene expression. In addition to these posttranscriptional functions, nuclear roles for miRNAs in regulating gene expression at the level of transcription have been shown. The brain-enriched miR-320 directs transcriptional silencing of POLR3D, a cell-cycle gene, directly at the promoter region by promoting association of AGO1, EZH2, a histone methyltransferase polycomb protein, and the histone repressor mark H3K27me3 (Kim et al. 2008). Gene-silencing effects at the transcriptional level have also been reported recently for other miRNAs (Benhamed et al. 2012).
lncRNA represents the other major category of ncRNA. They are a highly heterogeneous family of molecules that include enhancer RNAs (eRNAs), natural antisense transcripts (NATs), and intergenic RNA (Qureshi and Mehler 2012). lncRNAs are predominantly localized to the nucleus and enriched in the chromatin fraction of the cell consistent with potential epigenetic roles. Functional roles for most lncRNAs remain to be determined, and the reader is referred elsewhere for recent reviews on the topic (Ng et al. 2013; Schaukowitch and Kim 2014). Although lncRNAs can alter transcription in either direction, most functional work to date suggests that lncRNAs promote transcriptional silencing, although eRNA is thought to promote an open chromatin state amenable to transcription. More than 100,000 lncRNAs have been identified, although the number is likely to increase. Functional data on these remain scarce. Several lncRNAs display unique patterns of subfield expression and intracellular localization in the hippocampus (Mercer et al. 2008), and such precise spatiotemporal patterns of expression imply biological function.
MODULATION OF EPIGENETIC PATHWAYS IN EPILEPSY
Given the exquisite and elaborate complexity of the regulatory mechanisms governing the epigenome, it is unsurprising that insults to the brain severe enough to precipitate epilepsy produce major changes. Aberrant patterns of epigenetic modification could erroneously affect gene expression and contribute to disease pathogenesis or maintenance of the chronic epileptic state. Affected genes are likely to be involved in normal neuronal homeostasis, excitability, cell survival, and inflammatory processes (Lubin 2012; Roopra et al. 2012). Evidence has now emerged to support each of the major epigenetic processes as being altered in epilepsy. Research has also shown that these are causally important in some cases, contributing to both beneficial adaptive changes to reduce excitability as well as maladaptive pathogenic changes. It is notable that a number of genetic disorders that arise because of mutations in genes encoding epigenetic proteins have clinical phenotypes that include epilepsy. This includes Rett syndrome (mutations in MeCP2, a methylated DNA-binding protein), other methylated DNA-binding proteins (Williams et al. 2010), and a neuron-restrictive silencer factor (NRSF)-interacting protein PRICKLE1 (Bassuk et al. 2008).
DNA METHYLATION AND EPILEPSY
Evidence for a role of DNA methylation in the pathogenesis of seizures first came from in vitro studies demonstrating a decrease of spontaneous excitatory neurotransmission and network activity following 5-aza-cytidine or zebularine-mediated inhibition of DNMTs in hippocampal slices (Levenson et al. 2006) and hippocampal primary neurons (Nelson et al. 2008). A number of genes previously associated with neuronal hyperactivity and seizures in vitro and in vivo have been described with altered DNA methylation profiles in their promoters including Bdnf, Gria2, and Grin2b (Martinowich et al. 2003; Machnes et al. 2013; Ryley Parrish et al. 2013). There is further evidence for aberrant DNA methylation of the Reelin (RELN) promoter associated with granule cell dispersion, a frequent migration defect targeting the hippocampal granule cell layer, in temporal lobe epilepsy (TLE) patients with hippocampal sclerosis (Kobow et al. 2009). Most recently, increased carboxypeptidase A6 (CPA6) promoter methylation was observed in TLE patients with hippocampal sclerosis and a known history of febrile seizures (Belhedi et al. 2014). CPA6 is involved in the selective biosynthesis of neuroendocrine peptides, and loss-of-function mutations have been related to seizures and epilepsy (Sapio et al. 2012). Furthermore, increased DNMT1 and DNMT3A expression have been described in temporal neocortex samples obtained from TLE patients, which is in line with the localized DNA methylation changes described above, but could also point to even broader changes of DNA methylation in the pathogenesis of focal epilepsies (Kobow and Blumcke 2012; Zhu et al. 2012). In fact, methyl-CpG-capture-associated massive parallel sequencing (methyl-seq) as well as array-based analyses of genomic DNA methylation patterns in two different rodent seizure models identified genome-wide changes in DNA methylation following status epilepticus (SE) as well as epileptic tolerance (Miller-Delaney et al. 2012), and identified a methylation signature distinguishing chronic epileptic animals from healthy controls (Kobow et al. 2013). Although most genes in response to SE were characterized by hypomethylation events, more acquisition than loss of DNA methylation was reported in the pilocarpine-induced chronic rat epilepsy model. Here, >2500 individual loci showed differential DNA methylation targeting mainly CpG islands within genic and nongenic regions. About 250 gene loci (∼1% of all genes) showed a strong indirect correlation between DNA methylation and gene expression. This was particularly true when DNA methylation targeted the gene body, but not the promoter. Gene ontology and functional analysis revealed enrichment of genes implicated in cytoskeleton organization, immune response and inflammation, neuronal development and differentiation, cell adhesion, as well as cell projection. Furthermore, the observed changes affected genes involved in calcium signaling, DNA binding and transcription, programmed cell death, and synaptic transmission (Fig. 2) (Kobow et al. 2013).
Figure 2.

DNA methylation in epilepsy. (A) Biochemical pathway responsible for methylation and demethylation of DNA. (B) Heat map depicting hierarchical clustering of control and epileptic samples and genomic regions according to differential methylation profiles (yellow, methylation-up; red, methylation-down). (C) Bisulfite sequencing results showing differential methylation for two different genes. Open circles, Unmethylated CpGs; closed circles, methylated CpGs. (From Kobow et al. 2013, modified, with permission, from the authors.)
In another study, a global increase in hippocampal DNA methylation was correlated with an increase in DNMT activity, disruption of adenosine homeostasis, and spontaneous recurrent seizures in mice. Adenosine augmentation >10 days reversed DNA hypermethylation seen in the epileptic brain, inhibited hippocampal sprouting of mossy fibers, and prevented the progression of epilepsy for at least 3 months (Williams-Karnesky et al. 2013). However, pharmacological inhibition of DNA methylation was not found to alter epilepsy in another recent study (Ryley Parrish et al. 2013). Nevertheless, these data suggest that DNA methylation is involved in epileptogenesis and propagation of the chronic disease state, accounting for the synergistic misregulation of multiple genes. It needs to be noted that the number of genes targeted by DNA methylation seems to be restricted in all of these studies, but the specificity of the finding remains unresolved. The limited overlap in methylation patterns among models is a potential concern. It could be related to physiological differences with respect to species, strain, and method of seizure induction, or depend on the time point and tissue analyzed, as well as the platform technology used to screen for genomic DNA methylation. Future studies will need to prove whether specific DNA methylation events can serve as potential biomarkers for early detection of disease onset, prognosis, or monitoring of disease after therapy.
HISTONE MODIFICATIONS AND EPILEPSY
To date, only a small number of studies have investigated histone modifications in epilepsy. In one early study, increased histone H4 acetylation was reported at the BDNF gene locus after pilocarpine-induced seizures in rats (Huang et al. 2002). Increased H4 acetylation also occurs after electroconvulsive seizures (Tsankova et al. 2004). In other early work, a rapid increase in phosphorylation of histone H3 was reported after pilocarpine- and kainate-induced seizures in mice (Crosio et al. 2003). This may promote acetylation of nearby lysine residues and transcription of the immediate early gene (IEG) c-fos (Crosio et al. 2003). Sng and coworkers (2006) reported acetylation and phosphorylation changes to two core histone proteins after SE in rats (Fig. 3).
Figure 3.
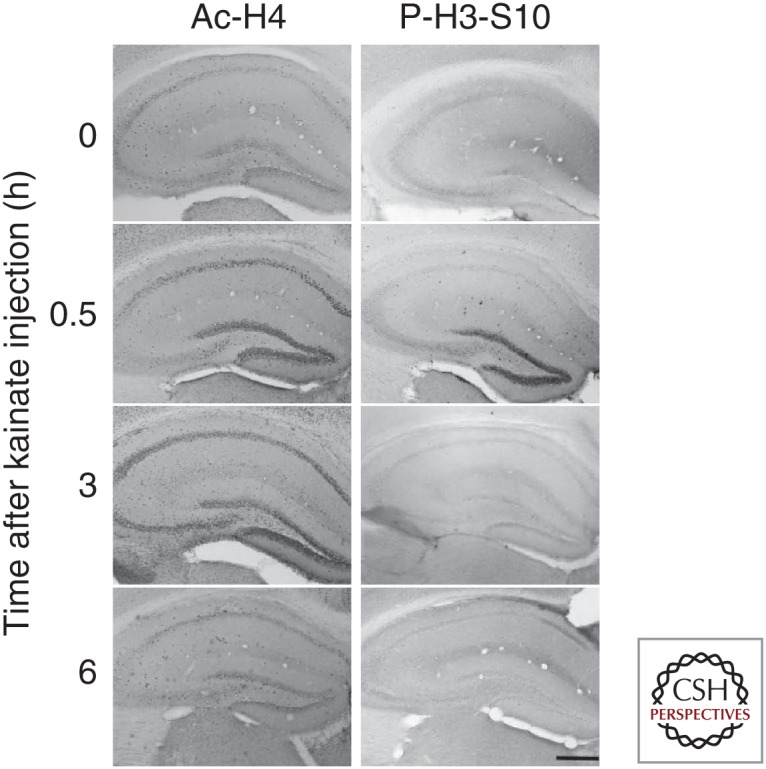
Histone modifications after seizures. Immunostaining showing the spatiotemporal profile of histone modifications Ac-H4 and phosphor (P)-H3-S10 after kainic acid–induced seizures in mice. Scale bar, 500 µm. (From Sng et al. 2006; adapted, with permission, from Wiley © 2006.)
In agreement with earlier work, H3 phosphorylation rapidly increased after kainate treatment and then decreased. This was restricted to dentate granule neurons, whereas acetylation of H4 appeared later and spread throughout the major hippocampal subfields. Further studies supported H4 acetylation associated with induction of the c-fos, but not c-jun (Sng et al. 2006). Other studies have reported PTM of histone variants following seizures in relation to DNA damage (Crowe et al. 2011). Expression of HDAC2 is up-regulated in experimental and human epilepsy (Huang et al. 2002, 2012), and predicted effects of changes in HDAC activity include transcriptional responses for several genes implicated in synaptic function, including ionotropic and metabotropic glutamate receptors (Huang et al. 1999; Kurita et al. 2012; Park et al. 2014).
A number of studies have explored the effects of HDAC inhibitors in epilepsy. The HDAC inhibitor trichostatin A (TSA) was found to prevent deacetylation of the Gria2 gene locus that encodes GluA2, a subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor that restricts calcium permeability and has postulated neuroprotective effects (Huang et al. 2002) and TSA increased basal acetylation and IEG expression (Sng et al. 2005). Pretreatment of mice with curcumin, which blocks acetylation, reduced the histone modifications and blocked c-fos induction indicating that histone PTMs directly influence gene expression after seizures (Sng et al. 2006). However, the drug also had an anticonvulsant effect, which, although potentially interesting, complicates interpretation of the curcumin effect on acetylation and gene expression. Sodium valproate (VPA) is a well-established antiepileptic drug (AED), which was also found to have HDAC inhibitory activity in addition to effects on ion channels and neurotransmission (Gottlicher et al. 2001). Interestingly, the anticonvulsant effects of VPA increase over time, which could be consistent with an epigenetic effect on gene transcription. However, unlike VPA, the more potent HDAC inhibitor TSA was not found to have anticonvulsant effects in the PTZ test suggesting that HDAC inhibition is unlikely to explain the antiseizure properties of VPA (Hoffmann et al. 2008). A lack of anticonvulsant effects of TSA has been reported by others (Taniura et al. 2006) and since has been confirmed for suberoylanilide hydroxamic acid (SAHA) (Rossetti et al. 2012). HDAC inhibitors may have other beneficial effects in epilepsy. HDAC inhibition reduces the aberrant neurogenesis, which may contribute to cognitive deficits in epilepsy (Jessberger et al. 2007) and neuroprotective effects of SAHA treatment were reported after SE (Rossetti et al. 2012). Taken together, histone modifications are seen as an important component of the epigenome changes associated with epilepsy and may represent potential therapeutic targets.
ncRNA
Short ncRNA
Several studies have now profiled miRNA expression in experimental models and human epileptic tissue (for a recent review, see Henshall 2014). These studies have identified changes to >100 different miRNAs in the epileptic rodent and human hippocampus. A similar number of miRNAs are altered in the acute wake of SE (Henshall 2013b). Cross-comparing these various studies reveal a number to be regulated in multiple models suggesting there are conserved “epilepsy miRNAs.” Experiments using small molecule miRNA inhibitors (chemically modified oligonucleotides called antagomirs) have shown functional roles for miR-34a, miR-132, and miR-184 in neuronal death after SE (Jimenez-Mateos et al. 2011; Hu et al. 2012; McKiernan et al. 2012b; Sano et al. 2012). Two additional miRNAs have been identified with effects on seizure thresholds and epilepsy. miRNA-134 is a brain-specific miRNA that targets the dendritic spine protein LIM kinase (Schratt et al. 2006) as well as cAMP response element-binding protein (CREB) (Gao et al. 2010), and miR-134 can also promote apoptosis (Zhang et al. 2014b). Expression of miR-134 is increased after SE and in experimental epilepsy (Jimenez-Mateos et al. 2012, 2014; Peng et al. 2013) and injection of antagomirs targeting miR-134 potently reduced seizures in mouse models of SE (Jimenez-Mateos et al. 2012, 2014). Injecting miR-134 antagomirs after SE also reduced the later development of epilepsy (Fig. 4) (Jimenez-Mateos et al. 2012).
Figure 4.

MicroRNA-134 as a target in epileptogenesis. (A) Neuronal localization of miR-134 in the mouse brain. (B) Status epilepticus (SE) in mice results in increased binding of miR-134 to Ago2, a component of the RNA-induced silencing complex. (C) Summary data on an electroencephalogram (EEG) and video monitoring of mice treated with antagomirs targeting miR-134 (Ant-134) or a nontargeting scrambled sequence (Scr). (Data from Jimenez-Mateos et al. 2012 and Henshall 2013a; modified, with permission.)
The mechanism of this effect is uncertain but spine density was altered in the mice and the antagomirs protected against excitotoxicity in vitro, which was LIM kinase-dependent (Jimenez-Mateos et al. 2012). More recently, another brain-enriched miRNA, miR-128, was identified to regulate seizures in mice (Tan et al. 2013). Loss of miR-128 led to fatal epilepsy, whereas ectopic expression of miR-128 reduced seizures triggered by kainic acid or picrotoxin (Tan et al. 2013). In a complementary finding to the miR-134 work, loss of miR-128 increased spine density (Tan et al. 2013), and miR-128 was linked to dysfunction of dopaminergic motor pathways rather than limbic/hippocampal circuitry. Together, these data indicate important roles for individual miRNAs in epilepsy.
Most of the in vivo targets of the miRNAs linked to epilepsy remain poorly defined. Protein levels of some of the validated targets of these miRNAs are changed in the predicted direction including LIM kinase 1, MAP3K9, CREB and components of the inflammatory pathways (Henshall 2013b), along with evidence for the direct loading of the miRNAs into the RISC after seizures (Jimenez-Mateos et al. 2011, 2012).
Other Links between miRNA and Epigenetics in Epilepsy
miRNAs have also been identified that target proteins with direct roles in epigenetics. NRSF/RE1-silencing transcription factor (REST) and its cofactors are targeted by miR-9, miR-124a, and miR-132 (Wu and Xie 2006), and each of these miRNAs has been found to be altered in epilepsy studies (Nudelman et al. 2010; Jimenez-Mateos et al. 2011; Pichardo-Casas et al. 2012; Peng et al. 2013; Risbud and Porter 2013). In turn, a number of cell-specific miRNAs have NRSF consensus sites in their promoter regions (Wu and Xie 2006), indicating that epigenetic silencing of miRNAs could lead to derepression of protein-coding genes in epilepsy. Transcription of several miRNAs is influenced by methylation of the miRNA transcriptional start sites (Bhadra et al. 2013). DNMTs are also direct targets of miRNAs, in particular the miR-29 family (Fabbri et al. 2007), which are also regulated in epilepsy (Hu et al. 2011b; Risbud and Porter 2013). Among miRNAs for which direct effects have been identified on transcription, let-7 family members, although not miR-320, have been found to be changed in a number of models/human epileptic tissue (Kan et al. 2012; McKiernan et al. 2012b; Risbud and Porter 2013). Also, loss of miR-101 was reported to result in increased expression of a histone-modifying enzyme (Varambally et al. 2008). Finally, a number of miRNAs localize to the nucleus of cells in human epileptic brain tissue, which are not there in control cells, suggesting a shift toward an increase in epigenetic functions for miRNAs in epilepsy (Kan et al. 2012). It is likely that examples of miRNA-epigenetic cross talk exist and will be of relevance to epilepsy. What remains unknown at this time is whether any of the miRNAs with direct epigenetic functions or which target proteins involved in epigenetic processes are functioning in an epigenetic manner in epilepsy.
lncRNA in Epilepsy
We know almost nothing about lncRNA expression in epilepsy, but research in other areas has suggested that important roles will emerge. The lncRNA Malat1 promotes dendritic spine density (Bernard et al. 2010) and loss of the lncRNA BC1 reduced convulsive thresholds (Zhong et al. 2009). Other lncRNAs have been implicated in the generation of GABAergic neurons (Qureshi and Mehler 2013) and a de novo mutation in an lncRNA (BX118339) was also recently implicated in a patient with West syndrome (Vandeweyer et al. 2012).
An emerging class of lncRNAs with potential relevance in epilepsy are NATs. The RNA transcript originates from the opposite strand of a sense (protein-coding) RNA transcript and remains associated with the transcriptional locus. The effect is usually to block expression of the coding gene by recruiting epigenetic gene-silencing machinery (Wahlestedt 2013). Recent work identified a NAT for the BDNF gene and showed that targeting the NAT using a 16-mer oligonucleotide antisense sequence up-regulated BDNF protein levels and exerted functional effects on neurogenesis in vivo (Modarresi et al. 2012). Because large numbers of genes may have associated NATs, this raises the potential to up-regulate certain genes in diseases of haplodeficiency, such as Dravet (Wahlestedt 2013).
A selection of the key findings on epigenetics in epilepsy are shown in Table 1.
Table 1.
Examples of epigenetic changes in in vivo experimental models and human epilepsy
| Epigenetic process | Model/tissue and species | Study/epigenetic change | Effects/other findings | References |
|---|---|---|---|---|
| DNA methylation | Hippocampus from TLE patients Rat model of SE (KA) Hippocampus from TLE patients Mouse model of SE (KA) Rat model of epilepsy (PILO) Rat model of epilepsy (KA) |
Increased RELN methylation Increased Grin2b methylation Decreased BDNF methylation Increased DNMT1/3a protein Genome-wide DNA methylation analysis Genome-wide DNA methylation and transcriptome analysis Increased methylation |
↓ Reelin expression ↓ Grin2b (NR2B) mRNA/protein ↑ BDNF mRNA/protein Hypomethylation of 275 genes Hypermethylation of 13 genes Hypomethylation of 1121 loci Hypermethylation of 1452 loci Adenosine augmentation opposed hypermethylation and prevented epilepsy progression |
Kobow et al. 2009 Ryley Parrish et al. 2013 Zhu et al. 2012 Miller-Delaney et al. 2012 Kobow et al. 2013 Williams-Karnesky et al. 2013 |
|
Histone modification |
Rat model of SE (PILO) Mouse model of SE (KA) Mouse model of SE (KA) Resected hippocampus from TLE patients and rat model of SE (PILO) |
Reduced acetylation of histone H4 Increased phosphorylation of histone H3 Increased acetylation of histone H4 and phosphorylation of H3 Increased HDAC2 expression |
↓ GluR2/Gria2 expression, which was prevented by HDAC inhibitor TCA Possible link to c-Fos induction Possible link to c-Fos induction Effects blocked by HAT inhibitor |
Huang et al. 2002 Crosio et al. 2003 Sng et al. 2006 Huang et al. 2012 |
|
Noncoding RNA |
Resected neocortex from TLE patients and model of SE (KA) Mouse model of miR-128 deficiency Mouse model of lncRNA BC1 deficiency West syndrome |
Increased miR-134 expression Loss of miR-128 Loss of BC1 lncRNA De novo mutation in lncRNA BX118339 |
Inhibition of miR-134 reduced post-SE development of epilepsy in mice Mice developed fatal epilepsy Lowered seizure threshold |
Jimenez-Mateos et al. 2012 Tan et al. 2013 Zhong et al. 2009 Vandeweyer et al. 2012 |
TLE, Temporal lobe epilepsy; KA, kainic acid; mRNA, messenger RNA; PILO, pilocarpine; SE, status epilepticus; lncRNA, long noncoding RNA.
OTHER EPIGENETIC FACTORS
A number of other epigenetic factors have been explored in epilepsy. Prominent among these is NRSF/REST (for a review, see Roopra et al. 2012). NRSF is implicated in silencing multiple genes via mechanisms that involve recruitment of MeCP2 and enhancement of DNA methylation. Protein levels of NRSF are very low in normal adult brain but increase rapidly after epileptogenic insults, such as SE, where it proceeds to promote silencing of key genes. Indeed, REST target genes are particularly overrepresented among down-regulated genes after SE. These include HCN1, an ion channel responsible for opposing dendritic excitability. Administration of an NRSF decoy oligonucleotide prevented REST-mediated down-regulation of HCN1 and restored electrophysiological properties in neurons (McClelland et al. 2011). It remains unknown what processes underlie the direction or silencing efficacy of REST among its many targets. Moreover, the assumption that NRSF-mediated gene repression promotes epileptogenesis was recently challenged by evidence that selective loss of NRSF from forebrain neurons accelerates kindling progression in mice (Hu et al. 2011b). Mixed results were reported when REST was knocked out of all neurons, with no differences reported for clonic components at low doses in the PTZ model but protection against tonic components and death at higher doses (Liu et al. 2012). Broad targeting of NRSF, therefore, may not be viable as a disease-modifying treatment in epilepsy.
MeCP2 is a key mediator of gene silencing through effects on DNA methylation, although it can also promote gene expression. Recent work showed that conditional mice lacking MeCP2 in the forebrain display seizures (Zhang et al. 2014a). This brings into line animal model data with the clinical phenotype of Rett syndrome, in which seizures are a common comorbidity. However, overexpression of MeCP2 may also disturb neuronal networks and predispose to seizures, indicating the need for a precise gene dosage for normal brain function (Bodda et al. 2013).
PERSPECTIVES, REMAINING CHALLENGES, AND FUTURE STUDIES ON EPIGENETICS IN EPILEPSY
Research into epigenetic processes in epilepsy is still in its infancy. Many enzymes linked to epigenetic changes, such as those that cause histone PTMs, are known to have other roles in cell physiology. Thus, changes in their expression and function in epilepsy cannot be always assumed to relate to their role in epigenetic processes. Some components of the epigenetic machinery have been found to localize outside the nucleus, including in synaptically localized mitochondria. This raises the possibility of epigenetic changes to the mitochondrial genome (Jakovcevski and Akbarian 2012). We have few data in epilepsy for many epigenetic processes. This includes whether epilepsy is associated with specific demethylation of DNA, nucleosome remodeling, lncRNA, or RNA editing. Another epigenetic process, genetic imprinting, is also poorly understood. This refers to the epigenetic process whereby one or other parental gene is deactivated and is believed to affect a hundred or more genes. This may be relevant to sex differences in epilepsy. Notably, several neurological disorders related to errors or failure of genetic imprinting feature high rates of epilepsy, including Angelman syndrome.
Interesting unanswered questions include whether conserved patterns of histone PTMs or DNA methylation exist in epilepsy that are unique or overlapping depending on etiology, and whether epigenetic markings changed in epilepsy overlap with any established in the aging brain. Unraveling the combinatorial complexity of the altered brain epigenome in epilepsy is a task of enormous complexity. These challenges are not unique to epilepsy and must be determined in the field of neuroepigenetics, along (Sweatt 2013) with the potential to realize therapeutic manipulation of epigenetics for brain disorders, such as epilepsy.
ACKNOWLEDGMENTS
D.C.H. thanks the following funding agencies for supporting work on epigenetic processes in epilepsy: Science Foundation Ireland (13/IA/1891, 12/COEN/18, 11/TIDA/B1988, 08/IN.1./B1875), Health Research Board (HRB/MRCG 2011/7, HRA-POR-2013-325), and National Institutes of Health (NIH/NINDS) (R56 073714). K.K. thanks EpiGENet (DFG Bl 421/3-1).
Footnotes
Editors: Gregory L. Holmes and Jeffrey L. Noebels
Additional Perspectives on Epilepsy: The Biology of a Spectrum Disorder available at www.perspectivesinmedicine.org
REFERENCES
- Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. 2001. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis 8: 784–791. [DOI] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T, Reinberg D. 2008. Epigenetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188. [DOI] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. 2009. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotech 27: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres R, Osler ME, Yan J, Rune A, Fritz T, Caidahl K, Krook A, Zierath JR. 2009. Non-CpG methylation of the PGC-1α promoter through DNMT3B controls mitochondrial density. Cell Metab 10: 189–198. [DOI] [PubMed] [Google Scholar]
- Bassuk AG, Wallace RH, Buhr A, Buller AR, Afawi Z, Shimojo M, Miyata S, Chen S, Gonzalez-Alegre P, Griesbach HL, et al. 2008. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Gen 83: 572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker PB, Horz W. 2002. ATP-dependent nucleosome remodeling. Ann Rev Biochem 71: 247–273. [DOI] [PubMed] [Google Scholar]
- Belhedi N, Perroud N, Karege F, Vessaz M, Malafosse A, Salzmann A. 2014. Increased CPA6 promoter methylation in focal epilepsy and in febrile seizures. Epilepsy Res 108: 144–148. [DOI] [PubMed] [Google Scholar]
- Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. 2012. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat Cell Biol 14: 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, Zhang MQ, Sedel F, Jourdren L, Coulpier F, et al. 2010. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J 29: 3082–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra T, Bhattacharyya M, Feuerbach L, Lengauer T, Bandyopadhyay S. 2013. DNA methylation patterns facilitate the identification of microRNA transcription start sites: A brain-specific study. PLoS ONE 8: e66722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N, Burns DM, Blau HM. 2011. DNA demethylation dynamics. Cell 146: 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodda C, Tantra M, Mollajew R, Arunachalam JP, Laccone FA, Can K, Rosenberger A, Mironov SL, Ehrenreich H, Mannan AU. 2013. Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. Am J Path 183: 195–210. [DOI] [PubMed] [Google Scholar]
- Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, Steinbusch HW, Coleman PD, Rutten BP, van den Hove DL. 2013. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging 34: 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Frommer M. 1995. CpNpG methylation in mammalian cells. Nat Genet 10: 20–27. [DOI] [PubMed] [Google Scholar]
- Crosio C, Heitz E, Allis CD, Borrelli E, Sassone-Corsi P. 2003. Chromatin remodeling and neuronal response: Multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci 116: 4905–4914. [DOI] [PubMed] [Google Scholar]
- Crowe SL, Tsukerman S, Gale K, Jorgensen TJ, Kondratyev AD. 2011. Phosphorylation of histone H2A.X as an early marker of neuronal endangerment following seizures in the adult rat brain. J Neurosci 31: 7648–7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulac C. 2010. Brain function and chromatin plasticity. Nature 465: 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. 2007. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci 104: 15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, et al. 2001. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci 21: 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. 2007. Phenotypic plasticity and the epigenetics of human disease. Nature 447: 433–440. [DOI] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. 2010. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13: 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, Mak G, Kim D, Su SC, Tsai LH. 2010. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466: 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al. 2001. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20: 6969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Kim D, Dobbin MM, Tsai LH. 2011. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev 91: 603–649. [DOI] [PubMed] [Google Scholar]
- Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, et al. 2012. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483: 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean V, Yaman R, Cuzin F, Rassoulzadegan M. 2007. Inheritance of an epigenetic mark: The CpG DNA methyltransferase 1 is required for de novo establishment of a complex pattern of non-CpG methylation. PLoS ONE 2: e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. 2005. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci 102: 9341–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, et al. 2011. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 14: 1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, Jui J, Jin SG, Jiang Y, Pfeifer GP, et al. 2013. Dynamics of 5-hydroxymethylcytosine and chromatin marks in mammalian neurogenesis. Cell Rep 3: 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauke J, Riessland M, Lunke S, Eyupoglu IY, Blumcke I, El-Osta A, Wirth B, Hahnen E. 2009. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet 18: 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. 2005. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352: 997–1003. [DOI] [PubMed] [Google Scholar]
- Henshall DC. 2013a. Antagomirs and microRNA in status epilepticus. Epilepsia 54: 17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall DC. 2013b. MicroRNAs in the pathophysiology and treatment of status epilepticus. Front Mol Neurosci 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall DC. 2014. MicroRNA and epilepsy: Profiling, functions and potential clinical applications. Curr Opin Neurol 27: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann K, Czapp M, Loscher W. 2008. Increase in antiepileptic efficacy during prolonged treatment with valproic acid: Role of inhibition of histone deacetylases? Epilepsy Res 81: 107–113. [DOI] [PubMed] [Google Scholar]
- Hu K, Zhang C, Long L, Long X, Feng L, Li Y, Xiao B. 2011a. Expression profile of microRNAs in rat hippocampus following lithium-pilocarpine-induced status epilepticus. Neurosci Lett 488: 252–257. [DOI] [PubMed] [Google Scholar]
- Hu XL, Cheng X, Cai L, Tan GH, Xu L, Feng XY, Lu TJ, Xiong H, Fei J, Xiong ZQ. 2011b. Conditional deletion of NRSF in forebrain neurons accelerates epileptogenesis in the kindling model. Cereb Cortex 21: 2158–2165. [DOI] [PubMed] [Google Scholar]
- Hu K, Xie YY, Zhang C, Ouyang DS, Long HY, Sun DN, Long LL, Feng L, Li Y, Xiao B. 2012. MicroRNA expression profile of the hippocampus in a rat model of temporal lobe epilepsy and miR-34a-targeted neuroprotection against hippocampal neurone cell apoptosis post-status epilepticus. BMC Neurosci 13: 115. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Huang Y, Myers SJ, Dingledine R. 1999. Transcriptional repression by REST: Recruitment of Sin3A and histone deacetylase to neuronal genes. Nat Neurosci 2: 867–872. [DOI] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R. 2002. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci 22: 8422–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhao F, Wang L, Yin H, Zhou C, Wang X. 2012. Increased expression of histone deacetylases 2 in temporal lobe epilepsy: A study of epileptic patients and rat models. Synapse 66: 151–159. [DOI] [PubMed] [Google Scholar]
- Hwang JY, Aromolaran KA, Zukin RS. 2013. Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology 38: 167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H. 2013. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res 41: 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. 2003. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 33: 245–254. [DOI] [PubMed] [Google Scholar]
- Jakovcevski M, Akbarian S. 2012. Epigenetic mechanisms in neurological disease. Nat Med 18: 1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. 2004. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 80: 1611–1617. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Nakashima K, Clemenson GD Jr, Mejia E, Mathews E, Ure K, Ogawa S, Sinton CM, Gage FH, Hsieh J. 2007. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J Neurosci 27: 5967–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Bray I, Sanz-Rodriguez A, Engel T, McKiernan RC, Mouri G, Tanaka K, Sano T, Saugstad JA, Simon RP, et al. 2011. miRNA Expression profile after status epilepticus and hippocampal neuroprotection by targeting miR-132. Am J Pathol 179: 2519–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Engel T, Merino-Serrais P, McKiernan RC, Tanaka K, Mouri G, Sano T, O’Tuathaigh C, Waddington JL, Prenter S, et al. 2012. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat Med 18: 1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Engel T, Merino-Serrais P, Fernaud-Espinosa I, Rodriguez-Alvarez N, Reynolds J, Reschke CR, Conroy RM, McKiernan RC, deFelipe J, et al. 2014. Antagomirs targeting microRNA-134 increase hippocampal pyramidal neuron spine volume in vivo and protect against pilocarpine-induced status epilepticus. Brain Struct Func 10.1007/s00429-014-0798-5. [DOI] [PubMed] [Google Scholar]
- Kan AA, van Erp S, Derijck AA, de Wit M, Hessel EV, O’Duibhir E, de Jager W, Van Rijen PC, Gosselaar PH, de Graan PN, et al. 2012. Genome-wide microRNA profiling of human temporal lobe epilepsy identifies modulators of the immune response. Cell Mol Life Sci 69: 3127–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Saetrom P, Snove O Jr, Rossi JJ. 2008. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci 105: 16230–16235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobow K, Blumcke I. 2012. The emerging role of DNA methylation in epileptogenesis. Epilepsia 53: 11–20. [DOI] [PubMed] [Google Scholar]
- Kobow K, Jeske I, Hildebrandt M, Hauke J, Hahnen E, Buslei R, Buchfelder M, Weigel D, Stefan H, Kasper B, et al. 2009. Increased reelin promoter methylation is associated with granule cell dispersion in human temporal lobe epilepsy. J Neuropathol Exp Neurol 68: 356–364. [DOI] [PubMed] [Google Scholar]
- Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, Fritzsche I, Hauke J, Hahnen E, Coras R, et al. 2013. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropath 126: 741–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Holloway T, Garcia-Bea A, Kozlenkov A, Friedman AK, Moreno JL, Heshmati M, Golden SA, Kennedy PJ, Takahashi N, et al. 2012. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci 15: 1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. 2010. Dynamic changes in the human methylome during differentiation. Genome Res 20: 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. 2006. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281: 15763–15773. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69: 915–926. [DOI] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. 2013. Global epigenomic reconfiguration during mammalian brain development. Science 341: 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Sheng Z, Cai L, Zhao K, Tian Y, Fei J. 2012. Neuronal conditional knockout of NRSF decreases vulnerability to seizures induced by pentylenetetrazol in mice. Acta Biochim Biophysica Sin 44: 476–482. [DOI] [PubMed] [Google Scholar]
- Lubin FD. 2012. Epileptogenesis: Can the science of epigenetics give us answers? Epilepsy Curr 12: 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnes ZM, Huang TC, Chang PK, Gill R, Reist N, Dezsi G, Ozturk E, Charron F, O’Brien TJ, Jones NC, et al. 2013. DNA methylation mediates persistent epileptiform activity in vitro and in vivo. PLoS ONE 8: e76299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. 2003. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science 302: 890–893. [DOI] [PubMed] [Google Scholar]
- Maxwell SS, Pelka GJ, Tam PP, El-Osta A. 2013. Chromatin context and ncRNA highlight targets of MeCP2 in brain. RNA Biol 10: 1741–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland S, Flynn C, Dube C, Richichi C, Zha Q, Ghestem A, Esclapez M, Bernard C, Baram TZ. 2011. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol 70: 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKiernan RC, Jimenez-Mateos EM, Bray I, Engel T, Brennan GP, Sano T, Michalak Z, Moran C, Delanty N, Farrell M, et al. 2012a. Reduced mature microRNA levels in association with dicer loss in human temporal lobe epilepsy with hippocampal sclerosis. PLoS ONE 7: e35921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKiernan RC, Jimenez-Mateos EM, Sano T, Bray I, Stallings RL, Simon RP, Henshall DC. 2012b. Expression profiling the microRNA response to epileptic preconditioning identifies miR-184 as a modulator of seizure-induced neuronal death. Exp Neurol 237: 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. 2012. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151: 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. 2008. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci 105: 716–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Delaney SF, Das S, Sano T, Jimenez-Mateos EM, Bryan K, Buckley PG, Stallings RL, Henshall DC. 2012. Differential DNA methylation patterns define status epilepticus and epileptic tolerance. J Neurosci 32: 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, van der Brug MP, Wahlestedt C. 2012. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotech 30: 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. 2008. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci 28: 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SY, Lin L, Soh BS, Stanton LW. 2013. Long noncoding RNAs in development and disease of the central nervous system. Trend Genet 29: 461–468. [DOI] [PubMed] [Google Scholar]
- Nudelman AS, DiRocco DP, Lambert TJ, Garelick MG, Le J, Nathanson NM, Storm DR. 2010. Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus 20: 492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257. [DOI] [PubMed] [Google Scholar]
- Park HG, Yu HS, Park S, Ahn YM, Kim YS, Kim SH. 2014. Repeated treatment with electroconvulsive seizures induces HDAC2 expression and down-regulation of NMDA receptor-related genes through histone deacetylation in the rat frontal cortex. Int J Neuropsychopharm 17: 1–14. [DOI] [PubMed] [Google Scholar]
- Peng J, Omran A, Ashhab MU, Kong H, Gan N, He F, Yin F. 2013. Expression patterns of miR-124, miR-134, miR-132, and miR-21 in an immature rat model and children with mesial temporal lobe epilepsy. J Mol Neurosci 50: 291–297. [DOI] [PubMed] [Google Scholar]
- Pichardo-Casas I, Goff LA, Swerdel MR, Athie A, Davila J, Ramos-Brossier M, Lapid-Volosin M, Friedman WJ, Hart RP, Vaca L. 2012. Expression profiling of synaptic microRNAs from the adult rat brain identifies regional differences and seizure-induced dynamic modulation. Brain Res 1436: 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Lukasiuk K. 2011. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol 10: 173–186. [DOI] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF. 2012. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci 13: 528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF. 2013. Understanding neurological disease mechanisms in the era of epigenetics. JAMA Neurol 70: 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. 2001. Epigenetic reprogramming in mammalian development. Science 293: 1089–1093. [DOI] [PubMed] [Google Scholar]
- Risbud RM, Porter BE. 2013. Changes in microRNA expression in the whole hippocampus and hippocampal synaptoneurosome fraction following pilocarpine-induced status epilepticus. PLoS ONE 8: e53464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopra A, Dingledine R, Hsieh J. 2012. Epigenetics and epilepsy. Epilepsia 53: 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti F, de Araujo Furtado M, Pak T, Bailey K, Shields M, Chanda S, Addis M, Robertson BD, Moffett M, Lumley LA, et al. 2012. Combined diazepam and HDAC inhibitor treatment protects against seizures and neuronal damage caused by soman exposure. Neurotoxicology 33: 500–511. [DOI] [PubMed] [Google Scholar]
- Rudenko A, Dawlaty Meelad M, Seo J, Cheng Albert W, Meng J, Le T, Faull Kym F, Jaenisch R, Tsai L-H. 2013. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 79: 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryley Parrish R, Albertson AJ, Buckingham SC, Hablitz JJ, Mascia KL, Davis Haselden W, Lubin FD. 2013. Status epilepticus triggers early and late alterations in brain-derived neurotrophic factor and NMDA glutamate receptor Grin2b DNA methylation levels in the hippocampus. Neuroscience 248C: 602–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, Reynolds JP, Jimenez-Mateos EM, Matsushima S, Taki W, Henshall DC. 2012. MicroRNA-34a upregulation during seizure-induced neuronal death. Cell Death Dis 3: e287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapio MR, Salzmann A, Vessaz M, Crespel A, Lyons PJ, Malafosse A, Fricker LD. 2012. Naturally occurring carboxypeptidase A6 mutations: Effect on enzyme function and association with epilepsy. J Biol Chem 287: 42900–42909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa S, Fuso A, D’Anselmi F, Cavallaro RA. 2003. Presenilin 1 gene silencing by S-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett 541: 145–148. [DOI] [PubMed] [Google Scholar]
- Schaukowitch K, Kim TK. 2014. Emerging epigenetic mechanisms of long non-coding RNAs. Neuroscience 264C: 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. 2006. A brain-specific microRNA regulates dendritic spine development. Nature 439: 283–289. [DOI] [PubMed] [Google Scholar]
- Sharma A. 2012. Genome-wide expression analysis in epilepsy: A synthetic review. Curr Top Med Chem 12: 1008–1032. [DOI] [PubMed] [Google Scholar]
- Sng JC, Taniura H, Yoneda Y. 2005. Inhibition of histone deacetylation by trichostatin A intensifies the transcriptions of neuronal c-fos and c-jun genes after kainate stimulation. Neurosci Lett 386: 150–155. [DOI] [PubMed] [Google Scholar]
- Sng JC, Taniura H, Yoneda Y. 2006. Histone modifications in kainate-induced status epilepticus. Eur J Neurosci 23: 1269–1282. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. 2013. The emerging field of neuroepigenetics. Neuron 80: 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD, Meany MJ, Nestler EJ, Akbarian S. 2013. Epigenetic regulation in the nervous system. Academic, San Diego. [Google Scholar]
- Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, et al. 2011. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 14: 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CL, Plotkin JL, Veno MT, von Schimmelmann M, Feinberg P, Mann S, Handler A, Kjems J, Surmeier DJ, O’Carroll D, et al. 2013. MicroRNA-128 governs neuronal excitability and motor behavior in mice. Science 342: 1254–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniura H, Sng JC, Yoneda Y. 2006. Histone modifications in status epilepticus induced by kainate. Histol Histopath 21: 785–791. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ. 2004. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci 24: 5603–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdinguio RG, Sanchez-Mut JV, Esteller M. 2009. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol 8: 1056–1072. [DOI] [PubMed] [Google Scholar]
- Vandeweyer G, Van der Aa N, Ceulemans B, van Bon BW, Rooms L, Kooy RF. 2012. A de novo balanced t(2;6)(p15;p22.3) in a patient with West syndrome disrupts an lncRNA. Epilepsy Res 99: 346–349. [DOI] [PubMed] [Google Scholar]
- Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al. 2008. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322: 1695–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. 2013. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 23: 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlestedt C. 2013. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Disc 12: 433–446. [DOI] [PubMed] [Google Scholar]
- Wang T, Pan Q, Lin L, Szulwach KE, Song CX, He C, Wu H, Warren ST, Jin P, Duan R, et al. 2012. Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum Mol Genet 21: 5500–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. 2010. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet 87: 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D. 2013. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest 123: 3552–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Xie X. 2006. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol 7: R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Peterson M, Beyer B, Frankel WN, Zhang ZW. 2014a. Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J Neurosci 34: 2754–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kim J, Mueller AC, Dey B, Yang Y, Lee DH, Hachmann J, Finderle S, Park DM, Christensen J, et al. 2014b. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ 21: 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Chuang SC, Bianchi R, Zhao W, Lee H, Fenton AA, Wong RK, Tiedge H. 2009. BC1 regulation of metabotropic glutamate receptor-mediated neuronal excitability. J Neurosci 29: 9977–9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Wang L, Zhang Y, Zhao FH, Luo J, Xiao Z, Chen GJ, Wang XF. 2012. Increased expression of DNA methyltransferase 1 and 3a in human temporal lobe epilepsy. J Mol Neurosci 46: 420–426. [DOI] [PubMed] [Google Scholar]
- Ziller MJ, Muller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. 2011. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet 7: e1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]