

Summary
Immunotherapy is a promising treatment modality for cancer as it can promote specific and durable anti-cancer responses. However, limitations to current approaches remain. Therapeutics administered as soluble injections often require high doses and frequent re-dosing, which can result in systemic toxicities. Soluble bolus-based vaccine formulations typically elicit weak cellular immune responses, limiting their use for cancer. Current methods for ex vivo T cell expansion for adoptive T cell therapies are suboptimal, and achieving high T cell persistence and sustained functionality with limited systemic toxicity following transfer remains challenging. Biomaterials can play important roles in addressing some of these limitations. For example, nanomaterials can be employed as vehicles to deliver immune modulating payloads to specific tissues, cells, and cellular compartments with minimal off-target toxicity, or to co-deliver antigen and danger signal in therapeutic vaccine formulations. Alternatively, micro-to macroscale materials can be employed as devices for controlled molecular and cellular delivery, or as engineered microenvironments for recruiting and programming immune cells in situ. Recent work has demonstrated the potential for combining cancer immunotherapy and biomaterials, and the application of biomaterials to cancer immunotherapy is likely to enable the development of effective next-generation platforms. This review discusses the application of engineered materials for the delivery of immune modulating agents to the tumor microenvironment, therapeutic cancer vaccination, and adoptive T cell therapy.
Keywords: Cancer Immunotherapy, Tumor Microenvironment, Therapeutic Vaccination, Adoptive T Cell Therapy, Nanoparticles, Porous Scaffolds
Graphical Abstract
1. Introduction
Cancers are among the leading causes of morbidity and mortality worldwide with incidence expected to rise by 70% over the next two decades [1]. Current treatments for cancer, such as chemotherapy and radiation therapy which are characterized by a lack of specificity and response durability, are insufficient. There is a need for therapies that can target cancer cells with a high degree of specificity, leading to lower treatment-related morbidity, and that can facilitate long-term remission.
Cancer immunotherapy refers to any intervention that leverages the immune system to eliminate a malignancy. Successful cancer immunotherapies generate an anti-cancer response that is systemic, specific, and durable, overcoming the primary limitations of traditional cancer treatment modalities. Recent progress in our understanding of the immune system has enabled the development of effective platforms for promoting anti-cancer immunity, particularly in the areas of biologics for reversing immunosuppression in the tumor microenvironment (TME) [2, 3], therapeutic cancer vaccines [4–6], and adoptive T cell therapies (ACT) [7–9]. For example, checkpoint inhibitors, monoclonal antibodies that block cell surface co-inhibitory receptors that disable the ability of T cells to destroy cancer cells, have shown unprecedented clinical success in a wide range of advanced stage malignancies [2, 3, 10–12]. To date, monoclonal antibodies for cytotoxic T-lymphocyte-associated protein 4 (CTLA-4; Ipilimumab) and programmed cell death protein 1 (PD-1; Pembrolizumab and Nivolumab) have been FDA approved for metastatic melanoma with the PD-1 antibodies given “breakthrough therapy” designation by the FDA. The discovery of key molecular players in the generation of immune responses, including pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and their respective ligands, has provided us with a vast toolbox of danger signals for precisely tuning the immune response. These cues, in combination with an improved understanding of dendritic cells (DCs), the most potent antigen-presenting cells (APCs), have enabled the design of promising therapeutic cancer vaccines, many of which are under investigation in various stages of clinical trials [4–6]. Importantly, these developments also lead to the first approval of a therapeutic cancer vaccine by the FDA in 2010 (Sipuleucel-T) [13]. Although Sipuleucel-T showed only modest therapeutic benefit and was associated with a high treatment cost, its approval set a precedence in the therapeutic cancer vaccine field and will likely lead to the development of more effective and efficient cancer vaccine approaches in the future. Advancements in our understanding of the cellular and molecular biology of T cells and antigen recognition has allowed for the development of highly efficacious ACTs, including tumor-infiltrating lymphocyte (TIL)-based therapies for advanced melanoma [9], and high-affinity/avidity T cell receptor (TCR)- [8] and chimeric antigen receptor (CAR)-transduced T cell-based therapies [7] for a wide range of hematologic malignancies.
Despite these advancements, drawbacks to current cancer immunotherapy strategies remain. Therapeutics are commonly administered as soluble injections, typically necessitating high doses and frequent re-dosing to achieve biologically relevant concentrations in target tissues, which often results in systemic toxicities [14, 15]. Soluble bolus-based vaccine formulations typically elicit weak cellular immune responses [16, 17], limiting their effective use for cancer. Current methods for ex vivo cell expansion for ACT are suboptimal and do not always facilitate the generation of high quality T cells [18, 19], and achieving high T cell persistence and sustained functionality with limited systemic toxicity following transfer remains challenging [20, 21]. The use of biomaterials as platforms for cancer immunotherapy could allow for some of these limitations to be overcome. Although beyond the scope of this review, there are also promising virus-based approaches for cancer vaccination being explored, and this is described elsewhere [22, 23].
To date, a wide range of material systems have been developed as molecular and cellular delivery vehicles in biomedical applications ranging from diagnostics [24–26] to therapeutics [27–29]. As delivery vehicles, biomaterials allow for a level of spatiotemporal control over payload delivery that is difficult to recapitulate with a bolus. For example, nanomaterials can be used to deliver diverse combinations of bioactive payloads to specific tissues [25, 30], cell types [31, 32], and intracellular compartments [33–35] in a controlled manner and with a high degree of specificity, curtailing off-target toxicity and allowing for dose-sparing. Micro- to macroscale materials can be designed as depots for the sustained local delivery of bioactive payloads with high spatiotemporal resolution [36–38], or as artificial cellular microenvironments displaying complex combinations of cues [39–43]. This review will point out some of the challenges associated with various current immunotherapy modalities, and will discuss how the application of biomaterials as delivery vehicles or engineered microenvironments can potentially aid in overcoming some of these challenges. Specifically, this review will discuss the use of nano- to microscale materials for modulation of immunosuppression in the TME, therapeutic vaccination, and for promoting in vitro and in vivo T cell survival and expansion, and the use of 3-D macroscale materials as engineered microenvironments for programming immune cells, and as cellular delivery devices.
2. Brief Review of Cancer and the Immune System
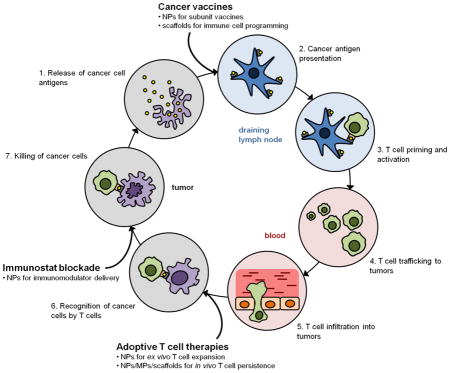
The generation of a productive anti-cancer immune response resulting in the elimination of cancer cells is dependent on a coordinated series of events that must take place in an iterative and self-sustaining manner (Fig. 1). This process, termed the “cancer-immunity cycle”, has been reviewed in detail elsewhere [44]. Briefly, antigens are released from cancer cells and captured by DCs, the primary mediators of adaptive immunity (step 1). DC activation, which is associated with the upregulation of cell surface co-stimulatory molecules and cytokine production, is necessary for efficient downstream priming of a T cell response, and may be promoted in the endogenous situation by factors released by dying cancer cells, broadly termed “danger associated molecular patterns”. DC activation facilitates efficient processing of the uptaken antigen and subsequent presentation of antigenic peptides on cell surface MHC molecules (step 2). In the draining lymph nodes, activated DCs present cancer antigens to naïve T cells, resulting in the priming and activation of cancer antigen-specific T cells, a subset of which will differentiate into long-lived memory cells (step 3). Activated T cells, in particular, effector CD8+ cytotoxic T lymphocytes (CTLs), subsequently traffic to (step 4) and infiltrate the tumor (step 5), recognize cancer cells presenting the cognate antigenic determinants (step 6), and kill the cancer cells (step 7).
Figure 1.

The Cancer-Immunity Cycle. Diagram illustrating the steps involved in the cancer-immunity cycle that take place in distinct spatiotemporal compartments in a sequential manner. Cancer immunotherapy approaches augment various steps in the cycle (bolded). Biomaterials can potentially be used to make these approaches more effective (bullet points). Adapted from [44].
In cancer patients, the cancer-immunity cycle is blocked at one or more of these steps, dampening the anti-cancer immune response and allowing for immune escape. Cancer immunotherapies seek to promote anti-cancer immunity by augmenting specific steps in the cycle (Fig. 1). For example, PD-1/PD-L1 axis inhibitors or other agents that seek to reverse immune dysregulation in the TME are aimed at improving the killing of cancer cells by pre-existing CTLs (step 7). Therapeutic vaccines target DCs to facilitate cancer antigen presentation (step 2) in order to promote more robust T cell priming and activation (step 3) and subsequent CTL effector function. In ACTs, large populations of cancer-specific CTLs are generated ex vivo that can efficiently recognize cancer cells (step 6) following transfer into the patient. Materials can be used to overcome some of the limitations of current cancer immunotherapy approaches, and could allow for the development of immunotherapies that fulfill their respective endpoints more effectively and efficiently.
3. Nanomaterials for Modulating Immune Dysregulation in the Tumor Microenvironment
The TME of most solid tumors is characterized by immune privilege and pro-tumorigenic inflammation [45, 46]. These properties are facilitated by the concerted activity of the cancer cells themselves [47], tumor-associated stromal cells such as cancer-associated fibroblasts (CAFs) [48], and tumor-infiltrating immune cells including regulatory T cells (Treg cells) [49, 50], tumor-associated macrophages (TAMs) [51], and myeloid-derived suppressor cells (MDSCs) [52, 53]. Through various mechanisms, these cells preferentially exclude effector T cells from the tumor parenchyma [53], inhibit local cytotoxic effector function [49], promote tumor growth and metastasis [46], and reinforce the tolerogenic bias [46, 54, 55]. Modulating this tumor-proximal immune dysregulation, for example, by inhibiting molecular or cellular tolerogenic mediators or activating local cytotoxic effector cells, can sometimes be sufficient to induce anti-cancer immunity [12], and will likely have significant synergy with approaches that promote the generation or expansion of cancer-specific T cells. Immunotherapies that reverse immunosuppression in the TME are likely to be particularly relevant in immunogenic cancer types in which a significant anti-cancer T cell population already exists but is functionally inhibited in the TME [55, 56]. Of note, although the TME represents the source of these tolerogenic mediators, they are often not confined to this site and can accumulate in sinks such as the tumor draining lymph nodes (tdLNs) [57, 58], and in some cases, lead to systemic effects in patients as well [59]. As such, addressing the tolerogenic effects of the tumor more broadly, for example, by targeting peripheral sinks or tolerogenic mediators in circulation, represent potential alternative treatment strategies. However, such approaches will have to be appropriately designed to minimize systemic inflammatory toxicities, particularly since many of the inflammatory mediators used in such approaches (e.g. cytokines) can induce potent and serious systemic side-effects [21]. In this review, we focus on addressing mechanisms of immune dysregulation in the TME, which leverages the extensive amount of research that has been done on the use of nanomaterials as drug delivery vehicles to the tumor.
Designing a system to address immune suppression in the TME presents numerous challenges for soluble bolus-based delivery approaches, specifically with respect to delivering combinations of immune modulating payloads in a bioactive state to the tumor site while minimizing systemic toxicity. To overcome these challenges, nanomaterials can be used to facilitate the preferential accumulation of associated payloads in the TME, by being designed to either passively accumulate in the TME via the enhanced permeability and retention (EPR) effect [60], or to actively target the TME via diverse targeting strategies including the use of peptides [30], aptamers [26, 61], small molecules [62], or single-chain variable fragments of antibodies (scFv) [32], that may either interact directly with tumor cells or other components of the TME. Alternatively, stimulus-responsive systems have been designed that release sequestered payloads in response to triggers that are enriched in the TME, such as moderately low pH [63] or proteases, such as the matrix metalloproteinases (MMP) MMP-2 and MMP-9 [64], or in response to exogenous triggers that are selectively directed at the tumor, for example light [65], magnetic field [66, 67], or heat [67].
In the design of such systems, it is important to consider the spatial and temporal pattern of delivery that would be optimal for the activity of the particular payload. For the delivery of immune modulators such as cytokines or antagonistic agents that target cell surface receptors, the vehicle should be designed to minimize interactions with phagocytic cells in order to release the payload in the TME extracellularly. For example such vehicles can be surface modified with bioinert polymeric coatings such as poly(ethylene) glycol (PEG) or PEG derivatives [68], or with anti-phagocytic signals such as CD47 [69]. Physical properties of the vehicle such as size [70], aspect ratio [71, 72], or stiffness [73, 74] can also be tuned to decrease cell uptake. Alternatively, for the delivery of immune modulators such as certain PRR ligands or siRNA targeting immune regulatory pathways, the vehicle should be designed to target the appropriate intracellular compartment. As examples, endolysomal targeting can be enhanced by tuning the surface [75] or physical properties [70–74] of the particle to promote endocytosis, or by modifying the particle surface with ligands that actively target endocytosis-associated receptors [76]. Release of payloads in the endolysosomal environment can be achieved, for example, through the use of a stimulus-responsive vehicle [77] or through conjugation of the payload to the vehicle via a stimulus-responsive linker (e.g. one responsive to acidic pH or proteases) [78]. Cytosolic targeting could be facilitated, for example, through modification of the particle surface with cell penetrating peptides [79], or polycationic polymers that act as proton sponges [80] and promote endosomal escape following endocytosis. The temporal pattern of payload release can be tuned by varying system parameters including the method of payload loading (e.g. encapsulation versus chemical conjugation), the porosity and degradability of the material, and the relative physicochemical properties of the payload and the vehicle.
Nanomaterials have been designed to deliver diverse immunomodulatory payloads to the TME in order to address various mechanisms of TME immunosuppression. One attractive therapeutic target that has been pursued is the transcription factor STAT3, which has been identified as a key regulator of immune dysregulation in the TME. More specifically, expression of STAT3 in tumor-infiltrating immune cells has been shown to promote Th2-biased inflammation, thereby inhibiting a Th1 response, and to promote the survival of Treg cells [81]. STAT3 inhibition in the TME was reported using systemically administered tumor-targeting liposome nanoparticles loaded with a hydrophobic small molecule STAT3 inhibitor [62]. Of note, such hydrophobic agents are rapidly removed when administered as a soluble bolus and are therefore challenging to efficiently deliver to a target tissue systemically in free form. The targeted nanoparticles facilitated the delivery of the STAT3 inhibitor to the tumor and promoted the upregulation of a large panel of Th1-associated proinflammatory cytokines, and the concomitant downregulation of Th2-biased cytokines. This translated to slower tumor growth in murine breast cancer models in prophylactic and therapeutic settings, with an enhanced response observed when the inhibitor was delivered in the targeted liposomes versus in free form or via non-targeted liposomes. Importantly, the targeted delivery of the STAT3 inhibitor to the TME was shown to improve the effects of a therapeutic cancer vaccine in a murine breast cancer model [62]. Similar studies have been performed using other methods of antagonizing the STAT3 pathway, including the use of STAT3 siRNA [82], and other small molecule inhibitors [83, 84].
Multifunctional nanomaterials have also been designed to simultaneously inhibit suppression and promote immunity in the TME. For example, 120 nm lipid-enveloped nanoparticles composed of PEG-crosslinked polylactide, termed “nanolipogels”, were designed to co-deliver a hydrophobic small molecule inhibitor of the immunosuppressive cytokine TGF-β, and the T cell mitogenic cytokine interleukin 2 (IL-2) [85] (Fig. 2a). Notably, these particles were specifically designed to facilitate the co-delivery of these payloads in a single vehicle, which is generally challenging due to the distinct physicochemical properties of these payloads. Nanolipogels facilitated the release of both payloads in a sustained manner for at least a week in vitro. Comparative biodistribution studies using nanolipogels bearing a fluorescein-labeled lipid shell and loaded with a rhodamine payload demonstrated that both vehicle and payload accumulated in subcutaneous tumors and lung metastases (Fig. 2b) following systemic administration. In a therapeutic murine model of metastatic melanoma, systemic administration of the TGF-β inhibitor and IL-2 co-loaded nanolipogels resulted in attenuated tumor growth and enhanced survival (Fig. 2c). Notably, co-delivery of both components in a single carrier resulted in improved outcomes over the delivery of each component alone in soluble or nanolipogel-associated form (Fig. 2c).
Figure 2.

Nanolipogels for modulating immunosuppression in the TME. (a) Diagram of nanolipogel formulation. (b) Analysis of healthy lung tissues (top row) or lung tissues bearing metastases (yellow arrows) at 2 h, 24 h, and 96 h following a single injection of nanolipogels loaded with rhodamine and containing fluorescein-labeled phospholipid in the lipid bilayer shell. Tissues are visualized under bright field (left) and fluorescence microscopy (right), showing the presence of both lipid carrier (green) and rhodamine payload (red) around individual lung tumors at these time points. (c) Survival of mice bearing melanoma lung metastases either untreated or treated via systemic injections of soluble TGF-β inhibitor (soluble SB), soluble TGF-β inhibitor with soluble IL-2 (sol SB + IL-2), nanolipogels loaded with TGF-β inhibitor (nLG-SB), nanolipogels loaded with IL-2 (nLG-IL-2), or nanolipogels loaded with both TGF-β inhibitor and IL-2 (nLG-SB+IL-2). Red arrows denote treatment days. From [85].
Alternatively, nanomaterials can also be designed to locally present agonistic cues to cell surface molecules, or to act as depots for sequestering payloads at the tumor site following administration into the vicinity of the tumor, enhancing the local bioavailability of the payload and minimizing systemic toxicity. For example, nanoscale liposomes were prepared that simultaneously displayed an agonistic CD40 antibody, and released unmethylated CpG oligodeoxynucleotides (CpG), a TLR ligand, in a sustained manner for over a week in vitro [86]. These agents have been shown to be have synergistic anti-tumorigenic effects in preclinical models [87, 88], but have been associated with off-target inflammatory effects following systemic over-exposure. In a murine melanoma model, anti-CD40 and CpG delivered intratumorally, either as a soluble bolus or co-delivered in the liposomal formulation, inhibited tumor growth to a comparable degree. Notably, however, serum levels of anti-CD40 and CpG were significantly attenuated when the agents were delivered in the liposomal formulation [86]. Consistent with this observation, compared to animals that received the agents as a soluble bolus, animals treated with the liposomal formulation exhibited minimal weight loss, and significantly decreased serum levels of hepatic ALT enzyme and the proinflammatory cytokines TNF-α and IL-6.
4. Nanomaterials as Therapeutic Cancer Vaccines
Nanomaterials are being developed as efficient and versatile mediators of therapeutic cancer vaccination. Fundamentally, an immune response is generated when DCs acquire antigen in the context of a danger signal such as a PRR ligand, and subsequently prime a cancer-specific T cell response. One family of PRRs that is particularly important in vaccine design is the TLRs, due to their frequent use as adjuvants in vaccine formulations. Examples of TLR ligands include lipopolysaccharide (LPS) and its derivate monophosphoryl lipid A (MPLA), found in the outer membrane of gram-negative bacteria, polyinosinic:polycytidylic acid (poly I:C), a synthetic mimic of double-stranded RNA found in some viruses, and CpG oligodeoxynucleotides, which resemble DNA motifs found in microbial genomes. A primary challenge in designing cancer vaccines is consolidating DCs, danger signal, and antigen into a single spatiotemporal compartment. To this end, nanomaterials can be employed as colloidal scaffolds to associate antigen with danger signal, and designed to target migratory peripheral or lymph node-resident DCs. Alternatively, adjuvanted nanomaterials can be designed to target DC-rich cancer antigen experienced sites such as the tdLNs or potentially the tumor itself.
4.1 Targeting the Draining Lymph Nodes
DCs are the most potent APCs for initiating an immune response [89, 90]. However, DCs are comprised of diverse subpopulations that vary with respect to their residence and function [91]. The discussion in this review will be limited to therapeutic vaccines that are administered in the skin. In this context, we draw a general and simplistic distinction between just two DC subpopulations: the migratory DCs that reside in peripheral tissues and migrate to the tissue-draining lymph nodes following activation, and the non-migratory lymphoid tissue-resident DCs. Of note, among the lymphoid tissue-resident DCs is a subset that expresses the cell surface marker CD8 in mouse and is specialized for cross-presentation, a process critical for CTL induction. As such, targeting the lymph nodes is, in general, of particular interest for nanoparticulate-based therapeutic vaccines.
Nanoparticulate vaccines that are administered in the skin, that is, either intradermally or subcutaneously, can be designed to either interact with migratory DCs at the injection site, or to drain passively to the lymph node for acquisition and presentation by lymph node-resident DCs. The former relies on cell trafficking to reach the draining lymph node, and takes place with relatively slower kinetics. The latter depends on lymphatic flow without the need for an active cell trafficking mechanism, and takes place with relatively faster kinetics. In general, the primary feature determining whether a colloid will spontaneously drain or be taken up by migratory DCs is size, although surface properties have been shown to play a role as well [92]. When trafficking of polystyrene particles between 20–2000 nm was evaluated following subcutaneous administration, it was observed that 20 nm particles rapidly and efficiently arrived in the draining lymph node two hours post-injection (Fig. 3a) with initial localization in the subcapsular sinus (Fig. 3b), a pattern consistent with lymphatic-mediated drainage [93]. In contrast, 1000 nm particles were not observable in the draining lymph node until 24–48 hours post-injection (Fig. 3a), and were excluded from the subcapsular sinus (Fig. 3b), suggesting a cell trafficking-mediated mechanism of lymph node accumulation. Consistent with this, depletion of DCs abrogated the trafficking of 500 nm particles to the draining lymph node but had minimal effects on the trafficking pattern of 20 nm particles. Taken together, these findings demonstrate that particles of 500 nm or larger likely depend on active DC-mediated trafficking to reach the lymph node. Notably, particle size and CD8+ DC uptake were found to be negatively correlated, with 20 nm particles found to be frequently associated with CD8+ DCs. A similar observation was made following intradermal administration of polypropylene sulfide (PPS) nanoparticles wherein 20 nm particles accumulated in draining lymph nodes more rapidly and at a higher magnitude than larger 100 nm particles (Fig. 3c). A significantly larger fraction of the small particles were also taken up by lymph node DCs (Fig. 3d).
Figure 3.

Effects of particle properties on lymph node accumulation. (a) Accumulation of 20 nm and 500 nm fluorescent polystyrene nanoparticles to the draining popliteal lymph nodes at 40 min, 2 h, 24 h, 48 h, and 8 days following subcutaneous injection in the footpad. Arrows point to injection site in footpad and arrowheads point to draining popliteal lymph nodes. (b) Localization of 20 nm and 1000 nm polystyrene nanoparticles (green) in the draining popliteal lymph node at 2 h, 48 h, and 8 days following subcutaneous injection in the footpad. B220 staining shows lymph node B cell follicles in red. (c) Accumulation of 100 nm and 25 nm fluorescent PPS nanoparticles (red) in the draining lymph node at 24 h after intradermal injection. Cell nuclei stained with DAPI are shown in blue. Scale bar = 200 μm. (e) Flow cytometry histograms showing CD11c+ DCs isolated from the draining lymph nodes after intradermal injection of fluorescent 100 nm and 25 nm particles (black) or mock (gray). a–c from [93]. d, e from [113].
4.2 Nanomaterials for Subunit Vaccines
A considerable focus in recent cancer vaccine development has been on subunit vaccines that contain purified tumor antigens or antigenic epitopes as an antigen source, often in combination with an adjuvant such as a TLR agonist. Although roles for Th1-biased anti-cancer antibodies [16] and cancer-specific CD4+ T cells [94] have been implicated, it is generally understood that the primary determinant of successful cancer immunotherapies is the generation of a large, functional cancer-specific CTL pool [95, 96]. However, soluble bolus-based subunit vaccines typically induce weak CTL responses [16, 17], which limit their utility for cancer, and this is likely due to the inefficient cross-presentation of soluble, nonimmunogenic antigens [97–101]. To overcome this, nanoscale colloids can be used to promote more efficient antigen presentation by acting as phagocytic substrates that physically associate antigen and adjuvant.
Nanoparticulate subunit vaccines that co-deliver these cues have been prepared using a wide range of synthetic [77, 102–104] and natural materials [105–107]. For example, virus-mimicking 25 nm protein dodecahedrons, based on the E2 subunit of pyruvate dehydrogenase, were immobilized with the TLR9 agonist CpG and a peptide antigen (Fig. 4a) [78]. Importantly, CpG was immobilized onto the capsules via acid-labile hydrazone bonds, facilitating its release in the endolysosomal compartment where TLR9 is localized. Attachment of CpG to the E2 particles enhanced the activation of primary bone marrow-derived dendritic cells (BMDCs) compared to free CpG, likely by enhancing cellular uptake of CpG. E2 particles presenting both CpG and peptide antigen facilitated more robust activation of antigen-specific T cells compared to the cues presented separately (Fig. 4b), illustrating the benefit of consolidating both antigen and danger signal on a single carrier.
Figure 4.

Nanoparticulate subunit vaccines. (a) TEM image of negatively-stained protein-based vaccines showing virus mimicking dodecahedral geometry. (b) Activation of B3Z antigen-specific T cell hybridoma cell line by BMDCs either unloaded (DC only), or loaded with E2 vehicle (E2), soluble CpG with E2 vehicle (CpG + E2), soluble peptide antigen (SIINFEKL), soluble CpG with soluble peptide antigen (CpG + S), soluble CpG with soluble peptide antigen and E2 vehicle (CpG + S + E2), soluble CpG with peptide antigen-conjugated E2 (CpG + S-E2), soluble peptide antigen with CpG-conjugated E2 (S + CpG-E2), or peptide antigen and CpG co-loaded E2 (CpG-S-E2). (c) Diagram showing OVA-loaded ICMV vaccine formulations containing MPLA only in the outer vesicle layer (ext-MPLA ICMVs) or containing MPLA throughout the multilayers (int-MPLA ICMVs). (d) Time-dependent quantification of antigen-specific CTLs in peripheral blood after subcutaneous vaccination of naive mice with soluble MPLA in combination with soluble OVA (MPLA), ICMVs loaded with the same amount of OVA and MPLA with the MPLA either in the outer vesicle layer only (ext-MPLA ICMVs) or distributed throughout the multilayers (int-MPLA ICMVs), or with soluble OVA in combination with a 10-fold higher dose of soluble MPLA. Black arrows represent vaccination timepoints. a, b from [78], c, d from [111].
Beyond solid colloids, subunit vaccines can also be prepared using polymersomes [108, 109] and liposomes [110]. However, a limitation to the use of standard liposomes for this purpose is their relatively low stability in serum. One approach that has been reported to improve the stability of liposomes is to prepare multilamellar liposomes with cross-linked layers, termed interbilayer-crosslinked multilamellar vesicles (ICMVs) [108, 111, 112]. PEGylated ICMV subunit vaccines were prepared that contained the model antigen ovalbumin (OVA) in the aqueous interior of the particles, and the lipophilic TLR 4 agonist MPLA embedded in the ICMV lipid walls (Fig. 4c). ICMV vaccines demonstrated enhanced extracellular stability compared to unilamellar and uncrosslinked multilamellar counterparts, but underwent rapid particle degradation and cargo release in response to enzymes present in the endolysosomal compartment.
When mice were vaccinated subcutaneously with the ICMV vaccine, robust induction of antigen-specific CD8+ T cells was observed. Using the optimal ICMV formulation, a maximum frequency of 28% antigen-specific T cells among total CD8+ T cells in the peripheral blood was generated (Fig. 4d). This was equivalent to a 14-fold enhancement compared to soluble OVA and MPLA at the same dose, and this magnitude of response could not be recapitulated with the soluble formulation even using a 10-fold higher MPLA dose, suggesting that there are inherent limitations to using soluble subunit vaccine formulations. The induced antigen-specific CTLs included a CD44+CD62L+ subset, which has been reported to be indicative of a memory phenotype. In addition, a significant portion of the antigen-specific CTLs were observed to be functional, producing the inflammatory Th1 cytokine IFNγ following in vitro peptide restimulation. To date, the magnitude of the antigen-specific CTL response induced using the ICMV system is one of the most potent achieved with a non-viral vaccine, albeit against a model antigen. Understanding the specific mechanisms by which the ICMVs, and similarly robust vaccines, derive their potent CTL-inducing activity, will be important for the design of even more effective systems in the future.
An alternative approach to using inert nanomaterials to simply consolidate antigen and adjuvant is to design the material itself to act as an immune directing cue. For example, OVA-conjugated αAl2O3 nanoparticles were found to target autophagosomes following uptake by BMDCs, promoting highly efficient cross-presentation of OVA-derived peptides via an autophagy-dependent mechanism [35]. In another example, degradable PPS nanoparticles were surface polyhydroxylated, which facilitated their opsonization by ubiquitous serum complement components, specifically, by C3b which typically reacts with hydroxyl groups on surface carbohydrates of pathogens [113]. Following intradermal administration, 25 nm polyhydroxylated nanoparticles were found to efficiently activate draining lymph node dendritic cells, whereas 25 nm polymethoxylated nanoparticles, which do not interact with complement, did not. In addition, OVA-conjugated polyhydroxylated nanoparticles promoted robust expansion of antigen-specific T cell in vivo whereas the polymethoxylated nanoparticles promoted only minimal expansion. In a follow-up study, the effect of surface chemistries on complement activation was further explored, specifically with respect to the ratio of surface deposited C3b to inactivated C3b (iC3b), which has been shown to have an immunosuppressive function [114]. It was shown that the C3b:iC3b ratio could be tuned by altering the ratio of carboxylated to hydroxylated groups on the nanoparticle surface. Consistent with the suppressive function of iC3b, a higher C3b:iC3b ratio was found to correlate with the induction of higher antigen-specific antibody titers and enhanced Th1-biased cytokine production following ex vivo peptide restimulation of splenocytes from vaccinated animals.
4.3 Adjuvanted Nanomaterials for Targeting Cancer Antigen Experienced Sites as Therapeutic Cancer Vaccines
Despite the promise of engineered subunit vaccines, there are limitations to such targeted approaches. First, targeted vaccines generally immunize against prototypic cancer antigens, which precludes their use in cancers for which a such a target has not been identified, or in patients refractory for the particular known target. Of note, however, recent work on the patient-specific identification of cancer-associated neoantigens [115, 116] may allow for the use of targeted vaccines as personalized therapies, and this is discussed later in this review. Second, targeted approaches do not capture the complex antigen repertoire of tumors, and suboptimal vaccines that do not induce efficient antigen spread can potentially facilitate cancer immunoediting favoring the selection of lowly immunogenic subpopulations, leading to immune escape [117]. Based on these observations, there is interest in the development of non-targeted vaccine approaches that immunize against a range of different tumor-associated antigens. Examples of such approaches include vaccines based on inactivated whole tumor cells, autologous tumor lysate, and endogenous cancer antigens. The latter, in which the presence of cancer antigens at antigen experienced sites is leveraged through in situ vaccination at those sites, is particularly attractive in that such approaches do not require biopsies or tumor samples, and do not involve the ex vivo manipulation or processing of cancer cells.
To this end, adjuvanted nanoparticles have recently been described for targeting the tdLNs, which are known to be cancer antigen experienced sites, for in situ vaccination against endogenous cancer antigens [57, 58]. In addition to being antigen experienced, tdLNs are unique in several other aspects that make them both attractive as well as challenging sites for successful vaccination. tdLNs are enriched in immunosuppressive factors from the TME, as indicated by the increase in PD-L1+ DCs and PD-1+CD8+ T cells in the tdLNs compared to non-tdLNs [58]. In addition, tdLNs generally have fewer T cells and CD8+ DCs [58]. However, the potential for the endogenous generation of cancer-specific T cells in the tdLNs is supported by the observation that there is a time-dependent increase in functional OVA-peptide specific CD8+ T cells in the tdLNs of E.G7-OVA (OVA-transduced lymphoma cell line) tumor-bearing mice [58]. Additionally, the pre-existence of endogenous cancer-specific T cells in untreated cancer patients is well established [95]. Collectively, it is unclear in general whether the tdLNs necessarily represent beneficial or detrimental sites of vaccination although the success of tdLN-targeting vaccines is likely to be dependent both on the progression and nature of the malignancy as well as the robustness of the vaccine.
The potential for inducing an anti-cancer immune response using adjuvanted nanoparticles targeting the tdLNs was recently interrogated using 30 nm pyridyl disulfide (PDS) conjugated to CpG via a reduction-sensitive bond (PDS-CpG) [57]. These particles were shown to activate DCs in vitro, owing to the endolysosomal-triggered release of the CpG payload, facilitating CpG-TLR 9 interactions, and to efficiently drain to the lymph nodes following intradermal administration. Induction of intradermal B16-F10 melanoma tumors on one side of the animal resulted in the selective drainage of tumor antigen exclusively to ipsilateral (i.l.), but not to contralateral (c.l.) lymph nodes, as evaluated based on the drainage pattern of intratumorally administered fluorescent dextran (Fig. 5a, b). Intradermal administration of the PDS nanoparticles i.l. or c.l. also resulted in drainage of the particles primarily to LNs on the respective side of the animal (Fig. 5a, c). Notably, treatment of mice i.l. with PDS-CpG led to significantly slower tumor growth compared to the same particle formulation administered c.l., which showed no therapeutic effect (Fig. 5d). Co-administration of PDS nanoparticles with free CpG either i.l. or c.l. also yielded no therapeutic effect. The therapeutic benefit of the i.l. administered PDS-CpG was associated with an increased frequency of CD40+CD11c+ cells in the tdLNs, that may represent DCs with an activated phenotype, as well as in the frequency of activated CD25+ CD8+ T cells and the CD8:CD4 T cell ratio. In addition, only i.l. treatment with PDS-CpG promoted an increase in the frequency of tumor-infiltrating CD8+ T cells specific for the melanoma-associated antigen TRP-2.
Figure 5.

Adjuvanted nanoparticles targeting the tdLNs as a therapeutic cancer vaccine. (a) Diagram showing location of tumor inoculation in relation to the i.l. (TDLN) and c.l. (NTDLN) axillary (A) and brachial (B) draining LNs. (b) Quantification of dextran in axillary and brachial draining LNs following intratumoral administration of dextran. (c) Quantification of PDS nanoparticles (NPs) in draining LNs following either i.l. or c.l. intradermal administration of PDS nanoparticles. (d) Quantification of tumor volume as a function of time post tumor allograft following treatment with PBS or PDS-CpG administered either i.l. or c.l.. From [57].
Interestingly, it was also recently shown that, in the context of both E.G7-OVA and wild-type B16-F10 melanoma tumor-bearing mice, nanoparticulate subunit vaccines were more effective in the therapeutic setting (i.e. after the tumor has affected the tdLNs) at slowing tumor growth and promoting survival when they were targeted to tdLNs versus non-tdLNs [58]. Consistent with this, it was observed that delivery of these vaccines to the tdLNs versus the non-tdLNs resulted in the generation of a greater cancer-specific CTL response both locally in the tdLNs as well as systemically. Collectively, these observations suggests that not only can appropriately designed vaccines overcome the immunosuppressive mechanisms in the tdLNs and thereby leverage the cancer antigen experience of these sites for vaccination, but also that vaccines that target the tdLNs and are able to overcome this immunosuppression could induce particularly potent effects. Based on this idea, the TME itself represents another potentially attractive cancer antigen experienced site for vaccination. However, overcoming the active immunosuppressive mechanisms in the TME will likely be more challenging than overcoming those at the tdLNs, requiring that such vaccines be highly potent, and possibly necessitating combinatorial treatment with TME immunomodulators.
5. Engineered Microenvironments for Dendritic Cell Programming as Therapeutic Cancer Vaccines
An alternative approach to preparing colloidal vaccines that target relatively infrequent populations of peripheral or draining lymph node DCs is to design micro- to macroscale materials to act as microenvironments that recruit large numbers of DCs to the material, and program them in situ. In such an approach, a biomaterial scaffold is designed to release a recruitment factor to promote mass immune cell trafficking to the scaffold site, where danger cues and antigen are presented locally within a microenvironment permissive for cell infiltration. Activated, antigen-loaded DCs can subsequently traffic to the draining lymph nodes to facilitate T cell priming and activation (Fig. 6). Compared to DC-based cancer vaccine approaches such as Sipuleucel-T, such material systems bypass the need for ex vivo cell culture. This may decrease the substantial financial and regulatory burdens typical for cell therapies, and also avoid inconsistent and suboptimal cell products.
Figure 6.

Porous scaffolds can act as microenvironments for programming dendritic cells in situ. Immature DCs accumulate within the porous scaffold in response to a recruitment factor gradient. Inside the scaffold microenvironment, immature DCs acquire cancer antigen and become activated by danger signals presented by the scaffold matrix. Activated DCs present cancer antigen-derived peptides on cell surface MHC molecules and upregulate surface co-stimulatory molecules. Activated DCs subsequently traffic from the scaffold to the draining lymph nodes where they can initiate a T cell response.
Based on this paradigm, highly porous bulk scaffolds fabricated from the degradable copolymer PLG, loaded with GM-CSF as an immune cell attractant, CpG as a danger signal, and tumor lysate as an antigen source were employed as in situ DC programming devices (Fig. 7a) [40, 41, 118]. PLG scaffolds released GM-CSF in a sustained manner, resulting in robust GM-CSF dose-dependent accumulation of CD11c+ DCs within the scaffold following subcutaneous implantation (Fig. 7b). Notably, DC numbers within the device were on the order of 106, comparable to the number of DCs commonly expanded and deployed in ex vivo protocols [118]. CpG nanoparticles presented locally within the PLG matrix promoted the activation of infiltrating DCs, facilitating their subsequent trafficking to the draining lymph nodes. In a murine melanoma model, vaccines promoted robust tumor-specific CTL responses (Fig. 7c), which translated to 90% survival in the prophylactic setting, and 50% survival in the therapeutic setting (Fig. 7d) [118]. The PLG vaccine was found to recruit diverse DC subsets to the scaffold site, including CD8+ DCs, which are important for cross-presentation, and plasmacytoid DCs, which produce type I interferons that could further promote local DC activation [118]. Importantly, vaccine persistence for greater than a week was found to be necessary for the induction of a durable response [119]. In follow-up studies, these matrices were alternatively used to deliver the cytokine Flt3L, the chemokine CCL20 [40], and the TLR ligands poly I:C and MPLA [41], demonstrating the versatility of this system as a platform approach that can be used for the controlled delivery of different immune modulating payloads.
Figure 7.

Porous PLG scaffolds as engineered microenvironments for therapeutic cancer vaccination. (a) SEM of porous PLG scaffold showing pores on the size scale of cells. Scale bar = 200 μm. (b) Quantification of CD11c+ DC accumulation in scaffolds at 14 days post-implantation in response to 0 ng, 400 ng, 3000 ng, and 7000 ng GM-CSF. (c) FACS plots of tumor antigen (TRP2)-specific CTLs in the spleens of mice on day 16 post-vaccination with blank PLG scaffolds, or PLG scaffolds containing 3000 ng GM-CSF, 100 μg CpG, and tumor lysate. (e) Survival of mice bearing subcutaneous melanoma tumors that were therapeutically vaccinated 9 days post-inoculation with blank PLG scaffolds (blank), 5×105 irradiated GM-CSF-transduced melanoma cells (GMB16), 3000 ng soluble GM-CSF with 100 μg soluble CpG (GM + CpG), or PLG vaccines containing 3000 ng GM-CSF, 100 μg CpG and tumor lysate either once (Vax, 1x) or twice (vaccinated day 9 and day 19; Vax, 2x). a from [119], b–d from [118].
This PLG scaffold-based vaccine, referred to as WDVAX, is currently being investigated in a phase I clinical trial for the treatment of advanced melanoma (NCT01753089). Nine patients have been treated at the time of this review. In general, erythema has been observed in the vicinity of the implant that has increased with subsequent implantations, suggesting general immune activity in response to the vaccine [120]. In addition, a post-treatment tumor biopsy has been obtained from one patient. Histological and flow cytometric analysis of the sample indicates a general increase in T cells in the TME relative to pre-treatment, associated with the upregulation of markers indicative of an activated phenotype. Although preliminary, the results thus far indicate that at the current dose, the vaccine is safe, and suggests that WDVAX may be contributing, at least in this particular patient, to the generation or expansion of a population of activated T cells in the TME. These early results support the potential of biomaterial scaffolds as in situ sites for DC recruitment and programming, and their use for therapeutic cancer vaccination.
The use of high aspect ratio mesoporous silica rods (MSRs) has recently also been reported as DC-programming scaffolds for cancer vaccination, based on the same paradigm [42] (Fig. 8a). In contrast to preformed bulk scaffolds that must be implanted, MSRs can be injected through a conventional needle, allowing for minimally invasive vaccine administration. Following injection, the MSRs nonspecifically assemble in situ to form structures with inter-particle spaces permissive for cell trafficking (Fig. 8a). The MSRs used in this study have nanopores on the order of approximately 10 nm, giving them an extremely high surface area for surface modification or payload adsorption. MSRs loaded with GM-CSF, CpG and OVA facilitated the sustained release of GM-CSF and CpG for upwards of 28 days in vitro, and enhanced the persistence of OVA compared to a soluble bolus in vivo. Notably, MSR vaccines were able to recruit on the order of 25×106 total cells by day 7, with roughly of 3×106 CD11c+ DCs, significantly more than the PLG-based vaccines. The MSR vaccines also promoted robust humoral and cellular anti-cancer immune responses, including immunoglobulin class switching to the Th1-like isotype IgG2a, which the conventional adjuvant alum was unable to facilitate (Fig. 8b), and the induction of antigen-specific CD8+ T cells. This translated to significant prophylactic protection from an OVA-expressing lymphoma cell line in a murine model (Fig. 8c).
Figure 8.

MSRs as engineered microenvironments for therapeutic vaccination. (a) Diagram illustrating mechanism of MSR in situ self assembly and in vivo function. (b) ELISA analysis of sera OVA-specific IgG2a after immunization with soluble OVA (bolus OVA), soluble GM-CSF, CpG, and OVA (bolus vaccine), soluble OVA with Imject Alum (Alum + OVA), MSRs with OVA (MSR + OVA), or MSR vaccines loaded with GM-CSF, OVA, and CpG (MSR vaccine). Black arrows indicate vaccination days. (c) Survival of naive mice or mice that were prophylactically vaccinated with 150 μg soluble OVA in combination with soluble GM-CSF and CpG (bolus vaccine), MSRs with OVA (MSR + OVA), or MSR vaccines loaded with GM-CSF, CpG, and either 50 μg or 150 μg OVA (MSR vaccine), and challenged with an OVA-expressing murine lymphoma model 10 days later. From [42].
6. Engineered Materials for Adoptive T Cell Therapy
In ACT, autologous T cells are isolated from the patient, either enriched for natural cancer-reactive clones or genetically modified with a cancer-specific TCR or CAR, and then transferred back into the patient to elicit an anti-cancer immune response. Naturally occurring polyclonal populations of cancer-reactive T cells can be enriched from various sources including peripheral blood and tumor biopsies. The isolation of cancer-reactive TILs from tumor samples represents a particularly promising approach for the treatment of various cancers [9, 121], and has shown exceptional efficacy in the clinic for the treatment of advanced melanoma [122]. However, the ex vivo culture of TILs still represents a bottleneck in TIL-based ACT, wherein, using standard methods, it is challenging to expand the cells in a rapid and cost-efficient manner while maintaining them in an undifferentiated and functional state, and in which a consistent product is obtained following the ex vivo culture protocol [18, 19]. In addition, challenges also remain with respect to maintaining cell persistence and sustained effector functionality following bolus cell administration, wherein lymphodepletion regimens and systemic co-administration of survival factors, which are associated with significant morbidity and inflammatory toxicities, respectively, are necessary for effective TIL-based ACT [20, 21].
TCR- and CAR-transduced T cell-based ACT has also shown extremely promising results in the clinic, in particular, for B cell malignancies, using transgenic T cells specific for CD19 [7], which is expressed by most malignant B cells. The use of such genetically engineered T cells for a range of other malignancies is also being actively explored [8, 123], although their widespread use has thus far been limited by their extreme potency, wherein the use of high-affinity/avidity TCR or CAR T cells specific for cancer antigens expressed even at low levels in normal tissues has resulted in significant, even fatal, off-tumor on-target toxicities [124, 125]. Consistent with this, treatment of patients with CD19-specific TCR- or CAR-transduced T cells also results in the T cell-mediated elimination of normal B cells, necessitating immunoglobulin infusions following therapy [126]. Also, despite these potent effects, similar to the use of TILs, genetically-transduced T cells can also suffer from limited survival, persistence, and sustained functionality following bolus transfer, typically necessitating preparative lymphodepletion regimens to be performed prior to cell transfer, and in some cases, systemic cytokine administration [127, 128].
Biomaterials can potentially help to overcome some of the challenges currently associated with ACT. Specifically, the following topics will be discussed: (1) the design of materials as biomimetic substrates for the efficient ex vivo expansion of “high quality” T cells, (2) engineered particles that support the in vivo expansion of transferred T cells, and (3) engineered microenvironments as cell delivery vehicles that support the in situ expansion and sustained functionality of transferred T cells.
Efficient ex vivo cell expansion is one of the primary bottlenecks in TIL-based ACT. Early approaches for ACT used autologous monocyte-derived APCs for T cell expansion [129]. However, the use of autologous APCs is associated with numerous limitations, specifically, (1) the autologous cells could be affected by the patient’s disease state or could respond to cues produced by the lymphocytes, resulting in the generation of a suboptimal and inconsistent T cell product [130, 131], and (2) the need to generate and culture these cells on a patient-specific basis makes the process cumbersome and costly. An alternative approach that is currently used in the clinical setting is the “rapid expansion protocol” (REP) which employs inactivated allogeneic feeder cells in the presence of a CD3 antibody and the mitogenic cytokine IL-2 [122]. However, the use of allogeneic cells can also lead to suboptimal and inconsistent T cell products [18]. Genetically engineered “artificial APCs” (aAPCs) have also been developed to facilitate more efficient ex vivo T cell expansion, and this is described elsewhere [132].
An alternative approach to the use of live cells as mediators for ex vivo T cell expansion is the use of synthetic aAPCs, acellular materials that present the cues for promoting T cell activation and proliferation. At a minimum, synthetic aAPCs must present two critical signals: (signal 1) TCR activation via peptide-MHC complexes (antigen specific) or TCR clustering using CD3 antibodies (antigen nonspecific), and (signal 2) co-stimulation via agonistic CD28 antibodies. The coordinated provision of both signals is important because signal 1 in the absence of signal 2 has been shown to induce T cell anergy [133]. Mitogenic cytokines such as IL-2, IL-15, or IL-21 can also be added, and have been shown to fulfill the role of a “signal 3” important for optimal T cell expansion [18, 134]. The benefits of using synthetic aAPCs for T cell expansion are the design versatility, reproducibility, cost-efficiency, and scalability of synthetic systems. Standard acellular protocols for the ex vivo expansion of T cells typically involve the use of commercial high-throughput automated systems employing synthetic anti-CD3/CD28-immobilized beads (e.g. Dynabeads). Although convenient, these systems provide a limited set of cues to the T cells, and therefore likely promote T cell expansion with suboptimal efficiency. In addition, recent studies have highlighted the fact that the quality of the resultant T cells, with respect to their persistence and sustained functionality following transfer, is highly dependent on the in vitro environment in which they are expanded, and that protocols based on standard anti-CD3/CD28 beads and REP fail to produce high quality CTLs [18]. This may be due to an effect of the expansion protocol on properties of the resultant population including CD4:CD8 ratio [135], differentiation state [136], or the “age” (telomere length) of the cells [127]. Synthetic aAPC systems that present the appropriate combinations of cues (signals 1–3) in a context that recapitulates natural APC-T cell interactions may more efficiently promote the expansion of high quality CTLs. Important contextual considerations that are discussed below include the spatial distribution of mitogen, and the avidity of the aAPC-T cell interaction, which is affected by the surface area of aAPC-T cell contact and the density of T cell stimuli.
During interactions between natural APCs and cognate T cells, APCs produce T cell mitogenic cytokines, such as IL-2, in a paracrine manner, which promote the expansion of reactive T cell clones. To mimic this process, synthetic aAPCs have been prepared using spherical PLG cores immobilized with either anti-CD3 or peptide-MHC, and anti-CD28, and loaded with soluble IL-2 for local release to interacting T cells [134] (Fig. 9a). These synthetic aAPCs were found to promote efficient expansion of T cells in vitro compared to the same cues in soluble form, and a 10-fold lower dose of encapsulated IL-2 was found to promote a comparable degree of T cell expansion as soluble factor (Fig. 9b). Based on a similar design, it could be possible to prepare synthetic aAPCs that release multiple soluble cues, including polarizing signals, such as IL-12 or IFNγ, which may enhance the quality of the resultant T cells [18]. It was additionally found with this system that spherical microbeads on the size scale of natural APCs were more effective aAPCs than spherical nanobeads bearing the same signals, likely due to the inability of T cells to form high surface area interfaces with the spherical nano-aAPCs due to their high angles of curvature. Related to this is the finding that the shape of the aAPC also impacts the effectiveness of T cell activation wherein higher aspect ratio ellipsoidal microparticle aAPCs promoted more efficient in vitro T cell expansion than lower aspect ratio or spherical counterparts of the same volume. Time-lapsed microscopy demonstrated this is likely due to a larger surface area of contact between the particles and the T cells along the long axis of the particles, as T cells were observed to actively reposition themselves along this axis and generate higher surface area contacts with the aAPCs [137].
Figure 9.

Material strategies for ex vivo T cell expansion. (a) Diagram of PLG bead-based aAPCs that are surface immobilized with either anti-CD3, for polyclonal T cell expansion, or peptide-presenting MHC dimers, for monoclonal T cell expansion. Beads also surface present anti-CD28 as a co-stimulatory signal, and can release soluble mitogenic factors in a controlled manner. (b) Flow cytometry histograms showing T cell expansion, as measured by CFSE dilution, when T cells are either left unstimulated, or stimulated with soluble anti-CD3 and anti-CD28 (soluble Ab), soluble anti-CD3 and anti-CD28 with 0.6 ng soluble IL-2 (Soluble Ab with IL-2), PLG microbeads presenting anti-CD3 and anti-CD28 (aAPC), PLG microbeads presenting anti-CD3 and anti-CD28 supplemented with either 0.6 ng or 6 ng soluble IL-2 (aAPC exogenous IL-2), or PLG microbeads presenting anti-CD3 and anti-CD28 and encapsulated with 0.6 ng IL-2 (aAPC encapsulated IL-2). The numbers represent the percentage of divided cells in that condition as defined by the gray gate. (c) Confocal (top row), and FRET efficiency images (bottom row) showing cell-scale antibody clusters on chemically activated carbon (AC), untreated bundled SWNTs (bSWNT), and chemically treated “functionalized” bundled SWNTs (f-bSWNT). Colored legend represents degree of FRET efficiency in lower row images. (d) Quantification of T cell activation based on maximal IL-2 production, as measured over a range of stimuli concentrations, following T cell culture for various amounts of time on anti-CD3- and anti-CD28-immobilized chemically activated carbon (AC), untreated bundled SWNTs (bSWNT), chemically treated “functionalized” bundled SWNTs (f-bSWNT), or hydroxylated polystyrene beads (PS-OH). a, b from [134]. c, d from [143].
Although solid microbead-based systems roughly mimic the size and geometry of natural APCs, the immobilization of surface cues on a static substrate precludes the molecular reorganization that takes place during natural APC-T cell interactions, in which an immunological synapse (IS) is formed. The formation of the IS facilitates the assembly of high-density TCR microdomains on the size scale of cells, and this process has been shown to be critical for efficient T cell signaling [138]. To more closely mimic this process, liposomes bearing MHC II-enriched lipid rafts [139] and 3D supported lipid bilayers incorporating purified MHC I molecules [140] have been prepared, which allow for dynamic reorganization of the respective surface-associated cues. An alternative approach is the use of aAPC “nanoworms” comprised of a semi-flexible 200 nm poly(isocyano peptide) polymer modified with anti-CD3 [141]. Anti-CD3-modified nanoworms activated T cells more efficiently than free or PLG microparticle-immobilized anti-CD3. This likely owes to the ability of the semi-flexible polymeric backbone to facilitate dynamic high avidity interactions with T cells.
An alternative approach for ex vivo T cell expansion is the use of high surface area materials, such as carbon nanotubes (CNTs), that can present T cell stimuli at extremely high local densities [142, 143]. Chemical treatment of CNTs can be used to introduce surface defects and improve protein adsorption, further increasing the density at which stimuli can be presented. Specifically, such treatment was found to facilitate the formation of local antibody clusters on the size scale of cells, approximately 5–6 μm in diameter, with an inter-antibody distance of approximately 4.5 nm, allowing for high avidity interactions with T cells over a contact surface area that has been shown to be in the range optimal for IS formation (Fig. 9c) [138]. Notably, based on IL-2 production, T cells cultured with chemically treated CNTs immobilized with anti-CD3 and anti-CD28 showed enhanced activation compared to untreated CNTs or polystyrene microbeads bearing the same cues (Fig. 9d).
Recently, an approach has been described for the generation of a carbon nanotube-polymer composite (CNP) that combines these T cell stimulatory CNTs with IL-2-releasing, magnetite-loaded PLG nanoparticles (Fig. 10a, b) [144]. By combining the high avidity antigen presentation afforded by the CNTs with the paracrine release of IL-2 facilitated by the PLG nanoparticles, extremely robust expansion of functional T cells was observed in vitro. In addition, the magnetite-loaded PLG nanoparticles allowed the CNPs to be efficiently separated from the expanded T cells prior to cell transfer, an important step for clinical translation. Following two-weeks of ex vivo culture, the magnitude of T cell expansion, and the effector function of the resultant T cells as evaluated by IFNγ and granzyme B production, were both significantly higher with CNPs than with CNTs, commercial Dynabeads, or soluble tetramer. This finding was similar with or without the addition of exogenous IL-2. Importantly, throughout the duration of the culture, CNPs facilitated the greatest maintenance of CD27+CD8+ T cells. CD27 is an early-differentiation T cell marker that has been strongly correlated with clinical response to ACT [136]. ACT using CNP-expanded CTLs significantly slowed tumor growth in a therapeutic murine melanoma model compared to mock treatment, and this effect was associated with an increase in tumor T cell infiltration. Notably, in order to obtain a comparable therapeutic effect with Dynabeads-expanded CTLs, a 1000-fold greater concentration of IL-2 during ex vivo culture was required. CNPs were also used for the expansion of human Epstein–Barr virus (EBV)-specific CD8+ T cells from healthy donor PBMCs or leukopaks using either EBV peptide-loaded MHC or anti-CD3. Strikingly, either CNP-mediated approach led to the more efficient expansion of EBV peptide-specific CD8+ T cells than EBV peptide-loaded primary DCs with exogenous IL-2, the clinical gold standard for T cell expansion (Fig. 10c). This work illustrates the potential of more closely recapitulating natural APC-T cell interactions through the design of systems that collectively provide multiple cues in an appropriate context. Specifically, this work demonstrates how the use of such composite systems can translate to the more efficient ex vivo expansion of higher quality T cells, and speaks to the potential of using materials to engineer novel synthetic approaches for ex vivo T cell expansion for ACT.
Figure 10.

Carbon nanotube-polymer composite (CNP) for ex vivo T cell expansion. (a) Schematic of CNP assembly. Neutravidin-adsorbed CNTs (NCNT) are associated with biotinylated T cell stimuli and biotin-presenting PLG nanoparticles encapsulating IL-2 and magnetite. (b) Schematic of CNP functionality. CNPs present high density surface-adsorbed T cell stimuli, facilitate paracrine IL-2 release, and can be separated from T cells using magnetic separation. (c) Flow cytometry plots showing enhanced expansion of human EBV-specific CD8+ human T cells using CNPs presenting EBV peptide in MHC or anti-CD3 (OKT-3), compared to primary DCs loaded with EBV peptide and cultured in soluble IL-2. From [144].
Limited cell persistence following bolus cell administration represents another challenge to effective ACT, and necessitates preparative lymphodepletion regimens prior to cell transfer, as well as the systemic co-administration of exogenous survival cues such as IL-2, leading to patient morbidity and systemic toxicities, respectively. Relevant to this, the use of synthetic aAPCs to improve in vivo T cell survival has also been explored. For example, subcutaneous administration of synthetic aAPCs based on PLG microparticles were reported to improve the outcome of prophylactic ACT in a murine melanoma model based on tumor growth kinetics and overall survival [137]. Interestingly, high aspect ratio ellipsoidal microparticle aAPCs were found to be more effective than their spherical counterparts at slowing tumor growth, consistent with the observation that elongated particles facilitate more efficient T cell expansion in vitro.
Although it has been shown that spherical cell-sized microparticles are more efficient aAPCs than spherical nanoscale counterparts, the generally unfavorable bioavailability profiles of microparticles following systemic administration makes the effective in vivo translation of microparticle- based aAPCs challenging. Based on these observations, nanoellipsoidal PLG aAPCs were prepared to leverage the improved bioavailability of nanoparticles following systemic administration, while achieving a larger radius of curvature to support a larger contact surface area with T cells [145]. Nanoellipsoidal aAPCs were found to resist uptake by macrophages in vitro to a greater extent, and to have a prolonged blood half-life following systemic administration compared to spherical counterparts. Nanoellipsoidal aAPCs were also observed to promote the in vitro expansion of responder T cells more efficiently than spherical counterparts loaded with the same amount of peptide-MHC and anti-CD28, demonstrating the beneficial effect of nano-aAPCs with larger radii of curvature. Notably, the systemic co-administration of nanoellipsoidal aAPCs with responder T cells into immunocompetent mice promoted the in vivo proliferation of the administered T cells, with enhanced persistence of the administered T cells observed in the blood, lymph nodes, and spleen at 10 days post-injection. In contrast, spherical nano-aAPCs showed no improvement with respect to in vivo T cell expansion compared to mock treatment. The nanoellipsoidal aAPCs could have significant synergy with current ACT protocols by enhancing the in vivo survival and persistence of administered cells, and could potentially also have utility as a therapeutic vaccine platform.
An alternative approach to improving cell persistence in ACT is the design of nanomaterials as cell-associated depots for the sustained pseudo-autocrine delivery of survival and proliferative cues [146]. In such an approach, stabilized multilamellar liposomes were attached to cell surfaces via endogenous cell-surface thiol groups. The degree of uptake of cell surface-associated particles was previously found to be dependent on the size and surface properties of the particle [147]. When 300 nm liposomes were attached to CD8+ T cells, it was observed that the particles remained at the cell surface, and that up to 100 particles could be conjugated to the T cells without affecting their overall proliferative capacity, cytotoxic effector functionality, or in vivo migratory potential. When these depots were loaded with IL-15 superagonist (IL-15Sa) and IL-21, which have been shown to promote T cell expansion and function in vivo, sustained release of the cytokines was observed over 7 days in vitro. In a therapeutic murine model of metastatic melanoma, the conjugation of cytokine-loaded particles to tumor-specific CTLs was found to significantly enhance their persistence compared to systemic cytokine administration, and a subset of the transferred T cells homed to the spleen and lymph nodes and adopted a central memory phenotype. Consistent with this, all mice that received T cells conjugated with the particulate cytokine depots exhibited long-term survival in this model. Such systems, broadly referred to as “cellular backpacks” have also been explored for the targeted delivery of cell-associated payloads to specific tissues, leveraging the carrier cells as tissue-homing vectors [148]. Such an approach could be similarly used with T cells for the delivery of microenvironment-polarizing factors to the TME that may enhance the activity of local effector cells, including the transferred cells themselves.
The accumulation of cancer-reactive T cells in the TME following systemic administration represents another challenge to effective ACT, and biomaterial scaffolds can be used to facilitate the sustained delivery of T cells to the vicinity of accessible or resected tumors. A macroporous alginate scaffold, modified with a lymphocyte adhesion peptide, and entrapping lipid-enveloped mesoporous silica microspheres loaded with soluble IL-15Sa and surface presenting anti-CD3 and anti-CD28 was recently described [43]. The scaffold facilitated time-dependent migration of seeded T cells out of the device (Fig. 11a) and promoted enhanced T cell expansion and survival compared to unloaded scaffolds in vitro (Fig. 11b). In therapeutic murine models of resected breast cancer and nonresectable ovarian cancer, delivery of tumor-targeting CTLs in the device (either primary 4T1 breast cancer-responsive T cells or NKG2D CAR-transduced T cells, respectively) promoted enhanced T cell persistence, likely due to time-dependent in vivo cell expansion, which took place over the course of at least 12 days (Fig. 11c). In contrast, bolus T cell delivery via various administration routes, with or without in vitro prestimulation and systemic exogenous mitogen administration, resulted in a time-dependent decrease in T cell load over the same timeframe (Fig. 11c). The enhanced T cell persistence translated to significantly enhanced survival in both tumor models (Fig. 11d). Of particular note is the observation that T cells deployed as a bolus directly into the tumor resection cavity showed limited proliferation and upregulated markers associated with exhaustion, whereas T cells delivered into the resection cavity via the scaffold device proliferated robustly, maintained a non-exhausted phenotype, and migrated from the implant into the peritumoral tissue. These observations highlight the critical importance of the local microenvironment for T cell function, and support the use of biomaterial scaffolds as cell delivery devices in ACT. The versatility of the general paradigm will allow for the potential development of next-generation systems, for example, based on injectable hydrogels [28, 149], or materials that deliver additional signals such as T cell polarizing factors, or chemokines for recruiting endogenous cells such as DCs or other myeloid cells that can favorably interact with the transplanted T cells within the scaffold.
Figure 11.

Biopolymer implants for in situ T cell expansion. (a) Top, schematic of the in vitro assay used to quantify cell migration out of the scaffold (purple) and into a collagen gel tissue mimetic (pink). Bottom, time-lapsed light micrographs showing lymphocyte migration out of adhesion peptide-modified scaffolds after one day. Scale bar = 100 μm. (b) Flow cytometry plots showing robust T cell proliferation, as measured by CFSE dilution, and maintenance of T cell viability, as measured by negative expression of the apoptosis marker annexin-V, after in vitro culture in microparticle-loaded but not blank scaffolds for 7 days. CFSE MFI is indicated on the left and percent of cells positive for annexin-V is indicated on the right for each plot. (c) In vivo bioluminescence imaging of breast tumor-specific T cells retrovirally transduced with CBR-luc 0 days, 4, days, and 12 days post-transfer in resected breast cancer model. 7×106 cells were administered intravenously, into the resection cavity (intracavitary) with or without in vitro prestimulation with IL-15Sa, anti-CD3, anti-CD28, and anti-CD137, or delivered via the scaffold device into the resection cavity. Colored legend indicates bioluminescence intensity. (d) Survival of mice in resected breast cancer model either untreated (no T cells) or treated with 7×106 T cells administered either intravenously, intracavitary, intracavitary following in vitro prestimulation as defined in (c), or via the scaffold device into the resection cavity. Mean survival of each group is indicated in the legend (ms). From [43].
7. Conclusion
The recent clinical successes of checkpoint inhibitor therapies in a wide range of solid and hematologic malignancies, and CAR T cell therapies in diverse hematologic malignancies, have significantly heightened the enthusiasm for cancer immunotherapy. However, despite these successes, the vast majority of immunotherapies in clinical trials fail to meet their endpoints. Further, even checkpoint inhibitor therapies, which have shown unprecedented clinical success for a wide range of cancers, are only effective to date for a minority of the patient population [10, 12, 150]. In order to design more effective cancer immunotherapies in the future, it is important to consider why current therapies fail, both in the context of therapies that elicit generally suboptimal effects, and in the context of immunotherapies that are highly efficacious for a subset of the patient pool, but show minimal effects in the remainder. Broadly, the reason cancer immunotherapies fail in either context is likely because, (1) the immunotherapy fails to elicit a robust enough response, or (2) the response elicited by the immunotherapy is irrelevant or suboptimal for the particular patient. Overcoming these limitations will require both an improved understanding of the immunobiology of cancer, as well as the design of therapies that allow for the more precise and robust induction of the desired response. In other words, we must be able to (1) accurately assess what type of response needs to be generated in the particular patient, (2) define specific immunotherapeutic subgoals (e.g. generating de novo cancer-specific T cells, expanding pre-existing cancer-specific T cell number, or enhancing the effector functions of cancer-specific T cells through regulation of tumor-imposed immunosuppression), and (3) design systems that can precisely and effectively achieve those subgoals. Engineered materials will likely play an important role in facilitating the latter.
Being able to define optimal immunotherapy regimens on a patient-specific basis has thus far proven challenging because, currently, we have a relatively limited understanding of the specific determinants that contribute to a robust anti-cancer response, and of the biomarkers that indicate what types of immune interventions are optimal for fulfilling those determinants. The significance of a large range of factors including the frequency, demographic, phenotype, and spatial distributions of tumor-infiltrating and peripheral immune cell populations has been demonstrated [95, 151], but the ability to translate these readouts into an efficacious therapeutic regimen remains elusive. A better understanding of the specific determinants underlying a robust anti-cancer response will inform the design of more effective immunotherapies by providing a more accurate set of design criteria. In addition, it is only when we are able to translate these determinants into specific immunotherapeutic subgoals that the spatiotemporal precision afforded by the use of engineered materials is likely to be exceptionally constructive. On the other hand, to be able to effectively apply materials for the development of cancer immunotherapies, it will be important to gain a better understanding of how material systems interact with the immune system. In this respect, a systematic characterization of how immunologic components interact with, and respond to material systems at relevant timescales, and how these interactions are affected by the physical, mechanical, and physicochemical properties of the particular material system, is necessary. Such characterization will need to be coupled with studies aimed at understanding the mechanisms by which these material-immune system interactions take place. Indeed, the development of such a framework represents the primary challenge to the establishment of a “materials toolbox” that will enable the widespread application of biomaterials in cancer immunotherapy design in the future.
In any case, the application of materials to the development of cancer immunotherapies holds significant promise, as demonstrated by the work highlighted in this review. Advancements in immuno-oncology will continue to fuel the development of new materials-based cancer immunotherapy strategies in the future. For example, targeted delivery strategies using nanomaterial vehicles could be used for the delivery of anti-CTLA-4 antibodies, which function by promoting indiscriminate T cell activation and expansion, often leading to systemic inflammatory toxicities. Indeed, the magnitude of the increase in peripheral T cell TCR diversity in patients following treatment with an anti-CTLA-4 antibody was shown to be correlated with patient inflammatory toxicity but not with clinical outcome [152]. Using materials to promote the preferential accumulation or sequestration of the antibody in target sites, such as the tdLNs or the TME, could curtail systemic toxicities and potentially increase the therapeutic window of the drug. Work is underway to develop agents that target other co-inhibitory [153] as well as co-stimulatory [154] pathways, and nanomaterial-mediated targeted delivery strategies could similarly be useful for some of these agents.
Recently, workflows have been described for the patient-specific identification of neoantigens [115, 116], antigens produced in tumors due to random nonsynonymous somatic mutations. Neoantigens represent particularly attractive vaccine targets because they are not subject to central tolerance mechanisms that promote the deletion of high-affinity reactive T cell clones, and the utility of such neoantigen-targeting vaccines has recently been demonstrated in preclinical models [155]. Material subunit vaccines could be tremendously effective mediators of neoantigen-based vaccination, particularly those that were developed using model antigens that are also not subject to central tolerance and may consequently be optimized for inducing responses against such antigens. The design of biomaterials for neoantigen vaccines is likely to be an extremely promising area in the future. Alternatively, materials-based approaches for promoting ex vivo T cell expansion and survival using antigen-presenting synthetic aAPCs could be employed, for example, for efficiently expanding neoantigen-reactive TILs for ACT.
As we gain a better understanding of the optimal conditions for expanding high quality T cells ex vivo, synthetic materials that can facilitate the combinatorial release or presentation of multiple cues in a spatiotemporally-defined manner are likely to become increasingly important. Future studies exploring the use of synthetic aAPCs for T cell expansion will likely focus on the quality of the resultant T cells following in vivo transfer rather than just the efficiency of ex vivo T cell expansion and function. The use of biomaterial scaffolds for the delivery of high-affinity/avidity TCR- and CAR-transduced T cells could potentially also be useful in the future not only as devices to promote the enhanced survival and expansion of these cells in vivo, as has already been demonstrated [43], but potentially also as physical microenvironments that sequester these T cells in the vicinity of the tumor, curtailing the off-tumor on-target toxicity that has largely precluded the use of these highly potent cells for the treatment of solid malignancies [124, 125].
In conclusion, biomaterials represent a broad and versatile toolbox with which to design tailored cancer immunotherapies that may overcome many limitations of current approaches. In particular, this review discussed the use of nano- to microscale materials as targeted delivery vehicles for immune modulating payloads, as colloidal scaffolds in subunit vaccine formulations, and as synthetic aAPCs for ex vivo T cell expansion. The use of macroscale materials as microenvironments for recruiting and programming cells in situ, and for facilitating the survival and expansion of adoptively transferred cells, was also discussed. The ways in which engineered materials can productively interface with cancer immunotherapy are vast, and as we gain a better understanding of cancer immunobiology and of material-immune system interactions, this interface is likely to become even more prolific.
Highlights.
Biomaterials can be used in cancer immunotherapy applications to overcome some of the limitations of current approaches
Nanomaterials can be used to deliver immune modulating payloads to the tumor microenvironment
Nanomaterials can be used to co-deliver antigen and danger signal for therapeutic cancer vaccination
Bulk scaffolds can be used as engineered microenvironments for in situ immune cell programming for therapeutic cancer vaccination
Nano- to microscale particles, and bulk scaffolds can be used to promote cell survival and expansion for adoptive T cell therapies
Acknowledgments
The authors acknowledge support from the NIH (R01 EB015498) and the Wyss Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.W.H. Organization. WHO. int. 2014 http://apps.who.int/bookorders/anglais/detart1.jsp.
- 2.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS. New England Journal of Medicine. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K. New England Journal of Medicine. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, Lally-Goss D, McGehee-Norman S, Paolino A, Reardon DA, Friedman AH. Molecular cancer therapeutics. 2009;8:2773–2779. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, Manson K, Panicali DL, Laus R, Schlom J. Journal of Clinical Oncology. 2010;28:1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R. Nature medicine. 2012;18:1254–1261. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 7.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DAN, Lanier BJ. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL. Journal of Clinical Oncology. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS. Science. 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC. New England Journal of Medicine. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheridan C. Nature biotechnology. 2014;32:297–299. doi: 10.1038/nbt0414-297. [DOI] [PubMed] [Google Scholar]
- 12.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS. New England Journal of Medicine. 2015 doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB. New England Journal of Medicine. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 14.Hussein M, Berenson JR, Niesvizky R, Munshi N, Matous J, Sobecks R, Harrop K, Drachman JG, Whiting N. haematologica. 2010;95:845–848. doi: 10.3324/haematol.2009.008003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouchlaka MN, Sckisel GD, Chen M, Mirsoian A, Zamora AE, Maverakis E, Wilkins DE, Alderson KL, Hsiao HH, Weiss JM. The Journal of experimental medicine. 2013;210:2223–2237. doi: 10.1084/jem.20131219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, Friedman HS, Bigner DD, Sampson JH. Clinical Cancer Research. 2003;9:4247–4254. [PubMed] [Google Scholar]
- 17.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, Warnick E, Whiteside T, Osborne J, Kim H. Cancer Immunology. Immunotherapy. 2005;54:254–264. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin C, Yu D, Hillerdal V, Wallgren A, Karlsson-Parra A, Essand M. Molecular Therapy—Methods & Clinical Development. 2014;1 doi: 10.1038/mtm.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng W, Su M, Anderson KS, Sasada T. Immunobiology. 2014;219:583–592. doi: 10.1016/j.imbio.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA. Journal of Clinical Oncology. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou SH, Shetty AV, Geng Y, Xu L, Munirathinam G, Pipathsouk A, Tan I, Morris T, Wang B, Chen A. Cancer Immunology. Immunotherapy. 2013;62:597–603. doi: 10.1007/s00262-012-1364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larocca C, Schlom J. Cancer journal (Sudbury, Mass) 2011;17:359. doi: 10.1097/PPO.0b013e3182325e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Nature Reviews Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 24.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS nano. 2008;2:889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian X, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, Yang L, Young AN, Wang MD, Nie S. Nature biotechnology. 2008;26:83–90. doi: 10.1038/nbt1377. [DOI] [PubMed] [Google Scholar]
- 26.Fan Z, Shelton M, Singh AK, Senapati D, Khan SA, Ray PC. ACS nano. 2012;6:1065–1073. doi: 10.1021/nn2045246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Guo R, Yang M, Jiang X, Liu B. Advanced Materials. 2007;19:2988–2992. [Google Scholar]
- 28.Koshy ST, Ferrante TC, Lewin SA, Mooney DJ. Biomaterials. 2014;35:2477–2487. doi: 10.1016/j.biomaterials.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiappini C, De Rosa E, Martinez J, Liu X, Steele J, Stevens M, Tasciotti E. Nature materials. 2015 doi: 10.1038/nmat4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JH, von Maltzahn G, Zhang L, Derfus AM, Simberg D, Harris TJ, Ruoslahti E, Bhatia SN, Sailor MJ. Small. 2009;5:694–700. doi: 10.1002/smll.200801789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Löwik CW, Ossendorp F. Journal of Controlled Release. 2014;192:209–218. doi: 10.1016/j.jconrel.2014.07.040. [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Mao H, Wang YA, Cao Z, Peng X, Wang X, Duan H, Ni C, Yuan Q, Adams G. Small. 2009;5:235–243. doi: 10.1002/smll.200800714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verma A, Uzun O, Hu Y, Hu Y, Han HS, Watson N, Chen S, Irvine DJ, Stellacci F. Nature materials. 2008;7:588–595. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marrache S, Tundup S, Harn DA, Dhar S. ACS nano. 2013;7:7392–7402. doi: 10.1021/nn403158n. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Li Y, Jiao J, Hu HM. Nature nanotechnology. 2011;6:645–650. doi: 10.1038/nnano.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macdonald ML, Samuel RE, Shah NJ, Padera RF, Beben YM, Hammond PT. Biomaterials. 2011;32:1446–1453. doi: 10.1016/j.biomaterials.2010.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fabiilli ML, Wilson CG, Padilla F, Martín-Saavedra FM, Fowlkes JB, Franceschi RT. Acta biomaterialia. 2013;9:7399–7409. doi: 10.1016/j.actbio.2013.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cezar CA, Kennedy SM, Mehta M, Weaver JC, Gu L, Vandenburgh H, Mooney DJ. Advanced healthcare materials. 2014;3:1869–1876. doi: 10.1002/adhm.201400095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gauvin R, Chen YC, Lee JW, Soman P, Zorlutuna P, Nichol JW, Bae H, Chen S, Khademhosseini A. Biomaterials. 2012;33:3824–3834. doi: 10.1016/j.biomaterials.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ali OA, Tayalia P, Shvartsman D, Lewin S, Mooney DJ. Advanced functional materials. 2013;23:4621–4628. doi: 10.1002/adfm.201203859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ali OA, Verbeke C, Johnson C, Sands RW, Lewin SA, White D, Doherty E, Dranoff G, Mooney DJ. Cancer research. 2014;74:1670–1681. doi: 10.1158/0008-5472.CAN-13-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, Mooney DJ. Nature biotechnology. 2015;33:64–72. doi: 10.1038/nbt.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT. Nature biotechnology. 2015;33:97–101. doi: 10.1038/nbt.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen DS, Mellman I. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 45.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Cancer cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coussens LM, Zitvogel L, Palucka AK. Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Proceedings of the National Academy of Sciences. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL. Proceedings of the National Academy of Sciences. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perret R, Sierro SR, Botelho NK, Corgnac S, Donda A, Romero P. Cancer research. 2013;73:6597–6608. doi: 10.1158/0008-5472.CAN-13-0875. [DOI] [PubMed] [Google Scholar]
- 50.Stewart CA, Metheny H, Iida N, Smith L, Hanson M, Steinhagen F, Leighty RM, Roers A, Karp CL, Müller W. The Journal of clinical investigation. 2013;123:4859. doi: 10.1172/JCI65180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. The Journal of experimental medicine. 2008;205:1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, Boulden J, Sutanto-Ward E, Soler AP, Laury-Kleintop LD. Cancer discovery. 2012;2:722–735. doi: 10.1158/2159-8290.CD-12-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M. The Journal of experimental medicine. 2011;208:1949–1962. doi: 10.1084/jem.20101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mantovani A, Allavena P, Sica A, Balkwill F. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 55.Mellman I, Coukos G, Dranoff G. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le HN, Lee NC, Tsung K, Norton JA. The Journal of Immunology. 2001;167:6765–6772. doi: 10.4049/jimmunol.167.12.6765. [DOI] [PubMed] [Google Scholar]
- 57.Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Biomaterials. 2014;35:814–824. doi: 10.1016/j.biomaterials.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Jeanbart L, Ballester M, De Titta A, Corthésy P, Romero P, Hubbell JA, Swartz MA. Cancer immunology research. 2014;2:436–447. doi: 10.1158/2326-6066.CIR-14-0019-T. [DOI] [PubMed] [Google Scholar]
- 59.von Bernstorff W, Voss M, Freichel S, Schmid A, Vogel I, Jöhnk C, Henne-Bruns D, Kremer B, Kalthoff H. Clinical Cancer Research. 2001;7:925s–932s. [PubMed] [Google Scholar]
- 60.Iyer AK, Khaled G, Fang J, Maeda H. Drug discovery today. 2006;11:812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 61.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Proceedings of the National Academy of Sciences. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao D, Liu Z, Wrasidlo WJ, Luo Y, Nguyen G, Chen T, Xiang R, Reisfeld RA. Cancer research. 2011;71:5688–5696. doi: 10.1158/0008-5472.CAN-11-1264. [DOI] [PubMed] [Google Scholar]
- 63.Dreaden EC, Morton SW, Shopsowitz KE, Choi JH, Deng ZJ, Cho NJ, Hammond PT. ACS nano. 2014;8:8374–8382. doi: 10.1021/nn502861t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh N, Karambelkar A, Gu L, Lin K, Miller JS, Chen CS, Sailor MJ, Bhatia SN. Journal of the American Chemical Society. 2011;133:19582–19585. doi: 10.1021/ja206998x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan NC, Cheng FY, Ho JaA, Yeh CS. Angewandte Chemie International Edition. 2012;51:8806–8810. doi: 10.1002/anie.201203339. [DOI] [PubMed] [Google Scholar]
- 66.Liu TY, Hu SH, Liu KH, Shaiu RS, Liu DM, Chen SY. Langmuir. 2008;24:13306–13311. doi: 10.1021/la801451v. [DOI] [PubMed] [Google Scholar]
- 67.Koppolu B, Bhavsar Z, Wadajkar AS, Nattama S, Rahimi M, Nwariaku F, Nguyen KT. Journal of biomedical nanotechnology. 2012;8:983–990. doi: 10.1166/jbn.2012.1465. [DOI] [PubMed] [Google Scholar]
- 68.Pignatello R, Leonardi A, Pellitteri R, Carbone C, Caggia S, Graziano ACE, Cardile V. Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2013;434:136–144. [Google Scholar]
- 69.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Foged C, Brodin B, Frokjaer S, Sundblad A. International journal of pharmaceutics. 2005;298:315–322. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 71.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Nature nanotechnology. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang X, Teng X, Chen D, Tang F, He J. Biomaterials. 2010;31:438–448. doi: 10.1016/j.biomaterials.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 73.Beningo KA, Wang Y-l. Journal of Cell Science. 2002;115:849–856. doi: 10.1242/jcs.115.4.849. [DOI] [PubMed] [Google Scholar]
- 74.Liu W, Zhou X, Mao Z, Yu D, Wang B, Gao C. Soft Matter. 2012;8:9235–9245. [Google Scholar]
- 75.Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Biochemical and biophysical research communications. 2007;353:26–32. doi: 10.1016/j.bbrc.2006.11.135. [DOI] [PubMed] [Google Scholar]
- 76.Kim HR, Gil S, Andrieux K, Nicolas V, Appel M, Chacun H, Desmaele D, Taran F, Georgin D, Couvreur P. Cellular and molecular life sciences. 2007;64:356–364. doi: 10.1007/s00018-007-6390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, Convertine A, Stayton PS. ACS nano. 2013;7:3912–3925. doi: 10.1021/nn305466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Molino NM, Anderson AK, Nelson EL, Wang SW. ACS nano. 2013;7:9743–9752. doi: 10.1021/nn403085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asai T, Tsuzuku T, Takahashi S, Okamoto A, Dewa T, Nango M, Hyodo K, Ishihara H, Kikuchi H, Oku N. Biochemical and biophysical research communications. 2014;444:599–604. doi: 10.1016/j.bbrc.2014.01.107. [DOI] [PubMed] [Google Scholar]
- 80.Chen CJ, Wang JC, Zhao EY, Gao LY, Feng Q, Liu XY, Zhao ZX, Ma XF, Hou WJ, Zhang LR. Biomaterials. 2013;34:5303–5316. doi: 10.1016/j.biomaterials.2013.03.056. [DOI] [PubMed] [Google Scholar]
- 81.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H. Cancer cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alshamsan A, Hamdy S, Samuel J, El-Kadi AO, Lavasanifar A, Uluda H. Biomaterials. 2010;31:1420–1428. doi: 10.1016/j.biomaterials.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 83.Molavi O, Mahmud A, Hamdy S, Hung RW, Lai R, Samuel J, Lavasanifar A. Molecular pharmaceutics. 2010;7:364–374. doi: 10.1021/mp900145g. [DOI] [PubMed] [Google Scholar]
- 84.Zhang X, Tian W, Cai X, Wang X, Dang W, Tang H, Cao H, Wang L, Chen T. PloS one. 2013;8:e65896. doi: 10.1371/journal.pone.0065896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Limon PL. Nature materials. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kwong B, Liu H, Irvine DJ. Biomaterials. 2011;32:5134–5147. doi: 10.1016/j.biomaterials.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. The Journal of Immunology. 2006;176:309–318. doi: 10.4049/jimmunol.176.1.309. [DOI] [PubMed] [Google Scholar]
- 88.Ahonen CL, Wasiuk A, Fuse S, Turk MJ, Ernstoff MS, Suriawinata AA, Gorham JD, Kedl RM, Usherwood EJ, Noelle RJ. Blood. 2008;111:3116–3125. doi: 10.1182/blood-2007-09-114371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Palucka K, Banchereau J. Nature Reviews Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paul WE. Cell. 2011;147:1212–1215. doi: 10.1016/j.cell.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 91.Steinman RM. Annual review of immunology. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 92.Helle M, Rampazzo E, Monchanin M, Marchal F, Guillemin F, Bonacchi S, Salis F, Prodi L, Bezdetnaya L. ACS nano. 2013;7:8645–8657. doi: 10.1021/nn402792a. [DOI] [PubMed] [Google Scholar]
- 93.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. European journal of immunology. 2008;38:1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 94.Bos R, Sherman LA. Cancer research. 2010;70:8368–8377. doi: 10.1158/0008-5472.CAN-10-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baumgaertner P, Jandus C, Rivals JP, Derré L, Lövgren T, Baitsch L, Guillaume P, Luescher IF, Berthod G, Matter M. International Journal of Cancer. 2012;130:2607–2617. doi: 10.1002/ijc.26297. [DOI] [PubMed] [Google Scholar]
- 97.Blander JM, Medzhitov R. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 98.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Science. 2003;299:1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- 99.Burgdorf S, Schölz C, Kautz A, Tampé R, Kurts C. Nature immunology. 2008;9:558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- 100.Rock KL. European journal of immunology. 1994;24:2421–2428. doi: 10.1002/eji.1830241024. [DOI] [PubMed] [Google Scholar]
- 101.Li M, Davey GM, Sutherland RM, Kurts C, Lew AM, Hirst C, Carbone FR, Heath WR. The Journal of Immunology. 2001;166:6099–6103. doi: 10.4049/jimmunol.166.10.6099. [DOI] [PubMed] [Google Scholar]
- 102.Zhang Z, Tongchusak S, Mizukami Y, Kang YJ, Ioji T, Touma M, Reinhold B, Keskin DB, Reinherz EL, Sasada T. Biomaterials. 2011;32:3666–3678. doi: 10.1016/j.biomaterials.2011.01.067. [DOI] [PubMed] [Google Scholar]
- 103.Lee IH, Kwon HK, An S, Kim D, Kim S, Yu MK, Lee JH, Lee TS, Im SH, Jon S. Angewandte Chemie. 2012;124:8930–8935. [Google Scholar]
- 104.Demento SL, Bonafé N, Cui W, Kaech SM, Caplan MJ, Fikrig E, Ledizet M, Fahmy TM. The Journal of Immunology. 2010;185:2989–2997. doi: 10.4049/jimmunol.1000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Borges O, Cordeiro-da-Silva A, Tavares J, Santarém N, de Sousa A, Borchard G, Junginger HE. European Journal of Pharmaceutics and Biopharmaceutics. 2008;69:405–416. doi: 10.1016/j.ejpb.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 106.Slütter B, Jiskoot W. Journal of Controlled Release. 2010;148:117–121. doi: 10.1016/j.jconrel.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 107.Bal SM, Slütter B, Verheul R, Bouwstra JA, Jiskoot W. European Journal of Pharmaceutical Sciences. 2012;45:475–481. doi: 10.1016/j.ejps.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 108.Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA, Hubbell JA. Biomaterials. 2012;33:6211–6219. doi: 10.1016/j.biomaterials.2012.04.060. [DOI] [PubMed] [Google Scholar]
- 109.Stano A, Scott EA, Dane KY, Swartz MA, Hubbell JA. Biomaterials. 2013;34:4339–4346. doi: 10.1016/j.biomaterials.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 110.Butts C, Maksymiuk A, Goss G, Soulières D, Marshall E, Cormier Y, Ellis PM, Price A, Sawhney R, Beier F. Journal of cancer research and clinical oncology. 2011;137:1337–1342. doi: 10.1007/s00432-011-1003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H. Nature materials. 2011;10:243–251. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Proceedings of the National Academy of Sciences. 2012;109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA. Nature biotechnology. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 114.Thomas SN, van der Vlies AJ, O’Neil CP, Reddy ST, Yu SS, Giorgio TD, Swartz MA, Hubbell JA. Biomaterials. 2011;32:2194–2203. doi: 10.1016/j.biomaterials.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 115.Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J. Blood. 2014;124:453–462. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 117.Schreiber RD, Old LJ, Smyth MJ. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 118.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Nature materials. 2009;8:151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ali OA, Doherty E, Mooney DJ, Emerich D. Biomatter. 2011;1:66–75. doi: 10.4161/biom.1.1.16277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hodi FS. Tumor Immunology: Multidisciplinary Science Driving Combination Therapy. Banff, Alberta, Canada: 2015. [Google Scholar]
- 121.Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, Dudley ME, Yang JC, Sherry RM, Kammula US. Journal of Clinical Oncology. 2015 doi: 10.1200/JCO.2014.58.9093. JCO. 2014.2058. 9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y. Clinical Cancer Research. 2013;19:4792–4800. doi: 10.1158/1078-0432.CCR-13-0380. [DOI] [PubMed] [Google Scholar]
- 123.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morgan RA, Chinnasamy N, Abate-Daga DD, Gros A, Robbins PF, Zheng Z, Feldman SA, Yang JC, Sherry RM, Phan GQ. Journal of immunotherapy (Hagerstown, Md: 1997) 2013;36:133. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R. Molecular Therapy. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grupp SA, Frey NV, Aplenc R, Barrett DM, Chew A, Kalos M, Levine BL, Litchman M, Maude SL, Rheingold SR. Blood. 2013;122:67–67. [Google Scholar]
- 127.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF. Journal of Clinical Oncology. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yee C, Thompson J, Byrd D, Riddell S, Roche P, Celis E, Greenberg P. Proceedings of the National Academy of Sciences. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP, Gabrilovich DI. Clinical Cancer Research. 2000;6:1755–1766. [PubMed] [Google Scholar]
- 131.Satthaporn S, Robins A, Vassanasiri W, El-Sheemy M, Jibril JA, Clark D, Valerio D, Eremin O. Cancer Immunology. Immunotherapy. 2004;53:510–518. doi: 10.1007/s00262-003-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Butler MO, Hirano N. Immunological reviews. 2014;257:191–209. doi: 10.1111/imr.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Appel H, Seth NP, Gauthier L, Wucherpfennig KW. The Journal of Immunology. 2001;166:5279–5285. doi: 10.4049/jimmunol.166.8.5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Steenblock ER, Fahmy TM. Molecular Therapy. 2008;16:765–772. doi: 10.1038/mt.2008.11. [DOI] [PubMed] [Google Scholar]
- 135.Peshwa MV, Benike C, Dupuis M, Kundu SK, Engleman EG, Merigan TC, van Schooten WC. Cell transplantation. 1998;7:1–9. doi: 10.1177/096368979800700103. [DOI] [PubMed] [Google Scholar]
- 136.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR. Clinical Cancer Research. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sunshine JC, Perica K, Schneck JP, Green JJ. Biomaterials. 2014;35:269–277. doi: 10.1016/j.biomaterials.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huppa JB, Davis MM. Nature Reviews Immunology. 2003;3:973–983. doi: 10.1038/nri1245. [DOI] [PubMed] [Google Scholar]
- 139.Ding Q, Chen J, Wei X, Sun W, Mai J, Yang Y, Xu Y. Pharmaceutical research. 2013;30:60–69. doi: 10.1007/s11095-012-0849-7. [DOI] [PubMed] [Google Scholar]
- 140.Goldstein SA, Mescher M. The Journal of Immunology. 1986;137:3383–3392. [PMC free article] [PubMed] [Google Scholar]
- 141.Mandal S, Eksteen-Akeroyd ZH, Jacobs MJ, Hammink R, Koepf M, Lambeck AJ, van Hest JC, Wilson CJ, Blank K, Figdor CG. Chemical Science. 2013;4:4168–4174. [Google Scholar]
- 142.Fadel TR, Steenblock ER, Stern E, Li N, Wang X, Haller GL, Pfefferle LD, Fahmy TM. Nano letters. 2008;8:2070–2076. doi: 10.1021/nl080332i. [DOI] [PubMed] [Google Scholar]
- 143.Fadel TR, Look M, Staffier PA, Haller GL, Pfefferle LD, Fahmy TM. Langmuir. 2009;26:5645–5654. doi: 10.1021/la902068z. [DOI] [PubMed] [Google Scholar]
- 144.Fadel TR, Sharp FA, Vudattu N, Ragheb R, Garyu J, Kim D, Hong E, Li N, Haller GL, Pfefferle LD. Nature nanotechnology. 2014;9:639–647. doi: 10.1038/nnano.2014.154. [DOI] [PubMed] [Google Scholar]
- 145.Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, Green JJ. Small. 2014 doi: 10.1002/smll.201402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Nature medicine. 2010;16:1035–1041. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Doshi N, Swiston AJ, Gilbert JB, Alcaraz ML, Cohen RE, Rubner MF, Mitragotri S. Advanced Materials. 2011;23:H105–H109. doi: 10.1002/adma.201004074. [DOI] [PubMed] [Google Scholar]
- 148.Anselmo AC, Gilbert JB, Kumar S, Gupta V, Cohen RE, Rubner MF, Mitragotri S. Journal of Controlled Release. 2015;199:29–36. doi: 10.1016/j.jconrel.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 149.Bencherif SA, Sands RW, Bhatta D, Arany P, Verbeke CS, Edwards DA, Mooney DJ. Proceedings of the National Academy of Sciences. 2012;109:19590–19595. doi: 10.1073/pnas.1211516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Lao CD. The Lancet Oncology. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 151.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B. Clinical Cancer Research. 2014;20:2424–2432. doi: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, Wang L. Cancer research. 2014;74:1933–1944. doi: 10.1158/0008-5472.CAN-13-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. The Journal of experimental medicine. 2014;211:715–725. doi: 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]