

Abstract
Mouse lines with dysferlin deficiency are accepted animal models for limb girdle muscular dystrophy 2B and Miyoshi myopathy, yet slow progression of pathology prevents rapid screening of potential therapies for this disease. Our goal was to define a functional signature for skeletal muscles that lack dysferlin. Force generation and susceptibility to eccentric contractile injury measurements were performed in isolated limb muscles and the diaphragm from 10- and 36-week-old A/J and age-matched control mice. Limb muscles had normal specific force at both 10 and 36 weeks, whereas the diaphragm had significant deficits in both specific force and susceptibility to eccentric contractile injury. Membrane ruptures in the diaphragm during eccentric contractions occurred predominantly in myosin heavy chain 2A-expressing fibers. Dysferlin content did not vary significantly between wildtype muscles, suggesting that there was no correlation between disease severity and normal endogenous levels of the protein. These studies show that, unlike limb muscles, the diaphragm from the A/J mouse displays early deficits in function that may lower the age needed for evaluating potential therapies for dysferlinopathies.
Keywords: muscle function, muscular dystrophy, mouse models, dysferlin, fiber type
Miyoshi myopathy and limb-girdle muscular dystrophy 2B (LGMD2B) are caused by autosomal recessive inheritance of mutations in the DYSF gene that lead to the absence of dysferlin.1,2 The rate of pro gression of LGMD2B/Miyoshi is slow compared to other types of muscular dystrophy, in which onset of muscle weakness and atrophy in the legs may not occur until late adolescence or adulthood. Muscle involvement begins distally in Miyoshi patients in the soleus and gastrocnemius, whereas LGMD2B patients exhibit primarily proximal weakness starting in the pelvic girdle.3 Most patients have significantly elevated serum creatine kinase, and many ultimately require wheelchairs. However, there is a wide spectrum of severity and progression among patients, even though all lack the dysferlin protein.
Mice with dysferlin deficiency have become accepted models for LGMD2B/Miyoshi myopathy and have been utilized to understand the progression of this disease. Natural mutations occur in the SJL/J and A/J mouse,4,5 and gene targeting has resulted in two additional murine models.4,6 In all animal models the absence of dysferlin causes a distinctive defect in membrane repair, which is evident upon physical or chemical disruption of the sarcolemma. The SJL/J mouse model exhibits poor resolution of acute muscle injury instigated by cardiotoxin.7 In addition, the mouse models have histologic indications of pathology associated with the inability to repair membranes, including significant fatty and fibrotic infiltration, an increased proportion of centrally nucleated fibers, and increased Evans blue dye uptake, which is most prominent in the rectus abdominus.4 Thus, abdominal muscles bear several key hallmarks of the disease; however, the fiber orientation and muscle architecture of the rectus abdominus precludes its utility for functional measurements; thus, there is no definitive measure of muscle weakness in the mouse. In addition, the disease phenotype progresses slowly in mice, similar to human patients, and inhibits rapid screening of potential therapies to counter loss of dysferlin. Therefore, new tools must be developed to accelerate preclinical assessment of treatments for dysferlinopathies. The goal of this study was to define a functional signature for dysferlin-deficient skeletal muscles of the mouse and to determine if symptomatic muscle weakness is also present in the murine muscles. We anticipate that these studies will provide a basis for evaluating potential therapies in terms of muscle strength and fragility in mouse models of dysferlinopathies.
MATERIALS AND METHODS
The university's Animal Care Committee approved the experiments in this study. Mouse strains included A/J, A/WySnJ, mdx, and C57Bl/6 (C57). The A/J mouse harbors an ETn retrotransposon insertion in intron 4 of the dysferlin gene, which disrupts dysferlin expression. The A/WySnJ mouse is a commercially available inbred strain that is a suitable control for A/J mice. The mdx mouse is a model for Duchenne muscular dystrophy, and the C57 mouse is a wildtype control. Both male and female mice were utilized for experiments.
Isolated Muscle Mechanics
Mice were anesthetized with ketamine/xylazine. Muscles were removed and placed in a bath of Ringer's solution gas-equilibrated with 95% O2 / 5% CO2. Sutures were attached to the distal and proximal tendons of the extensor digitorum longus (EDL) and soleus muscles and to the central tendon and rib of the diaphragm preparations. Muscles were subjected to isolated mechanical measurements using a previously described apparatus (Aurora Scientific, Ontario, Canada)8 and bathed in Ringer's solution gas-equilibrated with 95% O2 / 5% CO2 and containing 0.2% Procion orange.9 After determining optimum length (Lo) by supramaximal twitch stimulation, maximum isometric tetanus was measured in the muscles during a 500-ms stimulation. Upon completion of these measurements, muscles were then subjected to a series of five eccentric contractions with a 5-min rest between contractions. Muscles were stimulated for a total of 700 ms. For the EDL and diaphragm, muscles were stretched 10% Lo in the final 200 ms stimulation. For the soleus, muscles were stretch 17.5% Lo in the final 200 ms stimulation. The drop in force between the first and last contraction served as an index of susceptibility to eccentric contraction-induced injury. Additional sets of muscles (n = 3 per age and strain) were bathed in Ringer's solution containing 0.2% Procion orange for 20 min but were not subjected to contractile measurements. For morphological analysis, samples were rinsed in phosphate-buffered saline (PBS), blotted, weighed, covered in mounting medium (OCT) prior to freezing in melting isopentane, and stored at −80°C. Muscles utilized for hematoxylin and eosin staining were rinsed in PBS, incubated in 4% paraformaldehyde for 1 h, then incubated overnight in 20% sucrose. The following day, muscles were blotted and covered in OCT prior to freezing in melting isopentane. Muscle samples for RNA and protein were rapidly frozen and stored in liquid nitrogen. Muscle cross-sectional areas (CSAs) were determined using the following formula:
where m is muscle mass (m), Lo is muscle length, L/Lo is the ratio of fiber length to muscle length, and 1.06 is the density of muscle10: L/Lo was 0.45 for EDL, 0.69 for soleus, and 1.0 for diaphragm.
Procion Orange Uptake
Muscle cryosections (10 μm) were washed in PBS and covered in mounting media containing DAPI. Images were acquired on an epifluorescence microscope (Leica) and analyzed for the proportion of area of Procion orange fluorescence using image analysis software (Open-Lab, Improvision, UK). The threshold for damage was set to eliminate background fluorescence. The same technique was utilized to determine the functional CSAs of the diaphragms. The CSAs of diaphragm strips were corrected for the fiber damage incurred at the edges of the muscle preparation during dissection, which also fluoresced brightly.
Muscle Fiber Typing
The 10-μm frozen cross-sections taken from the midbelly of each muscle were subjected to immunohistochemistry for laminin (rabbit anti-laminin Ab-1, Neomarkers, Fremont, California) to outline the muscle fibers. Fiber typing was performed with antibodies that recognize myosin heavy chain (MHC) 2a (SC-71), MHC 2b (BF-F3), and MHC 1 (BAF-8) as previously described.11 Nuclei were counterstained with DAPI. Stained sections were visualized and analyzed as described above.
Immunoblotting
Muscles were homogenized in 10 volumes/muscle wet weight of modified lysis buffer (50 mM Tris-HCl, pH 7.4, 1% w/v Triton X-100, 150 mM NaCl, 1 mM EGTA plus protease and phosphatase inhibitors). Homogenates were centrifuged to pellet debris, and the total protein was measured in the supernatant (BioRad, Hercules, California). Equal amounts of protein from each muscle lysate were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (Immobilon-P, Millipore, Bedford, Massachusetts). Membranes were incubated in a blocking buffer (5% milk/TTBS) and then incubated in primary monoclonal anti-dysferlin (1:20, Neomarkers) or monoclonal anti-GAPDH (1:4,000, Sigma, St. Louis, Missouri) diluted in 5% milk/TTBS overnight at 4°C. Membranes were washed in 5% milk/TTBS and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody. Detection was performed using enhanced chemiluminescence (ECL), and analysis of band intensity was performed using image analysis software (Kodak mm4000).
Statistics
One-way analysis of variance (ANOVA) was utilized for comparisons of muscle measurements, followed by Tukey's multiple comparison tests to determine differences between groups. Unpaired t-tests were utilized to compare two conditions. Statistical significance was accepted for P < 0.05.
RESULTS
The goal of this study was to identify skeletal muscles in the dysferlin-deficient mouse that displayed functional deficits similar to that in Miyoshi/LGMD2B patients. Isolated muscle mechanics, including isometric tetanic force as well as susceptibility to eccentric contractile injury, were performed on the EDL, soleus, and diaphragm muscles, which are ideally suited for ex vivo functional measurements. Murine skeletal muscles from young (10 weeks old) and mature (36 weeks old) A/J and A/WySnJ mice were evaluated.
Strength was measured by specific force (force per cross-section area) for each muscle group. In all cases the specific forces tended to be lower in the A/J muscles compared to muscles from age-matched controls (Fig. 1). This resulted in significant differences in the EDL muscle specific force at 10 weeks of age. The most dramatic difference in strength was observed in the diaphragms, where specific force was ≈50% lower in A/J muscles compared to wildtype controls at both ages. Examination of diaphragm sections from young and mature animals (Fig. 2) showed little indication of pathology in muscles from 10-week-old A/J mice. However, by 36 weeks of age the diaphragms exhibited centralized nuclei, fatty infiltration, and rounded prenecrotic fibers. Thus, loss of muscle strength in the A/J diaphragm preceded histological signs of disease.
FIGURE 1.

Specific forces of isolated muscles from the A/J and A/WySnJ (WT) mice. Data are presented as mean ± SD. Top panel, EDL with n = 9 muscles for each experimental group; center panel, soleus with n = 8 muscles for each experimental group; lower panel, diaphragm with n = 5 muscles for each experimental group. Specific force was lower in EDL muscles from young A/J mice compared to age-matched controls. No significant differences were observed between the groups of soleus muscles. The specific forces of the diaphragms from both 10-week-old and 36-week-old A/J mice were significantly lower than those of the age-matched control mice (P < 0.001 by Tukey's post-hoc comparison after one-way ANOVA).
FIGURE 2.
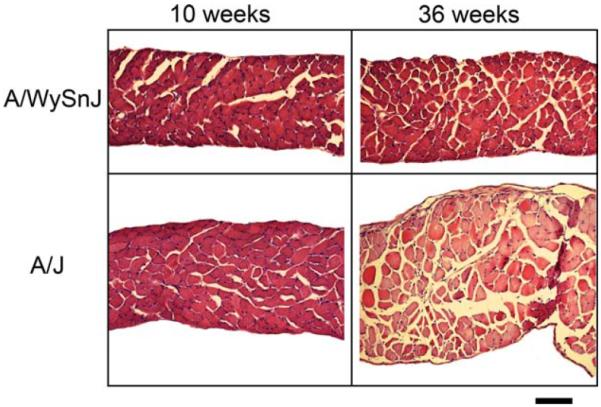
Muscle sections from diaphragms of young and mature A/J and wildtype (A/WySnJ) mice. Overt pathology is evident at 36 weeks of age, including rounded prenecrotic fibers, fatty infiltration, and central nuclei, whereas diaphragms from 10-week-old A/J mice appear relatively normal. Scale bar = 100 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Muscle fragility was evaluated using a series of eccentric contractions and measuring the drop in force production between the first and fifth contraction (Fig. 3). The EDL muscles of A/J mice showed no significant differences in the loss of force after eccentric contraction compared to the muscles from the strain match control mice. The soleus muscles from mature A/J mice exhibited a reduced drop in force after eccentric contractions, and the drop in force was not only lower than muscles from age-matched wildtype mice but also lower than muscles from young A/J animals. In contrast, diaphragms from 36-week-old A/J mice lost significantly more force after eccentric contractions than those of age-matched wildtype control mice. To determine if the drop in force was associated with sarcolemmal breaks, the proportion of Procion orange dye positive fibers was measured in cryosections obtained from muscles subjected to eccentric contraction9 (Fig. 4). EDL and soleus muscles showed no significant differences in Procion orange uptake. However, the greatest proportion of Procion orange dye-positive fibers was found in the diaphragms from 36-week-old A/J mice, similar to the heightened loss of force after eccentric contractions. To determine if Procion orange uptake was changed by eccentric contraction, muscles were incubated in Ringer's solution that contained the dye but did not undergo contractile measurements. In all wildtype muscles, and in young A/J muscles, dye penetration was less than 1% in the absence of contractile measurements (data not shown). In the muscles from mature A/J animals (Fig. 5), diaphragms had a higher proportion of Procion orange than the EDL muscle in unstimulated samples. However, all muscles showed a significantly higher proportion of Procion orange fibers after eccentric contraction compared to unstimulated controls. In addition, the diaphragm exhibited a much higher uptake of Procion orange dye after eccentric contractions than the EDL or soleus. Thus, dye uptake in A/J muscles was due to contractile damage and to damage that existed prior to eccentric contraction.
FIGURE 3.

The loss of force after a series of five eccentric contractions in isolated skeletal muscles of A/J and A/WySnJ (WT) mice. Data are presented as mean ± SD. Top panel, EDL with n = 9 muscles for each A/J group and n = 6 muscles for each WT group. Center panel, soleus with n = 8 muscles for each experimental group; lower panel, diaphragm with n = 5 muscles for each experimental group. No significant differences were observed between the groups of EDL. The drop in force in mature A/J soleus muscles was significantly less than soleus muscles from young A/J muscles and from mature WT mice. The drop in force in the diaphragms from 36-week-old A/J mice was significantly greater than those from the 10-week WT mice (P < 0.05 by Tukey's post-hoc comparison after one-way ANOVA).
FIGURE 4.
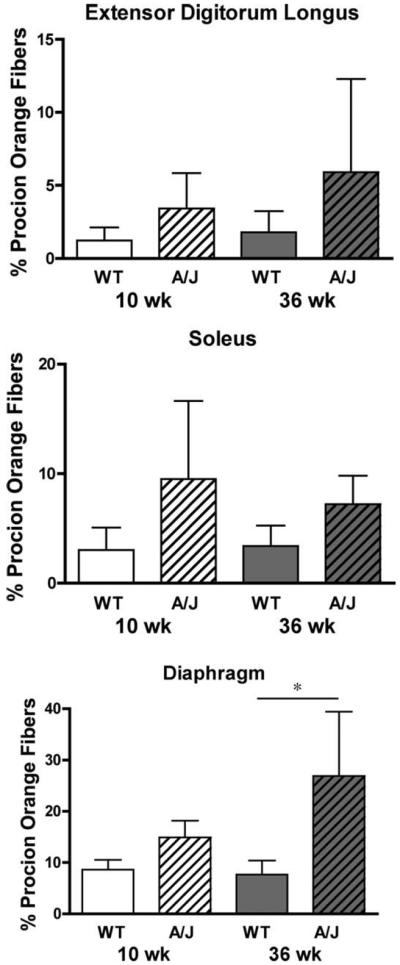
Procion orange uptake after five eccentric contractions in isolated mouse muscles reflects the drop in force. Data are presented as mean ± SD. Top panel, EDL with n = 9 muscles for each A/J group and n = 6 muscles for WT groups. Center panel, soleus with n = 8 muscles for each experimental group; lower panel, diaphragm with n = 5 muscles for each experimental group. No significant differences were observed between the groups of EDL or soleus muscles. The Procion orange uptake in the diaphragms from 36-week-old A/J mice was significantly greater than those from the age-matched WT mice (P < 0.05 by Tukey's post-hoc comparison after one-way ANOVA).
FIGURE 5.
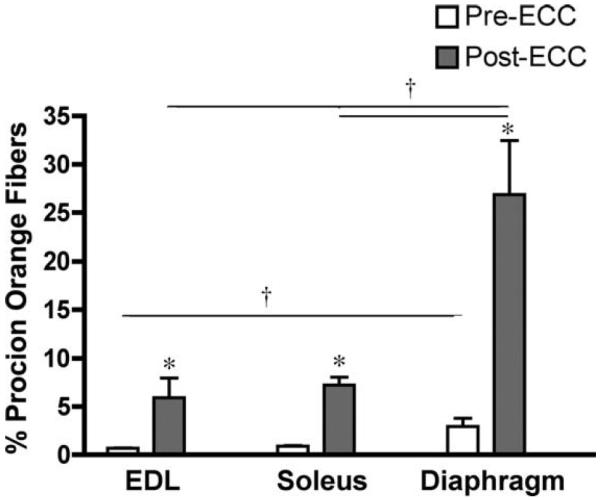
Procion orange uptake before and after eccentric contractions in muscles from mature A/J mice. Data are presented as mean ± SD for n = 3 muscles before eccentric contraction (pre-ECC) and n = 9 EDL, n = 8 soleus, and n = 5 diaphragms after eccentric contractions (post-ECC). The post-ECC groups are the same data as in Figure 4. Diaphragm muscles had elevated Procion orange uptake prior to eccentric contractions compared to EDL muscles. All muscle groups had significantly higher proportions of Procion orange fibers in post-ECC compared to pre-ECC. The proportion of Procion orange positive muscle in the post-ECC diaphragms exceeded that in the EDL and soleus.
The increased dye uptake in the diaphragm afforded an opportunity to examine the susceptibility of specific muscle fiber type to sarcolemmal tears during eccentric contractions. Serial sections were subjected to immunohistochemistry for MHC I/β, IIA, and IIB and compared to the Procion orange signal in adjacent sections (Fig. 6A). Fibers that were positive for Procion orange were identified in all sections. The Procion orange-positive fibers were predominantly MHC 2A (Fig. 6B) even though less than half of all muscle fibers were MHC 2A-positive.
FIGURE 6.

MHC 2A fibers are most susceptible to membrane rupture in the diaphragms of A/J mice. (A) Procion orange uptake (upper panel), and MHC 2A and laminin immunohistochemistry (lower panel) in serial sections from a diaphragm of an A/J mouse. Yellow x's in the lower panel indicate Procion orange-positive fibers that are above background fluorescence, and show that most of the Procion orange fibers are positive for MHC 2A. Scale bar = 100 μm. (B) Quantification of MHC 2A fibers in n = 5 diaphragms show that less than half of all fibers are positive for MHC 2A, whereas more than 80% of the Procion orange fibers are positive for MHC 2A. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
We extended the analysis of endogenous dysferlin content to several muscles from wildtype mice to determine if increased pathology in specific muscles in dysferlin null mice was associated with a normal high dysferlin content. Immunoblotting for dysferlin was performed on muscles removed from young wildtype mice (n = 3 muscles for each muscle). As shown in Figure 7A, there were variable levels of dysferlin in the muscles examined. Comparisons showed that quadriceps had significantly higher dysferlin content than diaphragm muscles. However, the three muscles utilized for functional analysis displayed no apparent differences in endogenous dysferlin. Even though there is a loss of force-generating capacity in the A/J diaphragms as well as histological pathology in the rectus abdominus,4 these muscles do not exhibit a greater endogenous dysferlin content in the wild-type mice. Thus, there is no apparent link between the level of dysferlin normally present and the extent of functional pathology in its absence.
FIGURE 7.

Dysferlin immunoblots. (A) A/WySnJ muscles have variable levels of dysferlin content among muscles (muscles from n = 3 animals). The quadriceps muscles have higher levels of dysferlin compared to diaphragms. (B) Muscles from mdx mice have higher dysferlin content than the same muscles from wildtype (C57) mice. Dysferlin levels are normalized to GAPDH for each sample. Data are presented as mean ± SD. Insets, Q, quadriceps; E, extensor digitorum longus; S, soleus; TA, tibialis anterior; R, rectus abdominus; D, diaphragm; A/J, TA from A/J mouse; c, C57; m, mdx.
Muscles from mdx mice, which serve as a genetic model for Duchenne muscular dystrophy, exhibit high susceptibility to eccentric contractile damage and increased cycles of degeneration and regeneration.12–14 In light of the impaired regeneration response found in dysferlin null animals,7 muscles of mdx mice and C57 controls were examined for dysferlin content by immunoblotting to determine if heightened membrane fragility and degeneration led to changes in increased dysferlin content. With the exception of the rectus abdominus, all muscles from the mdx mouse exhibited higher levels of dysferlin compared to the same muscles from C57 mice (Fig. 7B). The mechanism by which dysferlin levels were increased was not determined.
DISCUSSION
The goal of this study was to define a functional signature for dysferlin-deficient skeletal muscles of the mouse to gain a better understanding of the pathogenesis in Miyoshi myopathy and LGMD 2B, and to establish a platform for physiological evaluation of potential therapies for this disease. We examined the function of three murine skeletal muscles that are ideal for ex vivo measurements: the EDL, soleus, and diaphragm. These muscles span a range of fiber properties and pathologies in a number of muscular dystrophies, including dysferlin deficiency. We found that the diaphragm in the A/J mouse exhibits progressive loss of force-generating capacity as well as heightened susceptibility to eccentric contractile damage. These results support the use of the murine diaphragm as a way to test therapies for dysferlinopathies at a young age, prior to the onset of apparent histopathology.
The central role of dysferlin in membrane resealing has been firmly established.6 This property is apparent in all cell types tested, although it may hold particular relevance for skeletal muscle. Normal skeletal muscle activity can lead to small tears in the sarcolemma and acute muscle damage. Therefore, the ability to rapidly reseal the plasma membrane is the first defense in preventing progression from acute damage to muscle fiber necrosis. Initial demonstration that dysferlin was necessary for repair of the sarcolemma utilized mechanical and chemical rupture of either cultured muscles cells or isolated muscle fibers, and it was clear that the absence of dysferlin prevented membrane resealing.6 Skeletal muscles from dysferlin-deficient mice did not appear to be more susceptible to damage; however, the loss of dysferlin prevented rapid resealing of the tears associated with acute muscle damage, and as a consequence, acute damage could progress to fiber necrosis. The separation of susceptibility to damage and recovery from damage was recently demonstrated in an in vivo model of strain injury.15 The hindlimb muscles of A/J and A/WySnJ mice were subjected to a series of eccentric contractions, which resulted in indistinguishable drops in force production in the mouse strains. However, the recovery of force production was much slower in the A/J limb muscles compared to those in the strain-matched controls and was achieved through muscle regeneration rather than membrane resealing. Our measurements of isolated limb muscle function are consistent with these observations, where we observed no defect in the susceptibility to eccentric contraction damage in the EDL or soleus muscle.
In contrast to the results in limb muscles, the diaphragm in the A/J mouse was not only weaker than those from strain-matched controls, but it was also significantly more susceptible to loss of force after eccentric contractions (Figs. 3, 4). Functional deficits in the diaphragm occurred early (by 10 weeks of age) and persisted through 36 weeks of age. Loss of strength preceded histological evidence of pathology. This suggests that the muscle fibers in dysferlin null diaphragms are weaker than wildtype fibers because there was little fibrosis in the young A/J mouse diaphragms. Patients suffering from Miyoshi myopathy and LGMD2B do not exhibit significant pathology in the respiratory muscles, and so the dysferlin-deficient mouse does not mimic the same progression of disease. However, the basis for pathology in the murine diaphragm may be similar to the mechanisms associated with pathology in the human limbs. A cage-bound mouse does not have significant power output in the limbs, unlike an ambulatory human. In contrast, the murine diaphragm undergoes the greatest work of any skeletal muscle in the animal because of its respiratory rate and high elastic recoil, and it serves as a good indicator of susceptibility to activity-related muscle disease. The basal work rate in the murine diaphragm is more than 4-fold higher than the resting respiratory work rate in a 70-kg human.16 Historically, the diaphragm was found to be the most profoundly affected muscle in the mdx mouse even though all muscles were susceptible to contractile damage13 because the high respiratory duty cycle exacerbated muscle damage. In the wildtype and A/J mice, high respiratory workloads may cause similar levels of small tears in the sarcolemma, but the cumulative inability to repair those tears in the absence of dysferlin leads to a loss of force and may compound the susceptibility to additional contractile damage. Although not directly tested in our study, histopathology in the abdominal muscles in dysferlin-deficient mice4 may also be caused by a similar mechanism.
If skeletal muscle requires dysferlin for acute membrane repair, then one would expect increased dysferlin levels in muscles that undergo more frequent damage. For example, the abdominal muscles and the diaphragm exhibit functional and histological deficits in the dysferlin-deficient mouse, and so it would be likely that more dysferlin would be present to counter damage in normal muscles. We addressed this possibility by measuring dysferlin protein in a range of murine skeletal muscles in wildtype mice. Counter to our expectations, the rectus abdominus and the diaphragm from wildtype mice had similar dysferlin levels to those in the limb muscles (Fig. 7A). Thus, there was no correlation between functional deficits observed in the absence of dysferlin and the endogenous content. We extended measurements of dysferlin content to muscles from mdx mice and found that for all muscles (except the rectus abdominus) there was an increase in dysferlin compared to wildtype muscle (Fig. 7B). In addition to membrane fragility, mdx muscles also exhibit the secondary consequences of heightened degeneration/regeneration cycles and inflammation,17 and these factors could also contribute to increased dysferlin. There does not appear to be a direct role for dysferlin in mediating muscle fiber formation during regeneration after injury. The related protein, myoferlin, regulates myoblast fusion after cardiotoxin injection, and its absence is not compensated by dysferlin.18 However, the impaired regeneration observed in the SJL/J muscles supports the need for dysferlin in resolving muscle damage.7 The model put forth by Chiu et al.7 proposes that reduced fusion of cytokinefilled vesicles in the absence of dysferlin leads to an impairment of inflammatory cell recruitment needed for muscle remodeling. Thus, the increase in dysferlin in mdx muscles may be compensatory not only for acute membrane repair, but also for heightened inflammation. A previous survey of biopsies from LGMD and DMD patients found normal dysferlin levels in all muscles except in the LGMD2B patients,19 which suggests that the mdx mouse phenotype may not recapitulate DMD patients in this aspect. These possibilities will require future studies to determine the roles dysferlin plays in diseased tissues.
Using the diaphragm, we investigated the susceptibility of specific muscle fiber types to membrane tears in the absence of dysferlin and found that most Procion orange-positive fibers were type MHC 2A (Fig. 6), even though less than half of the fibers were MHC 2A-positive. Given that there was no overall difference in dysferlin content across normal muscle groups, it was surprising that one muscle fiber type was particularly susceptible to membrane rupture. Further, there was no correlation between Procion orange-positive fibers and the size of the damaged fibers nor was there a correlation in dysferlin levels per fiber (data not shown). Large fibers generate more force and power than small fibers, and the mechanical force developed per unit surface area is larger, which ultimately predisposes large-diameter fibers to sarcolemmal tears.20 However, this cannot explain the susceptibility of fiber damage in the diaphragm. This suggests that other factors may contribute to the susceptibility to damage in dysferlin-deficient muscle in addition to fiber size and endogenous dysferlin content. The most likely explanation is that this fiber type is primarily responsible for the bulk of work performed during murine respiration. The identification of potential factors will require further study.
In conclusion, we anticipate that these studies will provide a basis for evaluating potential therapies in terms of muscle strength and fragility in mouse models of dysferlinopathies. The murine diaphragm may be the best indicator of pathology, because the loss of dysferlin results in deficits in strength and resistance to contractile damage at a relatively young age. Our results suggest that evaluation of potential therapies in the A/J mouse should include the diaphragm to assess functional recovery.
Acknowledgments
Supported by the Paul Wellstone Muscular Dystrophy Cooperative Research Center (U54 AR052646 to H.L.S.), and a Pennsylvania Muscle Institute Training grant (AR053461 to B.K.B.). We thank Z. Tian and E. Blanco for technical expertise in the Physiological Assessment Core for performing functional measurements.
Abbreviations
- CSA
cross-sectional area
- EDL
extensor digitorum longus
- LGMD2B
limb girdle muscular dystrophy 2B
- Lo
optimum muscle length
- MHC
myosin heavy chain
- PBS
phosphate-buffered saline
REFERENCES
- 1.Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 3.Urtizberea JA, Bassez G, Leturcq F, Nguyen K, Krahn M, Levy N. Dysferlinopathies. Neurol India. 2008;56:289–297. doi: 10.4103/0028-3886.43447. [DOI] [PubMed] [Google Scholar]
- 4.Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H, et al. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet. 2004;13:1999–2010. doi: 10.1093/hmg/ddh212. [DOI] [PubMed] [Google Scholar]
- 5.Bittner RE, Anderson LV, Burkhardt E, Bashir R, Vafiadaki E, Ivanova S, et al. Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat Genet. 1999;23:141–142. doi: 10.1038/13770. [DOI] [PubMed] [Google Scholar]
- 6.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC. Williamson R, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 7.Chiu YH, Hornsey MA, Klinge L, Jorgensen LH, Laval SH, Charlton R, et al. Attenuated muscle regeneration is a key factor in dysferlindeficient muscular dystrophy. Hum Mol Genet. 2009;18:1976–1989. doi: 10.1093/hmg/ddp121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barton ER, Morris L, Kawana M, Bish LT, Toursel T. Systemic administration of L-arginine benefits mdx skeletal muscle function. Muscle Nerve. 2005;32:751–760. doi: 10.1002/mus.20425. [DOI] [PubMed] [Google Scholar]
- 9.Petrof BJ, Stedman HH, Shrager JB, Eby J, Sweeney HL, Kelly AM. Adaptations in myosin heavy chain expression and contractile function in dystrophic mouse diaphragm. Am J Physiol. 1993;265:C834–841. doi: 10.1152/ajpcell.1993.265.3.C834. [DOI] [PubMed] [Google Scholar]
- 10.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schiaffino S, Salviati G. Molecular diversity of myofibrillar proteins: isoform analysis at the protein and mRNA level. In: Sweeney HL, editor. Methods in muscle biology. Vol. 52. Academic Press; New York: 1997. pp. 349–369. [DOI] [PubMed] [Google Scholar]
- 12.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 14.Vilquin JT, Brussee V, Asselin I, Kinoshita I, Gingras M, Tremblay JP. Evidence of mdx mouse skeletal muscle fragility in vivo by eccentric running exercise. Muscle Nerve. 1998;21:567–576. doi: 10.1002/(sici)1097-4598(199805)21:5<567::aid-mus2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 15.Roche JA, Lovering RM, Bloch RJ. Impaired recovery of dysferlin-null skeletal muscle after contraction-induced injury in vivo. Neuroreport. 2008;19:1579–1584. doi: 10.1097/WNR.0b013e328311ca35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stahl WR. Scaling of respiratory variables in mammals. J Appl Physiol. 1967;22:453–460. doi: 10.1152/jappl.1967.22.3.453. [DOI] [PubMed] [Google Scholar]
- 17.Peterson JM, Guttridge DC. Skeletal muscle diseases, inflammation, and NF-kappaB signaling: insights and opportunities for therapeutic intervention. Int Rev Immunol. 2008;27:375–387. doi: 10.1080/08830180802302389. [DOI] [PubMed] [Google Scholar]
- 18.Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, et al. Normal myoblast fusion requires myoferlin. Development. 2005;132:5565–5575. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vainzof M, Anderson LV, McNally EM, Davis DB, Faulkner G, Valle G, et al. Dysferlin protein analysis in limb-girdle muscular dystrophies. J Mol Neurosci. 2001;17:71–80. doi: 10.1385/JMN:17:1:71. [DOI] [PubMed] [Google Scholar]
- 20.Karpati G, Carpenter S. Small-caliber skeletal muscle fibers do not suffer deleterious consequences of dystrophic gene expression. Am J Med Genet. 1986;25:653–658. doi: 10.1002/ajmg.1320250407. [DOI] [PubMed] [Google Scholar]