

ABSTRACT
The membrane-proximal region of murine leukemia virus envelope (Env) is a critical modulator of its functionality. We have previously shown that the insertion of one amino acid (+1 leucine) within the membrane-spanning domain (MSD) abolished protein functionality in infectivity assays. However, functionality could be restored to this +1 leucine mutant by either inserting two additional amino acids (+3 leucine) or by deleting the cytoplasmic tail domain (CTD) in the +1 leucine background. We inferred that the ectodomain and CTD have protein interfaces that have to be in alignment for Env to be functional. Here, we made single residue deletions to the Env mutant with the +1 leucine insertion to restore the interface alignment (gain of functionality) and therefore define the boundaries of the two interfaces. We identified the glycine-proline pairs near the N terminus (positions 147 and 148) and the C terminus (positions 159 and 160) of the MSD as being the boundaries of the two interfaces. Deletions between these pairs restored function, but deletions outside of them did not. In addition, the vast majority of the single residue deletions regained function if the CTD was deleted. The exceptions were four hydroxyl-containing amino acid residues (T139, T140, S143, and T144) that reside in the ectodomain interface and the proline at position 148, which were all indispensable for functionality. We hypothesize that the hydroxyl-containing residues at positions T139 and S143 could be a driving force for stabilizing the ectodomain interface through formation of a hydrogen-bonding network.
IMPORTANCE The membrane-proximal external region (MPER) and membrane-spanning domains (MSDs) of viral glycoproteins have been shown to be critical for regulating glycoprotein fusogenicity. However, the roles of these two domains are poorly understood. We report here that point deletions and insertions within the MPER or MSD result in functionally inactive proteins. However, when the C-terminal tail domain (CTD) is deleted, the majority of the proteins remain functional. The only residues that were found to be critical for function regardless of the CTD were four hydroxyl-containing amino acids located at the C terminus of the MPER (T139 and T140) and at the N terminus of the MSD (S143 and T144) and a proline near the beginning of the MSD (P148). We demonstrate that hydrogen-bonding at positions T139 and S143 is critical for protein function. Our findings provide novel insights into the role of the MPER in regulating fusogenic activity of viral glycoproteins.
INTRODUCTION
Enveloped viruses require membrane-spanning cell surface glycoproteins to coordinate fusion between viral and host cell membranes. Retroviral envelope (Env) glycoproteins are produced as precursors that undergo trimerization in the endoplasmic reticulum (1–3). Subsequently, the precursor trimer is cleaved into two subdomains: the receptor-binding surface domain (SU) and the fusion-promoting transmembrane domain (TM). This cleavage process is mediated by a host-cell furin, or furin-like protease (4, 5). In the case of alpha-, gamma-, and deltaretroviruses, SU and TM are covalently linked through a single disulfide bond (6–13), whereas lentiviruses and betaretroviruses are noncovalently associated (14, 15).
In the case of murine leukemia virus (MLV) Env, fusogenicity is tightly controlled by a short, 16-amino-acid peptide (R-peptide) in the cytoplasmic tail domain (CTD), which negatively regulates fusogenicity (16–21). Cleavage of the R-peptide by the viral protease during or shortly after viral assembly potentiates receptor-mediated isomerization of the disulfide bond between SU and TM, subsequently activating fusogenic activity (16–18, 21, 22). Recent cryo-electron microscopy (EM) reconstruction data show that the ectodomain of fusogenically inactive full-length MLV Env is held in a tight conformation in the presence of the R peptide. However, fusogenically active (ΔR peptide) Env reconstructions depict a structure wherein the TM legs are splayed (23). Thus, R peptide cleavage is thought to potentiate fusogenic activation through molecular rearrangements in the membrane-spanning domain (MSD) and in the extracellular region of MLV Env. Similarly, the membrane-proximal external region (MPER) and MSD of HIV-1 Env have been shown to be crucial for promoting fusogenicity. Disruption of hinge regions or the tryptophan-rich sequence in the MPER of HIV-1 Env, as well as polar residues within the MSD, all result in nonfunctional glycoproteins that are fusogenically inactive (24–29). The potential role for the MSDs of MLV and HIV-1 Env in potentiating molecular rearrangements necessary for fusogenic activity would be analogous to the MSDs of voltage-gated ion channels and G-protein-coupled receptors (GPCRs). Conformational changes in the transmembrane domains of GPCRs allows for transmission of signals from the extracellular space to the intracellular environment (30–34).
Recently, we demonstrated that leucine insertions in the MSD of MLV Env have a significant effect on glycoprotein functionality (35). Insertion of one (+1L) or two (+2L) leucines in the MSD abrogated function, whereas insertion of three (+3L) leucines restored transduction capacity to levels comparable to wild-type Env (35). Interestingly, when the CTD was truncated by 23 amino acids or more in the +1L background, the transduction capacity was restored. Further study revealed that the loss of transduction capacity in the +1L Env mutant was due to disruption of the fusion mechanism and that +3L restored fusogenicity and became constitutively active (35). All leucine insertion mutants were incorporated into viral particles at levels similar to that of the wild type, and all were processed properly into SU and TM domains in cell lysates (35). In agreement with a previous study (36), we propose that the CTD forms a coiled-coil interface and that if it is out of phase with the trimer interface of the ectodomain it results in disruption of Env functionality. This hypothesis is supported by the cryo-electron microscopy reconstruction data that suggest the CTD interface holds the glycoprotein in a tight conformation. Therefore, the +1L insertion would distort the helical orientation of the ectodomain and CTD by approximately 103°, whereas the insertion of +3L would restore the orientation by adding almost a complete α-helical turn (Fig. 1). Restoration of function of the +1L Env in the context of the CTD truncation suggested that removal of the CTD interface eliminated the alignment conflict with the ectodomain, allowing for restoration of function. Here, we sought to define the borders of these two interfaces by identifying gain-of-function mutations that restored Env functionality in the +1L background.
FIG 1.

Diagram of predicted trimer interfaces. (Left) Depiction of predicted ectodomain and CTD trimer interfaces highlighted as white segments within the trimer core of wild-type MLV Env. (Middle) Depiction of the +1L insertion within the MSD and the resulting disruption of the CTD interface by ∼103°. (Right) Depiction of the +3L insertions within the MSD and the subsequent restoration of orientation of the CTD interface. The introduction of three leucines nearly restores alignment of the CTD interface within the trimer core.
MATERIALS AND METHODS
Plasmids and cell culture.
The amino acid deletions and residue substitutions were created by using oligonucleotide-mediated mutagenesis. Constructs expressing the truncated version of MLV Env were created by introducing a stop codon after RLVQFVK, which removes 25 residues from the CTD. Unless indicated, all MLV Env expression constructs were cytomegalovirus (CMV) promoter driven and contained a hemagglutinin (HA) tag (YPYDVPDYA) in a proline-rich region in SU (35). For creation of stable cell lines, indicated deletion mutants with the truncated CTD and +1 leucine insertion were cloned into a retroviral vector which contained a puromycin gene upstream of the MLV Env coding region driven by a CMV promoter. To ensure each protein was expressed independently, a self-cleaving T2A peptide was inserted between the open reading frames of the puromycin and MLV Env genes (37, 38). This construct also contained a green fluorescent protein (GFP) tag inserted into a proline-rich region in SU (39). For all HIV-1 infectivity experiments, an NL4-3-derived HIV-CMV-GFP proviral vector, defective for Vif, Vpr, Vpu, Nef, and Env, was used (Vineet Kewal-Ramani, National Cancer Institute). This construct has a CMV immediate-early promoter driving a GFP reporter in place of Nef. The gene encoding the tTA (tet-off) protein was produced from a CMV expression vector. The TRE-driven Gaussia luciferase (Gluc)-inducible expression system was created by introduction of the Gluc gene into the Retro-Tight-X-Hygro retroviral transfer vector (Clontech). MLV infectivity was carried out using the MLV packaging construct CMV-MLV-GagPol (Walter Mothes, Yale University) and reporter plasmid pQCXIP-GFP.
HEK-293FT (Invitrogen), 293T mCAT-1 (Walter Mothes, Yale University), and 293T mCAT-1 cells stably expressing Gluc under the control of a tet-off driven (TRE) promoter (35) were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 1 mM sodium pyruvate, 10 mM nonessential amino acids, and 1% minimal essential medium vitamins.
Infectivity assay.
For HIV-1 infectivity experiments, 293FT cells were transfected in a six-well plate with 500 ng of HIV-CMV-GFP and 500 ng of Env expression plasmid. For MLV infectivity experiments, cells were transfected with 300 ng of CMV-MLV-GagPol, 200 ng of pQCXIP-GFP, and 500 ng of Env expression plasmid. Transfections were performed using 3 μg of polyethyleneimine (PEI) per μg of DNA (40). The medium was changed at 6 to 12 h posttransfection to remove residual transfection reagent. Supernatant medium was collected 24 h after medium exchange and frozen at −80°C for at least 3 h to lyse any cells contained within the supernatant medium. After the supernatants were thawed at 37°C in a water bath, the samples were centrifuged at 1,500 × g for 5 min to pellet any cellular debris. Then, 500 μl of the supernatant was added to target cells for 48 h. Cells were collected at 48 h, fixed with 4% paraformaldehyde, and analyzed using an Accuri C6 flow cytometer.
Western blotting.
293FT cells stably expressing the indicated Env proteins were transfected with 500 ng of HIV-CMV-GFP plasmid in a six-well plate using the procedure described the for infectivity assays. Viral samples were pelleted through a 20% sucrose cushion for 2 h at 20,000 × g at 4°C. Residual supplement medium and sucrose were aspirated off the sample pellet, and samples were resuspended in 6× SDS-PAGE loading buffer. The equivalent of 1 ml of viral supernatant was analyzed by 10% discontinuous SDS-PAGE. Cell samples were detached using 10 mM EDTA/phosphate-buffered saline (PBS) solution and pelleted at 500 × g for 10 min. Pellets were resuspended in radioimmunoprecipitation assay buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl), and then 5 to 10% of the lysate was combined with 6× SDS-PAGE loading buffer and analyzed by 10% discontinuous SDS-PAGE. Proteins were transferred onto a 0.45-μm-pore-size polyvinylidene difluoride membrane. The membrane was blocked with 5% nonfat dried milk in PBS containing Tween 20 (PBS-T) and probed with rabbit anti-GFP antibody diluted 1:5,000 (Sigma) and mouse anti-HIV p24 hybridoma medium diluted 1:500 (AIDS Research and Reference Program, Division of AIDS, National Institute of Allergy and Infectious Disease, National Institutes of Health; HIV-1 p24 hybridoma [183-H12-5C]). Primary antibody incubations were performed overnight, on a shaker, at 4°C. Blots were washed with PBS-T and then probed with horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse antibodies diluted 1:10,000 (Sigma) for probing Env and p24, respectively. Visualization of the membranes was performed using Luminata Classico and Cresendo Western HRP chemiluminescence reagents. Imaging was performed using a LAS3000 image analyzer from Fujifilm.
Cell-to-cell fusion assay.
293FT cells stably expressing the indicated Env protein were transfected with 500 ng of tet-off expression plasmid in a six well plate. AT 24 h after transfection, the cells were cocultured with an equal number of 293T mCAT-1 TRE Gluc cells for 48 h (35). Then, 20-μl portions of sample supernatant from the cocultured cells were assayed in duplicate for Gluc content with 50 μl of 10 μM coelenterazine in 0.1 M Tris (pH 7.4) and 0.3 M sodium ascorbate.
Surface labeling.
293FT cells stably expressing the indicated Env protein were detached using 10 mM EDTA-PBS. Cells were centrifuged at 500 × g for 10 min at 4°C and resuspended in 1% bovine serum albumin-PBS blocking solution for 20 min. After 20 min of blocking, the cells were centrifuged at 500 × g for 10 min at 4°C and resuspended in 10 mM EDTA-PBS, 1% goat serum, and 1:1,000 primary anti-GFP Alexa-Fluor 647 antibody (Life Technologies) for 1 h. After primary antibody incubation, the cells were centrifuged at 500 × g for 10 min at 4°C, resuspended in 4% paraformaldehyde, and fixed for 20 min. The cells were then centrifuged at 500 × g for 10 min at 4°C, resuspended in PBS, and analyzed using an Accuri C6 flow cytometer.
Statistical analysis.
Each experiment was performed at least three independent times with similar results. All statistics and graphs were prepared using GraphPad Prism4 software.
RESULTS
Identification of interface boundaries.
We observed previously that proper helical orientation of MLV Env trimers is crucial for functionality (35). Disruption of the spacing between the ectodomain and CTD caused by amino acid insertions within the MSD resulted in loss of transduction capacity (35). We hypothesized that we could determine the boundaries of these interfaces by identifying gain-of-function mutations that restored helical alignment and, thus, function. In our previous study, we demonstrated that the insertion of a leucine (+1L) between positions L154 and L155 in the MSD resulted in a nonfunctional glycoprotein (Fig. 1) (35). If our hypothesis is correct and we perform a deletion scan across the indicated regions of the ectodomain, MSD, and CTD (positions 135 to 176) in the context of full-length +1L Env, we could identify residues that restore transduction capacity (Fig. 2A). If a residue is located between the ectodomain and CTD interfaces, removing it would offset the addition of the single leucine and restore transduction capacity; however, if the residue is positioned within or beyond the interfaces themselves, a single residue deletion would not be expected to restore functionality because it would not be able to restore alignment, or it could potentially remove a critical residue necessary for forming the interface itself. Furthermore, if the CTD interface is destroyed, whether through truncation of the CTD or deletion of critical interfacial residues, we would also expect to see restoration of function because conflicting interfaces would no longer exist. As shown in Fig. 2B, deletion of residues in the +1L Env (HA-tagged) background upstream of P148, which is positioned within the MSD, did not restore transduction capacity. However, deletion of residues between P148 and P160 restored some or all functionality compared to the +1L Env control. Further, deletion of residues positioned downstream of P160, within the CTD, generally did not restore function. The exceptions were deletion of amino acids K171 or D172 in the CTD which partially restored transduction capacity. We have previously shown that point mutations in this region of the CTD could partially restore function to a protein with the +1L in the MSD (35). Therefore, it is reasonable to infer that these residues are critical to the CTD interface and that deleting them is functionally similar to making truncations in the CTD (35).
FIG 2.

Infectivity of MLV Env deletion scan in the +1L background. (A) Diagram of the MPER, MSD, and CTD in MLV Env targeted for the −1 deletion scan. The arrow indicates the insertion of +1L within the MSD. The predicted α-helical structure of this region is depicted below (secondary structure prediction constructed using Phyre2 protein fold recognition software). (B) The infectivity of the full-length Env with +1L insertion at position 154 is shown as a control, and its insertion position within the membrane-spanning domain is indicated with an arrow. All filled bars are full-length (HA-tagged) Env with the +1L insertion and the indicated residue deleted. The hashed open bar is full-length wild-type Env shown as a positive control (5.6 × 105 IU/ml). All data are normalized relative to the wild-type HA-tagged Env control. The dashed line shown across the data indicates the relative level of infectivity of the +1L Env. The data shown here are the averages of three independent experiments.
Insertion of +3 leucine residues outside of the glycine-proline pairs do not recover function.
If our hypothesis is correct and the glycine-proline pairs designated the end and beginning of the ectodomain and CTD interfaces, respectively, then +3 leucine insertions upstream or downstream of these glycine-proline pairs would not recover function. To test this, we introduced +1, +2, or +3 leucines before, between, and after the glycine-proline pairs and tested for transduction capacity (Fig. 3). As we found previously, insertion of +1 or +2 leucines between the glycine-proline pairs in the MSD abrogates functionality; however, a construct with +3 leucines in the same region remains functional (Fig. 3; MSD insertions). However, introduction of +1, +2, or +3 leucines to the N terminus or the C terminus of the glycine-proline pairs (G147/P148 and G159/P160) abrogated function and did not display retention of function when +3 leucines were added (Fig. 3; N-MSD and CTD insertions). These data suggest that the region between G147/P148 and G159/P160 does not have an interface critical for function and can therefore tolerate the introduction of a turn of an α-helix.
FIG 3.

Leucine insertions upstream and downstream of the respective glycine-proline pairs do not display a phasing effect. (Top) Diagram of the leucine insertions upstream of the glycine-proline pair positioned at the N terminus of the membrane-spanning domain and downstream of the glycine-proline pair positioned at the C terminus of the membrane-spanning domain. Leucine insertions in the middle of the MSD, between the glycine-proline pairs, are shown as a control. (Bottom) Infectivity of leucine insertion mutants shown relative to wild-type MLV Env (HA tag, 4.9 × 105 IU/ml). The data shown in this figure are the average of three independent experiments.
Discovery of residues in the MPER and MSD crucial for functionality.
We previously showed that deletion of the CTD restored functionality to MLV Env mutants with both +1L and +2L MSD insertions (35). Therefore, we sought to determine which, if any, of the residues within the MPER and MSD were crucial for function even in the absence of the complete CTD interface. To accomplish this, we made truncation mutants lacking the final 25 residues of the cytoplasmic tail (Δ25CTD) to a subset of the clones analyzed in Fig. 2. Previously, we demonstrated that removal of the 25 C-terminal residues in the CTD restored function to the +1L Env mutant (35). The mutants with deletions between P148 and L157 were not tested because these clones were already functional with the full-length CTD. Truncation of the CTD resulted in restoration of function for all of the deletion mutants examined except for the hydroxyl-containing amino acids T139, T140, S143, T144, and P148 (Fig. 4). If the CTD had an interfacial surface, making large truncations in this region would eliminate the conflict between trimer interfaces because only the MPER interface would remain intact. As expected, all of the deletion residues in the CTD displayed restored transduction capacity when the last 25 amino acids were removed. To ensure that the phenotypes observed in Fig. 2 and 4 were not due to pseudotyping MLV Env mutants with HIV particles, we directly tested transduction capacity of a panel of full-length and Δ25CTD deletion mutants comparing MLV to HIV cores. The observed trend was the same between MLV and HIV cores for all full-length and Δ25CTD deletion mutants tested (data not shown).
FIG 4.

Truncation of the CTD restores abrogated infectivity of certain deletion mutants. The CTD of the specified deletion mutants, in the context of the +1L glycoprotein background, was truncated (Δ25CTD), and the infectivity was assessed. Filled bars indicate full-length glycoprotein with +1L at position 154 along with indicated single amino acid deletions. Open bars indicate the same glycoproteins with the last 25 residues of the CTD deleted. The hashed open bar is wild-type Env Δ25CTD (HA tagged) shown as a positive control (3.0 × 105 IU/ml). The dashed line between positions P148 and L157 indicates that these residues were not tested in this experiment. All data are normalized relative to the wild-type Env Δ25CTD control. The data shown here are the averages of three independent experiments.
Hydroxyl-containing residues in the MSD are crucial for fusogenicity.
Our finding that P148 was crucial for function was expected since this residue most likely caps the end of the N-terminal interface and acts as a molecular hinge within the trimer; however, it was unexpected that the hydroxyl residues were also critical amino acids in the MPER and the MSD. The loss of functionality resulting from deletion of these residues could be caused by many factors, therefore, we focused on determining the source of the defect. To avoid inconsistencies in the expression levels of Env, we stably expressed the hydroxyl-residue deletion mutants with CTD truncations in 293FT cells. We also engineered a green fluorescent protein (GFP) tag in the variable proline-rich region of SU for these constructs. It has been shown that introduction of GFP, or other sequences, into this proline-rich variable region in SU is well tolerated with no discernible effects on Env function (39, 41, 42). In addition, deletions, insertions, or substitutions in, or near, the MSD of Env can result in improper processing and aberrant trafficking (42). The GFP tag allowed for visual confirmation of mutant Env expression and provided a conformation-independent epitope for surface expression experiments. Surface expression studies revealed that all of the truncated hydroxyl deletion mutants in the +1L background were expressed on the surface at similar levels as the truncated wild-type Env positive control (Fig. 5A).
FIG 5.
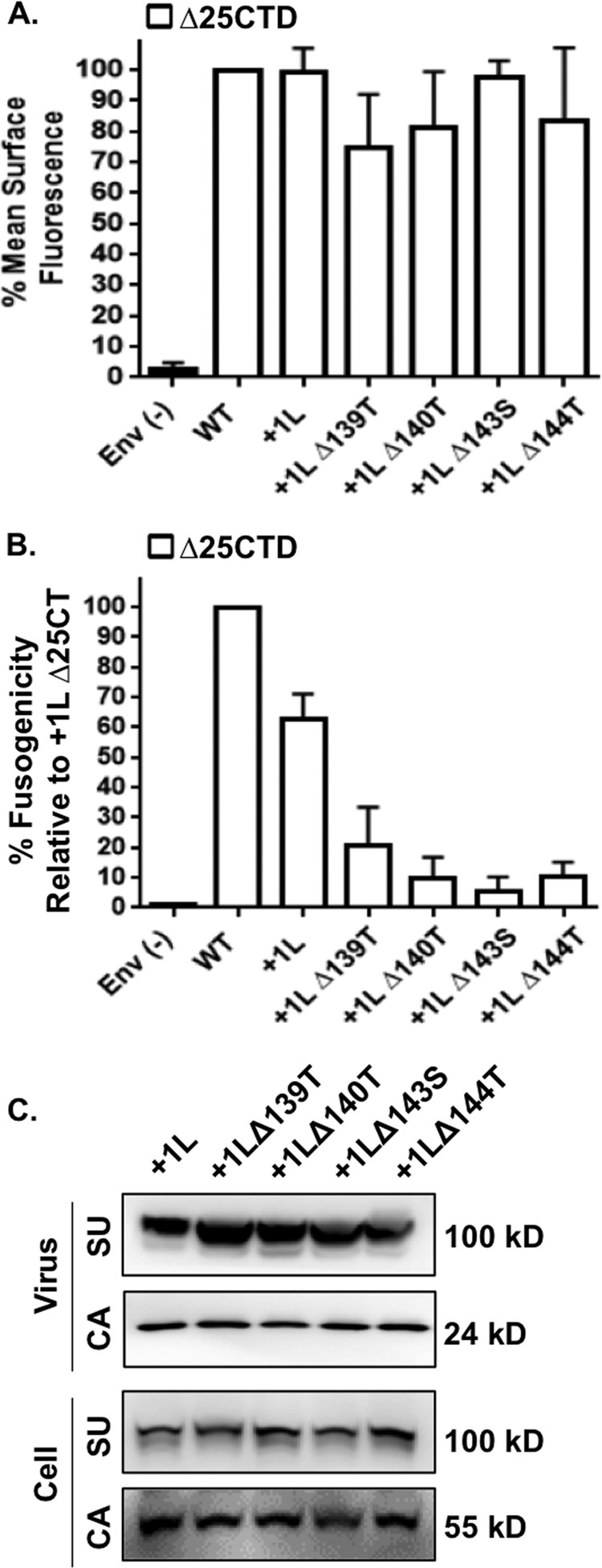
The hydroxyl-containing amino acids in the MPER and MSD are crucial for promoting cell-to-cell fusion. (A) Stable cell lines were created in 293FT cells expressing constructs for the indicated Env mutants (GFP tagged). Cells were surface labeled with an anti-GFP Alexa Fluor 647 antibody (Sigma) and analyzed via flow cytometry. The mean fluorescence intensity was normalized to the WT Δ25CTD control. 293FT cells not expressing MLV Env were included as a negative control in the labeling process and are indicated as Env(−). (B) Stable cell lines were assayed for cell-to-cell fusogenicity. Stable cells lines were transfected with a tet-off expression plasmid and cocultured with a permissive TRE-Gluc cell line. The results are depicted as relative light units normalized to the WT Δ25CTD control. 293FT cells not expressing MLV Env were transfected with the tet-off expression plasmid and were included in the cell-to-cell fusion assay as a negative control [Env(−)]. (C) Western blot analysis of Env incorporation into viral particles. Stable cell lines were transfected with HIV GagPol, and supernatants and cell lysates were analyzed for Env incorporation and cellular expression, respectively.
Next, we wanted to determine whether these mutants retained fusogenic activity. Because these Env mutants had a truncated CTD (Δ25CTD), they were missing the R peptide and could be tested for fusogenicity. Each cell line stably expressing the indicated Env mutant was transfected with an expression construct containing the transcriptional activator tet-off (35). At 24 h after transfection, an equal number of cells from each mutant cell line were cocultured 1:1 with receptor containing 293T mCAT-1 cells expressing TRE-driven Gaussia luciferase (Gluc). If the Env mutants are fusogenic, the transfected cells fuse with the receptor-expressing cells, and tet-off-dependent induction of Gluc occurs (35). Reporter induction can then be compared between mutant Env and wild-type controls to assess relative fusogenic activity. Interestingly, our results indicated that all four of the hydroxyl deletion mutants displayed reduced cell-to-cell fusion activity (Fig. 5B). To determine whether loss of fusogenicity was the sole basis for the loss of transduction capacity, we examined incorporation of these mutants into HIV particles. As shown in Fig. 5C, all of the hydroxyl deletion mutant glycoproteins were incorporated into HIV particles to a similar level compared to the truncated +1L control, indicating loss of transduction capacity was due to loss of fusogenicity. In addition, all of the mutant glycoproteins accumulated to similar levels in the stable cell lines compared to the wild-type Δ25CTD and +1L Δ25CTD control (Fig. 5C; Cell). Taken together, the results depicted in Fig. 5 indicated that the loss of transduction capacity was due to the loss of fusogenicity and not due to gross defects in Env processivity, trafficking, or incorporation into viral particles.
Hydrogen-bonding requirements and steric constraints of the hydroxyl-containing residues.
Polar residues located in MSDs of transmembrane helices have been shown to be important contributors to protein packing, oligomerization, and TM stabilization (43–48). To determine whether the hydrogen-bonding capacity of the residues we identified in the ectodomain interface was important for function, we made alanine substitutions to the wild-type protein (HA tagged) in both the presence or absence of the full-length cytoplasmic tail. We were surprised to observe that only two of the residues, T139 and S143, displayed abrogated functionality when substituted by alanine (Fig. 6A).
FIG 6.

Amino acid substitutions in the critical hydroxyl positions display defective functionality. (A) Infectivity of alanine substitutions in the context of full-length HA-tagged (filled bars) and truncated HA-tagged (open bars) Env relative to wild-type (2.1 × 105 IU/ml). (B) Infectivity of amino acid substitutions in the context of full-length (HA-tagged) Env relative to wild-type Env (6.9 × 105 IU/ml). The hydroxyl-containing positions were substituted to N (asparagine), Q (glutamine), Y (tyrosine), A (alanine), or F (phenylalanine), and the infectivity was assessed. The asterisks indicate a significant difference between the infectivity of tyrosine and phenylalanine substitutions at all four positions tested. (***, P < 0.001; **, P < 0.01).
We hypothesized that these residues were positioned in, or near, the core of the trimer interface and that their ability to hydrogen bond was crucial for monomer orientation or trimer stabilization. If this hypothesis was correct, it would also suggest that positions T139 and S143 would have greater steric restriction to amino acid side chain size because of their location toward the trimer core. Conversely, positions T140 and T144 would be able to tolerate amino acid side chains with greater bulk size because of their orientation toward the outside of the trimer. To test this hypothesis, we made substitutions at these positions with amino acid side chains that were still polar but had increasing bulk size (asparagine, glutamine, and tyrosine). As shown in Fig. 6B, increasing amino acid side chain size at positions T139 and S143 resulted in a corresponding decrease in transduction capacity. However, increasing amino acid side chain size at positions T140 and T144 showed a minimal effect on transduction capacity, suggesting that these positions have less steric restriction (Fig. 6B). It is important to highlight that substitution of T139 and S143 with hydrogen-bonding side chains (asparagine, glutamine, and to a lesser extent, tyrosine) retained function; however, substitution to a residue with no polar side chain (alanine, and phenylalanine) resulted in severely reduced transduction capacity. This suggests that the polarity of the side chain at these positions is important for function.
Finally, substitution of phenylalanine at positions T139, T140, S143, and T144 resulted in severely abrogated functionality. The most direct way to address the importance of the hydroxyl group was to compare the phenylalanine substitution to the tyrosine substitution. The only molecular difference between these two amino acid side chains is the presence of the hydroxyl group on the benzyl ring of tyrosine. Importantly, as highlighted in Fig. 6B, tyrosine substitution at each position significantly retained transduction capacity compared to the phenylalanine substitution. These data strongly suggest that hydrogen-bonding at these positions is crucial for function.
DISCUSSION
We have previously shown that the spacing of the MPER and CTD interfaces is critical for glycoprotein functionality (35). Disruption of the relative helical orientation by the insertion of +1L within the MSD resulted in conflicting alignment of the ectodomain and CTD interfaces (Fig. 1). Truncation of the CTD restored functionality of the +1L Env by relieving the conflict between interfaces through removal of a majority of the CTD. This study explored the boundaries of the ectodomain and CTD interfaces as well as probed the role of this region in promoting fusogenic activity.
Identifying the boundaries of the ectodomain and the CTD.
We identified the glycine-proline pairs positioned within the MSD as boundaries between the end of the ectodomain interface and the start of the predicted interface located in the CTD. This observation is consistent with proline being able to induce helical kinks, as well as previous studies that examined positioning of glycine-proline pairs within MSDs and their roles in forming molecular hinges and inducing α-helical kinks (30, 32, 44, 49, 50). A previous study reported that substitution of proline at position 148 with other residues resulted in Env proteins that were processed properly and incorporated into viral particles but had greatly diminished, or undetectable, fusogenic activity and infectivity (51). However, it is also possible that the CTD interface extends beyond the glycine-proline pair at positions 159 and 160 into the MSD. This would be consistent with the single-residue deletion scan data that displays lower recovery compared to other deletions in this region (Fig. 2B). Alternatively, it is also plausible that these resides are important for an independent function and that deleting them diminishes infectivity regardless of helical alignment. Mutations to introduce +1, +2, or +3 leucines either upstream or downstream of the glycine proline pairs did not display a restoration of function with the insertion of +3 leucine residues (Fig. 3). This suggests that insertion of +3 leucines, and consequently nearly a complete turn of an α-helix, between the glycine-proline pairs is tolerated because no critical interfacial surface exists. However, +3L insertions in the ectodomain and CTD are not tolerated because it may disrupt contacts between residues in these domains (Fig. 3).
Hydroxyl-containing residues in the ectodomain are critical for fusogenicity.
To understand why the hydroxyl-containing residues were crucial for Env function, we probed the role of these residues by creating substitutions that abrogated hydrogen bonding or introduced steric hindrance (Fig. 6). We were surprised to find that only two of the positions showed decreased function when hydrogen bonding was disrupted (Fig. 6A, T139A and S143A). Interestingly, these two positions also displayed the greatest sensitivity to polar side chain substitution with increasing bulk size. These results are consistent with the hypothesis that positions T139 and S143 are part of the trimer core, whereas T140 and T144 are peripheral. This interpretation could also explain why the deletion of residues L141 and I142 displayed restored functionality when the CTD was truncated. Deleting either of these residues would retain the hydrogen-bonding network by shifting T144 toward the core. A previous study identified residues in the CTD involved in forming a trimeric coiled-coil interface (36). Because the trimer forms a coiled-coil domain from the beginning of the MSD to the end of the CTD, we can use the predicted trimer interface located in the CTD to predict where T139 and S143 would position relative to the coiled-coil interface. Based on that prediction, residues T139 and S143 would reside in close proximity to the predicted coiled-coil trimer interface (Fig. 7).
FIG 7.

Predicted hydrogen-bonding network in the core of the MLV Env trimer. MLV Env monomer structural predictions were generated using Phyre2 protein fold recognition software. The monomers were assembled into trimers using the molecular modeling program Chimera based on our prediction. The trimer is depicted from both the side and the top. A space-filling model is shown to highlight the bulky side chains of the tyrosine substitutions. Black dotted lines in the first panel indicate theoretical hydrogen-bonding between polar residues T139 and S143 in the predicted trimer core.
We suggest that residues T139 and S143 not only contribute to the fusion mechanism but may also stabilize the TM trimer or direct monomer orientation through the formation of an interhelical hydrogen-bonding network (Fig. 7). A recent study using a random peptide library produced in Escherichia coli identified two consensus motifs of SxxxSSxxT and SxxSSxxT that drove helical oligomerization of transmembrane peptides (43). The hydroxyl-containing residues at the C terminus of the MPER and at the N terminus of the MSD appear to be part of a consensus sequence SxxxTTxxST, which may participate in oligomerization of the TM domain of MLV Env. Other γ-retroviral glycoproteins such as feline leukemia virus and gibbon ape leukemia virus also have similar consensus sequences, suggesting it may be a conserved mechanism used to drive trimer oligomerization in some gamma-retroviral Envs. Interestingly, vesicular stomatitis virus glycoprotein has a similar sequence of SSxxSSxxS at the external MPER-MSD interface, suggesting that this mechanism may not be exclusive to only gamma-retroviral glycoproteins. Although HIV Env does not have serine-threonine clustering in this region, it does have a GxxxG helical packing motif in the MSD. Together, these data suggest that the glycine-proline pairs cap the ectodomain and CTD interfaces and that the hydroxyl-containing residues within the ectodomain interface contribute significantly to the cell-to-cell fusion mechanism.
ACKNOWLEDGMENT
This study was supported by U.S. Public Health Service grant GM110776.
REFERENCES
- 1.Kamps CA, Lin Y-C, Wong PKY. 1991. Oligomerization and transport of the envelope protein of Moloney murine leukemia virus-TB and of ts1, a neurovirulent temperature-sensitive mutant of MoMuLV-TB. Virology 184:687–694. doi: 10.1016/0042-6822(91)90438-H. [DOI] [PubMed] [Google Scholar]
- 2.Einfeld D, Hunter E. 1988. Oligomeric structure of a prototype retrovirus glycoprotein. Proc Natl Acad Sci U S A 85:8688–8692. doi: 10.1073/pnas.85.22.8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Earl PM, Moss B, Doms R. 1991. Folding, interaction with GRP78-BiP, assembly, transport of the human immunodeficiency virus type 1 envelope protein. J Virol 65:2047–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallenberger SB, Angliker VH, Shaw E, Klenk H, Gerten W. 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360:358–361. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- 5.Anderson ET, Hayflick LJ, Thomas G. 1993. Inhibition of HIV-1 gp160-dependent membrane fusion by a furin-directed α 1-antitrypsin variant. J Biol Chem 268:. [PubMed] [Google Scholar]
- 6.Fass D, Harrison S, Kim P. 1996. Retrovirus envelope domain at 1.7 angstrom resolution. Nat Struct Biol 3:465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- 7.Johnston E, Radke K. 2000. The SU and TM envelope protein subunits of bovine leukemia virus are linked by disulfide bonds, both in cells and in virions J Virol 74:2930–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leamnson R, Halpern M. 1976. Subunit structure of the glycoprotein complex of avian tumor virus. J Virol 18:956–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Opstelten D, Wallin M, Garoff H. 1998. Moloney murine leukemia virus envelope protein subunits, gp70 and Pr15E, form a stable disulfide-linked complex. J Virol 72:6537–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinter A, Fleissner E. 1977. The presence of disulfide-linked gp70-p15(E) complexes in AKR murine leukemia viruses. Virology 83:417–422. doi: 10.1016/0042-6822(77)90187-8. [DOI] [PubMed] [Google Scholar]
- 11.Pinter A, Lieman-Hurwitz J, Fleissner E. 1978. The nature of the association between the murine leukemia virus envelope proteins. Virology 91:345–351. doi: 10.1016/0042-6822(78)90382-3. [DOI] [PubMed] [Google Scholar]
- 12.Leamnson RN, Shander MHM, Halpern MS. 1977. A structural protein complex in Moloney leukemia virus. Virology 76:437–439. doi: 10.1016/0042-6822(77)90318-X. [DOI] [PubMed] [Google Scholar]
- 13.Witte ON, Tsukamoto-Adey A, Weissman IL. 1977. Cellular maturation of oncornavirus glycoproteins: topological arrangement of precursor and product forms in cellular membranes. Virology 76:539–553. doi: 10.1016/0042-6822(77)90236-7. [DOI] [PubMed] [Google Scholar]
- 14.Henzy J, Coffin J. 2013. Betaretroviral envelope subunits are noncovalently associated and restricted to the mammalian class. J Virol 87:1937–1946. doi: 10.1128/JVI.01442-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh W, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. 1987. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 237:. [DOI] [PubMed] [Google Scholar]
- 16.Loving R, Li K, Wallin M, Sjoberg M, Garoff H. 2008. R-peptide cleavage potentiates fusion-controlling isomerization of the intersubunit disulfide in Moloney murine leukemia virus Env. J Virol 82:2594–2597. doi: 10.1128/JVI.02039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ragheb J, Anderson W. 1994. pH-independent murine leukemia virus ecotropic envelope-mediated cell fusion: implications for the role of the R peptide and p12E TM in viral entry. J Virol 68:3220–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubo Y, Tominaga C, Yoshii H, Kamiyama H, Mitani C, Amanuma H, Yamamoto N. 2007. Characterization of R-peptide of murine leukemia virus envelope glycoprotein in syncytium formation and entry. Arch Virol 152:2169–2182. doi: 10.1007/s00705-007-1054-6. [DOI] [PubMed] [Google Scholar]
- 19.Li M, Li Z, Yao Q, Yang C, Steinhauer D, Compans R. 2006. Murine leukemia virus R peptide inhibits influenza virus hemagglutinin-induced membrane fusion. J Virol 80:6106–6114. doi: 10.1128/JVI.02665-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green N, Shinnick T, Witte O, Ponticelli A, Sutcliffe J, Lerner R. 1981. Sequence-specific antibodies show that maturation of Moloney leukemia virus envelope polyprotein involves removal of a COOH-terminal peptide. Proc Natl Acad Sci U S A 78:6023–6027. doi: 10.1073/pnas.78.10.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henderson L, Sowder R, Copeland T, Smythers G, Oroszlan S. 1984. Quantitative separation of murine leukemia virus proteins by reverse-phase high-pressure liquid chromatography reveals newly described gag and env cleavage products. J Virol 52:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rein A, Mirro J, Haynes J, Ernst S, Nagashima K. 1994. Function of the cytoplasmic domain of a retroviral transmembrane protein: p15E-p2E cleavage activates the membrane fusion capability of the murine leukemia virus Env protein. J Virol 68:1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loving R, Wu S, Sjoberg M, Lindqvist B, Garoff H. 2012. Maturation cleavage of the murine leukemia virus Env precursor separates the transmembrane subunits to prime it for receptor triggering. Proc Natl Acad Sci U S A 109:7735–7740. doi: 10.1073/pnas.1118125109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Owens R, Burke C, Rose J. 1994. Mutations in the membrane-spanning domain of the human immunodeficiency virus envelope glycoprotein that affect fusion activity. J Virol 68:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Apellániz B, Rujas E, Serrano S, Morante K, Tsumoto K, Caaveiro J, Jiménez M, Nieva J. 2015. The atomic structure of the HIV-1 gp41 transmembrane domain and its connection to the immunogenic membrane-proximal external region. J Biol Chem 290:12999–13015. doi: 10.1074/jbc.M115.644351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Z, Cheng Y, Kim M, Song L, Choi J, Kudahl U, Brusic V, Chowdhury B, Yu L, Seaman M, Bellot G, Shih W, Wagner G, Reinherz E. 2014. Disruption of helix-capping residues 671 and 674 reveals a role in HIV-1 entry for a specialized hinge segment of the membrane proximal external region of gp41. J Mol Biol 426:1095–1108. doi: 10.1016/j.jmb.2013.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi H, Diaz-Rohrer B, Saminathan P, Jacobs A. 2015. The membrane proximal external regions of gp41 from HIV-1 Strains HXB2 and JRFL have different sensitivities to alanine mutation. Biochemistry 54:1681–1693. doi: 10.1021/bi501171r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salzwedel K, West J, Hunter E. 1999. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol 73:2469–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Apellaniz B, Rujas E, Carravilla P, Requejo-Isidro J, Huarte N, Domene C, Nieva J. 2014. Cholesterol-dependent membrane fusion induced by the gp41 membrane-proximal external region-transmembrane domain connection suggests a mechanism for broad HIV-1 neutralization. J Virol 88:13367–13377. doi: 10.1128/JVI.02151-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordes F, Bright J, Samson M. 2002. Proline-induced distortions of transmembrane helices. J Mol Biol 323:951–960. doi: 10.1016/S0022-2836(02)01006-9. [DOI] [PubMed] [Google Scholar]
- 31.Jardetzky O. 1996. Simple allosteric model for membrane pumps. Nature 211:696–670. [DOI] [PubMed] [Google Scholar]
- 32.Sansom M, Weinstein H. 2000. Hinges, swivels & switches: the role of prolines in signaling via transmembrane α-helices. Trends Pharmacol Sci 21:445–451. doi: 10.1016/S0165-6147(00)01553-4. [DOI] [PubMed] [Google Scholar]
- 33.Govaerts C, Blanpain C, Deupi X, Ballet S, Ballesteros JA, Wodak SJ. 2001. The TXP motif in the second transmembrane helix of CCR5: a structural determinant of chemokine-induced activation. J Biol Chem 276:13217–13225. doi: 10.1074/jbc.M011670200. [DOI] [PubMed] [Google Scholar]
- 34.Ri Y, Ballesteros J, Abrams C, Oh S, Verselis V, Weinstein H, Bargiello T. 1999. The role of a conserved proline residue in mediating conformational changes associated with voltage gating of Cx32 gap junctions. Biophys J 76:2887–2898. doi: 10.1016/S0006-3495(99)77444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janaka S, Gregory D, Johnson M. 2013. Retrovirus glycoprotein functionality requires proper alignment of the ectodomain and the membrane-proximal cytoplasmic tail. J Virol 87:12805–12813. doi: 10.1128/JVI.01847-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor G, Sanders A. 2003. Structural criteria for regulation of membrane fusion and virion incorporation by the murine leukemia virus TM cytoplasmic domain. Virology 312:295–305. doi: 10.1016/S0042-6822(03)00297-6. [DOI] [PubMed] [Google Scholar]
- 37.Kim J, Lee S, Li L, Park H, Park J, Lee K, Kim M, Shin B, Choi S. 2011. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trichas G, Begbie J, Srinivas S. 2008. Use of the viral 2A peptide for bicistronic expression in transgenic mice. BMC Biol 6:40. doi: 10.1186/1741-7007-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erlwien O, Buchholz C, Schnierle B. 2003. The proline rich region of ecotropic Moloney murine leukemia virus envelope protein tolerates the insertion of the green fluorescent protein and allows the generation of replication-competent virus. J Gen Virol 84:369–373. doi: 10.1099/vir.0.18761-0. [DOI] [PubMed] [Google Scholar]
- 40.Boussif O, Lezoualc'h F, Zanta M, Scherman D, Demeneix B, Behr J. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethyleneimine. Proc Natl Acad Sci U S A 92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu B, Lu J, Gallaher T, Anderson W, Cannon P. 2000. Identification of regions in the Moloney murine leukemia virus SU protein that tolerate the insertion of an integrin-binding peptide. Virology 269:7–17. doi: 10.1006/viro.2000.0201. [DOI] [PubMed] [Google Scholar]
- 42.Rothenberg S, Olsen M, Laurent L, Crowly R, Brown P. 2001. Comprehensive mutational analysis of the Moloney murine leukemia virus envelope glycoprotein. J Virol 75:11851–11862. doi: 10.1128/JVI.75.23.11851-11862.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dawson J, Weinger J, Engleman D. 2002. Motifs of serine and threonine can drive association of transmembrane helices. J Mol Biol 316:799–805. doi: 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- 44.Deupi X, Olivella M, Govaerts C, Ballesteros J, Campillo M, Pardo L. 2004. Ser and Thr residues modulate the conformation of Pro-kinked transmembrane alpha-helices. Biophys J 86:105–115. doi: 10.1016/S0006-3495(04)74088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.del Val C, White S, Bondar A. 2012. Ser/Thr motifs in transmembrane proteins: conservation patterns and effects on local protein structure and dynamics. J Membr Biol 245:717–730. doi: 10.1007/s00232-012-9452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurochkina N. 2007. Amino acid composition of parallel helix-helix interfaces. J Theor Biol 247:110–121. doi: 10.1016/j.jtbi.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Bowie J. 2011. Membrane protein folding: how important are hydrogen bonds? Curr Opin Struct Biol 21:42–49. doi: 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gray T, Matthews B. 1984. Intrahelical hydrogen bonding of serine, threonine and cysteine residues within alpha-helices and its relevance to membrane-bound proteins. J Mol Biol 175:75–81. doi: 10.1016/0022-2836(84)90446-7. [DOI] [PubMed] [Google Scholar]
- 49.Tieleman P, Shrivastava I, Ulmschneider M, Sansom M. 2001. Proline-induced hinges in transmembrane helices: possible roles in ion channel gating. Proteins 44:63–72. doi: 10.1002/prot.1073. [DOI] [PubMed] [Google Scholar]
- 50.Adamian L, Liang J. 2001. Helix-helix packing and interfacial pairwise interactions of residues in membrane proteins. J Mol Biol 311:891–907. doi: 10.1006/jmbi.2001.4908. [DOI] [PubMed] [Google Scholar]
- 51.Taylor G, Sanders A. 1999. The role of the membrane-spanning domain sequence in glycoprotein-mediated membrane fusion. Mol Biol Cell 10:2803–2815. doi: 10.1091/mbc.10.9.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]