

Abstract
Activated T cells rely on aerobic glycolysis and glutaminolysis in order to proliferate and differentiate into effector cells. Therefore, intervention in these metabolic pathways inhibits proliferation. The aim of the present study was to evaluate the effects of Krebs' cycle inhibition at the level of malate dehydrogenase-2 (MDH2) in human activated T cells using the MDH2 inhibitor LW6. Activated T cells from healthy volunteers were cultured in the presence or absence of LW6 and cytotoxicity, cell proliferation and the expression levels of hypoxia-inducible factor (HIF)-1α, c-Myc, p53, cleaved caspase-3 and certain enzymes involved in glucose metabolism and glutaminolysis were evaluated. The results revealed that LW6 was not toxic and decreased apoptosis and the levels of the pro-apoptotic tumor suppressor p53. In addition, LW6 inhibited T-cell proliferation and decreased the levels of c-Myc, HIF-1α, glucose transporter-1, hexokinase-II, lactate dehydrogenase-A and phosphorylated pyruvate dehydrogenase. By contrast, LW6 increased the levels of pyruvate dehydrogenase. These alterations may lead to decreased production of pyruvate, which preferentially enters into the Krebs' cycle. Furthermore, LW6 decreased the levels of glutaminase-2, while increasing those of glutaminase-1, which may preserve glutaminolysis, and possibly pyruvate-malate cycling, potentially protecting the cells from energy collapse. In summary, the inhibition of MDH2 in activated T cells abrogates proliferation without adversely affecting cell survival. Adaptations of cellular glucose and glutamine metabolism may prevent energy collapse.
Keywords: malate dehydrogenase, T cell, proliferation, apoptosis, glucose metabolism, glutaminolysis
Introduction
Activated T cells rely on aerobic glycolysis and glutaminolysis in order to proliferate and differentiate into effector T cells (1,2). Intervention in these metabolic pathways inhibits the proliferation of activated T cells and their differentiation to effector cells. For example, treatment of activated T cells with 2-deoxy-D-glucose abrogates their proliferation and differentiation into effector T cells by inhibiting glycolysis at the level of hexokinase II (HKII) (3,4). A similar effect is yielded by treatment with dichloroacetate, which activates pyruvate dehydrogenase (PDH) through the inhibition of pyruvate dehydrogenase kinase (PDK) (3,5). The PDH dehydrogenase regulates the influx of pyruvate into the mitochondria, increasing the ratio of glucose oxidation to glycolysis. Notably, indoleamine 2,3-dioxygenase, a key immunomodulatory enzyme, exerts its suppressive effect at least in part by inhibiting aerobic glycolysis and glutaminolysis in activated T cells (6–8).
To date, the effect of Krebs' cycle inhibition on the proliferation of activated T cells has not been fully evaluated. For this purpose, the present study was devised to investigate the activity of the 3-(aryloxyacetylamino)-4-hydroxybenzoic acid derivative LW6. LW6 was originally considered to be an inhibitor of hypoxia-inducible factor-1α (HIF-1α) (9). However, a further study by the same investigators revealed that LW6 is a specific malate dehydrogenase-2 (MDH2) inhibitor (10). Furthermore, the inhibition of HIF-1α is secondary and results from its degradation due to increased intracellular O2 induced by LW6. By inhibiting the Krebs' cycle, LW6 decreases the production of reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which are electron donors required for oxidative phosphorylation, and thus reduce O2 consumption (10). The specific Krebs' cycle inhibitor LW6 was selected because suppression at the MDH2 level may prevent total cell energy collapse due to pyruvate-malate cycling. Accumulated malate exits the mitochondria and enters the cytoplasm, where it is converted by malic enzyme to pyruvate, which may re-enter the mitochondria and the Krebs' cycle (11).
The aim of the present study was to evaluate the effects of the MDH2 inhibitor LW6 on human activated T-cell proliferation and survival. In addition, the effects of LW6 on the levels of certain transcription factors involved in cell proliferation, apoptosis and metabolism, and certain enzymes associated with glucose metabolism and glutaminolysis were evaluated, and Krebs' cycle activity was assessed.
Materials and methods
Subjects
Blood samples were collected from 8 healthy volunteers (4 men and 4 women; 33±8 years old). Informed consent was obtained from each individual enrolled in the study, and the study protocol was approved by the Ethics Committee of the Medical School, University of Thessaly (Larissa, Greece).
Isolation of T cells and culture conditions
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by Ficoll-Hypaque density gradient centrifugation (Histopaque-1077; Sigma-Aldrich, St. Louis, MO, USA). T cells were isolated from PBMCs. This comprised the indirect magnetic labeling of non-T cells by using a cocktail of biotin-conjugated monoclonal antibodies and their depletion using a Pan T cell Isolation kit (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Negatively selected T cells were counted using an optical microscope Axiovert 40 C, (Carl Zeiss Light Microscopy, Göttingen, Germany) on a Neubauer plaque. Cell viability was assessed via trypan blue assay (Sigma-Aldrich). T cells were cultured in RPMI-1640 medium with L-glutamine and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), supplemented with 10% fetal bovine serum (Sigma-Aldrich) and antibiotic-antimycotic solution (Sigma-Aldrich). All culturing was performed at 37°C in a humidified atmosphere containing 5% CO2.
Unstimulated T cells were used as the control cells, whereas activated T cells were induced by stimulation with anti-CD2, anti-CD3 and anti-CD28-conjugated beads using the T-Cell Activation/Expansion kit (Miltenyi Biotec GmbH), with a bead-to-cell ratio of 1:2. Stimulated T cells were cultured in the presence or absence of LW6 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at a concentration of 30 µM for 72 h.
Evaluation of LW6 toxicity and its effect on the proliferation of activated T cells
Control, stimulated or stimulated and LW6 (30 µΜ)-treated T cells were cultured in 96-well plates (1×105 cells/well) for 72 h. LW6 toxicity in the activated T cells was evaluated by a lactate dehydrogenase (LDH) release assay using a CytoTox Non-Radioactive Cytotoxic assay kit (Promega Corporation, Madison, WI, USA) according to the instructions provided by the manufacturer. Cytotoxicity was calculated using the following equation: Cytotoxicity (%) = (LDH in the supernatant/Total LDH) × 100.
Control, stimulated or LW6-treated stimulated T cells were cultured in 96-well plates (1×105 cells/well) for 72 h. At the end of the 72-h period, cell proliferation was assessed via Cell Proliferation ELISA (Roche Diagnostics, Indianapolis, IN, USA) using bromodeoxyuridine (BrdU) labeling and immunoenzymatic detection according to the manufacturer's instructions. The proliferation index was calculated as the ratio of the optical density (OD) derived from the stimulated or the stimulated and LW6-treated T cells to the OD derived from the control T cells.
Experiments were performed in T cells derived from the blood of 8 individuals. In each experiment, three measurements were made for the T cell samples obtained from each individual, and the results are expressed as the mean of the three measurements.
Assessment of the effect of LW6 on HIF-1α, c-Myc, p53, cleaved caspase-3 and certain enzymes of glucose metabolism and glutaminolysis in activated T cells
Control, stimulated or stimulated and LW6 (30 µΜ)-treated T cells were cultured in 12-well plates (1×106 cells/well) for 72 h. Subsequently, T cells were lysed using the T-PER tissue protein extraction reagent (Pierce Biotechnology, Inc., Rockford, IL, USA) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich and Roche Diagnostics, respectively). Protein was quantified using a Bradford assay (Sigma-Aldrich) and western blot analysis was performed. Equal quantities of protein extracts (50 µg) from each sample were loaded for electrophoresis in sodium dodecyl sulfate (SDS) polyacrylamide gels (Invitrogen Life Technologies, Carlsbad, CA, USA). Subsequently, proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Invitrogen Life Technologies). Blots were incubated with primary antibody for 16 h, followed by anti-rabbit IgG, HRP-linked secondary antibody (cat. no., #7074; dilution, 1/1000); Cell Signaling Technology, Inc., Danvers, MA, USA) for 30 min. A benchmark pre-stained protein ladder (Invitrogen Life Technologies) was used as a marker. Bands were visualized via enhanced chemiluminescent detection using a LumiSensor Plus Chemiluminescent HRP Substrate kit (GenScript Corporation, Piscataway, NJ, USA). In cases of reprobing PVDF blots, the previous primary and secondary antibodies were safely removed using Restore Western Blot Stripping Buffer (Thermo Fisher Scientific, Rochford, IL, USA) according to the manufacturer's instructions. The PVDF blot was then reused and western blot analysis was resumed as previously described, using a different primary antibody. Analysis of the bands was performed using Image J software (National Institute of Health, Bethesda, MD, USA).
The primary antibodies used in western blot analysis were specific for HIF-1α (cat. no., #3716; dilution, 1/500), c-Myc (cat. no., #5605; dilution, 1/500), p53 (cat. no., #9282; dilution, 1/500), cleaved caspase-3 (cat. no., #9664; dilution, 1/500; Cell Signaling Technology, Inc.), glucose transporter-1 (GLUT1; (cat. no., sc-7903; dilution, 1/200; Santa Cruz Biotechnology, Inc.), hexokinase II (HKII; (cat. no., #2867; dilution, 1/1000; Cell Signaling Technology, Inc.), lactate dehydrogenase-A (LDH-A; cat. no., #2012; dilution, 1/1000; Cell Signaling Technology, Inc.), pyruvate dehydrogenase (PDH; cat. no., #2784; dilution, 1/1000; Cell Signaling Technology, Inc.), PDH phosphorylated at serine 393 (p-PDH; cat. no., orb6670; dilution, 1/100; Biorbyt, Ltd., San Francisco, CA, USA), glutaminase (GLS)-1 and GLS2 (cat. nos., #AP18036PU-N and AP17426PU-N, respectively; dilutions, 1/100; Acris Antibodies GmbH, Herford, Germany) and β-actin (cat. no., #4967; dilution, 1/2500; Cell Signaling Technology, Inc.).
Assessment of the effect of LW6 on Krebs' cycle activity in activated T cells
Control, stimulated or stimulated and LW6 (30 µΜ)-treated T cells were cultured in 96-well plates (1×105 cells/well) for 72 h. Krebs' cycle activity was assessed by colorimetric measurement of the reduction of the yellow tetrazolium salt sodium 2,3-bis (2-methoxy-4-nitro-5-sulfo-phenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium (XTT), to an orange formazan product. XTT is reduced in the plasma membrane by trans-plasma membrane electron transport via the electron carrier, 1-methoxy-5-methyl-phenazinium methyl sulfate (1-methoxy-PMS), in which case the cellular reductant is reduced NADH. NADH is derived primarily from the Krebs' cycle in the mitochondria (12). For this purpose, a TACS XTT assay kit (Trevigen, Gaithersburg, MD, USA) was used according to the instructions provided by the manufacturer. An ELISA reader (Microplate Reader PR2100; Sanofi Diagnostics Pasteur Inc., Redmond, WA, USA) was used to determine the results of the XTT assay. Experiments were performed in T cells derived from the blood of 8 individuals. In each experiment three measurements were made for each of the 8 individuals and the results are expressed as the mean of the three measurements.
Statistical analysis
The normality of the evaluated variables was assessed and confirmed by the one-sample Kolmogorov-Smirnov test. For comparison of mean values between two conditions, a paired t-test was used. For comparison of mean values among more than two conditions, the sphericity assumption was evaluated using Mauchly's test and, if violated, degrees of freedom were corrected using Greenhouse-Geisser or Huynh-Feldt estimates of sphericity. Comparison of mean values was assessed by one-way repeated-measures analysis of variance, followed by the Bonferroni's correction test. SPSS 13.0 software for Windows (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. Results are expressed as the mean ± standard deviation (SD) and P<0.05 was considered to indicate a statistically significant difference.
For the western blot analysis, results are expressed as optical densities (OD) derived from analysis of the bands using Image J software (National Institute of Health). P-values were calculated by comparing the mean values of OD. Statistical analysis after adjustment to the control OD values was avoided to prevent violation of the prerequisite for normal distribution of the compared variables when applying parametric statistical tests. Results are presented according to the OD, and the error bars correspond to the SD. For convenience, in the text the results are expressed after the normalization of mean values for the control group. Similarly, the results of the XTT assay are expressed as the OD values derived using the ELISA reader.
Representative western blotting results of two out of the eight experiments performed are displayed in Fig. 1.
Figure 1.

Effects of the malate dehydrogenase-2 inhibitor LW6 on the levels of evaluated proteins. Western blot lanes correspond to two of the eight performed experiments. HIF-1α, hypoxia inducible factor-1α; GLUT1, glucose transporter-1; HKII, hexokinase II; LDH-A, lactate dehydrogenase-A; PDH, pyruvate dehydrogenase; pPDH, PDH phosphorylated at serine 393; GLS1, glutaminase-1; GLS2, glutaminase-2; Ctrl, resting control T-cells; Act, activated T-cells; Act + LW6, LW6-treated activated T cells.
Results
Western blotting
Representative western blotting results of two of the eight performed experiments are displayed in Fig. 1.
LW6 is not toxic and decreases the levels of the pro-apoptotic protein p53 and activated cleaved caspase-3
LDH release assay revealed a cytotoxicity of 14.0±1.46% in unstimulated T cells, 13±1.36% in stimulated T cells and 10.75±1.44% in stimulated T cells treated with LW6. Comparison of the control and stimulated T cells indicated that stimulation was not cytotoxic (P=0.534). Concurrent treatment of T cells with stimulation and LW6 decreased the cytotoxicity compared with that of the stimulated T cells (P=0.034; Fig. 2A).
Figure 2.
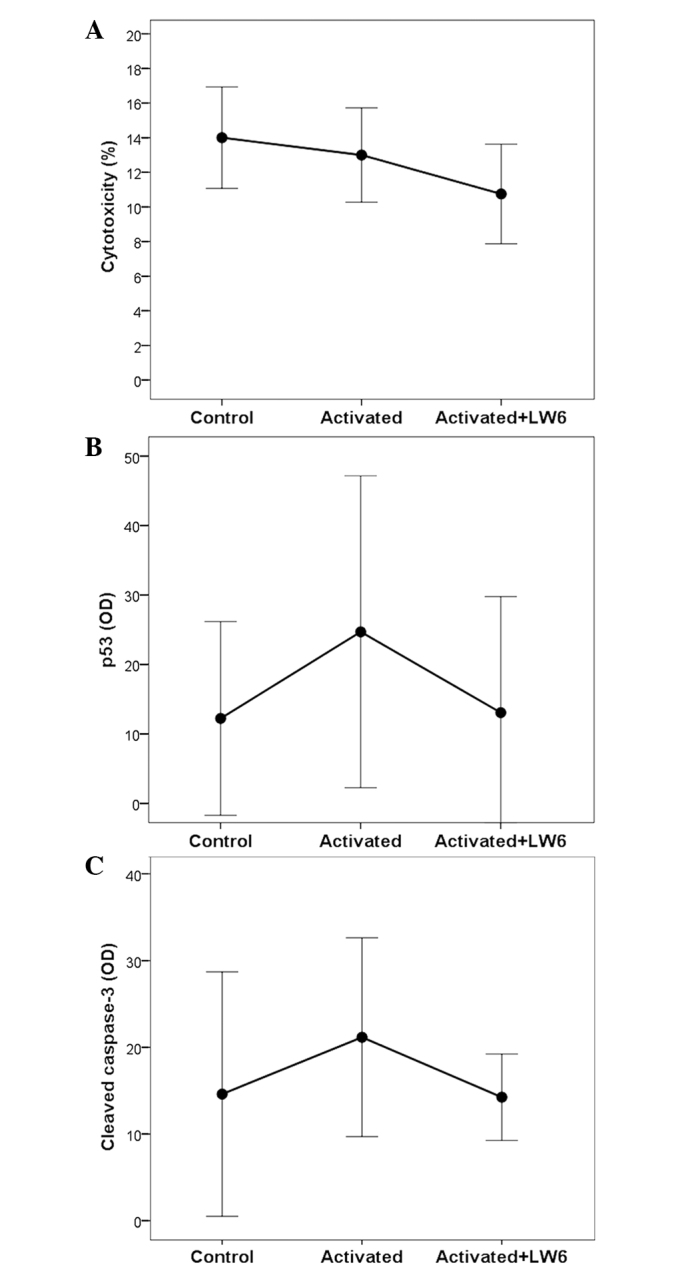
Cytotoxicity and the effect of LW6 on the apoptosis of activated T cells. (A) LW6 was not toxic for the activated T cells. (B) Stimulation increased the level of the pro-apoptotic transcription factor p53, whereas treatment with LW6 decreased the p53 level. (C) Stimulation increased the level of activated cleaved caspase-3, whereas treatment with LW6 resulted in decreased levels. Assays were conducted in control (resting), activated and LW6-treated activated T cells. OD, optical density.
When compared with the unstimulated T cells, stimulation increased the level of the pro-apoptotic transcription factor p53 by a factor of 2.31±0.64 (P<0.001), whereas stimulation combined with LW6 treatment increased the level of p53 by a factor of 1.42±0.86, which was not statistically significant (P=1.000). In stimulated LW6-treated T cells, the level of p53 decreased significantly compared with that of the stimulated T cells (P=0.039; Figs. 1 and 2B).
The level of activated caspase-3, which is central to the execution phase of cell apoptosis, was increased by T-cell stimulation by a factor of 1.58±0.41 compared with that of the unstimulated T cells (P=0.026), whereas T-cell stimulation combined with LW6 treatment increased the level of cleaved caspase-3 by a factor of 1.09±0.29, which was not statistically significant (P=1.000). Consistent with p53, LW6-treated stimulated T cells exhibited a significant reduction in the levels of cleaved caspase-3 (P=0.008) compared with those of stimulated T cells (Figs. 1 and 2C).
LW6 inhibits the proliferation of activated T cells and decreases the level of c-Myc
The BrdU assay revealed a proliferation index of 5.47±0.42 for stimulated T cells. Concurrent treatment with LW6 decreased the proliferation index significantly to 1.56±0.32 (P<0.001; Fig. 3A).
Figure 3.

Effect of LW6 on the proliferation of activated T cells. (A) LW6 inhibits the proliferation of stimulated T cells. (B) Activation increased the level of c-Myc, a transcription factor required for cell proliferation, whereas treatment with LW6 decreased c-Myc levels. Assays were conducted in control (resting), activated and LW6-treated activated T cells. OD, optical density.
The level of c-Myc, a transcription factor required for cell proliferation, was significantly increased by stimulation, rising by a factor of 1.73±0.36 compared with that of unstimulated T cells (P<0.001). Concurrent treatment of T cells with stimulation and LW6 decreased c-Myc levels by a factor of 0.84±0.33, which was statistically insignificant (P=0.527). When the stimulated T cells and LW6-treated stimulated T cells were compared, the c-Myc levels were decreased significantly in the latter cells (P=0.002; Figs. 1 and 3B).
LW6 decreases the levels of HIF1α, GLUT1, HKII, LDH-A and pPDH, while increasing those of PDH
As shown in Figs. 1 and 4A, stimulation increased the level of the transcription factor HIF-1α significantly by a factor of 2.06±0.99 compared with that in the unstimulated T cells (P<0.001). However, concurrent stimulation and treatment of the T cells with LW6 decreased the level of HIF-1α by a factor of 0.59±0.19 (P=0.005). Regarding the levels of HIF-1α in stimulated T cells compared with stimulated LW6-treated T cells, a significant reduction was attained in the latter case (P<0.001).
Figure 4.

Effect of LW6 on HIF-1α, GLUT1 and certain enzymes involved in glucose metabolism in activated T cells. (A) Stimulation of T cells increased the levels of the transcription factor HIF-1α, whereas treatment with LW6 decreased HIF-1α levels. A similar pattern was observed in the case of (B) GLUT1, (C) HKII and (D) LDH-A. (E) Stimulation of T-cells did not affect the level of PDH, whereas treatment with LW6 increased PDH levels. (F) Stimulation of T-cells did not affect the level of pPDH phosphorylated at serine 393, whereas treatment with LW6 returned pPDH to its basal level. HIF-1α, hypoxia-inducible factor-1α; OD, optical density; GLUT1, glucose transporter-1; HKII, hexokinase II; LDH-A, lactate dehydrogenase-A; PDH, pyruvate dehydrogenase; pPDH, PDH phosphorylated at serine 393. Assays were conducted in control (resting), activated and LW6-treated activated T cells.
With regard to the pathway of glycolysis, the stimulation of T cells significantly increased the levels of GLUT1 and HKII by factors of 2.08±1.39 (P<0.048) and 2.43±1.32 (P<0.001), respectively, compared with the respective levels in the unstimulated T cells. Treatment of T cells with stimulation and LW6 resulted in a moderate increase in GLUT1 and HKII levels, by factors of 1.11±0.51 and 1.19±0.84, respectively, neither of which was statistically significant (P=1.000). However, the comparison of stimulated T cells with LW6-treated stimulated T cells indicated that the addition of LW6 markedly reduced GLUT1 and HK2 levels (P=0.002 and P<0.001, respectively; Figs. 1, 4B and C).
With regard to the pathway of glucose conversion, stimulation of T cells increased the levels of LDH-A significantly, by a factor of 4.01±2.92 (P=0.007) compared with those of the unstimulated T cells, while stimulation combined with LW6 exerted a 2.53±1.56-fold increase in LDH-A, which did not reach statistical significance (P=0.085). In the LW6-treated stimulated T cells, the LDH-A levels were significantly decreased compared with those in the stimulated T cells (P=0.026; Figs. 1 and 4D).
In addition to LDH-A, which is responsible for pyruvate conversion to lactate, PDH, which converts pyruvate to acetyl-CoA, was also assessed. No significant difference was observed in the levels of PDH between stimulated and unstimulated cells (1.15±0.36, P=1.000). However, treatment with stimulation and LW6 induced a 5-fold increase in PDH levels (5.34±6.39, P=0.006). In LW6-treated stimulated T cells, the PDH levels were significantly increased compared with those in the stimulated T cells (P=0.002; Figs. 1 and 4E).
Stimulation increased the levels of pPDH significantly by a factor of 1.28±0.19 (P=0.015) compared with those of the unstimulated cells, whereas concurrent treatment of T cells with LW6 and stimulation decreased pPDH levels by a factor of 0.81±0.21, which was statistically insignificant (P=0.137). In the LW6-treated stimulated T cells, the pPDH levels were decreased significantly compared with those in the stimulated T cells (P<0.001; Figs. 1 and 4F).
LW6 decreases the levels of GLS2, while markedly increasing those of GLS1
The effect of T-cell stimulation on the two isoforms of glutaminase, GLS1 and GLS2, was investigated. Stimulation increased the levels of both isoforms 2-fold compared with those of the unstimulated cells (2.03±0.74, P=0.001 and 1.89±0.27, P<0.001, respectively). In the stimulated T cells treated with LW6, GLS1 levels were increased 3-fold (3.25±1.55, P<0.001), whereas GLS2 levels remained comparable to those observed in the unstimulated cells (1.04±0.62, P=1.000). Furthermore, by comparing glutaminase expression in the cell groups that were stimulated in the presence and absence of LW6, it was revealed that LW6 further increased the levels of the GLS1 enzyme (P<0.001; Figs. 1 and 5A), while the expression of the GLS2 isoform significantly declined in the presence of LW6 (P=0.029; Figs. 1 and 5B).
Figure 5.
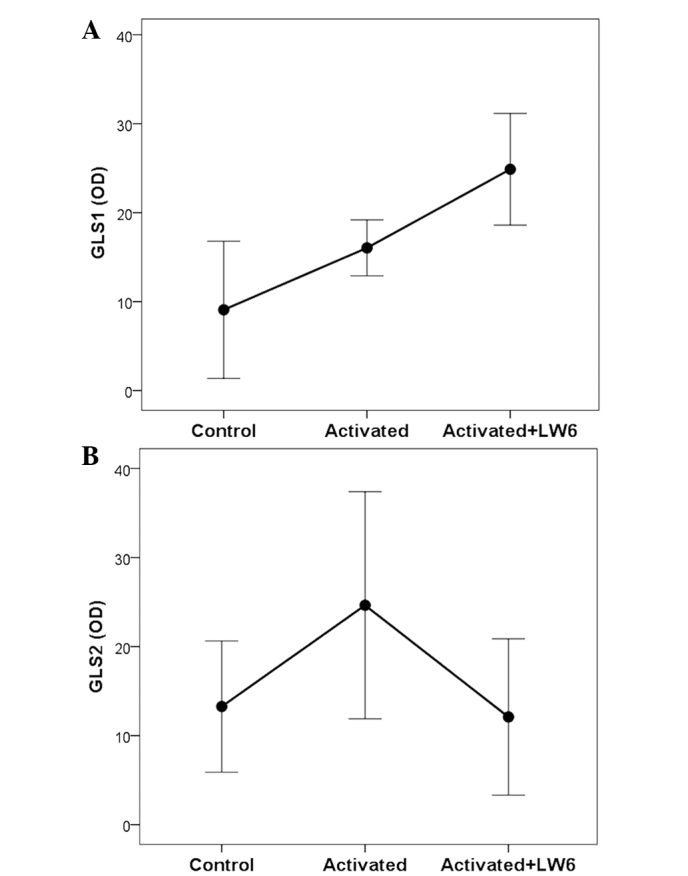
Effect of LW6 on the two glutaminase isoforms in stimulated T cells. (A) Stimulation of T cells increased GLS1 levels, and concurrent treatment with LW6 increased GLS1 levels further. (B) Stimulation of T-cells increased the levels of GLS2, whereas treatment with LW6 decreased GLS2 levels. Assays were conducted in control (resting), activated and LW6-treated activated T cells. GLS1, glutaminase-1; GLS2, glutaminase-2; OD, optical density.
LW6 significantly decreases, but does not completely prevent, Krebs' cycle activity in activated T cells
The XTT assay determined an OD of 60.83±1.72 for the unstimulated control T cells. Activation of the T cells increased the OD significantly to 169.17±12.89 (P<0.001). Adding LW6 to the activated T cells decreased the OD to 113.67±7.94 (P=0.001); however, the value remained significantly higher than that for the control T cells (P<0.001). Hence, in activated T cells LW6 suppresses significantly, but not absolutely, the production of NADH by the Krebs' cycle (Fig. 6).
Figure 6.

Effect of LW6 on Krebs' cycle activity in stimulated T cells. Assessment of NADH production by the Krebs' cycle via XTT assay revealed that stimulation of T cells increased Krebs' cycle activity. Treatment with the MDH2 inhibitor LW6 decreased Krebs' cycle activity, which however remained at a markedly higher level compared with that of unstimulated control T-cells. Assays were conducted in control (resting), activated and LW6-treated activated T cells. MDH2, malate dehydrogenase-2; NADH, nicotinamide adenine dinucleotide; XTT, sodium 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium; OD, optical density.
Discussion
In the present study, the effects of the MDH2 inhibitor LW6 on proliferation, apoptosis and the expression levels of certain enzymes of glucose metabolism and glutaminolysis were evaluated in the context of activated T cells. Specific enzyme and glucose transporter isoforms that are known to be upregulated upon T-cell activation were selected for evaluation (1) and Krebs' cycle activity was assessed.
It is known that following stimulation of T cells their proliferation and differentiation to effector cells are coincident with an increase of HIF-1α (1,13). In the present study, the treatment of stimulated T cells with LW6 decreased HIF-1α levels. It has been demonstrated in a cell type other than that employed in the present study that the inhibition of the Krebs' cycle and consequently of oxidative phosphorylation by LW6 results in decreased intracellular O2 consumption and increased HIF-1α degradation (10).
The proliferation of stimulated T cells in the present study was accompanied by increased levels of c-Myc, which is expected because c-Myc is upregulated once T-cell receptor activation is initiated and is required for cell proliferation (1,14). Treatment of stimulated T cells with LW6 decreased c-Myc levels and proliferation. Although it is known that c-Myc may increase HIF-1α levels, whether HIF-1α has a reciprocal effect has not yet been confirmed (15–17). In cell types other than that employed in the present study, it has been demonstrated that HIF-1α decreases c-Myc expression or function (18–20). The mechanisms involved in the observed LW6-induced reduction of c-Myc activity remain to be elucidated.
Treatment of stimulated T cells with LW6 revealed no signs of toxicity, suggesting that this compound may possess pharmacological potential. Notably, stimulation of T cells increased the expression of the pro-apoptotic transcription factor p53. Increased HIF-1α may contribute to p53 upregulation via a number of pathways (21), and may be an intrinsic cell mechanism for controlling proliferation. In primary embryo fibroblasts c-Myc activates the p19ARF-Mdm2-p53 tumor suppressor pathway (22). The reduction of p53 levels in stimulated LW6-treated T cells may be explained by the reductions in the HIF-1α and c-Myc levels that LW6 induces. Activated cleaved caspase-3 is the central caspase in the execution phase of cell apoptosis and consequently its level may be used as a marker of apoptosis. The fluctuation of cleaved caspase-3 levels, which occurs concurrently and with the same trend as the changes in pro-apoptotic p53 levels indicates that decreased p53 levels may be responsible for reduced apoptosis in LW6-treated stimulated T cells (23,24).
Regarding glucose metabolism, stimulation of T cells increased GLUT1 and HKII levels, increasing glucose influx into the cells and accelerating glycolysis, which is required for these rapidly proliferating cells. Treatment of stimulated T cells with LW6 decreased GLUT1 and HKII levels and thus inhibited glucose influx and decelerated glycolysis. The decreased levels of HIF-1α and c-Myc in LW6-treated T cells may be responsible for the reduced GLUT1 and HKII levels, as these transcription factors control their expression (1,25–27).
Once formed, pyruvate may be further converted to lactate by LDH-A. In the stimulated T cells in the present study, LDH-A levels increased markedly; the effect of this would be to enhance the conversion of pyruvate to lactate, the aerobic glycolysis that characterizes activated T cells. Increased HIF-1α and c-Myc levels may be responsible for LDH-A upregulation, as these transcription factors control the expression of this enzyme (1,25,28,29). Treatment of stimulated T cells with LW6 decreased the levels of LDH-A, which may be attributable to the reduced levels of HIF-1α and c-Myc in the LW6-treated T cells.
An alternative pathway that pyruvate formed by glycolysis may take is conversion to acetyl-CoA by PDH and entry into the more energetically efficient Krebs' cycle. PDH activity is controlled by PDK, which phosphorylates and inactivates it (30). In the stimulated T cells in the present study, the PDH levels remained unaffected. Although it is known that HIF-1α upregulates PDK and inactivates PDH (31), T-cell stimulation moderately increased pPDH levels. The increased influx of glucose and the consequently increased pyruvate production in stimulated T cells, in addition to the moderate increase of pPDH, may explain the increased Krebs' cycle activity observed in activated T cells in the present study. Treatment of stimulated T cells with LW6 moderately decreased pPDH levels, while increasing the levels of PDH. The reduction of HIF-1α levels observed in LW6-treated T cells may be responsible for the reduced pPDH levels (30), whereas the cause of the observed PDH upregulation remains to be elucidated. Notably, the XTT assay revealed that treatment of stimulated T cells with LW6 decreased Krebs' cycle activity, apparently as a result of the direct inhibition of a single step of the cycle, although decreased glucose influx may contribute. However, Krebs' cycle activity in LW6-treated stimulated T cells was not completely abrogated, and remained markedly increased compared with that of unstimulated T cells. It is possible that by increasing PDH expression, LW6 facilitates the entry of pyruvate into the Krebs' cycle, which is energetically more effective compared with the conversion of pyruvate to lactate. This may be useful in the context of the low intracellular glucose conditions induced by LW6.
The process by which sufficient Krebs' cycle activity is maintained to prevent energy collapse in LW6-treated T cells remains unclear. In addition, the mechanism by which the obstacle set by the MDH2 inhibitor is bypassed requires clarification. A potential explanation involves the stage at which the Krebs' cycle is inhibited. The accumulated malate resulting from MDH2 inhibition may be shuttled from the mitochondria into the cytoplasm, converted to pyruvate by malic enzyme and then re-enter into the mitochondria and the Krebs' cycle. Pyruvate-malate cycling was initially described in liver cells (11), and subsequently in various primary cell types from fibroblasts to B cells, in addition to cancer cells (32–34). Notably, in activated T cells, malic enzyme is upregulated (1). Thus, the pyruvate-malate cycle may prevent total cell energy collapse and apoptosis following the treatment of stimulated T cells with LW6.
Another possible source of energy for cells under conditions of low intracellular glucose is glutaminolysis. The initial enzymes in this pathway are the two isoforms of glutaminase, GLS1 and GLS2. Glutaminases convert glutamine to glutamate. Glutamate is converted to α-ketoglutarate, which may enter into Krebs' cycle a number of steps prior to the stage at which MDH2 intervenes (35). In stimulated T cells in the present study, GLS1 and GLS2 were upregulated and favor glutaminolysis, which is known to occur in these rapidly proliferating cells (1,36). Increased levels of c-Myc may be responsible for GLS1 upregulation (37), whereas increased c-Myc and p53 levels may be responsible for enhanced GLS2 expression (1,38,39). Treatment of stimulated T cells with LW6 decreased GLS2 levels, which may be attributed to the reduced levels of c-Myc and p53. However, glutaminolysis may be preserved in LW6-treated T cells as a result of the markedly increased GLS1 levels. This is controversial, as decreased c-Myc is expected to downregulate GLS1 transcription. Alternative mechanisms, such as anaphase-promoting complex/cyclosome (APC/c)-cdh1-mediated GLS1 degradation, remain to be evaluated (36). However, it remains possible that the preservation of glutaminolysis in low intracellular glucose conditions may rescue the cell from energy collapse.
In conclusion, the inhibition of MDH2 in human activated T cells abrogates proliferation without adversely affecting cell survival. Adaptations of cellular glucose and glutamine metabolism may prevent energy collapse. Further studies involving interference with T-cell metabolism may reveal novel drug targets, which could include MDH2.
References
- 1.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev. 2012;249:14–26. doi: 10.1111/j.1600-065X.2012.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostroukhova M, Goplen N, Karim MZ, Michalec L, Guo L, Liang Q, Alam R. The role of low-level lactate production in airway inflammation in asthma. Am J Physiol Lung Cell Mol Physiol. 2012;302:L300–L307. doi: 10.1152/ajplung.00221.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Antoniadis N, Liakopoulos V, Stefanidis I. Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. J Basic Clin Physiol Pharmacol. 2013;24:271–276. doi: 10.1515/jbcpp-2013-0001. [DOI] [PubMed] [Google Scholar]
- 6.Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Liakopoulos V, Dafopoulou K, Pournaras S, Koukoulis G, Stefanidis I. The indoleamine 2,3-dioxygenase inhibitor 1-methyl-tryptophan suppresses mitochondrial function, induces aerobic glycolysis and decreases interleukin-10 production in human lymphocytes. Immunol Invest. 2012;41:507–520. doi: 10.3109/08820139.2012.682244. [DOI] [PubMed] [Google Scholar]
- 7.Eleftheriadis T, Pissas G, Yiannaki E, Markala D, Arampatzis S, Antoniadi G, Liakopoulos V, Stefanidis I. Inhibition of indoleamine 2,3-dioxygenase in mixed lymphocyte reaction affects glucose influx and enzymes involved in aerobic glycolysis and glutaminolysis in alloreactive T-cells. Hum Immunol. 2013;74:1501–1509. doi: 10.1016/j.humimm.2013.08.268. [DOI] [PubMed] [Google Scholar]
- 8.Eleftheriadis T, Pissas G, Antoniadi G, Spanoulis A, Liakopoulos V, Stefanidis I. Indoleamine 2,3-dioxygenase increases p53 levels in alloreactive human T cells and both indoleamine 2,3-dioxygenase and p53 suppress glucose uptake, glycolysis and proliferation. Int Immunol. 2014;26:673–684. doi: 10.1093/intimm/dxu077. [DOI] [PubMed] [Google Scholar]
- 9.Lee K, Kang JE, Park SK, Jin Y, Chung KS, Kim HM, Lee K, Kang MR, Lee MK, Song KB, et al. LW6, a novel HIF-1 inhibitor, promotes proteasomal degradation of HIF-1alpha via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol. 2010;80:982–989. doi: 10.1016/j.bcp.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 10.Lee K, Ban HS, Naik R, Hong YS, Son S, Kim BK, Xia Y, Song KB, Lee HS, Won M. Identification of malate dehydrogenase 2 as a target protein of the HIF-1 inhibitor LW6 using chemical probes. Angew Chem Int Ed Engl. 2013;52:10286–10289. doi: 10.1002/anie.201304987. [DOI] [PubMed] [Google Scholar]
- 11.Friedman B, Goodman EH, Jr, Saunders HL, Kostos V, Weinhouse S. Estimation of pyruvate recycling during gluconeogenesis in perfused rat liver. Metabolism. 1971;20:2–12. doi: 10.1016/0026-0495(71)90055-2. [DOI] [PubMed] [Google Scholar]
- 12.Berridge MV, Herst PM, Tan AS. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol Annu Rev. 2005;11:127–152. doi: 10.1016/S1387-2656(05)11004-7. [DOI] [PubMed] [Google Scholar]
- 13.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, Zhang H, Huppa JB, Tsai YH, Lobry C, et al. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol. 2013;14:262–270. doi: 10.1038/ni.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doe MR, Ascano JM, Kaur M, Cole MD. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012;72:949–957. doi: 10.1158/0008-5472.CAN-11-2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Cai S, Wang G, Cao X, Yang X, Luo X, Feng Y, Hu J. c-Myc enhances colon cancer cell-mediated angiogenesis through the regulation of HIF-1α. Biochem Biophys Res Commun. 2013;430:505–511. doi: 10.1016/j.bbrc.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Podar K, Anderson KC. A therapeutic role for targeting c-Myc/Hif-1-dependent signaling pathways. Cell Cycle. 2010;9:1722–1728. doi: 10.4161/cc.9.9.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong KS, Park JI, Kim MJ, Kim HB, Lee JW, Dao TT, Oh WK, Kang CD, Kim SH. Involvement of SIRT1 in hypoxic down-regulation of c-Myc and β-catenin and hypoxic preconditioning effect of polyphenols. Toxicol Appl Pharmacol. 2012;259:210–218. doi: 10.1016/j.taap.2011.12.025. [DOI] [PubMed] [Google Scholar]
- 19.Huang LE. Carrot and stick: HIF-alpha engages c-Myc in hypoxic adaptation. Cell Death Differ. 2008;15:672–677. doi: 10.1038/sj.cdd.4402302. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Kluz T, Sun H, Costa M. Mechanisms of c-myc degradation by nickel compounds and hypoxia. PloS One. 2009;4:e8531. doi: 10.1371/journal.pone.0008531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164. doi: 10.1038/cddis.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brady CA, Attardi LD. p53 at a glance. J Cell Sci. 2010;123:2527–2532. doi: 10.1242/jcs.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 27.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valera A, Pujol A, Gregori X, Riu E, Visa J, Bosch F. Evidence from transgenic mice that myc regulates hepatic glycolysis. FASEB J. 1995;9:1067–1078. doi: 10.1096/fasebj.9.11.7649406. [DOI] [PubMed] [Google Scholar]
- 29.Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc Natl Acad Sci USA. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem. 2001;276:37223–37229. doi: 10.1074/jbc.M103069200. [DOI] [PubMed] [Google Scholar]
- 31.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006;2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56:414–424. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mates JM, Segura JA, Martin-Rufián M, Campos-Sandoval JA, Alonso FJ, Marquez J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med. 2013;13:514–534. doi: 10.2174/1566524011313040005. [DOI] [PubMed] [Google Scholar]
- 36.Colombo SL, Palacios-Callender M, Frakich N, De Leon J, Schmitt CA, Boorn L, Davis N, Moncada S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc Natl Acad Sci USA. 2010;107:18868–18873. doi: 10.1073/pnas.1012362107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:7461–7466. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]