

ABSTRACT
Airway epithelium is the primary target of many respiratory viruses. However, virus induction and antagonism of host responses by human airway epithelium remains poorly understood. To address this, we developed a model of respiratory syncytial virus (RSV) infection based on well-differentiated pediatric primary bronchial epithelial cell cultures (WD-PBECs) that mimics hallmarks of RSV disease in infants. RSV is the most important respiratory viral pathogen in young infants worldwide. We found that RSV induces a potent antiviral state in WD-PBECs that was mediated in part by secreted factors, including interferon lambda 1 (IFN-λ1)/interleukin-29 (IL-29). In contrast, type I IFNs were not detected following RSV infection of WD-PBECs. IFN responses in RSV-infected WD-PBECs reflected those in lower airway samples from RSV-hospitalized infants. In view of the prominence of IL-29, we determined whether recombinant IL-29 treatment of WD-PBECs before or after infection abrogated RSV replication. Interestingly, IL-29 demonstrated prophylactic, but not therapeutic, potential against RSV. The absence of therapeutic potential reflected effective RSV antagonism of IFN-mediated antiviral responses in infected cells. Our data are consistent with RSV nonstructural proteins 1 and/or 2 perturbing the Jak-STAT signaling pathway, with concomitant reduced expression of antiviral effector molecules, such as MxA/B. Antagonism of Jak-STAT signaling was restricted to RSV-infected cells in WD-PBEC cultures. Importantly, our study provides the rationale to further explore IL-29 as a novel RSV prophylactic.
IMPORTANCE Most respiratory viruses target airway epithelium for infection and replication, which is central to causing disease. However, for most human viruses we have a poor understanding of their interactions with human airway epithelium. Respiratory syncytial virus (RSV) is the most important viral pathogen of young infants. To help understand RSV interactions with pediatric airway epithelium, we previously developed three-dimensional primary cell cultures from infant bronchial epithelium that reproduce several hallmarks of RSV infection in infants, indicating that they represent authentic surrogates of RSV infection in infants. We found that RSV induced a potent antiviral state in these cultures and that a type III interferon, interleukin IL-29 (IL-29), was involved. Indeed, our data suggest that IL-29 has potential to prevent RSV disease. However, we also demonstrated that RSV efficiently circumvents this antiviral immune response and identified mechanisms by which this may occur. Our study provides new insights into RSV interaction with pediatric airway epithelium.
INTRODUCTION
Airway epithelium is an extremely important barrier to respiratory pathogens. It is also the primary infection target for many respiratory viruses. Elucidating the interactions between respiratory viruses and airway epithelium is fundamental to understanding aspects of their pathogenesis. We recently developed and characterized models of respiratory syncytial virus (RSV) infection based on well-differentiated pediatric primary airway epithelial cells derived from pediatric bronchial (WD-PBECs) or nasal (WD-PNECs) brushings (1, 2). RSV is the primary viral cause of infant hospitalizations in the first year of life and is capable of repeated infections throughout life (3). Despite its original isolation in 1957 (4), no effective RSV therapies or vaccines are available. The mechanisms by which RSV causes disease and is capable of repeated infections in humans remain an enigma. Our models reproduce several hallmarks of RSV infection in vivo, suggesting that they provide authentic surrogates with which to study RSV-induced innate immune responses and interaction with human airway epithelium (1, 2).
We previously reported secretion of high levels of CXCL10, an interferon (IFN)-stimulated gene (ISG) product, from RSV-infected WD-PBECs (1, 2). This is consistent with the induction of an IFN-mediated antiviral response to infection. IFNs are a heterogeneous family of cytokines with well-characterized capacities to induce antiviral states in cells (5–7). They include types I (IFN-α/β), II (IFN-γ), and III (IFN-λs), only the first and last of which are expressed by airway epithelial cells upon appropriate stimulation (8–10). Despite considerable CXCL10 secretion, we detected no IFN-α/β secretion from RSV-infected WD-PBECs, while little or no IFN-α/β was detected in nasal or bronchoalveolar lavages from RSV-infected infants (1, 2, 11–13). In contrast, little is known about IFN-λs responses to RSV infection of airway epithelium in vitro and especially in vivo. IFN-λs comprise three closely related molecules designated IFN-λ1 (IL-29), IFN-λ2 (IL-28A) and IFN-λ3 (IL-28B) (IFN-λ2 and -λ3 share 96% identity) (14). They are induced by viruses, such as influenza A virus and rhinovirus, and inhibit replication of HCV and HIV. Interestingly, we and others recently demonstrated that IFN-λs, rather than IFN-α/β, were induced following RSV infection of primary monolayer or well-differentiated nasal and bronchial epithelial cell cultures (2, 15). This suggested a role for IFN-λs in RSV-induced antiviral responses. IFN-λs induce antiviral states by signaling through a heterodimeric receptor complex composed of IL-28Rα and IL-10RA (14). This activates signal transduction through the Jak/STAT pathway in a manner that is virtually identical to IFN-α/β, resulting in phosphorylation of mainly signal transducer and activator of transcription 1 (STAT1) (pSTAT1) and STAT2 (pSTAT2) and, to a lesser extent, STAT3, STAT4, and STAT5. This is followed by pSTAT homo- or heterodimerization, complexing with IFN regulatory factor 9 (IRF9), nuclear translocation and induction of numerous ISGs, such as CXCL10 or the potent antiviral protein MxA (14, 16–18).
In the current study, we explored the induction of antiviral responses following RSV infection of WD-PBECs. We found that RSV induced a potent antiviral state against the related Sendai virus (SeV), which was mediated, in part at least, by secreted factors, including IFN-λ1/IL-29. We also demonstrated that RSV-induced IL-29 secretion from WD-PBECs reflected IL-29 secretions in lower airway samples from RSV-infected infants. These RSV-induced secreted factors also demonstrated prophylactic antiviral activity against RSV, albeit with lower potency than against SeV. Pretreatment of WD-PBEC cultures with a high dose of IL-29 reduced RSV replication, suggesting the prophylactic potential of IL-29. Interestingly, RSV nonstructural proteins NS1 and NS2, which we and others have previously shown to antagonize IFN-α/β-mediated antiviral responses (19–21), were critical for RSV growth in WD-PBECs and antagonism of IL-29-induced antiviral responses. We also found that in WD-PBECs, RSV-infected cells had significantly reduced MxA/B and pSTAT2 expression levels compared to surrounding noninfected cells, indicating active antagonism of antiviral responses by RSV but restriction of this antagonism to infected cells. In summary, our data provide novel insights into the induction and antagonism of antiviral responses, and in particular IL-29, following RSV infection of human airway epithelium.
MATERIALS AND METHODS
Cell lines and viruses.
HEp-2 and Vero cell lines were cultured as previously described (22). The origin and characterization of the clinical isolate RSV BT2a were previously described (23). Recombinant RSV expressing enhanced green fluorescent protein (eGFP) (rA2-eGFP) was generated by cloning a cassette consisting of the eGFP ORF-NS1 gene end signal-NS1 gene start signal into an antigenomic cDNA (D53) of RSV at the position of the NS1 ATG. This scheme inserts an additional transcription unit encoding eGFP at the first position in the genome, preserving the RSV sequence from the leader through the NS1 5′ untranslated region. The eGFP containing D53 was then used to recover recombinant RSV in BSR-T7 cells as described previously (20, 24, 25). The production of recombinant RSV expressing eGFP in place of NS1 and NS2 (rA2-ΔNS1/2-eGFP) has been described (26). rA2-eGFP and rA2-ΔNS1/2-eGFP stock production and titrations were performed in Vero cells. Virus stocks were harvested and stored as previously described (27). The rescue, characterization, stock production, and titration of rSeV/eGFP were previously described (22).
WD-PBEC culture and infection.
WD-PBEC culture was described previously (28). Briefly, primary PBECs were obtained by bronchial brushings from healthy children undergoing elective surgery, expanded in culture flasks, and seeded onto collagen-coated Transwell inserts (6.5-mm diameter, 0.4-μm pore size). Once confluence was reached, apical medium was removed to create an air-liquid interface (ALI) and trigger differentiation into pseudostratified mucociliary epithelium. Cultures were infected 3 to 4 weeks after ALI, as previously described (28), with the multiplicities of infection (MOIs) specified in the figure legends. Virus stocks were diluted in Dulbecco modified Eagle medium (DMEM) where necessary. Inoculum or DMEM only was added to the apical surface, and the cultures were incubated for 1.5 h at 37°C in 5% CO2, followed by five rinses with 500 μl of DMEM. The last rinse was retained as the 2-h virus titration point. Every 24 h thereafter, apical rinses and basal medium were collected to determine virus growth kinetics and IFN responses, respectively. Infected and control cultures were monitored daily by light and UV microscopy, where appropriate (Nikon Eclipse TE-2000U). eGFP quantification in infected cultures was performed by using ImageJ software (http://rsbweb.nih.gov/ij/).
Immunofluorescence staining and enzyme-linked immunosorbent assay (ELISA).
Immunostaining of the cultures was previously described (28). Briefly, WD-PBECs were rinsed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 20 min. Cultures were washed and stored in PBS at 4°C until used. For immunofluorescence staining, cultures were permeabilized with PBS plus 0.2% Triton X-100 (vol/vol) for 2 h and blocked with 0.4% (wt/vol) bovine serum albumin in PBS for 30 min. RSV-infected cells were detected by using an anti-RSV F-specific mouse monoclonal antibody (MAb; clone 133-1H conjugated with Alexa 488, 1:200, Chemicon, USA). MxA/B was detected using an anti-human Mx mouse MAb (Santa-Cruz, clone C-1, 1:200). pSTAT1 and pSTAT2 were detected using mouse anti-human pSTAT1 (pY701) MAb (BD Biosciences, 1:200) and rabbit anti-human pSTAT2 (Tyr690) polyclonal antibody (Antibodies-Online, Inc., GA; 1:200), respectively. The mouse anti-Mx and pSTAT1 (pY701) MAbs were detected with Alexa-568-conjugated goat anti-mouse IgG1 (Invitrogen, 1:500), while rabbit polyclonal antibodies were detected with Alexa-568-conjugated goat anti-rabbit IgG(H+L) (Invitrogen, 1:500) polyclonal antibodies. Inserts were mounted on microscopy slides and nuclei were counterstained using DAPI (4′,6′-diamidino-2-phenylindole) mounting medium (Vectashield). Fluorescence was detected by confocal laser scanning microscopy (TCS SP5; Leica). MX and p-STAT fluorescence intensities were determined by using confocal images of infected cultures in ImageJ by dividing the raw integrated density by the area of each individual cell. This was done for >120 RSV-infected and noninfected cells.
IL-29 (IFN-λ1), pan-IFN-α, and IFN-β concentrations in WD-PBEC basal media and in clinical samples were measured using human IFN-λ1 ELISA kits (eBioscience, United Kingdom), human IFN-β ELISA kits (R&D Systems), and human pan-IFN-α ELISA kits (Mabtech, Sweden). All ELISAs were undertaken according to the manufacturers' instructions. IL-28A (IFN-λ2) was detected by using a custom Milleplex kit, according to the manufacturer's instructions (Merck-Millipore, United Kingdom).
Superinfection, conditioned medium, and IFN-λ treatment.
Superinfection experiments were undertaken by infecting WD-PBECs with RSV BT2a (MOI ≈ 4) for 72 h and superinfecting them with rSeV/eGFP (MOI ≈ 0.1), as described previously, for 144 h (28). Conditioned medium (CM) experiments were performed by transferring basal medium from mock (CMCON)-infected or RSV-infected WD-PBEC cultures incubated with (CMRSV+αIL-29) or without (CMRSV) neutralizing antibody against IL-29 (R&D Systems; MAb 15981, 10 μg/ml) at 72 h postinfection (hpi) into the basal compartment of fresh cultures derived from the same individuals. After 24 h, conditioned cultures were infected with rSeV/eGFP. To assess the antiviral effects of IFN-λ1/IL-29 against RSV, rSeV/eGFP, rA2-eGFP, and rA2-ΔNS1/2-eGFP, fresh WD-PBECs or Vero cells were treated before or after infection with IFN-λ1/IL-29 (Peprotech, United Kingdom). Antiviral effects were determined by measuring eGFP fluorescence and/or virus growth kinetics.
Lower airway sampling.
Samples were obtained from 10 infants (mean age, 0.31 years; range, 0.06 to 1.3 years) with severe RSV disease who were treated in the pediatric intensive care unit of the Royal Belfast Hospital for Sick Children. Samples were direct tracheobronchial aspirates or deeper suction samples obtained after the instillation of 2 ml of saline. All sampling was clinically indicated and not performed for research purposes. Fourteen uninfected and otherwise healthy children (mean age, 1.7 years; range, 1 to 2.4 years) acted as controls with blind nonbronchoscopic bronchoalveolar lavage samples obtained (after the instillation of 10 ml of saline) at the time of intubation for an elective surgical procedure (29). All RSV-infected infants were confirmed as having mono-infections by using a multiplex virus reverse transcriptase-PCR (RT-PCR) strip for 12 respiratory viruses, as previously described (30).
Statistical analyses.
Data obtained in vitro are presented as means ± the standard errors of the mean (SEM), and skewed data were log transformed before comparisons were made by using the Student paired t test or by comparing the areas under the curves using GraphPad Prism 5.0. The data from RSV-infected and control children were compared by using a Mann-Whitney t test and GraphPad Prism 5.0. A P value of <0.05 was considered statistically significant.
Ethics.
This study was approved by The Office for Research Ethics Committees Northern Ireland (ORECNI). Written informed parental consent was obtained.
RESULTS
RSV infection induces an antiviral state in WD-PBECs.
RSV infection of WD-PBECs generally occurred in noncontiguous or small clusters of ciliated epithelial cells (1, 2). This suggested the possibility that infection induced an antiviral state in neighboring noninfected cells that limited viral spread. To detect the induction of antiviral responses, we used rSeV/eGFP, since SeV replicates efficiently in WD-PBECs but is restricted in human cells pretreated with human IFN (22, 28, 31). WD-PBECs (n = 3 donors) were mock infected or infected with RSV BT2a (MOI∼4). After 72 h, the cultures were superinfected with rSeV/eGFP (MOI ≈ 0.1). Fluorescence was monitored, and apical washes were collected for virus titration every 24 h postinfection with rSeV/eGFP for 144 h. Preinfection with RSV potently inhibited rSeV/eGFP replication, as evidenced by greatly diminished eGFP expression and rSeV/eGFP growth kinetics (Fig. 1). Thus, RSV infection induced a strong antiviral state in WD-PBECs.
FIG 1.

Preinfection of WD-PBECs with RSV induces an antiviral effect. WD-PBECs (n = 3 donors) were mock infected or infected with RSV BT2a (MOI ≈ 4). At 72 hpi, RSV- and mock-infected cultures were superinfected with rSeV/eGFP (MOI ≈ 0.1). (A) RSV-infected and mock-infected cultures were monitored by UV microscopy every 24 h (original magnification, ×4). These photos are representative of duplicate cultures from three individual donors. (B) eGFP expression was quantified every 24 h by measuring the percentage of green pixels in three different microscope fields. (C) Virus growth kinetics were determined by titrating rSeV/eGFP in apical washes at 24-h intervals after infection. The data are presented as means ± the SEM log10 fluorescent focus units (FFU)/ml.
IFN-λ1/IL-29 is the predominant IFN following RSV infection in vivo and in vitro.
Previous work from us and others reported little or no IFN-α/β secretion following RSV infection of infants or airway epithelial cells in vitro (1, 32). In contrast, IL-29 was evident after RSV infection of primary airway (nasal) epithelial cells in vitro (2, 15). To confirm and extend these findings, we determined the IFN-α/β, IL-28A, and IL-29 concentrations in lower airway samples from infants hospitalized with severe RSV infections (n = 8 to 10) and uninfected controls (n = 11 to 14). Apart from one RSV-infected individual with low levels of IFN-α, no IFN-α/β was detected in lower airway samples from infected infants (Fig. 2A and B). In contrast, IL-29 was significantly elevated in lower airway samples from RSV-infected patients compared to controls (Fig. 2C), although IL-28A was not (Fig. 2D). Furthermore, IL-29 concentrations in basolateral medium from RSV-infected WD-PBECs were significantly increased compared to controls and were similar to those in lower airway samples (Fig. 2E). Thus, our RSV/WD-PBEC model reproduced IL-29 responses to RSV infection in vivo. Similarly, CMRSV from 2/5 RSV-infected WD-PBECs had only low levels of IL-28A at 96 hpi (Fig. 2F). The cumulative data suggest that IL-29 is an important IFN protagonist induced by RSV infection of infants and WD-PBECs and consequently might be responsible for the RSV-induced antiviral responses. Whether IFN-α/β are implicated in these antiviral responses, in contrast, is unlikely, although this remains to be definitively confirmed.
FIG 2.

Type III IFN, but not type I IFN, is detected after RSV infection in vivo and in vitro. Lower airway samples from infants hospitalized for severe RSV infection (n = 8 to 10 donors) and healthy controls (n = 11 to 14 donors) were analyzed for pan-IFN-α (A), IFN-β (B), IL-29 (C), or IL-28A (D) by ELISA. The data are presented as means ± the SEM. Statistical analyses were undertaken using a nonparametric t test, followed by a Mann-Whitney test. ***, P = 0.0005. WD-PBECs (n = 5 donors) were infected with RSV (MOI ≈ 4). IL-29 (E) or IL-28A (F) secretions in the basolateral medium of RSV- and mock-infected cultures harvested at 24 and 96 hpi were measured. Since the medium was replaced every day, the data correspond to IFN secretions within the preceding 24 h. Values are means ± the SEM. **, P < 0.01.
Secreted factors, including IL-29, are implicated in the RSV-induced antiviral state.
To determine whether secreted factors, including IL-29, were implicated in the antiviral state induced in RSV-infected WD-PBECS, cultures (n = 2 to 3 donors) were mock infected or infected with RSV BT2a (MOI ≈ 1 or 4). At 72 hpi, CMRSV (with or without anti-IL-29 [10 μg/ml]) and CMCON were transferred to uninfected cultures derived from the same individuals. The cultures were infected 24 h later with rSeV/eGFP (MOI ≈ 0.1). eGFP expression (Fig. 3A and B) and rSeV/eGFP growth kinetics (Fig. 3C) indicated that factors secreted from RSV BT2a-infected WD-PBECs were responsible, in part, for the RSV-induced antiviral effects. Importantly, when CMRSV was preincubated with anti-IL-29 (CMRSV + αIL-29), the ability of CMRSV to abrogate rSeV/eGFP infection was significantly reduced, suggesting an important role for IL-29 in the antiviral effect of CMRSV.
FIG 3.

RSV-induced antiviral effect is partially mediated by IL-29. WD-PBECs (n = 2 to 4 donors) were infected with RSV BT2a (MOI ≈ 4) or mock infected. At 72 hpi, the basal medium of mock (CMCON)- or RSV-infected cultures incubated with (CMRSV+αIL-29) or without (CMRSV) neutralizing antibody against IL-29 (10 μg/ml) was transferred into the basal compartment of fresh cultures from the same individual donor. Twenty-four hours later, the cultures were infected with rSeV/eGFP (MOI ≈ 0.1). (A) eGFP expression in CMCON-, CMRSV+αIL-29-, and CMRSV-treated rSeV/eGFP-infected WD-PBECs over time (original magnification, ×4). (B) eGFP expression was quantified every 24 h by measuring the percentage of green pixels in five random microscopic fields. (C) Virus growth kinetics were determined by titrating rSeV/eGFP in apical washes at 24-h intervals after infection. The data are presented as means ± the SEM log10 FFU/ml. Areas under the curves were calculated and compared by using an unpaired Student t test. **, P < 0.01; ***, P < 0.001.
IL-29 attenuates rSeV/eGFP replication in WD-PBECs.
To confirm that IL-29 has antiviral activities in airway epithelium, WD-PBECs (n = 2 to 3 donors) were pretreated for 24 h with 100 or 1,000 pg of IL-29/ml before infection with rSeV/eGFP (MOI ≈ 0.1). These concentrations represented high physiological and superphysiological concentrations of IL-29, respectively, relative to IL-29 concentrations evident in CMRSV and lower airway samples. IL-29 demonstrated a dose-dependent suppression of eGFP expression following rSeV/eGFP infection, although both doses resulted in substantial reductions in eGFP expression relative to controls (Fig. 4A and B). Furthermore, rSeV/eGFP growth kinetics were significantly reduced following pretreatment with both IL-29 doses (Fig. 4C), although these reductions, even at the higher dose, were lower than those evident after CMRSV pretreatment (Fig. 3C). The data demonstrated that IL-29 has antiviral activities in airway epithelium and may account for some, but not all, of the antiviral activity associated with CMRSV.
FIG 4.

Influence of IL-29 pretreatment on rSeV/eGFP growth in WD-PBECs. WD-PBECs (n = 2 to 3 donors) were pretreated with IL-29 (100 or 1,000 pg/ml) or mock treated. Twenty-four hours later, cultures were infected with rSeV/eGFP at an MOI ≈ 0.1. (A) UV microphotographs were taken every 24 hpi. Original magnification, ×4. (B) eGFP expression was quantified every 24 hpi by measuring the percentage of green pixels in three random microscopic fields. Virus growth kinetics were determined by titrating rSeV/eGFP in apical washes at 24-h intervals after infection (C). The data are presented as means ± the SEM log10 FFU/ml. Areas under the curves were calculated and compared by using an unpaired Student t test. *, P < 0.05.
CMRSV and IL-29 prophylaxis attenuates RSV growth in WD-PBECs.
To assess CMRSV antiviral activity against RSV, WD-PBECs cultures (n = 2 donors) were mock-infected or infected with RSV BT2a (MOI ≈ 4). At 72 hpi, CMCON and CMRSV were transferred to uninfected cultures derived from the same individuals. CM-treated cultures were infected with rA2-eGFP (MOI ≈ 0.1) 24 h later, and the fluorescence was monitored for 96 h (Fig. 5A). The eGFP expression data demonstrated a similar, albeit lower, antiviral effect of CMRSV against RSV compared to rSeV/eGFP (Fig. 5A). This was also reflected in significantly reduced rA2-eGFP growth kinetics in CMRSV-treated compared to CMCON-treated WD-PBEC cultures (Fig. 5B).
FIG 5.
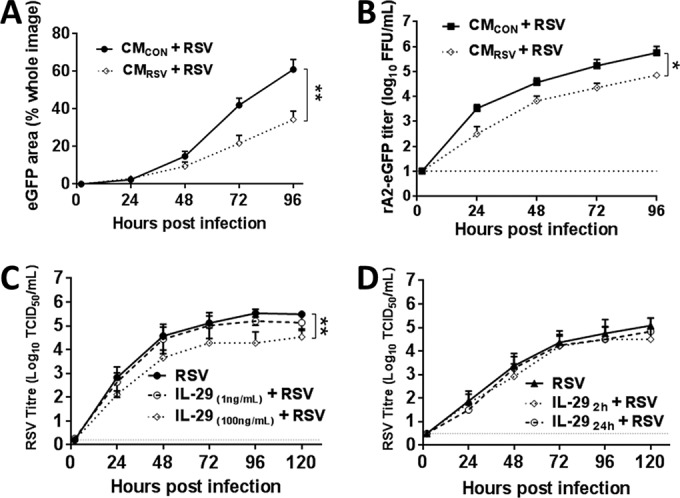
CMRSV and IL-29 pretreatment attenuates RSV growth in WD-PBECs. WD-PBECs (n = 2 donors) were infected in duplicate with RSV BT2a (MOI ≈ 4) or mock infected. At 72 hpi, the basal medium of mock (CMCON)- or RSV-infected (CMRSV) cultures was transferred into the basal compartment of fresh cultures from the same individual donor. Twenty-four hours later, the cultures were infected with rA2-eGFP (MOI ≈ 0.1). (A) eGFP expression was quantified every 24 h by measuring the percentage of green pixels in three different microscopic fields. (B) Virus growth kinetics were determined by titrating rA2-eGFP in apical washes at 24-h intervals after infection. WD-PBECs (n = 2 to 3 donors) were pretreated with IL-29 (1 or 100 ng/ml). (C) Twenty-four hours later, the cultures were infected with RSV BT2a at MOI ≈ 0.01. WD-PBECS (n = 3 donors) were infected with RSV BT2a at an MOI of 0.01. (D) At 2 or 24 hpi, infected cultures were treated with IL-29 (100 ng/ml). The virus growth kinetics presented in panels C and D were determined by titrating RSV in apical washes every 24 h after infection. The data are presented as means ± the SEM log10 TCID50/ml. Areas under the curves were calculated and compared by using an unpaired Student t test. *, P < 0.05; **, P < 0.01.
To assess whether IL-29 had prophylactic or therapeutic potential against the clinical isolate RSV BT2a, cultures (n = 2 to 3 donors) were untreated or treated with IL-29 for 24 h before infection (MOI ≈ 0.01; 1 or 100 ng/ml) or at 2 or 24 hpi (100 ng/ml). Pretreatment with 100 ng of IL-29/ml significantly reduced RSV replication (P < 0.05), whereas pretreatment with 1 ng of IL-29/ml did not (Fig. 5C). In contrast, IL-29 did not demonstrate therapeutic potential against RSV under our experimental conditions (Fig. 5D). The cumulative data from Fig. 3 and 5 suggested that RSV is more resistant to CMRSV and IL-29 than rSeV/eGFP and that the antiviral activity of CMRSV against RSV in WD-PBECs is similar to that evident following pretreatment with 100 ng of IL-29/ml. In view of the considerably lower levels of IL-29 evident in CMRSV than those used in these experiments, IL-29 is unlikely to be solely responsible for the limited spread of RSV in WD-PBECs.
RSV NS proteins antagonize IL-29-mediated antiviral effects.
The relative resistance of RSV to IL-29 suggested efficient antagonism of the IFN-λ signaling pathway in WD-PBECs. We and others have shown that RSV NS1 and NS2 are responsible for antagonizing IFN-α/β signaling through degradation of pSTAT2 (19, 33). However, the capacity of RSV NS1/2 to antagonize IFN-λ-mediated innate immune responses is unknown. To determine whether these proteins were implicated in antagonizing IL-29-induced responses in WD-PBECs, cultures were initially infected with recombinant RSV expressing eGFP, either wild-type (rA2-eGFP) or lacking NS1 and NS2 (rA2-ΔNS1/2-eGFP) (MOI ≈ 0.1). Although rA2-eGFP grew efficiently in WD-PBECs, replication of the NS1/2-deleted mutant was virtually abrogated (Fig. 6), indicating that NS1 and/or NS2 were critical for RSV replication in WD-PBECs. This result precluded the use of WD-PBECs to address antagonism of IL-29-mediated antiviral responses by RSV. Since both the mutant and the wild-type viruses grew efficiently in Vero cells and these cells were sensitive to IFN-λs stimulation (34, 35), we addressed the role of RSV NS1/2 in IL-29 antagonism in Vero cells. The cells were pretreated with IL-29 (1 or 100 ng/ml) or mock treated for 24 h before infection with either rA2-eGFP or rA2-ΔNS1/2-EGFP (MOI ≈ 0.1). Treated and control cultures were subsequently incubated for 72 h in the presence or absence of IL-29, respectively. Pretreatment with 1 ng of IL-29/ml had no effect on rA2-eGFP replication, as indicated by eGFP expression kinetics, whereas 100 ng/ml did have an effect (Fig. 7A and C). In comparison, pretreatment with either dose of IL-29 had much more dramatic effects on rA2-ΔNS1/2-eGFP replication than rA2-eGFP (Fig. 7B and D). Thus, RSV NS1/2 proteins are implicated in antagonizing IL-29-mediated antiviral responses.
FIG 6.
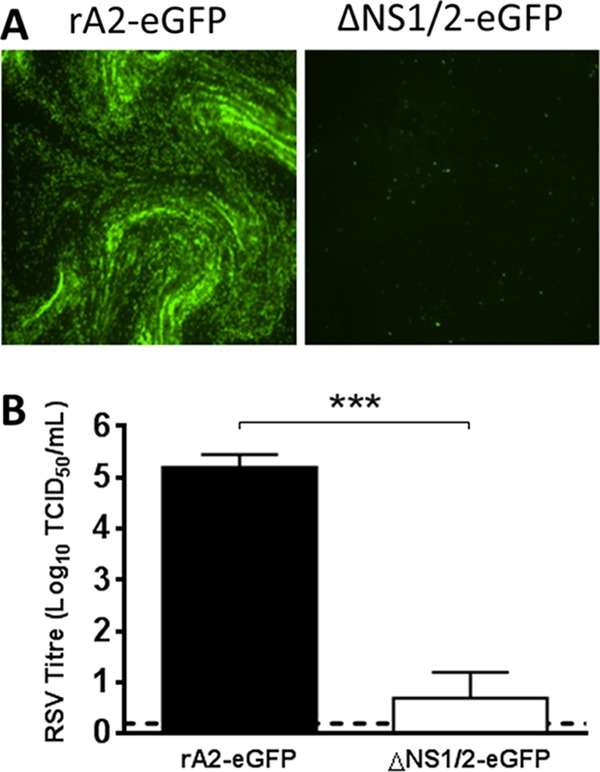
rA2-ΔNS1/2-eGFP does not infect WD-PBECs efficiently. WD-PBEC cultures were infected in duplicate with either rA2-eGFP or rA2-ΔNS1/2-eGFP (MOI ≈ 1) for 96 h. (A) Representative fluorescence pictures were taken at 96 hpi. (B) rA2-eGFP or rA2-ΔNS1/2-eGFP titers at 96 hpi were measured. The data are presented as means ± the SEM log10TCID50/ml. ***, P < 0.001.
FIG 7.
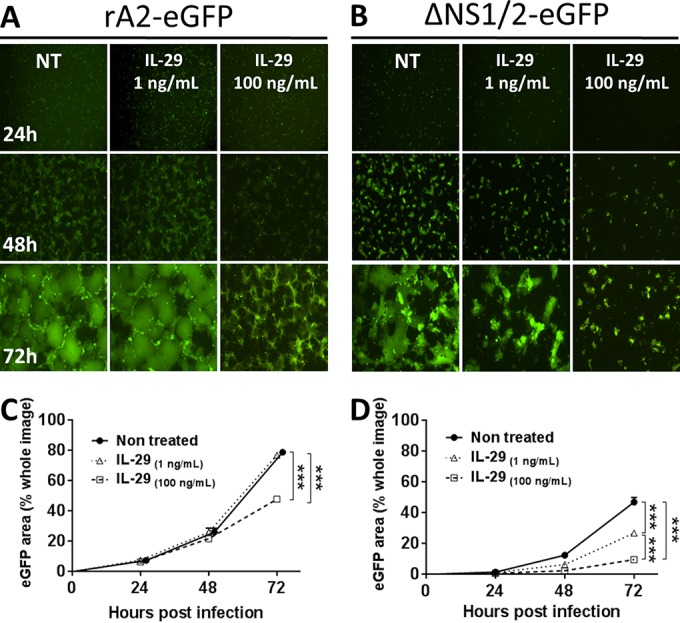
RSV NS1/2 partially counteract IL-29-mediated antiviral effects. Vero cells were pretreated with IL-29 (1 or 100 ng/ml) or nontreated. At 24 h posttreatment, the cells were infected with rA2-eGFP (A) or rA2-ΔNS1/2-eGFP (B) (MOI = 0.1). Cultures were monitored by UV microscopy every 24 hpi (original magnification, ×4). (C and D) Infections were undertaken in triplicate, and a surrogate for virus replication kinetics was quantified by measuring the percentage of eGFP coverage of three to five microscope fields at each time point. The data are presented as means ± the SEM eGFP area (percentage of the whole image) and are representative of three independent experiments in triplicate. Areas under the curves were calculated and compared by using an unpaired Student t test. ***, P < 0.001.
RSV antagonizes pSTAT2 and MxA/B expression in WD-PBECs.
To gain insight into the mechanisms behind RSV antagonism of the antiviral responses induced in WD-PBECs, we looked at the capacity of RSV to antagonize Jak/STAT signaling and MxA/B expression on a single cell basis within individual infected and surrounding noninfected cells. Since MxA/B expression is a reliable marker of IFN bioactivity (36), we initially confirmed that RSV infection, IL-29 treatment, or CMRSV treatment induced MxA/B expression in WD-PBECs (Fig. 8A and B). Furthermore, when CMRSV was pretreated with anti-IL-29, MxA/B expression was virtually abrogated, demonstrating that MxA/B induction following CMRSV treatment was in large part mediated by IL-29 (Fig. 8B and C). We and others previously reported that RSV NS proteins block the IFN-α/β-stimulated Jak/STAT signaling pathway by targeting pSTAT2 for proteasomal degradation (19, 37). To study RSV antagonism of Jak/STAT signaling and MxA/B expression in WD-PBECs, cultures (n = 3 to 4 donors) were infected with RSV (MOI ≈ 0.1), subjected to daily apical rinses and basolateral medium changes, fixed, permeabilized, and costained for RSV F and either pSTAT1, pSTAT2, or MxA/B at 144 hpi (Fig. 9). The pSTAT1 fluorescence intensities were similar in both infected and noninfected cells (Fig. 9E and F). In contrast, the pSTAT2 protein was significantly diminished (Fig. 9C and D), while the MxA/B proteins were virtually absent (Fig. 9A and B), in RSV-infected compared to surrounding noninfected cells. Thus, antagonism of the RSV-induced IFN-mediated antiviral responses was restricted to infected cells and implicated the perturbation of the Jak/STAT signaling pathway by pSTAT2 downregulation.
FIG 8.

MxA/B expression is induced following RSV infection and IL-29 or CMRSV treatment of WD-PBECs. (A) WD-PBECs were either mock infected, RSV BT2a infected (MOI ≈ 0.1) for 96 h, or treated with IL-29 (1 ng/ml) for 24 h. Cultures were fixed and stained for MxA/B (red), and nuclei were counterstained with DAPI (blue). Confocal images show typical staining in WD-PBECs from three individual donors (original magnification, ×63). (B and C) MxA/B expression after treatment with CMRSV is mediated in large part by IL-29. WD-PBECs (two independent cultures from one donor) were incubated with either CMRSV or CMCON alone or combined with a neutralizing antibody against IL-29 (10 μg/ml) for 24 h. Cultures were fixed and stained for MxA/B, and nuclei were counterstained with DAPI. The MxA/B signal was quantified in five individual fields from each culture, and the mean fluorescence was calculated and plotted. **, P < 0.01; ***, P < 0.001.
FIG 9.

RSV blocks the expression of MxA/B, pSTAT2 but not pSTAT1 in infected WD-PBECs. WD-PBECs were either mock- or RSV-infected (MOI ≈ 0.1) for 96 h. Cultures were fixed and stained for either MxA/B (A), pSTAT1 (C), or pSTAT2 (E) (red); RSV was detected using a fluorescein isothiocyanate (FITC)-conjugated anti-RSV-F-specific antibody (green). Confocal images show typical staining from WD-PBECs derived from three different individual donors (original magnification, ×63). The fluorescence of MxA/B (B), pSTAT1 (D), and pSTAT2 (F) in RSV-infected and uninfected cells within RSV-infected WD-PBEC cultures was quantified by dividing the raw integrated density by the area of >120 individual cells. The fluorescence was quantified by using ImageJ software. The data are presented as means ± the SEM. ***, P < 0.0005.
DISCUSSION
Innate immune responses are critical first lines of defense against virus infection (38, 39). The capacity for viruses to antagonize or circumvent these responses is essential for their successful replication. Our RSV/WD-PBEC model provided a unique opportunity to study the induction and antagonism of innate antiviral immune responses by RSV in a morphologically and physiologically authentic model of pediatric bronchial epithelium. We exploited the fact that RSV infection does not lead to gross destruction of WD-PBECs to establish superinfection experiments with rSeV/eGFP. rSeV/eGFP was particularly useful for these experiments since it replicates very efficiently in untreated WD-PBECs but is sensitive to human IFN (28, 31). These characteristics provided the unique opportunity to establish a novel bioassay to study RSV-induced antiviral responses in WD-PBECs based on SeV-derived eGFP expression and SeV growth kinetics.
Our rSeV/eGFP superinfection bioassay unambiguously demonstrated that RSV induced a potent antiviral response in WD-PBECs and that basolaterally secreted factors were, in part, responsible for this antiviral activity. Importantly, IFN-λs, particularly IL-29, but not IFN-α/β, were detected in RSV-infected WD-PBECs. This is consistent with Okabayashi et al. (15), who showed a predominance of IL-29 secretion from RSV-infected primary and immortalized nasal epithelial cell monolayers. A major finding of our study is the preponderance of IL-29 and the absence of detectable IFN-α/β in lower airway samples from RSV-infected infants, suggesting that IFN-λs, and in particular IL-29, are the principal IFNs responding to RSV infection in vitro and in vivo. The lack of detectable IFN-α/β in CMRSV, as reported previously (1), and in lower airway samples from infants hospitalized with RSV, as reported here, is consistent with earlier clinical observations (11–13). It is unclear whether this lack of detectable IFN-α/β in CMRSV is due to a failure to stimulate these responses and/or active antagonism of induction by RSV NS1/2. Indeed, using immortalized cell lines, RSV NS proteins were shown to antagonize IFN-α/β induction by interacting with RIG-I, disrupting association of IRF-3 with CBP and, thereby, IRF-3 binding to the IFN-β promoter, and suppressing activation and nuclear translocation of IRF-3 (19, 40, 41), They also antagonized IFN-α/β signaling by inducing proteasome-mediated degradation of pSTAT2 (19, 37). In addition, RSV NS1 and, to a lesser extent, NS2 were shown to decrease cellular levels of TRAF3 and IKKϵ, both key members of the IFN response pathway (42). However, Killip et al. recently demonstrated that even when viral IFN-α/β antagonists were deleted, the paramyxovirus PIV5 failed to activate the IFN-β promoter, suggesting that members of the Paramyxoviridae are very inefficient at inducing IFN-α/β responses (43). The corollary, however, is that the IFN antagonistic capacities of these viruses likely evolved to cope with IFN-λs, rather than IFN-α/β, responses.
Therefore, we evaluated the capacity of RSV NS1/2 to antagonize IFN-λ1/IL-29-mediated antiviral effects. We demonstrated that NS1 and/or NS2 were essential for RSV resistance to IL-29-mediated antiviral activity in Vero cells and for replication of RSV in WD-PBECs. At a cellular level in WD-PBECs, we showed that RSV infection inhibited the IFN-inducible Jak/STAT pathway through pSTAT2 suppression, with concomitant reduction of the expression of the IFN-induced antiviral GTPases, MxA/B (18). However, this inhibition in WD-PBECs was restricted to infected cells. Since MxA and MxB are reliable markers of IFN bioactivity and IL-29 was the only IFN detected in CMRSV, our cumulative data are consistent with a model in which RSV infection induces IL-29-mediated antiviral activity in WD-PBECs but that RSV NS1/2 proteins efficiently antagonize these responses only in infected cells through pSTAT2 degradation. Although IFN-α/β were not detected in our assays, the possibility remains that very low biologically active levels were present. Further work is therefore needed to definitively exclude their role in RSV-induced antiviral responses in WD-PBECs.
There is an increasing body of evidence confirming the capacity of IL-29 to induce antiviral states in infected cells (5, 8, 44). Our data extend this IL-29 capacity to SeV and RSV. We found no evidence that IL-29 has therapeutic potential against RSV infection. However, we present evidence that IL-29 has prophylactic potential against RSV, as demonstrated by retarded RSV growth kinetics in IL-29 pretreated WD-PBECs compared to untreated controls. These data provide the rationale for further studies on IL-29 prophylaxis to modulate RSV pathogenesis. This is of particular interest for individuals at risk for severe illness due to RSV infection, although more work is needed to better understand IL-29 responses in vivo. Moreover, the IL-29-mediated antiviral effects against RSV, combined with the tissue restriction of the type III IFN receptor to epithelial cells, the liver, and some leukocytes, suggest that IL-29 prophylaxis may result in limited toxicities that are typical of type I IFN therapy (45). Indeed, early data from clinical trials using IFN-λ to treat chronic hepatitis C virus support this possibility (46).
Our evidence suggests the exciting prospect of potentially novel potent antiviral molecules in CMRSV that are not explained by its IL-29 content alone. Neutralizing IL-29 eliminated a large portion of the antiviral activity of CMRSV. However, CMRSV demonstrated greater antiviral potency than 1 ng of recombinant IL-29/ml, which represents an ∼25-fold increase relative to the mean IL-29 concentration in CMRSV. Therefore, it is possible that other molecules in CMRSV act in synergy with IL-29 to exert its impressive antiviral activities. Indeed, such synergistic antiviral activity was previously reported for IFN-α/β and IFN-γ against herpes simplex virus 1, hepatitis C virus, and severe acute respiratory syndrome-associated coronavirus (47–49). However, further work is required to identify such molecules and determine whether such synergy is evident in our WD-PBEC model.
Finally, there is increasing molecular diagnostic evidence demonstrating concomitant dual or multiple respiratory viral infections in individuals (50, 51). Debate is ongoing as to whether such dual/multi-infections result in exacerbated disease compared to mono-infections. Extrapolation of our data to the clinic suggests that a primary infection with RSV would result in the induction of an antiviral state in the airway epithelium that may greatly compromise the capacity of subsequent viruses to infect and replicate, unless the second virus was adept at circumventing the pre-established innate immune responses. This is consistent with a recent study showing that infection with multiple respiratory viruses correlated with less severe disease (52).
In conclusion, our study significantly advances our understanding of RSV induction and antagonism of type III IFN responses in human airway epithelium. Importantly, it provides the rationale for dissecting the molecular mechanisms by which these occur and the possible exploitation of IL-29 as a novel RSV prophylactic, either alone or in combination with other, yet-to-be discovered CMRSV antiviral molecules.
ACKNOWLEDGMENTS
We are grateful to the children and parents who consented to participate in the present study. M.N.T. thanks Peter Collins (NIAID) for the use of the RSV reverse genetics system.
Funding was provided by the Public Health Agency HSC Research and Development Division (Northern Ireland), the European Social Fund, Northern Ireland Chest Heart and Stroke, and the Royal Belfast Hospital for Sick Children.
REFERENCES
- 1.Villenave R, Thavagnanam S, Sarlang S, Parker J, Douglas I, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2012. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc Natl Acad Sci U S A 109:5040–5045. doi: 10.1073/pnas.1110203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo-Parke H, Canning P, Douglas I, Villenave R, Heaney LG, Coyle PV, Lyons JD, Shields MD, Power UF. 2013. Relative respiratory syncytial virus cytopathogenesis in upper and lower respiratory tract epithelium. Am J Respir Crit Care Med 188:842–851. doi: 10.1164/rccm.201304-0750OC. [DOI] [PubMed] [Google Scholar]
- 3.Glezen WP, Taber LH, Frank AL, Kasel JA. 1986. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child 140:543–546. [DOI] [PubMed] [Google Scholar]
- 4.Chanock R, Roizman B, Myers R. 1957. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). I. Isolation, properties and characterization. Am J Hyg 66:281–290. [DOI] [PubMed] [Google Scholar]
- 5.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. 2003. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 6.Zorzitto J, Galligan CL, Ueng JJM, Fish EN. 2006. Characterization of the antiviral effects of interferon-alpha against a SARS-like coronavirus infection in vitro. Cell Res 16:220–229. doi: 10.1038/sj.cr.7310030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samuel CE. 2001. Antiviral actions of interferons. Clin Microbiol Rev 14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. 2006. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol 80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jewell NA, Cline T, Mertz SE, Smirnov SV, Flaño E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, Durbin JE. 2010. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol 84:11515–11522. doi: 10.1128/JVI.01703-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS, Bakaletz LO, Durbin RK, Flaño E, Durbin JE. 2007. Differential type I interferon induction by respiratory syncytial virus and influenza A virus in vivo. J Virol 81:9790–9800. doi: 10.1128/JVI.00530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall CB, Douglas RG Jr, Simons RL, Geiman JM. 1978. Interferon production in children with respiratory syncytial, influenza, and parainfluenza virus infections. J Pediatr 93:28–32. doi: 10.1016/S0022-3476(78)80594-0. [DOI] [PubMed] [Google Scholar]
- 12.Melendi GA, Coviello S, Bhat N, Zea-Hernandez J, Ferolla FM, Polack FP. 2010. Breastfeeding is associated with the production of type I interferon in infants infected with influenza virus. Acta Paediatr 99:1517–1521. doi: 10.1111/j.1651-2227.2010.01862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scagnolari C, Midulla F, Pierangeli A, Moretti C, Bonci E, Berardi R, De Angelis D, Selvaggi C, Di Marco P, Girardi E, Antonelli G. 2009. Gene expression of nucleic acid-sensing pattern recognition receptors in children hospitalized for respiratory syncytial virus-associated acute bronchiolitis. Clin Vaccine Immunol 16:816–823. doi: 10.1128/CVI.00445-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witte K, Witte E, Sabat R, Wolk K. 2010. IL-28A, IL-28B, and IL-29: promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev 21:237–251. doi: 10.1016/j.cytogfr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Okabayashi T, Kojima T, Masaki T, Yokota S-I, Imaizumi T, Tsutsumi H, Himi T, Fujii N, Sawada N. 2011. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res 160:360–366. doi: 10.1016/j.virusres.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Donnelly RP, Kotenko SV. 2010. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res 30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, Yao L, Liu H, Barahmand-pour F, Sivakumar P, Chan C, Birks C, Foster D, Clegg CH, Wietzke-Braun P, Mihm S, Klucher KM. 2006. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 18.Haller O, Kochs G. 2011. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J Interferon Cytokine Res 31:79–87. doi: 10.1089/jir.2010.0076. [DOI] [PubMed] [Google Scholar]
- 19.Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, Buick R, Stevenson NJ, Touzelet O, Gadina M, Power UF, Johnston JA. 2007. Respiratory syncytial virus NS1 protein degrades STAT2 by using the elongin-cullin E3 ligase. J Virol 81:3428–3436. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ling Z, Tran KC, Teng MN. 2009. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J Virol 83:3734–3742. doi: 10.1128/JVI.02434-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spann KM, Tran K-C, Chi B, Rabin RL, Collins PL. 2004. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J Virol 78:4363–4369. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Touzelet O, Loukili N, Pelet T, Fairley D, Curran J, Power UF. 2009. De novo generation of a non-segmented negative-strand RNA virus with a bicistronic gene. Virus Res 140:40–48. doi: 10.1016/j.virusres.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 23.Villenave R, O'Donoghue D, Thavagnanam S, Touzelet O, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2011. Differential cytopathogenesis of respiratory syncytial virus prototypic and clinical isolates in primary pediatric bronchial epithelial cells. Virol J 8:43. doi: 10.1186/1743-422X-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Techaarpornkul S, Barretto N, Peeples ME. 2001. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J Virol 75:6825–6834. doi: 10.1128/JVI.75.15.6825-6834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins PL, Hill MG, Camargo E, Grosfeld H, Chanock RM, Murphy BR. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine. Proc Natl Acad Sci U S A 92:11563–11567. doi: 10.1073/pnas.92.25.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webster Marketon JI, Corry J, Teng MN. 2014. The respiratory syncytial virus (RSV) nonstructural proteins mediate RSV suppression of glucocorticoid receptor transactivation. Virology 449:62–69. doi: 10.1016/j.virol.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Power UF, Plotnicky-Gilquin H, Huss T, Robert A, Trudel M, Stahl S, Uhlen M, Nguyen TN, Binz H. 1997. Induction of protective immunity in rodents by vaccination with a prokaryotically expressed recombinant fusion protein containing a respiratory syncytial virus G protein fragment. Virology 230:155–166. doi: 10.1006/viro.1997.8465. [DOI] [PubMed] [Google Scholar]
- 28.Villenave R, Touzelet O, Thavagnanam S, Sarlang S, Parker J, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2010. Cytopathogenesis of Sendai virus in well-differentiated primary pediatric bronchial epithelial cells. J Virol 84:11718–11728. doi: 10.1128/JVI.00798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heaney LG, Stevenson EC, Turner G, Cadden IS, Taylor R, Shields MD, Ennis M. 1996. Investigating paediatric airways by non-bronchoscopic lavage: normal cellular data. Clin Exp Allergy 26:799–806. doi: 10.1046/j.1365-2222.1996.d01-385.x. [DOI] [PubMed] [Google Scholar]
- 30.Coyle PV, Ong GM, O'Neill HJ, McCaughey C, De Ornellas D, Mitchell F, Mitchell SJ, Feeney SA, Wyatt DE, Forde M, Stockton J. 2004. A touchdown nucleic acid amplification protocol as an alternative to culture backup for immunofluorescence in the routine diagnosis of acute viral respiratory tract infections. BMC Microbiol 4:41. doi: 10.1186/1471-2180-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bousse T, Chambers RL, Scroggs RA, Portner A, Takimoto T. 2006. Human parainfluenza virus type 1 but not Sendai virus replicates in human respiratory cells despite IFN treatment. Virus Res 121:23–32. doi: 10.1016/j.virusres.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Taylor CE, Webb MS, Milner AD, Milner PD, Morgan La, Scott R, Stokes GM, Swarbrick aS, Toms GL. 1989. Interferon alfa, infectious virus, and virus antigen secretion in respiratory syncytial virus infections of graded severity. Arch Dis Child 64:1656–1660. doi: 10.1136/adc.64.12.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lo MS, Brazas RM, Holtzman MJ. 2005. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol 79:9315–9319. doi: 10.1128/JVI.79.14.9315-9319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin H, Zhou H, Cheng X, Tang R, Munoz M, Nguyen N. 2000. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2-2 genes are attenuated in vitro and in vivo. Virology 273:210–218. doi: 10.1006/viro.2000.0393. [DOI] [PubMed] [Google Scholar]
- 35.Stoltz M, Klingström J. 2010. Alpha/beta interferon (IFN-alpha/beta)-independent induction of IFN-lambda1 (interleukin-29) in response to Hantaan virus infection. J Virol 84:9140–9148. doi: 10.1128/JVI.00717-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova J-L, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol 81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. 2006. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 344:328–339. doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 40.Ren J, Liu T, Pang L, Li K, Garofalo RP, Casola A, Bao X. 2011. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J Gen Virol 92:2153–2159. doi: 10.1099/vir.0.032987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spann KM, Tran KC, Collins PL. 2005. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-κB, and proinflammatory cytokines. J Virol 79:5353–5362. doi: 10.1128/JVI.79.9.5353-5362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swedan S, Musiyenko A, Barik S. 2009. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J Virol 83:9682–9693. doi: 10.1128/JVI.00715-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Killip MJ, Young DF, Ross CS, Chen S, Goodbourn S, Randall RE. 2011. Failure to activate the IFN-β promoter by a paramyxovirus lacking an interferon antagonist. Virology 415:39–46. doi: 10.1016/j.virol.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte J-M, Diebold J, Diepolder H, Adler B, Auernhammer CJ, Göke B, Dambacher J. 2005. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am J Physiol Gastrointest Liver Physiol 289:G960–G968. doi: 10.1152/ajpgi.00126.2005. [DOI] [PubMed] [Google Scholar]
- 45.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog 4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donnelly RP, Dickensheets H, O'Brien TR. 2011. Interferon-lambda and therapy for chronic hepatitis C virus infection. Trends Immunol 32:443–450. doi: 10.1016/j.it.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larkin J, Jin L, Farmen M, Venable D, Huang Y, Tan S-L, Glass JI. 2003. Synergistic antiviral activity of human interferon combinations in the hepatitis C virus replicon system. J Interferon Cytokine Res 23:247–257. doi: 10.1089/107999003321829962. [DOI] [PubMed] [Google Scholar]
- 48.Sainz B, Halford WP. 2002. Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J Virol 76:11541–11550. doi: 10.1128/JVI.76.22.11541-11550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scagnolari C, Trombetti S, Alberelli A, Cicetti S, Bellarosa D, Longo R, Spanò A, Riva E, Clementi M, Antonelli G. 2007. The synergistic interaction of interferon types I and II leads to marked reduction in severe acute respiratory syndrome-associated coronavirus replication and increase in the expression of mRNAs for interferon-induced proteins. Intervirology 50:156–160. doi: 10.1159/000098242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huguenin A, Moutte L, Renois F, Leveque N, Talmud D, Abely M, Nguyen Y, Carrat F, Andreoletti L. 2012. Broad respiratory virus detection in infants hospitalized for bronchiolitis by use of a multiplex RT-PCR DNA microarray system. J Med Virol 84:979–985. doi: 10.1002/jmv.23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richard N, Komurian-Pradel F, Javouhey E, Perret M, Rajoharison A, Bagnaud A, Billaud G, Vernet G, Lina B, Floret D, Paranhos-Baccalà G. 2008. The impact of dual viral infection in infants admitted to a pediatric intensive care unit associated with severe bronchiolitis. Pediatr Infect Dis J 27:213–217. doi: 10.1097/INF.0b013e31815b4935. [DOI] [PubMed] [Google Scholar]
- 52.Martin ET, Kuypers J, Wald A, Englund JA. 2012. Multiple versus single virus respiratory infections: viral load and clinical disease severity in hospitalized children. Influenza Other Respir Viruses 6:71–77. doi: 10.1111/j.1750-2659.2011.00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]