

Abstract
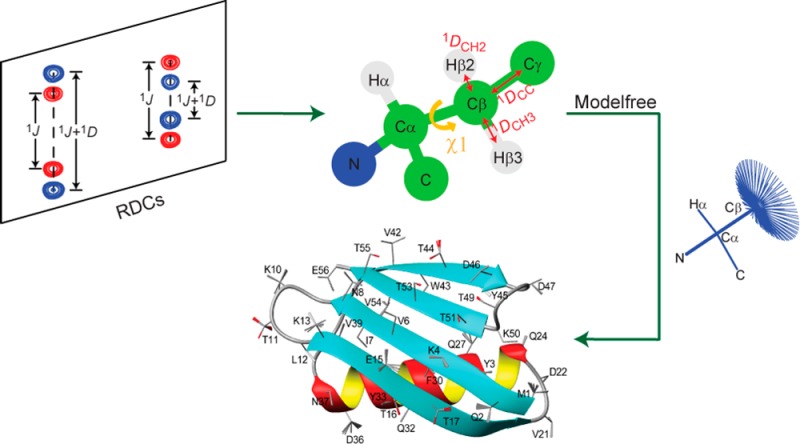
Accurate quantitative measurement of structural dispersion in proteins remains a prime challenge to both X-ray crystallography and NMR spectroscopy. Here we use a model-free approach based on measurement of many residual dipolar couplings (RDCs) in differentially orienting aqueous liquid crystalline solutions to obtain the side chain χ1 distribution sampled by each residue in solution. Applied to the small well-ordered model protein GB3, our approach reveals that the RDC data are compatible with a single narrow distribution of side chain χ1 angles for only about 40% of the residues. For more than half of the residues, populations greater than 10% for a second rotamer are observed, and four residues require sampling of three rotameric states to fit the RDC data. In virtually all cases, sampled χ1 values are found to center closely around ideal g–, g+ and t rotameric angles, even though no rotamer restraint is used when deriving the sampled angles. The root-mean-square difference between experimental 3JHαHβ couplings and those predicted by the Haasnoot-parametrized, motion-adjusted Karplus equation reduces from 2.05 to 0.75 Hz when using the new rotamer analysis instead of the 1.1-Å X-ray structure as input for the dihedral angles. A comparison between observed and predicted 3JHαHβ values suggests that the root-mean-square amplitude of χ1 angle fluctuations within a given rotamer well is ca. 20°. The quantitatively defined side chain rotamer equilibria obtained from our study set new benchmarks for evaluating improved molecular dynamics force fields, and also will enable further development of quantitative relations between side chain chemical shift and structure.
Introduction
It is well recognized that proteins are subject to extensive conformational fluctuations, and much research over the past two decades has focused on linking this structural and dynamic heterogeneity to function.1−4 Recent advances in the analysis of X-ray crystallographic data now make it possible to reliably fit protein X-ray electron density maps, acquired at high resolution, to ensembles of structures that correlate with the order parameters derived from NMR relaxation data.5 Such analyses frequently reveal the presence of multiple different side chain conformations in any given crystal, permitting identification of distinct allosteric networks.6 Unfortunately, collection of high quality X-ray data often requires the use of cryogenic temperatures, which results in extensive remodeling of many of the side chains, even when samples are flash frozen,7 possibly providing a distorted view of the functionally relevant ensemble that exists at room temperature.
Historically, NMR spectroscopy has played a dominant role in studying internal dynamics of proteins. In particular, 15N relaxation measurements are used ubiquitously for defining the amplitudes and time scales of rapid backbone motions.8−11 Side chain dynamics, in particular methyl-bearing side chains, have been extensively studied by 13C relaxation measurements,12−14 and 2H relaxation rates of deuterated methyl groups also have proven to be particularly robust parameters for quantifying such dynamics.14,15 In addition, dipole–dipole cross-correlated relaxation rates were shown to be excellent reporters on methylene group motions,16 with the restriction on all of these relaxation methods being that only motions faster than the overall tumbling time of the protein can be accurately detected.17 Motions on the ms−μs time scale can be studied by NMR relaxation dispersion methods, providing access to conformational rearrangements that often are of key importance to biological function.18−20 However, unless the chemical shift changes underlying relaxation dispersion can be interpreted in structural terms, relaxation dispersion is often limited to providing time scales and populations of the dynamic process. As a complement to these measurements, the conformational range sampled by proteins across the entire NMR time scale from ps to ms can be defined by RDCs measured under at least three linearly independent alignment conditions.3,21−23 However, when the motional amplitudes involved are modest, as often is the case for backbone dynamics in well-ordered proteins, quantitative interpretation of the RDCs in terms of motions demands very high measurement precision, a goal that can be difficult to reach under the required three or more alignment conditions.24,25
In the past, side chain dynamics have been studied mostly in terms of the distributions of their rotameric states, most commonly analyzed by measurement of extensive sets of 3J couplings, possibly in conjunction with 1H–1H nuclear Overhauser enhancements (NOEs).26−28 Interpretation of these data in terms of dynamics was shown to be rather sensitive to the distributional model underlying such analyses,29 and clearly is also highly sensitive to the parametrizations of the Karplus equations used. Because energetically favorable side chain conformations typically fall close to staggered rotameric states, their dynamics in terms of changes in bond vector orientation often is of much larger amplitude than for the protein backbone, making them highly suitable for study by RDCs.30 Analysis of the 13Cβ–1Hβ RDCs measured for the B1 domain of protein L using two quite different liquid crystalline alignments yielded up to four very accurate parameters to define the χ1 angle for side chains with Cβ methylene groups, and two parameters for residues such as Val, Ile, and Thr, with only a single 13Cβ–1Hβ RDC.30 For residues with methylene groups, this permitted definition of both the populations and χ1 values of up to two side chain rotamers that are in rapid exchange with one another or, for both residues with methylene or methine Cβ carbons this could be interpreted in terms of populations of three rotamers (two independent parameters), assuming these are ideally staggered.
Here, we expand on the approach introduced by Mittermaier and Kay, and measure side chain RDCs for six differently oriented samples of the third Igg-binding domain of protein G (GB3 for short), and in addition to the 13Cβ–1Hβ RDCs we also measure 13Cβ–13Cγ RDCs for residues with nonprotonated Cγ (Asn, Asp, and aromatic residues) and for residues with 13CγH3 groups (Ile, Val, Thr), thereby yielding up to 18 observed parameters. Effectively, the number of independent experimental parameters is somewhat lower, however, as only five of the six protein alignments can be linearly independent of one another. Nevertheless, the now much larger set of experimental parameters allows definition of both the populations and χ1 values of all three side chain rotamers for the vast majority of residues. In fact, for residues with a complete or nearly complete set of couplings, we show it is possible to determine a continuous distribution of χ1 values, albeit that the latter analysis requires a “maximum entropy” term to smooth the distributions. Thus, our analysis provides a model-free evaluation of the χ1 values sampled by each side chain, without the restriction that these cluster in two or three distinct rotameric states.
Protein alignment conditions were varied by using different liquid crystalline suspensions30,31 as well as by introducing conservative charge mutations that alter the protein’s alignment in a medium containing a liquid crystalline suspension of the filamentous bacteriophage Pf1.32 The large amount of backbone RDC data obtained for these samples also permitted generation of an average backbone structure that predicts Cα–Cβ bond vector orientations at an accuracy that is considerably increased over that of the X-ray structure, as judged by agreement between observed and predicted 13Cα–13Cβ RDCs. High accuracy of these vector orientations is a prerequisite for quantitative RDC analysis of the χ1 values sampled by the different side chains.30
For nearly all solvent-exposed residues we find population of two and sometimes even three sets of χ1 angles that typically cluster close to ideally staggered rotameric states. By contrast, the relatively small number of buried side chains in GB3 for the most part can be well fit by single, narrow clusters of χ1 values. For the vast majority of residues, population of these clusters, as well as their centers, can be defined at high accuracy. Although the width of the clusters, i.e., the amplitude of the dynamics within a given rotameric well, is poorly defined by the RDC data, comparison between observed and predicted 3JHαHβ values indicates that the root-mean-square amplitude of χ1 angle fluctuations within a given rotamer well is ca. 20°.
Materials and Methods
Protein Preparation
The sequence of wild-type GB3 used in our study is MQYKLVINGK TLKGETTTKA VDAETAEKAF KQYANDNGVD GVWTYDDATK TFTVTE. Samples of the following three mutants also were generated: (1) K4A/K19E/V42E/CHis6; (2) K19A/V42E/D47K; and (3) K4A/K19E/V42E. For mutant (1), CHis6 refers to a set of six His residues, immediately following C-terminal residue E56. Proteins were expressed and purified as described previously,33 and selection of the three mutants was based on those previously studied by Yao et al. in Pf1, which had shown backbone perturbations caused by the mutations to be minimal.32 Samples of uniformly 99%-enriched 13C/15N protein were generated by growing BL21-DE3 Escherichia coli cells in M9 medium containing 1.0 g/L 15NH4Cl and 2.0 g/L 13C6-glucose, supplemented by 0.5 g/L U–13C/15N Isogro rich medium (Sigma-Aldrich). Measurements were also carried out for randomly fractionally (ca. 75%) deuterated protein, obtained by growing the cells in M9 medium, including 1 g/L 15NH4Cl and 3.0 g/L 13C6/2H7-glucose in 75% D2O/25% H2O, and for samples with full perdeuteration of the nonexchangeable hydrogens by growing the cells in a medium containing M9 medium, including 1 g/L 15NH4Cl and 3.0 g/L 13C6/2H7-glucose in 99% D2O, followed by back exchange of the labile hydrogens in H2O for at least 24 h at 37 °C.
NMR Measurements
An extensive set of two-dimensional (2D) and three-dimensional (3D) heteronuclear NMR spectra was recorded to collect 15N–1H, 13C–1H, and 13Cα–13Cβ and 13Cβ–13Cγ one-bond J couplings and RDCs on isotropic and six differently aligned protein samples. A nearly complete set of 3JHαHβ couplings was also obtained from a 3D HA[HB,HN](CACO)NH spectrum.34 The different types of spectra recorded are summarized in Supporting Information (SI) Table S1, and the compositions of the different samples are summarized in Table S2. Spectra were recorded on Bruker Avance-III spectrometers operating at 1H frequencies of 600 and 900 MHz, both equipped with triple resonance cryogenic probeheads and z-axis pulsed field gradient accessories. Details regarding the NMR measurements are presented in the SI Text Section.
All spectra were processed using the NMRPipe software system35 and analyzed with the Sparky program.36
Backbone Refinement of GB3
The structure of GB3 was refined starting from the coordinates of the PDB deposition 2OED,33 against an extensive set of N–HN, Cα–Hα, C′–Cα, C′–N, HN–Hα, and Cα–Cβ RDCs using a variable alignment tensor treatment as described previously, to reach working Q factors ranging between 0.05 and 0.11.37 In addition, 3JHNHα and 3JC′C′ couplings reported by Li et al.37 were fitted with force constants that yielded rmsd values of 0.29 and 0.1 Hz, respectively. For the PDB-deposited ensemble obtained from our study (PDB entry 2N7J), the protein structure was refined as if its backbone and Cβ atomic positions are fully static, i.e., as previously done for ubiquitin,25 thereby aiming to give the best representation of the time-averaged backbone structure.
Results and Discussion
Analogous to prior studies of protein backbone dynamics,21,22,24,38 we measured RDCs for a wide range of different protein orientations. The GB3 domain, used in our study, has a quite prolate shape, making it challenging to find truly orthogonal alignment conditions.33 Here, we rely on a combination of the two approaches previously used for this purpose. First, we collected RDC data in three liquid crystalline media that differentially align the protein relative to the magnetic field: a positively charged phospholipid bicelle suspension,39,40 a polyethylene glycol (PEG) based liquid crystal,41 and a suspension of the negatively charged filamentous phage Pf1.42 Second, we used conservative mutagenesis of charged surface residues, which can be used to alter the alignment of the protein relative to the negatively charged Pf1 liquid crystal, without significantly impacting the backbone structure of the protein.32 Three such mutants were prepared and studied in Pf1. As the current study focuses on side chain conformations rather than backbone, it is important to ensure that the use of mutants does not significantly perturb the side chain conformations either. Here, we simply exclude data for residues in the mutants that have significant chemical shift differences for 13Cβ (≥0.2 ppm) or 1Hβ (≥0.1 ppm) relative to the wild type protein. This only concerns a small number of residues (SI Tables S3 and S4; SI Figure S3), most significantly residue E15 which exhibits a >0.6 ppm 13Cβ chemical shift change in the K4A mutants, presumably because a side chain salt bridge interaction between K4 and E15, seen in the X-ray structure of the wild type protein,43 is abolished in the mutant. Although RDC data were collected under six different alignment conditions (three different media, plus three mutants in Pf1), analysis of the corresponding alignment tensors using singular value decomposition and SECONDA analysis44 indicates that one of the five possible orthogonal alignments is only weakly sampled (SI Figure S4). However, in contrast to the study of backbone motions by RDCs, where often only a single 15N–1H RDC is used to define the orientation of the peptide plane relative to the liquid crystal director, for our study of side chain conformations two very different orientations, 13Cβ–1Hβ2 and 13Cβ–1Hβ3 are sampled if their separate RDCs can be measured. For residues with methine Cβ carbons, i.e., Thr, Val and Ile, 13Cβ–13Cγ RDCs were measured through the 13Cγ methyl resonance, and for aromatic and Asp/Asn residues 13Cβ–13Cγ RDCs could also readily be measured (see Materials and Methods and SI). Therefore, for each alignment condition, two or three RDCs are available for defining the χ1 angle of most residues.
Measurement of Side Chain 1H–13C RDCs
The accuracy of the experimental RDCs is paramount when using these to define a dynamic structural ensemble. Indeed, it is primarily the inconsistency between the RDCs and a single, unique χ1 angle that necessitates their analysis in terms of an ensemble. However, random or systematic measurement errors will also result in inconsistencies, and it is therefore critical to minimize such errors.
A 1H–13C RDC can be derived from the change in 1JCH splitting observed in either the 1H or 13C dimension of a multidimensional NMR spectrum. Measurement in the 1H dimension often is adversely affected by the presence of multiple unresolved 1H–1H RDCs, resulting in extensive broadening of the apparent 1H line width and thereby decreasing the accuracy of extracted couplings. For this reason, measurement of heteronuclear RDCs is usually carried out in the dimension of the low-γ nucleus, where the effect of remote 1H nuclei is far less severe.45 On the other hand, for methylene sites the coupled 13C–{1H} spectrum yields a doublet of doublets multiplet structure, for which the center components are usually unresolvable, therefore only providing access to the sum of the two 1JCβHβ couplings, i.e., to the sum of the corresponding RDCs. In our study, 13C–1H RDCs were measured using two independent methods. In one set of experiments, the summed 1Hβ–13Cβ RDCs were simply measured from a 3D HN(COCA)CB spectrum, recorded without 1H decoupling in the 13Cβ dimension (SI Figure S5). Separate 13Cβ–1Hβ2 and 13Cβ–1Hβ3 RDCs were measured also in the 13C dimension but for a sample that had been randomly deuterated at the 75% 2H level. For this sample, ca. 38% of the 13Cβ sites are CHD isotopomers and therefore, at least in principle, allow measurement of individual 13Cβ–1Hβ2 and 13Cβ–1Hβ3 RDCs from a constant-time 2D 1H–13C HSQC spectrum recorded without 1H decoupling in the 13C dimension, provided that 1Hβ2 and 1Hβ3 have different chemical shifts (Figure 1A). In practice, however, the ca. 3-fold smaller fraction of 13CβH2 isotopomers in such a fractionally deuterated sample gives rise to weak correlations that are partially overlapping with the 13CβHD correlations. Relative to 13CβH2, 13CβHD correlations are shifted upfield in both the 1H and 13C dimensions by the 2H isotope effect, but insufficiently to yield well-resolved resonances (Figure 1A). Instead, we therefore developed a DEPT-filtered46 1H–13C CT-HSQC pulse scheme (SI Figure S2), which actively suppresses 13CH2 correlations (Figure 1B). For the vast majority of methylene sites, this experiment allows separate measurement of the 13Cβ–1Hβ2 and 13Cβ–1Hβ3 RDCs. To validate the accuracy of these couplings, we compare their summed values with the result measured from the 3D HN(COCA)CB spectrum. Note that the latter are measured in H2O solution, whereas the DEPT-filtered HSQC measurement is carried out on the fractionally deuterated sample in D2O solution, and a scaling factor is needed to account for the slightly different alignment strengths of the two samples. After scaling, the excellent agreement seen in Figure 1C (pairwise root-mean-square difference (rmsd) of 2 Hz; Pearson’s correlation coefficient, RP = 0.99) validates the accuracy of both measurements.
Figure 1.
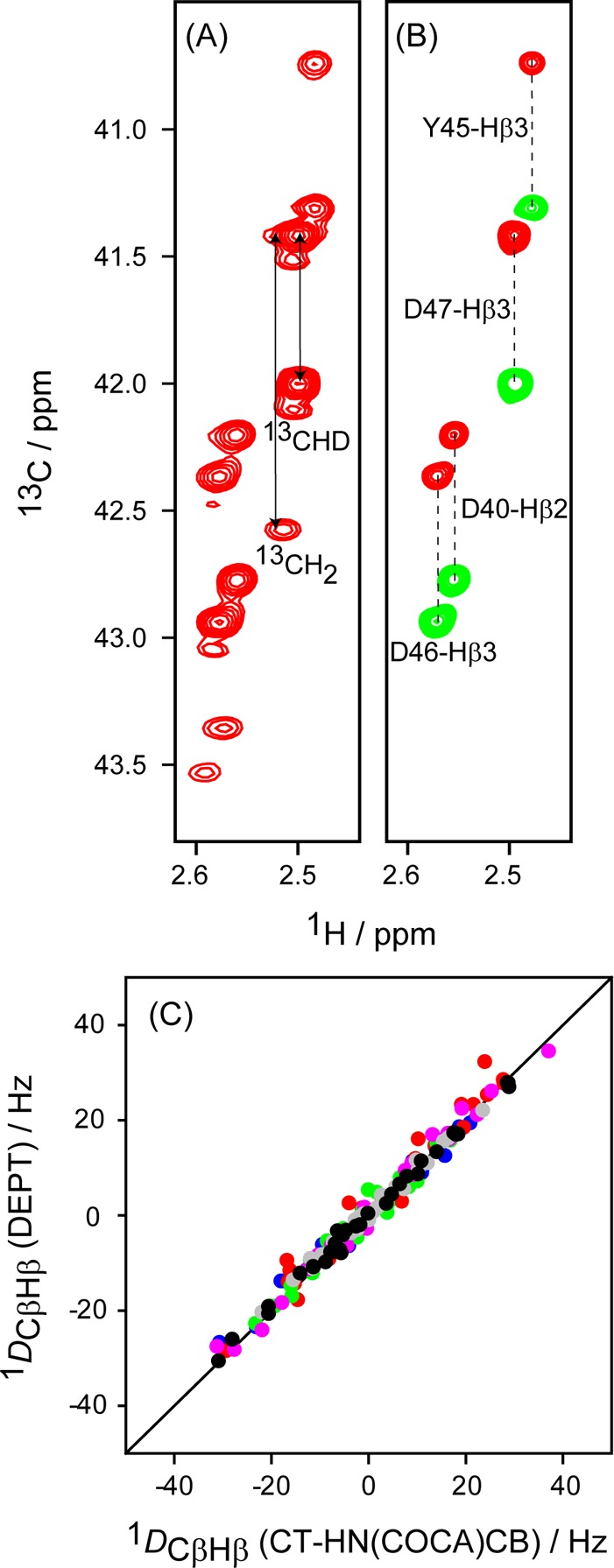
Enhanced accuracy of 1DCβHβ couplings through spectral editing of the 1H–13C CT-HSQC spectrum of randomly fractionally (75%) deuterated GB3. (A) Small region of the regular 1H–13C HSQC spectrum, 1H-coupled in the 13C dimension, showing the signals of both 13CH2 and 13CHD isotopomers, the latter upfield shifted due to the deuterium isotope effect. (B) Same spectral region, recorded with the DEPT-filtered 1H–13C CT-HSQC experiment (SI Figure S2), which selects 13CH signals and effectively suppresses 13CH2 isotopomers. (C) Comparison of the sum of 13Cβ–1Hβ2(Dβ3) and 13Cβ–Hβ3(Dβ2) couplings derived from the DEPT-filtered HSQC spectra of wild-type GB3 in Pf1 and isotropic solutions, with the corresponding summed couplings derived from CT-HN(COCA)CB spectra. Blue: K4A/K19E/V42E-CHis6 in Pf1; red: K19A/V42E/D47K in Pf1; green: K4A/K19E/V42E in Pf1; pink: wild-type GB3 in bicelle; gray: wild-type GB3 in PEG; black: wild-type GB3 in Pf1. Spectra were recorded at 900 MHz 1H frequency in 99.8% D2O (1H–13C HSQC) and at 600 MHz in 95% H2O for the CT-HN(COCA)CB experiment.
The same set of DEPT-filtered CT-HSQC spectra used above for measurement of Cβ–Hβ RDCs also yielded accurate Cα–Hα couplings. A large set of these was measured previously when the structure of the protein was first refined against RDCs,33 but values measured for the three mutants in Pf1 medium (see Materials and Methods) now add new data to this set. Moreover, with the previous and present measurements being independent of one another, contributions from small random measurement errors partially cancel when both sets are used, and the new data therefore were used jointly with the earlier data as input for all structure calculations.
Measurement of 13C–13C RDCs
As mentioned above, 13Cβ–13Cγ RDCs also contain valuable information on the χ1 angle, and are particularly useful for Val, Thr, and Ile residues for which only a single Cβ–Hβ RDC is available. Their values were derived from the difference in 1JCβCγ splitting between aligned and isotropic samples, observed in regular (non-CT) 1H-decoupled 1H–13C HSQC spectra (Figure 2A). For aromatic and Asp/Asn residues, 13Cβ–13Cγ RDCs were obtained from 1H–13C CT-HSQC spectra47 in which no decoupling of the downfield 13Cγ nuclei was employed, and the 13C 180° pulse applied during the 56 ms CT-evolution period was REBURP-shaped48 to invert only aliphatic carbon spins, thereby removing the 1JCαCβ splitting and leaving clean 1JCβCγ doublets (Figure 2B).
Figure 2.

Examples of spectral quality used for deriving 1DCβCγ couplings. (A) Small section of the methyl region of the 1H–13C HSQC spectrum of wild-type GB3 in Pf1 medium, recorded with regular (non-CT) 13C evolution (red), superimposed on the corresponding spectrum recorded under isotropic conditions (blue). (B) Small region of the 1Hβ–13Cβ region of the 1H–13C CT-HSQC spectrum, recorded with a REBURP 180° pulse covering only the 13C aliphatic region during the CT 13C evolution of the aliphatic region of wild-type GB3 in Pf1 medium (red) superimposed on the corresponding spectrum recorded under isotropic conditions (blue). The spectra were recorded with a double constant-time duration (56 ms) at 900 MHz. (C) Comparison of 1DCαCβ and the sum of the three other Cβ–related 1D couplings, which all have been scaled to 1DaCH (measurements for wild type GB3: pink, in bicelle; blue, in PEG; green, in Pf1).
In addition to the one-bond Cβ–Cγ RDCs (1DCβCγ) that contain information on χ1, complete sets of 1DCαCβ couplings (with the exception of C-terminal residue E56) were obtained from 3D HNCOCA spectra recorded on fully deuterated samples of the wild-type GB3 sample in Pf1, bicelle and PEG media. These data were used to further refine the backbone structure of the protein, and also served as a measure to evaluate the accuracy of Cα–Cβ bond vector orientations in structures derived without including this data, i.e., to establish a measure for the “structural noise”.49 Scaling factors for these values to account for the small changes in liquid crystal concentrations used in the different samples were established independently by comparing 1DNH values. After normalizing the alignment strengths of the different samples used for measurement of 1DCαCβ, 1DCβCγ, 1DCβHβ2, and 1DCβHβ3 values, and further accounting for the intrinsic difference between 1DCC and 1DCH couplings due to differences in bond length and gyromagnetic ratio,45 in the absence of measurement error the sum of these four couplings must be zero under the approximation of ideal tetrahedral geometry at Cβ. Indeed, for residues for which all four one-bond RDCs to Cβ were measured, a very good correlation between 1DCαCβ and the sum of the three other, normalized couplings is observed (Figure 2C). For several residues, the residual nonzero values of these sums (SI Table S5) are somewhat larger than expected based on propagation of the estimated uncertainties in the RDC measurements, which may reflect small deviations from idealized tetrahedral geometry at Cβ. Indeed, quantum calculations suggest, for example, that the Hβ2–Cβ–Hβ3 angle can deviate by up to ∼5° from the tetrahedral 109.4° value in a χ1-dependent manner. Deviations from ideal 109.4° values, outside of the statistical uncertainty, for the Cα–Cβ–Cγ angle are also commonly seen in atomic resolution X-ray data.50
Generation of Side Chain Ensembles
The earlier structure refinement (PDB entry 2OED) was based on an extensive set of backbone RDCs, measured in different liquid crystalline and stretched acrylamide gel media.33 In addition to the RDC restraints used previously, the newly recorded RDC data add orientations enabled by the use of surface charge mutations while using Pf1 as the alignment medium.32 The data also include 13Cα–13Cβ RDCs measured in three media for fully deuterated protein, which results in very high resolution in the 13C dimension, thereby increasing the accuracy of their measurements (SI Table S6). Use of perdeuterated protein also allows the use of rather strong alignment,51 which is particularly beneficial for measuring the intrinsically small 1DCC values. These 13Cα–13Cβ RDCs directly impact the orientations of the corresponding bond vectors, whose accuracy is key when defining the distributions of χ1 rotations around these vectors on the basis of 13Cβ–1Hβ and 13Cβ–13Cγ RDCs.
The 13Cα–13Cβ RDCs also are very useful for validation purposes, establishing the accuracy of the Cα–Cβ vectors in structures calculated without using any 1DCαCβ restraints. The averaged QfreeCα-Cβ factors for data measured in Pf1, bicelle and PEG media were ∼10%, corresponding to an uncertainty of 3.1 ± 0.6° (structural noise) in their time-averaged orientations.49 The actual structures used for all χ1 analyses and side chain ensemble generation included the 1DCαCβ restraints, and therefore should be of slightly higher accuracy than structures used for 1DCαCβ validation. Populations of the side chain conformers in the 20 structures deposited in the PDB (PDB entry 2N7J; Figure 3) then were randomly selected by sampling the distributions observed in our study (Table 1), followed by a Monte Carlo minimization of the clash score using the χ2, χ3, and χ4 distributions from the backbone-independent rotameric library.52 During this process the backbone and the atoms defining the χ1 torsion angles remained fixed, thus allowing rearrangement of the atoms beyond Cγ, but without consideration of side chain H-bonding or other potentially relevant physical interactions. Structural statistics are reported in SI Table S7.
Figure 3.
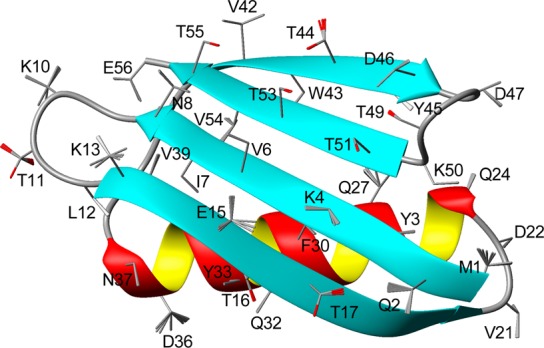
Structure of GB3, derived from NMR data. The backbone coordinates are presented as a ribbon diagram, with side chains depicted as sticks for the Cα–Cβ and Cβ–Cγ1 (Cβ–Oγ for Thr) bonds. For residues with the RDC-derived χ1 rotamer distributions, shown positions of the side chains (stick models) are obtained by 20 random samplings from the models reported in Table 1. Spread within each rotamer corresponds to the Monte Carlo-derived rms uncertainty of the best-fitted χ1 values as reported in Table 1.
Table 1. Analysis of χ1 Rotamer Distributions in GB3.
| residue | NRDCa | modelb | Pc | ρ(1)d | σρ(1)e | χ̅1(1)f | σχ1(1)g | ρ(2)d | χ̅1(2)f | σχ1(2)g | ρ(3)d | χ̅1(3)f |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1 | 11 | 2 | <10–4 | 0.53 | 0.04 | 306 | 12 | 0.47 | 203 | 15 | – | – |
| Q2 | 10 | 2 | <10–4 | 0.69 | 0.02 | 176 | 5 | 0.31 | 265 | 11 | – | – |
| Y3 | 15 | 1 | 0.59 | 1.0 | – | 298 | 1 | – | – | – | – | – |
| K4 | 8 | 2 | <10–4 | 0.67 | 0.02 | 183 | 3 | 0.33 | 293 | 5 | – | – |
| L5 | 7 | 1 | 0.98 | 1.0 | – | 173 | 1 | – | – | – | – | – |
| V6 | 11 | 1 | 0.89 | 1.0 | – | 178 | 1 | – | – | – | – | – |
| I7 | 10 | 1 | 0.39 | 1.0 | – | 299 | 1 | – | – | – | – | – |
| N8 | 12 | 2 | <10–4 | 0.58 | 0.01 | 190 | 2 | 0.42 | 296 | 3 | – | – |
| K10 | 5 | 3 | h | 0.58 | 0.04 | 293 | 3 | 0.23 | 187 | 3 | 0.19 | 57 |
| T11 | 10 | 3 | 0.09 | 0.63 | 0.03 | 54 | 3 | 0.28 | 282 | 7 | 0.09 | 162 |
| K13 | 10 | 3 | 0.005 | 0.67 | 0.02 | 302 | 2 | 0.16 | 62 | 12 | 0.16 | 160 |
| E15 | 8 | 3 | 0.004 | 0.44 | 0.01 | 285 | 3 | 0.34 | 40 | 4 | 0.22 | 163 |
| T16 | 10 | 2 | <10–4 | 0.74 | 0.01 | 186 | 2 | 0.26 | 37 | 8 | – | – |
| T17 | 10 | 2 | <10–4 | 0.76 | 0.01 | 65 | 1 | 0.24 | 195 | 7 | – | – |
| T18 | 10 | 1 | 0.10 | 1.0 | – | 197 | 1 | – | – | – | – | – |
| V21 | 13 | 2i | 0.0002 | 0.60 | 0.05 | 302 | 3 | 0.40 | 68 | 4 | – | – |
| D22 | 15 | 1 | 1.0 | 1.0 | – | 61 | 1 | – | – | – | – | – |
| E24 | 10 | 2 | <10–4 | 0.53 | 0.02 | 284 | 6 | 0.47 | 182 | 5 | – | – |
| E27 | 10 | 2 | 0.07 | 0.93 | 0.03 | 185 | 3 | 0.07 | 286 | 34 | – | – |
| K28 | 5 | 1 | 1.0 | 1.0 | – | 305 | 3 | – | – | – | – | – |
| F30 | 15 | 1 | 0.35 | 1.0 | – | 289 | 1 | – | – | – | – | – |
| K31 | 10 | 2 | <10–4 | 0.84 | 0.01 | 285 | 2 | 0.16 | 181 | 9 | – | – |
| Q32 | 5 | 2 | 0.05 | 0.72 | 0.04 | 174 | 6 | 0.28 | 297 | 15 | – | – |
| Y33 | 14 | 1j | 0.009 | 1.0 | – | 179 | 1 | – | – | – | – | – |
| N35 | 15 | 2 | <10–4 | 0.52 | 0.01 | 184 | 2 | 0.48 | 288 | 3 | – | – |
| D36 | 15 | 2 | 0.04 | 0.95 | 0.11 | 290 | 4 | 0.05 | 188 | 40 | – | – |
| N37 | 15 | 1 | 1.0 | 1.0 | – | 285 | 1 | – | – | – | – | – |
| V39 | 15 | 2 | <10–4 | 0.83 | 0.02 | 178 | 2 | 0.17 | 324 | 9 | – | – |
| D40k | 15 | 2 | <10–4 | 0.69 | 0.13 | 179 | 11 | 0.31 | 237 | 15 | – | – |
| V42 | 9 | 2 | 0.0005 | 0.83 | 0.02 | 171 | 1 | 0.17 | 35 | 6 | – | – |
| W43 | 11 | 1 | 1.0 | 1.0 | – | 287 | 1 | – | – | – | – | – |
| T44 | 10 | 2 | 0.001 | 0.84 | 0.02 | 48 | 1 | 0.16 | 270 | 6 | – | – |
| Y45 | 15 | 1 | 1.0 | 1.0 | – | 178 | 2 | – | – | – | – | – |
| D46 | 15 | 2 | 0.0007 | 0.90 | 0.03 | 179 | 1 | 0.10 | 30 | 17 | – | – |
| D47 | 15 | 2 | <10–4 | 0.82 | 0.02 | 189 | 3 | 0.18 | 98 | 12 | – | – |
| T49 | 10 | 1 | 1.0 | 1.0 | – | 63 | 2 | – | – | – | – | – |
| K50 | 10 | 1 | 1.0 | 1.0 | – | 299 | 1 | – | – | – | – | – |
| T51 | 5 | 1 | 0.92 | 1.0 | – | 298 | 1 | – | – | – | – | – |
| F52 | 15 | 1 | 1.0 | 1.0 | – | 287 | 1 | – | – | – | – | – |
| T53 | 10 | 2 | 0.06 | 0.90 | 0.03 | 301 | 2 | 0.10 | 86 | 25 | – | – |
| V54 | 15 | 2 | 0.0003 | 0.81 | 0.04 | 57 | 2 | 0.19 | 205 | 7 | – | – |
| T55 | 6 | 1 | 0.06 | 1.0 | – | 303 | 1 | – | – | – | – | – |
| E56 | 9 | 2 | <10–4 | 0.56 | 0.02 | 286 | 7 | 0.44 | 184 | 8 | – | – |
NRDC is the total number of experimental RDCs available for side chain rotamer analysis, with a maximum of 5 per bond vector, as the 6th RDC is a linear combination of the other 5.
Model refers to the number of discrete χ1 rotamers required to fit the data.
P refers to the probability, extracted from F statistics. For cases where model 1 is selected, P refers to the probability that model 1 is more applicable than model 2; for cases where model 2 is selected, P refers to the probability that model 2 is not better than model 1; for cases where model 3 is selected, P refers to the probability that model 3 is not better than model 2.
Fractional population of rotamer (n).
Uncertainty in fractional population of rotamer 1.
χ1 angle of rotamer (n), degrees.
Uncertainty in χ1 angle of rotamer (n), degrees.
With Hβ2 and Hβ3 having the same chemical shifts, only 6 RDCs for the sum of the Cβ–Hβ couplings were available. Because these RDCs were incompatible with 1- or 2-rotamer distributions with energetically favorable χ1 angles, a 3-rotamer fit was carried out while restricting the χ1 angles to fall within 10° from 60, 180 and 300°.
Although F statistics suggests model 3, model 2 is in better agreement with 3JHαHβ than model 3.
Although F statistics suggests the presence of a 7% populated g+ rotamer, g+ would result in severe steric clashing and is therefore excluded.
Although it seemed likely that side chain angles of residues with fluctuating χ1 rotamer populations will not change fully independently of one another, as some combinations of χ1 would result in a steric clash of side chains, relatively few clear-cut examples of such concerted motions were evident in our analysis. In the deposited ensemble, structures with such clashes are simply absent due to the high energy associated with distances between atoms that are separated less than the sum of their scaled van der Waals radii during the Monte Carlo clash minimization protocol.
Evaluation of χ1 as Continuous Distributions
The amount of RDC data available to define the χ1 angle distribution varied strongly from residue to residue. For 33 out of 46 non-Gly/Ala residues, ten or more experimental RDCs were available to define χ1 (Table 1). On the other hand, six or fewer couplings could be measured reliably for seven residues: For T25, which exhibits strong 13Cα–13Cβ coupling due to a very small 13Cα–13Cβ chemical shift difference, no reliable side chain RDCs were obtained. For K19, which has two equivalent 1Hβ chemical shifts and is the site of mutation in three of the samples studied, only three RDCs were available. Both K19 and T25 therefore were excluded, as was L12 because its high amplitude backbone dynamics24,53 was incompatible with our analysis method. Other residues with very small numbers of RDCs included K10, K28, Q32 and T55 and T51. For K10, K28 and Q32 the equivalent chemical shifts of their Hβ protons only permitted measurement of the sum of their 1DCβHβ. Several of the other missing RDCs result from structural perturbations caused by the mutations of K4, K19 and V42, used to alter GB3′s alignment, as judged by 13Cβ or 1Hβ chemical shift changes >0.2 or >0.1 ppm, respectively. Moreover, the difficulty in measuring 13Cβ–13Cγ RDCs at sufficient precision for several residues further decreased the number of observable parameters. Nevertheless, with the backbone and thereby Cα–Cβ orientations tightly defined, at least in principle only two RDCs are needed to define χ1 under the assumption that the side chain conformation is static. Therefore, even for residues with only very few RDCs, the lack of consistency between the RDCs and a single, static χ1 value can reveal χ1 dynamics.
The actual search for the ensemble of χ1 conformers that best agrees with the experimental data is performed by means of the previously introduced variable weight fit (VW-Fit) program.25 VW-Fit uses a simulated annealing protocol to select from an ensemble of conformers the population that best matches the experimental data. For the present case, a 36-member ensemble was simply generated as that of the (static) backbone structure, with the 36 ensemble members corresponding to χ1 values ranging from 0 to 350°, regardless of steric clashing or other energetic considerations. Populations of the 36 different side chain conformers (P0, P10, ..., P350; ∑nPn = 1) correspond to 35 independent variables, which in all cases exceeds the number of measured RDCs, making the solution under-determined. This problem becomes even more pronounced when dividing the conformers into narrower bins of, say, 2° each. For this case, it is readily seen that, for example, equal 33.3% populations of P298, P300, and P302 give essentially indistinguishable RDCs relative to the case where only P300 is populated. We solve this problem by adding a very weak “entropy” term, −θ ∑n[Pn log(Pn)], to the X2 penalty score that is minimized by VW-Fit:
| 1 |
Here, RDCjexp corresponds to one of the M experimental RDCs available to define any given χ1 angle, σj is its estimated measurement uncertainty, and RDCj(χ1n) is the corresponding RDC value calculated for χ1 rotamer n, using the alignment tensor obtained from the backbone 1DNH and 1DCαHα RDCs. Note that in order to avoid confusion with the χ1 angle, we use X rather than the more common χ symbol to describe the residual in the fit. The entropy term, −θ ∑n[Pn log(Pn)], would be at a maximum when all populations Pn are equal (i.e., 2.78% for 36 conformers), but the weight factor θ is adjusted to a very low value, such that the RDC contribution to X2 increases by only 2% over the minimal value reached without inclusion of the entropy term. We find that this criterion for choosing θ is suitable for reaching convergence while only minimally broadening the distributions, which are typically centered around the staggered rotamers. Note that the entropy term aims to make the population of all χ1 bins equal, regardless of whether or not a bin corresponds to an energetically preferred staggered rotamer, and with the very low value of θ this method essentially yields a model-free depiction of the distribution of χ1 angles best compatible with the experimental data. Note, however, that for all residues a comparably low X2 value can be reached by considering the distribution to consist of a small (≤3) number of discrete rotamers (see below).
Figure 4 shows the χ1 distributions observed for the three Asn residues, which are representative for what is observed for the full set of residues (presented in SI Figure S6 and S7). Whereas for helical C-cap residue N37 a narrow χ1 distribution centered at 285° (i.e., equivalent to χ1 = −75°) is observed (Figure 4C), equal populations of the t and g– rotamers are obtained for N35, with both rotamers centered close to the ideal staggered conformations of χ1 = 180° and χ1 = 300° (Figure 4B). For N8, located at the end of strand β1, again population of both t and g– rotamers is observed, but with a higher population of t (Figure 4A). The excellent correlations between observed and predicted RDCs for these residues (Figure 4D–F) highlight the close agreement between experimental data and the obtained rotamer distributions. Nevertheless, the width of the rotamer distributions is artificially increased by the maximum entropy term and, in particular for N8, results in non-negligible population of the energetically unfavorable χ1 values near 240 and 0°. Another small problem that arises sometimes results from the degeneracy of RDCs with respect to bond vector orientation, i.e., an RDC cannot distinguish between an orientation and its inverse. Assuming tetrahedral geometry at Cβ, a 180° χ1 rotation brings the Cβ–Hβ and Cβ–Cγ vectors close, within 39°, to their inverted orientation, sometimes creating a shallow false minimum, which then results in a false, low population at the true χ1 plus 180° (see, e.g., the nonzero population at χ1 = 110° in Figure 4C). Below we therefore will also analyze the best fits obtained when using a small number of discrete rotameric states.
Figure 4.

Distributions of χ1 torsion angles for the three Asn residues in GB3, derived from RDC data, (A) Asn8, (B) Asn35, (C) Asn37 obtained with the program VW-Fit from an ensemble of 36 conformers, with identical backbone coordinates but the side chain χ1 angles ranging from 0° to 350° in 10° increments. Corresponding distributions for all residues are presented in SI Figure S6. The positive weight of the entropy term, θ, was adjusted such as to increase the total RDC component of the error function (eq 1) by 2% over not using this term, resulting in a smoothing of the distribution profile (maximum entropy) and improved convergence of the simulated annealing protocol. The correlations between observed and predicted RDCs for these residues are shown in panel (D) Asn8, (E) Asn35, (F) Asn37.
Stereospecific Hβ and Cγ Assignment
Traditionally, stereospecific assignments of Hβ resonances have been made through a joint analysis of NOE and 3JHαHβ information, which generally suffices to assign such protons for approximately half the cases.54 Stereospecific assignment of Val Cγ methyl groups, which is needed when exploiting 1DCβCγ RDCs for determining Val χ1 distributions, most commonly is accomplished by fractional 13C incorporation.55 Alternatively, complete stereospecific Hβ and Cγ assignments can also be obtained by using selectively deuterated amino acids in a cell-free growth medium (SAIL labeling),56 but this approach can be technically challenging and expensive in terms of the required selectively deuterated amino acid precursors.
In our above χ1 analysis, we followed the procedure used by Mittermaier and Kay,30 and simply tried both sets of Hβ (or Cγ for Val) assignments. For four residues (K10, K19, K28 and Q32) the near-identical chemical shifts of Hβ2 and Hβ3 obviated their stereospecific assignments. For the vast majority of the remaining residues one of the two assignments yielded a much better fit to the RDC data than the other. Moreover, we found that when using the wrong stereospecific assignment, the χ1 populations generally no longer clustered around those of the ideally staggered rotameric states, g–, g+, and t (SI Figure S8). For M1, E15, E24 and E56, the above considerations alone were insufficient for making confident stereospecific assignments. These residues showed clear indications of extensive side chain dynamics, as reflected in small ranges for the observed RDCs, small chemical shift differences (≤0.2 ppm) between Hβ2 and Hβ3, and broad distributions of χ1 angles obtained with the VW-Fit program when fitting the RDC data to the ensemble of 36 χ1 conformers (SI Figure S6). However, as described below, for all but E15 the stereospecific assignment could be completed when fitting the RDCs to a small number of discrete rotamers. Parenthetically, we note that a small chemical shift difference between Hβ2 and Hβ3 alone is not a sufficient indication of rotamer averaging as there are also residues in GB3 with very small |δHβ2 – δHβ3| differences (<0.1 ppm) that are well fit by single, narrow χ1 distributions (e.g., K50, F52).
Discrete Rotamer Analysis
From the above model-free VW-Fit analysis it is clear that for the vast majority of residues χ1 angle distributions cluster around one or two values (SI Figure S6). We therefore also carried out fits of the RDC data to models where either a single χ1 rotamer is populated (one variable in the fit, χ1), a fit with two χ1 rotamers (three independent variables: two χ1 values and one independent population), and a fit to three χ1 rotamers (five independent variables: three χ1 values and two independent populations). In principle, the width of the distribution around each χ1 maximum could also be used as an adjustable parameter,30 increasing the number of adjustable parameters by 1, 2, or 3 for the 1-, 2-, and 3-rotamer models. However, in relatively few of these cases a statistically meaningful improvement in the fit (P < 0.05, Student t test, see below) was obtained over the assumption of a fixed standard deviation, σ = 10°, of a Gaussian distribution centered around the optimized χ1 value(s). The σ = 10° value was obtained from a shallow X minimum when systematically varying σ (SI Figure S12), and was therefore used in all 1-, 2-, and 3-rotamer-fit analyses.
Evaluating which of the three models is applicable was carried out by standard F statistics, where the validity of decreasing the degrees of freedom (NA) in the fit to model A over a more complex model B (NB) for a given number of experimental observables (NRDC) was calculated from
| 2 |
and the conditional probability that a model with NB degrees of freedom is chosen erroneously over the simpler model is defined by P(F, NA, NB).57 In our analysis we have chosen a relatively high cutoff value of P = 0.1, but it is worth noting that even if, for example, the 2-rotamer is chosen erroneously over the 1-rotamer model, the population of the second rotamer generally will be low. We also note that the most populated rotamer chosen for residues with 0.01 ≤ P ≤ 0.1 invariably moves closer to an ideally staggered conformation than when using the single rotamer model and yields better agreement with the 3JHαHβ data (see below). Other than the experimental data there is no term driving such a selection, suggesting that the high P cutoff is physically reasonable. In practice, the presence of a second rotamer at a population higher than ca. 10% is detected quite unambiguously, in particular when 10 or more RDCs are available for such a residue (Table 1). Only four residues (K10, T11, K13, and E15), all located on the face of the protein involved in IgG-binding, show population of three rotameric states. The improvement in the RDC fit for K13, obtained when using the 1-, 2-, and 3-rotamer models is illustrated in Figure 5. The presence of three rotameric states for K10 is not mandated by F statistics, but was selected because the simpler 2-rotamer model yields highly skewed, unrealistic χ1 angles. Because the two Hβ protons are magnetically equivalent, only six RDCs are available to define the χ1 angle of this residue, and we therefore restricted the VW-Fit search to 9 conformers, representing the ideally staggered g–, g+, and t conformers, plus the 6 conformers that deviate by ±10° from these perfectly staggered conformations. For residue V21, the improvement in a 3-rotamer RDC fit over the 2-rotamer fit is acceptable based on F-statistics, but agrees less well with the experimental 3JHαHβ = 4.1 Hz value. It also results in a minor steric clash for one of the three rotamers, and the 2-rotamer solution is therefore selected for this residue. V21 shows somewhat elevated backbone dynamics (S2 = 0.74, based on RDC analysis24) and it is likely that the 2-state χ1 rotamer averaging for this loop residue is correlated with small changes in the backbone torsion angles (so-called “backrub motion”58), which somewhat lowers the quality of the RDC fit in our 2-rotamer analysis.
Figure 5.

Best fits between measured and predicted RDCs values for K13 under six different protein orientations for (A) single-rotamer model, (B) 2-rotamer model, (C) 3-rotamer model. All RDCs are normalized relative to the 13C–1H dipolar couplings, i.e., 1DCβCγ values are scaled up 10-fold, and all alignment strengths are scaled to DaNH = 10 Hz for the backbone RDCs. Colors correspond to the differently aligned samples (red: K19A/V42E/D47K GB3 in Pf1; green: K4A/K19E/V42E GB3 in Pf1; pink: wild-type GB3 in bicelles; gray: wild-type GB3 in PEG; black: wild-type GB3 in Pf1). Circles correspond to 1DCβHβ couplings. (D–F) The best-fitted K13 χ1 rotamers for the 1-, 2-, and 3-rotamer models.
The continuous χ1-population model discussed above did not allow us to make reliable stereospecific assignment for four residues: M1, E15, E24 and E56. When fitting the RDCs of these residues with the discrete rotamer model, while taking into account that significant populations of severely skewed rotamers (deviating >30° relative to the ideally staggered conformers) are unfavorable and that Cγ atoms should not make a serious steric clash with any backbone or Cβ atom, this permitted additional stereospecific assignment of all but E15 (SI Table S4).
Uncertainty in ⟨χ1⟩ and Populations of Rotamers
As mentioned above, the width of the χ1 distribution within a given rotamer well is generally ill-defined by the RDC data. For example, assuming a Gaussian χ1 distribution within each rotamer well of standard deviations σ yielded very similar fit qualities for any σ ≤ 15°, with a very shallow minimum for σ ≈ 10° (see SI Figure S12). The quality of the fit, however, is quite sensitive to population of the rotameric wells and to their average ⟨χ1⟩ values. To evaluate how tightly these latter two parameters are defined by the experimental data we resorted to a Monte Carlo analysis.59
Starting from the best-fit χ1 result obtained from the experimental data by the multiple-rotamer fit program (Table 1), synthetic RDC values are first generated for this best-fit solution. Because the errors in the experimental RDC data were simply based on lower limit estimates, made on the basis of line width and signal-to-noise ratio,45 Gaussian noise is added to the synthetic RDC data, with its amplitude adjusted such that the error in the best fit to these synthetic, noise-contaminated data equals that observed in the original fit of the experimental data. This level of noise is added in 1000 separate runs of the fitting program, with the distribution in the output parameters then being representative of their uncertainty. Below, we illustrate the results of this error analysis for three typical cases: K4, K13 and K50 (Figure 6). Fits to the experimental data indicate a single χ1 = 299° rotamer for K50. For K4, a dominant (67%) population centered at χ1 = 183 ± 3° is seen, and a minor population centered at χ1 = 293 ± 5°. For K13, a major g– rotamer (χ1 = 302 ± 2°) populated at 67% is accompanied by g+ and t rotamers, each populated at ca. 16%. The Monte Carlo derived uncertainties in the populations of the χ1 rotamers are quite small for most residues, but the uncertainty in the actual χ1 values increases roughly inversely with their population (Table 1).
Figure 6.
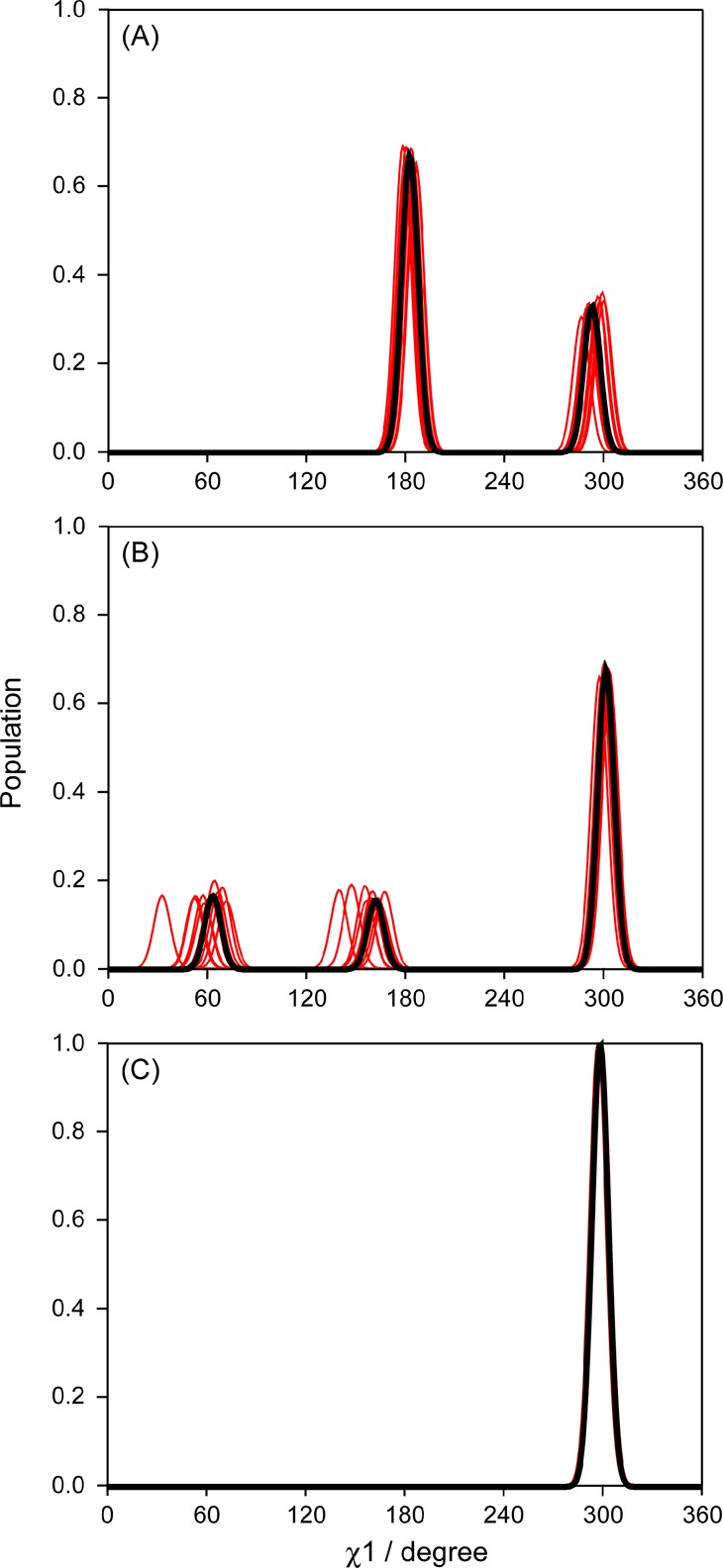
Effect of uncertainty in the structure and side chain RDC data on the χ1 distributions, obtained by Monte Carlo rotamer analysis, of (A) K4, (B) K13 and (C) K50. The heavy line corresponds to the results of the fits to the experimental data, using the newly refined backbone structure and 13Cα–13Cβ orientations. The thin lines (obscured by the black line for K50) correspond to 10 separate fits obtained when perturbing the 13Cα–13Cβ orientations by 3.2° structural noise, and using the predicted RDCs of the initial fit to which Gaussian noise has been added that results in the same quality of fit as obtained for the experimental data (see text), presenting a visualization of the uncertainty in the derived rotamer populations. A full set is presented in SI Figure S9.
Comparison to X-ray Structures
Numerous high resolution X-ray structures are available for GB3 and its closely homologous first IgG binding domain, GB1, all showing very similar backbone coordinates. Below, we will focus on comparison with the X-ray structures determined by Derrick and Wigley at room temperature (PDB entry 1IGD)43 and a subsequent study that used anisotropic temperature factors during refinement (PDB entry 2IGD).60 Note that both 1IGD and 2IGD include five additional N-terminal residues, but the protein studied here by NMR is identical in sequence to these earlier structures from residue Y3 onward. Comparison to the other closely related structures is included in SI Table S8.
With backbone RDC Q-factors of ca. 18% for the N–H and Cα–Hα RDCs for both 1IGD and 2IGD, the backbone of these two X-ray structures agrees as well as, or better than, seen for any other protein for which RDCs and X-ray coordinates are available. However, as was seen previously for ubiquitin,61 side chain RDCs are predicted much less accurately by the X-ray coordinates (Q factors of 55 and 51% for 1IGD and 2IGD). To a large extent, this worse agreement for side chains is dominated by those residues that adopt multiple rotameric states in our NMR study, whereas in the X-ray study the default analysis uses a single rotameric state to fit the electron density. For example, the electron density used for generating the side chain of E24 in the 1IGD model is in good agreement with the equilibrium of g– and t observed in our RDC study. On the other hand, the electron density of V42 is in good agreement with the g– rotamer of its X-ray coordinates, which strongly disagrees with both the observed RDCs and the large 3JHαHβ value, and which is considered to be energetically unfavorable in β-sheet structures.62 Remarkably, the predominant t rotamer seen in our NMR analysis can be accommodated in the crystal lattice without causing steric clashing. It therefore appears likely that less favorable hydration of the t rotamer within the confines of the crystalline lattice is responsible for the difference between crystal and solution states for this residue. This result highlights that even in the absence of direct crystal contacts side chain orientations may well differ between the solution and crystalline states.
Multiconformer refinement is possible for very high resolution X-ray data, and four residues in the 2IGD X-ray structure of GB3 show significantly different pairs of χ1 values.60 All but one of these (I7) correspond to residues for which our RDC analysis also finds multiple conformers, mostly with very similar χ1 values and populations as the X-ray structure. For example, for V21 2IGD reports χ1 = 295/75°, with populations of 62/38%, versus χ1 = 302/68° and populations of 60/40% in our RDC analysis (Table 1). Similarly, for N35 2IGD shows χ1 = 189/291°, with populations of 50/50%, versus χ1 = 184/288°, with populations of 52/48% in our RDC analysis.
For 20 of the GB3 residues, our above RDC analysis shows a dominant (≥90%) population of a single narrow distribution of χ1 angles. For all but one (T55) of these 20 residues, their χ1 angle corresponds to the rotameric state observed in the X-ray structure, and for a subset of 16 the rmsd relative to the values seen in the 1.1-Å X-ray structure (PDB entry 1IGD(43)) is only 4°. For T55, 1IGD shows a t rotamer, whereas our NMR results indicate g–, also seen in the X-ray structures of seven high resolution X-ray structures of closely related mutants (SI Table S8). For residues I7, Y33 and K50 the NMR χ1 angle also differs substantially from 1IGD (by ca. 14°), but these residues show much above average variation in the X-ray structures too. For about half of all non-Gly, non-Ala residues we find that the side chain shows significant (≥10%) population of more than one χ1 rotameric state. Although in most cases the most populated rotamer corresponds to the one seen in the X-ray structure, for surface-exposed residues N8 and N35 the X-ray observed χ1 rotamer is populated just below 50% in solution.
Fitting of 3JHαHβ to RDC-Derived χ1 Values
The importance of 3JHαHβ couplings for evaluating side chain conformations in peptides and proteins has long been recognized.27,63−65 However, optimal parametrization of the corresponding Karplus equations has proven to be challenging because the assumption of static, accurately known χ1 angles often does not apply. A number of elegant solutions have been put forward to address this problem,28,29,66 but the challenge in empirically optimizing this relation for an intrinsically flexible system remains. On the other hand, Altona and co-workers have convincingly demonstrated for a wide range of rigid, small organic molecules that correction terms for electronegativity of substituents and their chirality can greatly improve the accuracy of the relation between 3JHH couplings and the intervening dihedral angle.67
Here, we compare 3JHαHβ couplings predicted by the modified Karplus equations of Haasnoot et al.67 for our discrete rotamer analysis (Table 1) with experimental values obtained from a 3D HA[HN,HB](CACO)NH spectrum34 (SI Table S9; SI Figure S10). For peptides and proteins, six different substituent patterns apply for 3JHαHβ, and the corresponding Karplus equations are summarized in SI Table S10. Comparison of observed with predicted 3JHαHβ couplings shows a tight correlation (RP = 0.96), but a slope that is distinctly smaller than 1 (Figure 7). A reduction in the slope of the correlation is consistent with the presence of fluctuations of each rotamer around its average χ1 value.68 The Haasnoot-modified Karplus relations are written as67
| 3 |
with the coefficients for the different types of couplings presented in SI Table S10. In the presence of normally distributed χ1 fluctuations with standard deviation σ, these equations can be rewritten as
| 4 |
with A′ = A exp(−2σ2); B′ = B exp(−σ2/2); C′ = C exp(−2σ2); and D′ = D + (A/2)[1 – exp(−2σ2)]. 3JHαHβ Karplus curves, generated by eq 4 for different values of σ show the expected decrease in the amplitude of the Karplus curve.68 Comparison of the 3JHαHβ values predicted by eq 4 when using σ = 20° against those predicted for the corresponding static (σ = 0°) Karplus curve (SI Figure S11), yields nearly the same slope and intercept as observed experimentally, and an rmsd of 0.75 Hz. This result suggests that the smaller range of observed 3JHαHβ couplings relative to that expected based on the static Haasnoot-modified Karplus equations is consistent with rapid χ1 fluctuations of substantial amplitude (σ ≈ 20°). This σ amplitude is somewhat larger than the σ ≈ 10° obtained from the shallow minimum when systematically stepping σ as a global parameter (applied to all residues) in the fitting of RDCs to discrete rotamers (see SI Figure S12). However, it should be noted that the σ value obtained from RDC fitting reflects the increase in χ1 fluctuations over the analogous fluctuations that exist in the protein backbone, whose RDCs were used to define the alignment tensor strength. By contrast, the σ value obtained from the J coupling analysis reflects the amplitude of dynamics relative to the conformationally highly restrained, and therefore quite rigid, small molecules used by Altona and co-workers to define their Karplus equations. Therefore, the larger σ value seen in the analysis of the 3JHαHβ couplings is not surprising.
Figure 7.
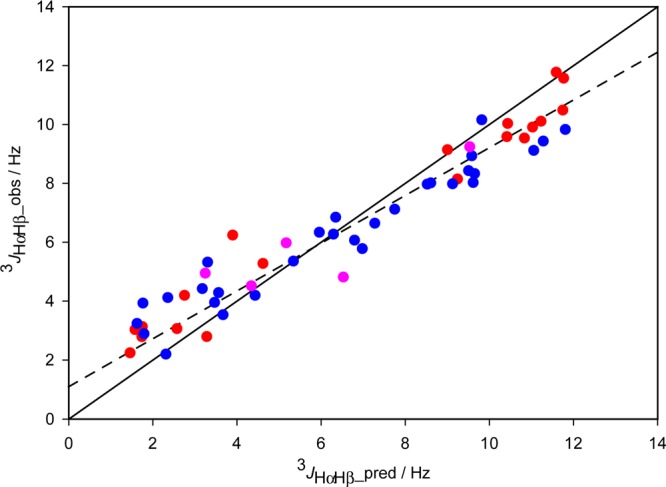
Comparison of observed 3JHαHβ couplings with those predicted by the substituent-specific Karplus equations of Haasnoot et al.67 The solid line corresponds to x = y; the dashed line corresponds to values predicted by these same equations after adding Gaussian fluctuations (cf. eq 4) with a standard deviation of σ = 20° to the χ1 angles reported in Table 1. Note that to a very good approximation, the effect of σ = 20° simply scales the difference in the predicted 3JHαHβ relative to a value of ca. 6 Hz (Figure S11). The rmsd between observed and predicted 3JHαHβ values (using the σ = 20° motional model) is 0.75 Hz. 3JHαHβ values (Table S16) for residues with a single χ1 rotamer are in red, two χ1 rotamers in blue, and three rotamers in pink.
When the Haasnoot-modified Karplus equations without any motional correction are used to predict 3JHαHβ values for the continuous, model-free χ1 distributions, better agreement (rmsd 0.93 Hz) is obtained than for the discrete rotamer model (1.15 Hz), presumably because the effect of the Gaussian fluctuations around the discrete rotameric states is already somewhat reflected in the model-free χ1 distributions.
Concluding Remarks
The presence of extensive χ1 rotamer averaging for many residues in folded proteins has long been recognized, mostly from the analysis of 3JHH couplings28,65 but also from analysis of NMR relaxation data of side chain 13C and 2H nuclei,15,16,69 and from the observation that most side chain RDCs are smaller in solution than predicted from a static X-ray structure.61,30 Protein X-ray structures collected at high resolution and signal-to-noise ratios also reveal the presence of extensive side chain conformational heterogeneity.58,70,71,45 Commonly only the electron density of the most populated side chain conformers is represented in the deposited coordinates, and the presence of residual electron density reflecting alternate conformations becomes embedded in elevated temperature or B factors.4,72 Interestingly, side chain conformational heterogeneity present at room temperature may be reduced by widely used cryo-cooling methods used for collecting X-ray diffraction data.7
Our study provides an exceptionally large set of precise experimental data for defining the χ1 distribution of nearly all residues in a small protein. For slightly less than half of all non-Gly/Ala residues, we find that the χ1 angle predominantly (≥90%) samples a single, relatively tight distribution. The width of these distributions is not very well-defined by the RDC data, but when this width is used as a global fitting parameter the RDC data suggest that the amplitude of the χ1 fluctuations is ca. 10° larger than for the protein backbone. Comparison of the observed 3JHαHβ couplings with those predicted on the basis of substituent-specific Karplus equations indicates χ1 fluctuations that are ca. 20° larger than the angular fluctuations in the conformationally highly restrained small cyclic molecules, used by Altona and co-workers for parametrizing these Karplus equations.67 We also note that the fit between experimental 3JHαHβ and Altona-Karplus predicted values improves more than 2-fold when using our RDC analysis compared to structures obtained from the advanced eNOE analysis (PDB entry 2LUM,73 averaged over its 60 ensemble members), even though these eNOE-derived structures, determined without RDCs, validate considerably better against backbone RDCs (QCαCβ = 28%) than those from standard NOE analysis (QCαCβ = 42% for PDB entry 1GB1(74)).
The majority of the residues in GB3 show significant, ≥ 10% sampling of at least two conformers (Table 1). In part, this relatively high fraction may be related to the small size of the protein, which results in a higher surface to volume ratio compared to larger proteins. Indeed, the vast majority of multiconformer side chains correspond to surface residues, whereas most interior residues predominantly sample a single χ1 distribution. However, there are multiple exceptions to this rule: For example, solvent-exposed Asn37 shows a single, narrow χ1 cluster, related to its side-chain being involved in a C-cap H-bond. On the other hand, Val54 which is well buried in the hydrophobic interior of the protein shows a nearly 20% population of a 25°-skewed t rotamer, in rapid exchange with its predominant nearly ideal g+ rotamer. Only four residues are found to sample more than two χ1 rotamers, all of them located in the loop that connects strands β1 and β2.
As discussed above, the width of each χ1 rotameric well is not tightly restricted by the RDC data, but the average χ1 value of each well is sharply defined. Our analysis is completely model-free when calculating the χ1 distributions, but inspection of the results shows exceptionally tight clustering of the center of each most populated well to the ideal staggered rotamer values of 60 + N × 120°. The center of the most populated rotamer (χ1(1) column in Table 1) shows an rmsd of only 8° from the ideal staggered rotamer values. A previous analysis of X-ray structures indicated that the average χ1 deviation from ideal staggered rotamers for many residue types was a function of crystallographic resolution, a finding ascribed to the electron density of minor conformers skewing the average attributed to the major conformer.75 In the analysis of X-ray structures, the standard deviations of the rotamer distributions also were found to decrease from >20° for structures determined at a resolution >2.0 Å to ca. 15° for structures solved at higher resolution (≤2.0 Å). Our study shows a substantial further reduction in these standard deviations, at least when considering the most populated conformer for each residue (Table 1).
Many of the lowly populated side chain χ1 conformers identified in our study deviate substantially more from ideal staggered conformations (rmsd 22°) than the major χ1 conformer. The low values of their populations suggests that these conformers are in energetically less favorable environments, and steric clashing may cause their skewing away from perfectly staggered states. On the other hand, our error analysis also points to larger experimental uncertainty in the χ1 angles sampled by minor conformers (σχ1 in Table 1). For example, when considering Monte Carlo analysis of K13 (Figure 5B), the variance in the χ1 angle of the major g– conformer (χ1 = 302°) is only 2° whereas the minor t conformer (16% populated) shows a highly skewed χ1 angle of 160° but with a standard deviation of 11°.
It can be noted that even while the values of the fitted χ1 torsion angles as well as their total rotamer populations summed for given residue types agree well with the statistics exhibited by structural databases,62,52 fitted site-specific χ1 populations sampled by the individual residues differ significantly from the database-derived aggregate values. This observation indicates that selection of a given side chain rotamer is significantly influenced by its nonlocal tertiary contacts. It also is worth noting that essentially all residues located in the regions around residues L12 and G41, known to be the most dynamic based on the backbone 15N relaxation data, exhibit the sampling of more than one χ1 rotamer, consistent with the rotameric selection being affected by tertiary contacts.
It has long been recognized that RDC data collected for a protein under multiple different alignment conditions carries valuable information on the distribution of atomic coordinates sampled.76,77 For ordered regions in well-structured proteins, the amplitudes of angular excursions tend to be rather modest for the backbone N–H vectors, widely used for such studies, making accurate analysis of such data in terms of dynamics quite challenging.3,25,78−80 By contrast, the changes in side chain bond vector orientations associated with χ1 rotamer hopping are large and result in bond vector changes of ca. 110°, a value that maximizes its effect on the corresponding RDCs. Therefore, side chain RDCs are exquisitely well suited for analyzing the χ1 rotameric averaging process. Our data show that the most populated staggered conformer χ1 angles fall very close to those seen in atomic resolution X-ray structures of small peptides, and those of proteins when extrapolated to a resolution of 0.8 Å.75 By contrast, the RDC data are relatively insensitive to small excursions within each rotameric well, as was highlighted by the difference in well distributions obtained when adding a small entropy term to the data analysis. This difficulty in extracting the amplitude of fluctuations within each rotameric well is akin to the above-mentioned challenges faced when quantifying from 15N–1H RDCs the relatively small amplitudes of backbone motions typically seen in α-helices and β-sheet.
Although it may seem straightforward to extend our RDC χ1 analysis to any other protein, we need to point out that the very detailed analysis presented here requires an extraordinary amount of data, not easily collected for many other proteins. So, most side chain analyses by RDCs likely will remain more limited in scope, and simply aim to ascertain whether a single χ1 rotamer is populated, and which one. Our data provide strong evidence that if a single rotamer is populated, it will be very close to the ideally staggered conformation. This property allows a χ1 rotamer estimate to be made by simply comparing the 1DCβHβ or 1DCβCγ to the backbone 1DCαHα and 1DCαC′ couplings: For example, for Thr and χ1 = 300°, the Cβ–Hβ vector is antiparallel to Cα–Hα, and they will have very similar RDCs. Again for Thr, but for χ1 = 180°, the Cβ–Cγ vector is antiparallel to Cα–Hα and its normalized RDC (or the linearly related CγH3 methyl RDC81) will be close to 1DCαHα, while Cβ–Hβ will be antiparallel to the Cα–C′ vector, making their normalized RDCs equivalent. Similar analysis can be made for other residues, allowing straightforward evaluation of χ1, even without recourse to a backbone structure. However, if such a cursory analysis does not point to a single rotamer, establishing the rotameric equilibrium and deviations from perfectly staggered rotameric states rapidly requires far more data.
GB3 is sufficiently small to allow generation of very long molecular dynamics trajectories and the protein has been used to evaluate the quality of computational methods by validation against experimental results.82−85 Our new results complement these earlier experimental data by introducing detailed side chain information, greatly extending its use for force field development. Moreover, the model-free χ1 distributions derived in our study will enable improved parametrization of the wide range of different Karplus parametrizations that have been in common use for 3JNHβ, 3JC′Hβ, 3JC′Cγ, and 3JNCγ.28,29,86,87 Similarly, our data will provide a new benchmark for evaluating the impact of side chain conformations on chemical shifts, an area that remains under-developed but holds strong potential for increasing the level of structural detail that can be extracted from these readily accessible parameters.88−91
Acknowledgments
We thank Dr. Yang Shen for assistance in data analysis and Dennis Torchia for useful discussions. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and by the Intramural Antiviral Target Program of the Office of the Director, NIH. F. Li acknowledges financial support from the China Scholarship Council.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b10072.
Experimental procedures and additional data. (PDF)
Author Present Address
‡ Key Laboratory of Pesticides and Chemical Biology, College of Chemistry, Central China Normal University, Wuhan, Hubei 430079, People’s Republic of China.
The authors declare no competing financial interest.
Supplementary Material
References
- Gardner K. H.; Kay L. E. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 357–406 10.1146/annurev.biophys.27.1.357. [DOI] [PubMed] [Google Scholar]
- Furnham N.; Blundell T. L.; DePristo M. A.; Terwilliger T. C. Nat. Struct. Mol. Biol. 2006, 13, 184–185 10.1038/nsmb0306-184. [DOI] [PubMed] [Google Scholar]
- Lange O. F.; Lakomek N. A.; Fares C.; Schroder G. F.; Walter K. F. A.; Becker S.; Meiler J.; Grubmuller H.; Griesinger C.; de Groot B. L. Science 2008, 320, 1471–1475 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Woldeyes R. A.; Sivak D. A.; Fraser J. S. Curr. Opin. Struct. Biol. 2014, 28, 56–62 10.1016/j.sbi.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick R. B.; van den Bedem H.; Fraser J. S.; Wright P. E. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E445–E454 10.1073/pnas.1323440111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang P. T.; Holton J. M.; Fraser J. S.; Alber T. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 237–242 10.1073/pnas.1302823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J. S.; van den Bedem H.; Samelson A. J.; Lang P. T.; Holton J. M.; Echols N.; Alber T. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16247–16252 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay L. E.; Torchia D. A.; Bax A. Biochemistry 1989, 28, 8972–8979 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- Dayie K. T.; Wagner G.; Lefevre J. F. Annu. Rev. Phys. Chem. 1996, 47, 243–282 10.1146/annurev.physchem.47.1.243. [DOI] [PubMed] [Google Scholar]
- Fushman D.; Tjandra N.; Cowburn D. J. Am. Chem. Soc. 1999, 121, 8577–8582 10.1021/ja9904991. [DOI] [Google Scholar]
- d’Auvergne E. J.; Gooley P. R. J. Biomol. NMR 2003, 25, 25–39 10.1023/A:1021902006114. [DOI] [PubMed] [Google Scholar]
- Richarz R.; Nagayama K.; Wuthrich K. Biochemistry 1980, 19, 5189–5196 10.1021/bi00564a006. [DOI] [PubMed] [Google Scholar]
- Nicholson L. K.; Kay L. E.; Baldisseri D. M.; Arango J.; Young P. E.; Bax A.; Torchia D. A. Biochemistry 1992, 31, 5253–5263 10.1021/bi00138a003. [DOI] [PubMed] [Google Scholar]
- Lee A. L.; Flynn P. F.; Wand A. J. J. Am. Chem. Soc. 1999, 121, 2891–2902 10.1021/ja983758f. [DOI] [Google Scholar]
- Muhandiram D. R.; Yamazaki T.; Sykes B. D.; Kay L. E. J. Am. Chem. Soc. 1995, 117, 11536–11544 10.1021/ja00151a018. [DOI] [Google Scholar]
- Yang D. W.; Mittermaier A.; Mok Y. K.; Kay L. E. J. Mol. Biol. 1998, 276, 939–954 10.1006/jmbi.1997.1588. [DOI] [PubMed] [Google Scholar]
- Lipari G.; Szabo A. J. Am. Chem. Soc. 1982, 104, 4546–4559 10.1021/ja00381a009. [DOI] [Google Scholar]
- Mulder F. A. A.; Mittermaier A.; Hon B.; Dahlquist F. W.; Kay L. E. Nat. Struct. Biol. 2001, 8, 932–935 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- Palmer A. G. Chem. Rev. 2004, 104, 3623–3640 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Mittermaier A. K.; Kay L. E. Trends Biochem. Sci. 2009, 34, 601–611 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Peti W.; Meiler J.; Bruschweiler R.; Griesinger C. J. Am. Chem. Soc. 2002, 124, 5822–5833 10.1021/ja011883c. [DOI] [PubMed] [Google Scholar]
- Tolman J. R. J. Am. Chem. Soc. 2002, 124, 12020–12030 10.1021/ja0261123. [DOI] [PubMed] [Google Scholar]
- Tolman J. R.; Ruan K. Chem. Rev. 2006, 106, 1720–1736 10.1021/cr040429z. [DOI] [PubMed] [Google Scholar]
- Yao L.; Vogeli B.; Torchia D. A.; Bax A. J. Phys. Chem. B 2008, 112, 6045–6056 10.1021/jp0772124. [DOI] [PubMed] [Google Scholar]
- Maltsev A. S.; Grishaev A.; Roche J.; Zasloff M.; Bax A. J. Am. Chem. Soc. 2014, 136, 3752–3755 10.1021/ja4132642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzakula Z.; Westler W. M.; Edison A. S.; Markley J. L. J. Am. Chem. Soc. 1992, 114, 6195–6199 10.1021/ja00041a043. [DOI] [Google Scholar]
- Dzakula Z.; Edison A. S.; Westler W. M.; Markley J. L. J. Am. Chem. Soc. 1992, 114, 6200–6207 10.1021/ja00041a044. [DOI] [Google Scholar]
- Perez C.; Lohr F.; Ruterjans H.; Schmidt J. M. J. Am. Chem. Soc. 2001, 123, 7081–7093 10.1021/ja003724j. [DOI] [PubMed] [Google Scholar]
- Schmidt J. M. J. Biomol. NMR 2012, 54, 97–114 10.1007/s10858-012-9653-2. [DOI] [PubMed] [Google Scholar]
- Mittermaier A.; Kay L. E. J. Am. Chem. Soc. 2001, 123, 6892–6903 10.1021/ja010595d. [DOI] [PubMed] [Google Scholar]
- Ramirez B. E.; Bax A. J. Am. Chem. Soc. 1998, 120, 9106–9107 10.1021/ja982310b. [DOI] [Google Scholar]
- Yao L. S.; Bax A. J. Am. Chem. Soc. 2007, 129, 11326–11327 10.1021/ja073937+. [DOI] [PubMed] [Google Scholar]
- Ulmer T. S.; Ramirez B. E.; Delaglio F.; Bax A. J. Am. Chem. Soc. 2003, 125, 9179–9191 10.1021/ja0350684. [DOI] [PubMed] [Google Scholar]
- Lohr F.; Schmidt J. M.; Ruterjans H. J. Am. Chem. Soc. 1999, 121, 11821–11826 10.1021/ja991356h. [DOI] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. J. Biomol. NMR 1995, 6, 277–293 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Goddard T. D.; Kneller D. G.. Sparky 3; University of California: San Francisco, 2008. [Google Scholar]
- Li F.; Lee J. H.; Grishaev A.; Ying J.; Bax A. ChemPhysChem 2015, 16, 572–578 10.1002/cphc.201402704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakomek N. A.; Walter K. F. A.; Fares C.; Lange O. F.; de Groot B. L.; Grubmuller H.; Bruschweiler R.; Munk A.; Becker S.; Meiler J.; Griesinger C. J. Biomol. NMR 2008, 41, 139–155 10.1007/s10858-008-9244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra N.; Bax A. Science 1997, 278, 1111–4 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- Losonczi J. A.; Prestegard J. H. J. Biomol. NMR 1998, 12, 447–451 10.1023/A:1008302110884. [DOI] [PubMed] [Google Scholar]
- Ruckert M.; Otting G. J. Am. Chem. Soc. 2000, 122, 7793–7797 10.1021/ja001068h. [DOI] [Google Scholar]
- Hansen M. R.; Mueller L.; Pardi A. Nat. Struct. Biol. 1998, 5, 1065–1074 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- Derrick J. P.; Wigley D. B. J. Mol. Biol. 1994, 243, 906–918 10.1006/jmbi.1994.1691. [DOI] [PubMed] [Google Scholar]
- Hus J. C.; Peti W.; Griesinger C.; Bruschweiler R. J. Am. Chem. Soc. 2003, 125, 5596–5597 10.1021/ja029719s. [DOI] [PubMed] [Google Scholar]
- Bax A.; Kontaxis G.; Tjandra N. Methods Enzymol. 2001, 339, 127–174 10.1016/S0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- Doddrell D. M.; Pegg D. T.; Bendall M. R. J. Magn. Reson. 1982, 48, 323–327 10.1016/0022-2364(82)90286-4. [DOI] [Google Scholar]
- Vuister G. W.; Bax A. J. Magn. Reson. 1992, 98, 428–435 10.1016/0022-2364(92)90144-V. [DOI] [Google Scholar]
- Geen H.; Freeman R. J. Magn. Reson. 1991, 93, 93–141 10.1016/0022-2364(91)90034-Q. [DOI] [Google Scholar]
- Zweckstetter M.; Bax A. J. Biomol. NMR 2002, 23, 127–137 10.1023/A:1016316415261. [DOI] [PubMed] [Google Scholar]
- Engh R. A.; Huber R. Acta Crystallogr., Sect. A: Found. Crystallogr. 1991, 47, 392–400 10.1107/S0108767391001071. [DOI] [Google Scholar]
- Ward J. M.; Skrynnikov N. R. J. Biomol. NMR 2012, 54, 53–67 10.1007/s10858-012-9651-4. [DOI] [PubMed] [Google Scholar]
- Lovell S. C.; Word J. M.; Richardson J. S.; Richardson D. C. Proteins: Struct., Funct., Genet. 2000, 40, 389–408. [DOI] [PubMed] [Google Scholar]
- Hall J. B.; Fushman D. J. Biomol. NMR 2003, 27, 261–275 10.1023/A:1025467918856. [DOI] [PubMed] [Google Scholar]
- Guntert P.; Braun W.; Billeter M.; Wuthrich K. J. Am. Chem. Soc. 1989, 111, 3997–4004 10.1021/ja00193a036. [DOI] [Google Scholar]
- Neri D.; Szyperski T.; Otting G.; Senn H.; Wuthrich K. Biochemistry 1989, 28, 7510–7516 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- Kainosho M.; Torizawa T.; Iwashita Y.; Terauchi T.; Ono A. M.; Guntert P. Nature 2006, 440, 52–57 10.1038/nature04525. [DOI] [PubMed] [Google Scholar]
- Soper D. S.p-Value Calculator for an F-Test, http://danielsoper.com/statcalc3/calc.aspx?id=7 (accessed November 1, 2015).
- Davis I. W.; Arendall W. B.; Richardson D. C.; Richardson J. S. Structure 2006, 14, 265–274 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Bevington P. R.; Robinson D. K.. Data Reduction and Error Analysis for the Physical Sciences; McGraw-Hill: New York, 1992. [Google Scholar]
- Butterworth S.; Lamzin V. L.; Wigley D. B.; Derrick J. P.; Wilson K. P. Protein Data Bank, entry 2IGD, 1997; http://www.rcsb.org/pdb/files/2IGD.pdb.
- Bax A. Protein Sci. 2003, 12, 1–16 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack R. L.; Karplus M. Nat. Struct. Biol. 1994, 1, 334–340 10.1038/nsb0594-334. [DOI] [PubMed] [Google Scholar]
- Feeney J. J. Magn. Reson. 1976, 21, 473–478 10.1016/0022-2364(76)90052-4. [DOI] [Google Scholar]
- Demarco A.; Llinas M.; Wuthrich K. Biopolymers 1978, 17, 617–636 10.1002/bip.1978.360170307. [DOI] [Google Scholar]
- Kariminejad Y.; Schmidt J. M.; Ruterjans H.; Schwalbe H.; Griesinger C. Biochemistry 1994, 33, 5481–5492 10.1021/bi00184a017. [DOI] [PubMed] [Google Scholar]
- Schmidt J. M. J. Biomol. NMR 2007, 37, 287–301 10.1007/s10858-006-9140-8. [DOI] [PubMed] [Google Scholar]
- Haasnoot C. A. G.; Deleeuw F.; Altona C. Tetrahedron 1980, 36, 2783–2792 10.1016/0040-4020(80)80155-4. [DOI] [Google Scholar]
- Bruschweiler R.; Case D. A. J. Am. Chem. Soc. 1994, 116, 11199–11200 10.1021/ja00103a062. [DOI] [Google Scholar]
- Igumenova T. I.; Frederick K. K.; Wand A. J. Chem. Rev. 2006, 106, 1672–1699 10.1021/cr040422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. L.; Hendrickson W. A.; Honzatko R. B.; Sheriff S. Biochemistry 1986, 25, 5018–5027 10.1021/bi00366a008. [DOI] [PubMed] [Google Scholar]
- Lang P. T.; Ng H.-L.; Fraser J. S.; Corn J. E.; Echols N.; Sales M.; Holton J. M.; Alber T. Protein Sci. 2010, 19, 1420–1431 10.1002/pro.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyan J.; Petsko G. A.; Levy R. M.; Karplus M. J. Mol. Biol. 1986, 190, 227–254 10.1016/0022-2836(86)90295-0. [DOI] [PubMed] [Google Scholar]
- Vogeli B.; Kazemi S.; Guntert P.; Riek R. Nat. Struct. Mol. Biol. 2012, 19, 1053–1058 10.1038/nsmb.2355. [DOI] [PubMed] [Google Scholar]
- Gronenborn A. M.; Filpula D. R.; Essig N. Z.; Achari A.; Whitlow M.; Wingfield P. T.; Clore G. M. Science 1991, 253, 657–661 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- MacArthur M. W.; Thornton J. M. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 994–1004 10.1107/S0907444999002231. [DOI] [PubMed] [Google Scholar]
- Meiler J.; Prompers J. J.; Peti W.; Griesinger C.; Bruschweiler R. J. Am. Chem. Soc. 2001, 123, 6098–6107 10.1021/ja010002z. [DOI] [PubMed] [Google Scholar]
- Tolman J. R. Curr. Opin. Struct. Biol. 2001, 11, 532–539 10.1016/S0959-440X(00)00245-1. [DOI] [PubMed] [Google Scholar]
- Briggman K. B.; Tolman J. R. J. Am. Chem. Soc. 2003, 125, 10164–10165 10.1021/ja035904+. [DOI] [PubMed] [Google Scholar]
- Fenwick R. B.; Esteban-Martin S.; Richter B.; Lee D.; Walter K. F. A.; Milovanovic D.; Becker S.; Lakomek N. A.; Griesinger C.; Salvatella X. J. Am. Chem. Soc. 2011, 133, 10336–10339 10.1021/ja200461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvignies G.; Bernado P.; Meier S.; Cho K.; Grzesiek S.; Bruschweiler R.; Blackledge M. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13885–13890 10.1073/pnas.0505129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottiger M.; Bax A. J. Am. Chem. Soc. 1999, 121, 4690–4695 10.1021/ja984484z. [DOI] [Google Scholar]
- Olsson S.; Voegeli B. R.; Cavalli A.; Boomsma W.; Ferkinghoff-Borg J.; Lindorff-Larsen K.; Hamelryck T. J. Chem. Theory Comput. 2014, 10, 3484–3491 10.1021/ct5001236. [DOI] [PubMed] [Google Scholar]
- Granata D.; Camilloni C.; Vendruscolo M.; Laio A. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 6817–6822 10.1073/pnas.1218350110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Jing Q.; Yao L. J. Phys. Chem. B 2011, 115, 3488–3495 10.1021/jp108217z. [DOI] [PubMed] [Google Scholar]
- Markwick P. R. L.; Bouvignies G.; Blackledge M. J. Am. Chem. Soc. 2007, 129, 4724–4730 10.1021/ja0687668. [DOI] [PubMed] [Google Scholar]
- Bystrov V. F. Prog. Nucl. Magn. Reson. Spectrosc. 1976, 10, 41–81 10.1016/0079-6565(76)80001-5. [DOI] [Google Scholar]
- Salvador P.Dependencies of J-Couplings upon Dihedral Angles on Proteins. In Annual Reports on NMR Spectroscopy; Elsevier Ltd.: Amsterdam, 2014; Vol. 81, pp 185–227. [Google Scholar]
- London R. E.; Wingad B. D.; Mueller G. A. J. Am. Chem. Soc. 2008, 130, 11097–11105 10.1021/ja802729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. F.; Kay L. E. J. Am. Chem. Soc. 2011, 133, 8272–8281 10.1021/ja2014532. [DOI] [PubMed] [Google Scholar]
- Wishart D. S. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 62–87 10.1016/j.pnmrs.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Hansen D. F.; Neudecker P.; Kay L. E. J. Am. Chem. Soc. 2010, 132, 7589–7591 10.1021/ja102090z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.