

ABSTRACT
Hepatitis C virus (HCV) envelope glycoproteins E1 and E2 form a heterodimer and mediate receptor interactions and viral fusion. Both E1 and E2 are targets of the neutralizing antibody (NAb) response and are candidates for the production of vaccines that generate humoral immunity. Previous studies demonstrated that N-terminal hypervariable region 1 (HVR1) can modulate the neutralization potential of monoclonal antibodies (MAbs), but no information is available on the influence of HVR2 or the intergenotypic variable region (igVR) on antigenicity. In this study, we examined how the variable regions influence the antigenicity of the receptor binding domain of E2 spanning HCV polyprotein residues 384 to 661 (E2661) using a panel of MAbs raised against E2661 and E2661 lacking HVR1, HVR2, and the igVR (Δ123) and well-characterized MAbs isolated from infected humans. We show for a subset of both neutralizing and nonneutralizing MAbs that all three variable regions decrease the ability of MAbs to bind E2661 and reduce the ability of MAbs to inhibit E2-CD81 interactions. In addition, we describe a new MAb directed toward the region spanning residues 411 to 428 of E2 (MAb24) that demonstrates broad neutralization against all 7 genotypes of HCV. The ability of MAb24 to inhibit E2-CD81 interactions is strongly influenced by the three variable regions. Our data suggest that HVR1, HVR2, and the igVR modulate exposure of epitopes on the core domain of E2 and their ability to prevent E2-CD81 interactions. These studies suggest that the function of HVR2 and the igVR is to modulate antibody recognition of glycoprotein E2 and may contribute to immune evasion.
IMPORTANCE This study reveals conformational and antigenic differences between the Δ123 and intact E2661 glycoproteins and provides new structural and functional data about the three variable regions and their role in occluding neutralizing and nonneutralizing epitopes on the E2 core domain. The variable regions may therefore function to reduce the ability of HCV to elicit NAbs directed toward the conserved core domain. Future studies aimed at generating a three-dimensional structure for intact E2 containing HVR1, and the adjoining NAb epitope at residues 412 to 428, together with HVR2, will reveal how the variable regions modulate antigenic structure.
INTRODUCTION
Hepatitis C virus (HCV) infects between 150 million to 200 million people worldwide and is now the leading indicator for liver transplants in developed countries. While direct-acting antiviral drugs have elevated the sustained virological response rate, their high cost and the need to identify those people infected with HCV remain major impediments to their widespread use to eradicate HCV. Vaccines remain the most effective way to prevent the spread of infectious diseases, yet there is no prophylactic vaccine for HCV. One of the major limitations to the design of an HCV vaccine is the need to afford protection against the 7 circulating genotypes and the >67 subtypes, which differ by up to 30% and 20%, respectively, at the nucleotide level.
Neutralizing antibodies (NAbs) are key components of all available vaccines. Both polyclonal and monoclonal NAbs can prevent HCV infection of experimental animals and have been implicated in playing a major role in viral clearance in natural HCV infection (1–6). The major target of the antibody response to HCV infection is glycoprotein E2, which mediates direct protein-protein interactions with tetraspanin CD81 and scavenger receptor class B type I (7, 8). The receptor-binding domain (RBD) of E2 extends from HCV polyprotein residues 384 to 661 (E2661) (9) and contains 4 discrete regions involved in CD81 binding as well as three hypervariable regions (HVRs) (Fig. 1) (9). Hypervariable region 1 is located at the N terminus of E2 and elicits type-specific NAbs with little ability to cross-neutralize heterologous strains (10). A function of HVR1 may be to modulate the exposure of the CD81-binding site and the ability of antibodies to mediate the neutralization of HCV (11). Hypervariable region 2 (HVR2) and the intergenotypic variable region (igVR) form surface-exposed loops but do not represent targets of the NAb response (Fig. 1A and B). All three variable regions can be deleted from intact wild-type (WT) E2661 to yield a minimized form of the glycoprotein (Δ123) that retains NAb epitopes and the ability to bind CD81 (12) (Fig. 1A).
FIG 1.

(A) Schematic representation of full-length E2, E2661, and E2661 variants with deletions of HVR1 (Δ1), HVR2 (Δ2), the igVR (Δ3), or combinations thereof (Δ12, Δ13, Δ23, and Δ123). HVR2 and the igVR were replaced with a GSSG linker. Numbering is done according to the H77c prototype strain. Epitope I, II, and III regions are underlined on the E2 structure and overlap CD81 binding sites, shown in blue, orange, and green. A fourth region (yellow) is also implicated in CD81 interactions. Hypervariable region 1, HVR2, and the igVR are shown in red. The transmembrane domain and the C-terminal stem region are shown in black and gray, respectively, on the full-length E2 schematic. (B) Cartoon drawing of the E2 core domain with its surface overlaid (PDB accession number 4MWF) (13). Coloring is according to that described above for panel A. The predicted location of the region spanning residues 411 to 420 (purple) that overlaps epitope I and precedes HVR1 is shown.
Recently, two crystal structures of an E2 core domain in complex with monoclonal antibodies (MAb) were solved to reveal the conformation of the CD81 binding site on the neutralizing face of the glycoprotein. Unlike glycoprotein E of the phylogenetically related flaviviruses, HCV E2 does not have a three-domain architecture typical of class II fusion proteins. Instead, E2 adopts a compact globular immunoglobulin-like fold comprising a central β sandwich surrounded by short front and back layers comprising loops, short helices, and β sheets (13, 14). The immunoglobulin sandwich is formed by 4 β strands that form an inner sheet and 2 solvent-exposed β strands that comprise the outer sheet. A loop connecting the inner and outer sheets contains many of the key CD81 binding residues and is adjacent to the front layer, where additional surface-exposed CD81 contact residues are found (Fig. 1B). This region of E2 was termed the neutralizing face, as many of the NAbs directed toward E2 bind this region and have the ability to inhibit CD81 binding. The igVR forms a disulfide-constrained loop within a flexible region spanning residues 567 to 596. However, three-dimensional (3D) structural information was not obtained for HVR1 or the adjacent epitope spanning residues 412 to 421 recognized by broadly neutralizing E2-CD81-blocking antibodies. HVR2, also absent from the E2 core domain crystal structures, resides within a highly flexible region on the nonneutralizing face of E2. In the case of the region spanning residues 412 to 421, structural information has been obtained by using MAbs bound to peptide analogs, revealing that it can adopt multiple conformations and suggesting a degree of conformational flexibility (15–18).
We previously proposed that the three variable regions could shield the underlying E2 core domain, providing a mechanism whereby the virus can modulate the exposure of conserved neutralization epitopes (12). In this study, we produced and characterized a panel of MAbs to E2661 and Δ123 and used well-characterized human MAbs in order to examine how the variable regions modulate the exposure of both neutralizing and nonneutralizing epitopes on E2 and examined antigenic differences between intact E2 and the Δ123 core domain. Our results indicate that Δ123 has different antigenic properties from those of intact E2661, related to the absence of three variable regions, and suggest that the variable regions can occlude the underlying CD81 binding site on the conserved core domain. In addition, our data suggest that there is an interaction between HVR1, HVR2, and the igVR that is not predicted from the three-dimensional structure of the E2 core domain monomer. These data suggest that all three variable regions alter both the structure and the accessibility of the CD81 binding site on E2 and modulate the presentation of neutralizing and nonneutralizing epitopes in E2.
MATERIALS AND METHODS
Vectors.
The codon-optimized DNA sequence encoding H77c E2661 was synthesized (GeneArt, Invitrogen, CA, USA) and contained an N-terminal human trypsin leader sequence and a C-terminal 6×His tag. The H77c E2661 clones containing deletions of HVR1 (Δ1); HVR2 (Δ2); igVR (Δ3); HVR1 and HVR2 (Δ12); HVR1 and the igVR (Δ13); HVR2 and the igVR (Δ23); or HVR1, HVR2, and the igVR (Δ123) were constructed by using overlap extension PCR. The region encoding HVR1 (residues 387 to 408) was deleted, while the regions encoding HVR2 (residues 460 to 485) and the igVR (residues 570 to 580) were replaced with a GSSG linker sequence. A codon-optimized DNA sequence encoding H77c E2661 was used as the template, and the products were cloned into pcDNA3.1 and sequenced by using BigDye Terminator chemistry. The DNA sequences encoding E2661 of H77c (GenBank accession number AF009606) (genotype 1a [G1a]), Con1 (accession number AJ238799) (G1b), JFH1 (accession number AB047639) (G2a), J6 (accession number AF177036) (G2a), S52 (accession number GU814263) (G3a), ED43 (accession number GU814265) (G4a), SA13 (accession number AF064490) (G5a), and EUHK2 (accession number Y12083) (G6a) were synthesized (GeneArt, Invitrogen, CA, USA) and cloned into a pcDNA3 expression vector with an N-terminal leader sequence and a C-terminal 6×His tag, as described previously (19).
For epitope mapping using H77c E2661 proteins, 36 single amino acids (implicated in CD81 binding or NAb recognition) were mutated to alanine or another amino acid. Twenty-seven mutations were generated by overlap extension PCR using the codon-optimized DNA sequence encoding H77c E2661 to generate single point mutants of E2661 proteins, including Q412A, L413A, N415A, N417A, G418A, W420A, W420F, H421A, N423A, S424A, L427A, N430A, W437F, G523A, P525A, P525G, Y527A, W529A, W529F, G530A, D535A, N540A, W549A, W549F, Y613A, W616A, and W616F, and contained a C-terminal 6×His tag. Previously described E2661-Myc constructs with the mutations G436A, W437A, L438A, A439P, A439S, G440A, L441A, F442A, and Y443A were reamplified such that the C-terminal Myc epitope tag was replaced with a 6×His sequence and sequenced by using BigDye Terminator chemistry (20).
Expression vectors for the production of infectious retroviruses pseudotyped with E1E2 heterodimers (HCVpp) from G1a (pE1E2H77c) were constructed as described previously (21). Cell culture-derived HCV (HCVcc) was produced from full-length in vitro-transcribed RNA transfected into human hepatoma Huh7.5 cells from G3a [S52/JFH1(T2701G,A4533C)], G4a [(ED43)-RLucΔ40], G5a [(SA13)-RLucΔ40], G6a [(HK6a)-RLucΔ40], G7a [(QC69)-RLucΔ40] (22) (kind gifts from Jens Bukh), and G2a [pJC1FLAG2(p7-NS-GLUC2A)] (kind gift from Charles Rice) (23).
pcNL4.3GagPolVpuRRE was prepared as follows. The HindIII (nucleotide [nt] 530)-KpnI (nt 6343) fragment of pNL4.3 (GenBank accession number AF324493.2) (24), encompassing the gagpol-vif-vpr-vpu region, was ligated into the corresponding restriction sites of pcDNA3.1 to give pcNL4.3GagPolVVV. Three tandem termination codons were introduced immediately 3′ of the NdeI site present in vif (nt 5121 [underlined]) using pNL4.3 as the template and the primers 5′-TTCCATATGTAATGATAGAGGAAAGCTAAGGAC and 5′-TTTTCTGGATCCCTACAGATCATCAATATCCCAAGG in a PCR. The mutated PCR product (spanning the vif-vpr-vpu region) was end filled by using the Klenow fragment of Escherichia coli DNA polymerase I and ligated into the NdeI-EcoRV sites present in pcNL4.3GagPolVVV. A termination codon was then introduced immediately 3′ of the initiation codon of vpr in pcNL4.3GagPolVVV by the Quickchange XLII mutagenesis method (Agilent Technologies, CA, USA) using the primers 5′-GGAAACTGACAGAGGACAGATGTAATAAGCCCCAGAAGACCAAGGGCCAC and 5′-GTGGCCCTTGGTCTTCTGGGGCTTATTACATCTGTCCTCTGTCAGTTTCC to give pcGagPolVpu. Finally, the Rev-responsive element was PCR amplified by using pNL4.3 as the template and the primers 5′-AAAAGGCCTTCTAGACCAAGGCAAAGAG and 5′-AAAAGGCCTTCTAGAAGCATTCCAAGGCAC and ligated into the unique XbaI site of pcNL4.3GagPolVpu to give pcNL4.3GagPolVpuRRE.
The vector used to express the large extracellular loop (LEL) residues 113 to 201 of CD81 as a maltose-binding protein (MBP) fusion (MBP-LEL113–201) was previously described (19).
Cell lines, transfections, and protein expression.
Human embryonic kidney HEK293T cells were maintained in Dulbecco's modified Eagle medium (Invitrogen, CA, USA) containing 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen, CA, USA), 2 mM l-glutamine (GE Healthcare, United Kingdom), 1 M HEPES buffer solution (Invitrogen, MA, USA), 0.1 mg/ml gentamicin antibiotic (Invitrogen, CA, USA), and 1 μg/ml minocycline hydrochloride salt (Sigma-Aldrich, MO, USA) (DMF10). Huh7.5 cells were used for infection assays and maintained in DMF10 medium for HCVpp or in DMF10 medium supplemented with 10 mM nonessential amino acids (DMF10NEAA) for HCVcc. Cells were incubated at 37°C with 5% CO2. Transfections were performed by using Fugene 6 (Promega, Madison, WI, USA) or calcium phosphate for DNA or DMRIE-C (Life Technologies, CA, USA) for RNA, according to the manufacturer's recommendations.
Virus-like particles (VLPs) were produced by transfecting HEK293T monolayers with pE1E2H77c, pNL4.3GagPolVpuRRE, and pCMV-rev (NIH AIDS Reagent Program) at a 1:1:0.6 ratio of DNA using polyethylenimine (PEI; Polysciences, Inc., USA).
E2661 glycoproteins and variants of E2661 containing variable region deletions were expressed in HEK293T cells as described previously (12, 19, 20). Alternatively, glycoproteins were produced by transfecting 25 μg of plasmid DNA in 160 μl of 2 M calcium phosphate in HEPES-buffered saline (pH 7.0) and applied to 80% confluent HEK293T cells in 175-cm2 flasks. Tissue culture medium was replaced 24 h later with Opti-MEM (Life Technologies, CA, USA), and tissue culture fluid containing secreted glycoproteins was harvested daily for up to 5 days and concentrated in a Amicon YM30 ultrafiltration device (Merck-Millipore, Darmstadt, Germany). Alternatively, 105 μg PEI was mixed with 35 μg of DNA and applied to 80% confluent HEK293T cells in T175 flasks. The cell culture supernatant was replaced with Opti-MEM after 4 h of transfection. Tissue culture fluid containing the secreted glycoproteins were harvested daily for up to 5 days and concentrated with a 30,000-molecular-weight-cutoff (MWCO) centrifugal unit (Vivaproducts, MA, USA).
Antibodies.
MAbs H53, H52, and A4 were kind gifts from Jean Dubuisson and Harry Greenberg (25, 26). MAbs HC1 (27) and HC84.22 were kind gifts from Steven Foung (28). AR3C was a kind gift from Mansun Law (3). The HIV-1 p24 hybridoma (183-H12-5C) reagent (MAb183) was obtained from Bruce Chesebro through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (29).
The VH and VL domains of HC84.1 and HC84.27 were constructed by gene synthesis using sequences reported under PDB accession numbers 4JZN and 4JZO (30), respectively. The VL and VH regions were subcloned into pcDNA3-tPA-LC and pcDNA3-tPA-HC, respectively, for the expression of IgG1 under the direction of a tissue plasminogen activator (tPA) leader sequence. Antibodies were expressed by transfection of HEK293T cells and purified from the supernatant fluid by using protein G-Sepharose (PGS).
Anti-NS5A antibody from mouse hybridoma 9E10 was a kind gift from Charles Rice (31). Anti-Myc epitope tag antibody from clone 9E10 is commercially available. Rabbit anti-6×His antibody was purchased (Rockland Immunochemicals, Inc., PA, USA). Horseradish peroxidase-conjugated antibodies were purchased (Dako, Glostrup, Denmark). Fluorescently conjugated antibodies were purchased (Life Technologies, CA, USA).
Ethics.
All procedures were performed in accordance with animal ethics guidelines set out by the National Health and Medical Research Council of Australia under ethics approval numbers 996 (CSL Ltd. animal ethics committee) and E/0585/2007/F (Alfred Medical Research and Education Precinct animal ethics committee).
MAb production.
BALB/c mice were immunized with a single dose of 20 μg of H77c E2661 (MAb25 to MAb50) or Δ123 (MAb1 to MAb23) protein in Iscomatrix adjuvant. Animals were euthanized 2 weeks later, and spleens were collected to obtain lymphocytes for fusion with SP-2 myeloma cells. One MAb (MAb24) was generated by vaccinating BALB/c mice with 20 μg Δ123 in Iscomatrix adjuvant once, followed by two further 20-μg Δ123 vaccinations in alum. Three months later, the mice were injected in the tail vein once with 20 μg Δ123 in saline. One week later, the mice were euthanized, and spleens were removed for hybridoma production as described above. Primary cloning of positive hybridomas was performed by limiting dilution in media containing 100 μM hypoxanthine and 16 μM thymidine using a BALB/c feeder layer. Secondary cloning of positive hybridomas was then performed to generate 48 MAbs. Concentrated solutions of 18 MAbs were produced by using a MiniPerm apparatus. MAbs were isotyped by using the IsoStrip kit according to the manufacturer's recommendations (Roche, Mannheim, Germany).
Radioimmunoprecipitation of E1E2 and Western blotting.
Radioimmunoprecipitation (RIP) was performed as described previously (21). Briefly, HEK293T cells were transfected with plasmid pE1E2H77c and biosynthetically labeled 24 h later by using 150 μCi per well of Tran-35S-Label, and RIPs were performed as described previously (21). E1E2 glycoproteins were separated in 10 to 15% SDS-PAGE gels under reducing conditions, and radiolabeled proteins were visualized by phosphorimaging. Western blots were performed on reduced H77c E2661 run in SDS-PAGE gels and transferred onto nitrocellulose. Blots were probed with MAb, and bound antibody was detected with goat anti-mouse antibody conjugated to Alexa 680 (Life Technologies, CA, USA) by using a Li-COR Odyssey imaging system (Li-COR BioSciences).
Protein purification.
MAbs were purified by using PGS beads (Genscript, USA). MAbs bound to PGS beads were then washed three times with 1× wash buffer (0.5 M NaCl, 0.05 M Tris [pH 7.4], 1 mM EDTA, 0.02% sodium azide, and 1% Triton X-100) and three times with phosphate-buffered saline (PBS). MAbs were dissociated from the PGS beads by the addition of 0.1 M glycine buffer (pH 2.7) and immediately neutralized by the addition of 1 M Tris-HCl buffer (pH 8.0). The concentration of MAbs was determined by using the Micro-BCA protein estimation kit according to the manufacturer's recommendations (Thermo Scientific, Rockford, IL, USA).
Secreted E2661 protein in tissue culture fluid was purified by immobilized metal affinity chromatography using nickel Sepharose (GE Healthcare Life Sciences, United Kingdom) according to the manufacturer's instructions. Eluted proteins were dialyzed into PBS, and their concentrations were determined by using the Bradford assay with bovine serum albumin (BSA) as a standard curve (32). Recombinant dimeric MBP-LEL113–201 was purified as described previously (19).
For VLPs, tissue culture fluid was collected at 72 h posttransfection, and virions were pelleted through a 25% sucrose cushion and resuspended in PBS. VLPs were assessed via Western blotting after protein separation through a 7.5 to 15% polyacrylamide gradient gel using polyclonal HIV IgG purified from HIV-1-infected individuals, and HCV envelope protein expression was assessed by using anti-E1 (A4) and anti-E2 (H52). Immunoblots were developed by using IRDye 800CW-conjugated rabbit anti-human Ig or Alexa Fluor 680-conjugated goat anti-mouse Ig and scanned by using a Li-COR Odyssey infrared imager.
Direct binding enzyme-linked immunosorbent assays.
The reactivity of MAbs to E2 antigens was tested by enzyme-linked immunosorbent assays (ELISAs) using 96-well Maxisorb plates (Nunc, Roskilde, Denmark) coated with 5 μg/ml of Galanthus nivalis lectin (GNA-lectin; Sigma, St. Louis, MO, USA) and incubated overnight at 4°C. Unoccupied sites were blocked for 1 h at room temperature (RT) with blocking buffer (1% BSA [Sigma, St. Louis, MO, USA] in PBS). Glycoprotein E2 (5 μg/ml) was then added in diluent buffer (0.5% BSA in PBS containing 0.05% Tween 20 [BSA5PBST]) and incubated for 2 h at RT. After washing, serial dilutions of MAbs or rabbit anti-His tag antibody in BSA5PBST were applied for 1 h at RT. Bound MAbs were detected by using horseradish peroxidase (HRP)-labeled rabbit anti-mouse or HRP-labeled goat anti-rabbit in BSA5PBST for 1 h at RT, after which plates were developed with the 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Sigma, St. Louis, MO, USA). The optical densities at 620 nm and 450 nm were measured with a FLUOstar Optima microplate reader (BMG Lab Technologies, Germany).
For ELISAs using VLPs, ELISA plates were coated directly with VLPs in PBS overnight. Unoccupied sites were blocked with PBS containing 1% BSA and 1% skim milk for 1 h at RT. After washes with PBS, MAbs were applied, titrated 0.5 log10, and incubated for 2 h at RT. After washes with PBS, rabbit-anti mouse HRP-labeled antibody was added for 1 h at RT, washed with PBS, and then detected with TMB as described above. To detect capsid protein within VLPs, after VLPs were bound to plates, they were permeabilized with 1% Triton X-100 for 15 min. Capsid protein was detected with MAb183, and the ELISA experiment was completed as described above.
Overlapping synthetic 18-mer peptides were used to search for linear epitopes recognized by the MAbs. The 39 overlapping peptides spanning residues 384 to 662 (McKesson Bioservices Corporation, USA) overlap by 11 amino acids and correspond to the sequence of the H77 isolate. A synthetic peptide corresponding to the HVR1 sequence of the H strain, E384THVTGGSAGRTTAGLVGLLTPGAKQN410, was additionally used to search for anti-HVR1 MAbs (Auspep). ELISA plates were coated directly with each peptide overnight, followed by blocking for 1 h at RT. After four washes with PBST, a single dilution of MAb was added to each peptide-coated well and incubated for 1 h at RT. Bound MAb was detected by using HRP-labeled rabbit anti-mouse antibody and the TMB substrate as described above. Alanine scanning of the MAb24 epitope, covering E2 amino acid residues 411 to 428, was performed by using 18 peptides containing single alanine or serine (position 424) substitutions (Mimotopes, USA). The ELISA was performed as described above.
CD81-E2 binding and binding inhibition assay.
Solid-phase immunoassay plates were coated with the dimeric MBP-LEL113–201 protein (19) (5 μg/ml) in PBS overnight, followed by blocking buffer for 1 h at RT. For measurement of direct binding of E2 to CD81, E2661 glycoproteins were titrated 0.5 log10 before addition to ELISA plates coated with CD81 and incubated for 1 h at RT. In the case of CD81-E2 binding inhibition assays, MAbs were titrated 0.5 log10 in diluent and admixed with 50 ng E2661 glycoproteins, and the antibody-antigen mixtures were added to plates coated with CD81 and incubated for 1 h at RT. Bound E2 was detected by using rabbit anti-His tag antibody for 1 h at RT, followed by HRP-labeled goat anti-rabbit for 1 h at RT and the TMB substrate, as described above.
Neutralization assays.
The production of infectious HCVpp was performed as described previously (21). Briefly, HEK293T cells were cotransfected with 1 μg each of pE1E2H77c and pNL4-3.LUC.R-E. Seventy-two hours later, tissue culture fluid was collected and filtered through a 0.45-μm syringe filter. To perform NAb assays, serial dilutions of MAb were added to HCVpp and incubated for 1 h at 37°C before addition to Huh7.5 cells seeded 24 h earlier at 30,000 cells/well in 48-well plates. After 4 h of incubation at 37°C, the inoculum was removed and replaced with DMF10NEAA for 72 h. Cells were washed with PBS before lysis in cell culture lysis buffer (Promega, Madison WI, USA). Luciferase activity in clarified lysates was measured by using a luciferase substrate (Promega, Madison, WI, USA) and a FLUOstar Optima microplate reader fitted with luminescence optics (BMG Lab Technologies, Germany).
Infectious HCVcc were produced by transfecting Huh7.5 cells with in vitro-transcribed RNA as described previously (33). Transfection was performed by using DMRIE-C reagent (Invitrogen, CA, USA). Tissue culture fluid collected 96 h later was filtered through 0.45-μm syringe filters and stored at −80°C. Neutralization assays were performed by mixing HCVcc with an equal amount of serially diluted MAb. The virus-MAb mixture was incubated for 1 h at 37°C before addition to Huh7.5 cells, seeded 24 h earlier at 30,000 cells/well in 48-well plates, for 4 h. Cells were washed 4 times, replenished with fresh DMF10NEAA, and incubated for a further 48 to 72 h. Inhibition of HCVcc entry into target cells by MAbs was determined by measuring luciferase activity.
Sequencing of the IgG-Fv region.
Total RNA was extracted from ∼1 × 106 hybridoma cells by using an RNeasy kit (Qiagen, Limburg, Netherlands). cDNAs of heavy chain (VH) and light chain (VL) variable region genes were amplified by using a Superscript III One-Step reverse transcription-PCR (RT-PCR) system with Platinum Taq DNA polymerase (Invitrogen, MA, USA). The cDNA of the heavy chain variable region was amplified by using primers 5′-AG GTS MAR CTG CAG SAG TCW GG-3′ and 5′-TGA GGA GAC GGT GAC CGT GGT CCC TTG GCC CC-3′, and the light chain variable region was amplified by using primers 5′-GGT GCA TGC GGA TAC AGT TGG TGC AGC ATC-3′ and 5′-GG GAG CTC GAY ATT GTG MTS ACM CAR WCT MCA-3′. Amplified cDNA for both the VH and VL regions was used directly for sequencing. Amino acid alignment was performed with previously reported VH and VL sequences by using ClustalW2 on the EBI website (http://www.ebi.ac.uk/Tools/msa/clustalw2/), where framework regions (FWRs) and complementarity-determining regions (CDRs) were allocated accordingly with manual adjustment (34).
Surface plasmon resonance.
All experiments were performed by using a Biacore 2000 unit (GE Healthcare Life Sciences, United Kingdom). Protein ligands were buffer exchanged into 10 mM sodium acetate (pH 4.2) prior to immobilization on a CM5 chip (GE Healthcare Life Sciences, United Kingdom) via amine-coupling methods to obtain ∼800 response units (RU). Immobilizations were performed in 1× HBS-N buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA). For all experiments, a negative-control ligand was immobilized in the preceding flow cell, or alternatively, negative-control analyte injection was used to allow subtraction for nonspecific binding. Kinetic experiments were performed at a flow rate of 10 μl/min in HBS-EP buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, 0.05% Tween 20). Protein analytes were injected into the system at 4 to 5 serial 2-fold dilutions in HBS-EP buffer with a starting concentration of 0.1 mg/ml. Injections were performed for 250 s to obtain an association rate and were allowed to dissociate for 600 s. An independent loading control at the midpoint concentration was included to ensure concentration accuracy. Regeneration between each cycle was performed at a flow rate of 100 μl/min and involved two 15-μl pulses of 100 mM phosphoric acid.
RESULTS
To examine antigenic differences between E2661 and variants of E2661 wherein HVR1, HVR2, and/or the igVR was deleted, we used 18 novel murine MAbs raised to either E2661 or Δ123 and previously characterized MAbs isolated from HCV-infected humans.
Characterization of the epitopes recognized by murine antibodies.
We first characterized the 18 MAbs raised to either Δ123 or E2661. Initially, we examined the ability of the MAbs to immunoprecipitate E1E2 heterodimers from transfected HEK293T cells. The results show that all of the MAbs were able to immunoprecipitate E2 and coprecipitate multiple different glycoforms of E1 at similar ratios (Fig. 2A). The MAbs were also tested for their ability to recognize denatured E2661 in a Western blot. MAbs 6, 13, 22, 24, 26, 33, 36, 39, and 44 bound strongly to denatured E2661, suggesting that their epitopes are contained within a continuous E2 sequence, while a small degree of binding above background was detected for MAbs 14, 23, and 25 (Fig. 2B). A library of overlapping 18-mer peptides derived from genotype 1a strain H77 was used in an ELISA to map the continuous MAb epitopes. The results show that MAb24 binds to an epitope within residues 407 to 424; MAb25, -39, and -44 bind to an epitope within residues 512 to 529; and MAb26 binds to an epitope within residues 645 to 662 (Fig. 2C). An elongated 27-residue synthetic peptide for HVR1 (residues 384 to 410) corresponding to the H sequence (differs from H77 at N391S) was also used in a direct binding ELISA with MAbs elicited by WT E2661. Only MAb36 bound strongly to the 27-mer HVR1 peptide, indicating that its epitope is contained within an HVR1 sequence not represented by the shorter peptides used in this library (Fig. 2D).
FIG 2.

Characterization of MAbs. (A) Immunoprecipitation of 35S-labeled H77c E1E2 from lysates of transfected HEK293T cells using each MAb. Immunoprecipitates were run on reducing SDS-PAGE gels and phosphorimaged. Locations of E2 and multiple glycoforms of E1 are shown on the right. Positions of molecular weight markers (M) (in thousands) are shown on the left. Quantitation was performed by using imageQuant software, and results are the means ± standard deviations of data from three experiments (bottom). The antigens to which the MAbs were raised are indicated above the gel. (B) Analysis of the ability of MAbs to recognize denatured E2661. Nickel affinity-purified E2661 was subjected to reducing SDS-PAGE and transferred onto nitrocellulose. Strips were probed with each MAb, followed by detection with anti-mouse Alexa 680-labeled antibody and infrared analysis (Li-COR Odyssey). The antigens to which the MAbs were raised are indicated above the gel. (C) Overlapping-peptide scan of antibodies reactive to denatured E2661. Synthetic 18-mers, overlapping the H77c E2 sequence by 11 amino acids, were used in a direct binding ELISA. Binding to a peptide is shown in gray and represents at least 10 times the background level. (D) Ability of MAbs raised to WT E2661 to recognize an extended HVR1 peptide of strain H. Antibodies were applied to plates coated with peptide and titrated 0.5 log10.
Further epitope mapping was performed by constructing a library of E2661 mutants targeting residues within known CD81 binding regions or previously described antibody epitopes (Table 1). Three of these regions, spanning residues 411 to 428, 430 to 451, and 523 to 549, overlap previously described antigenic regions and were designated epitope I, epitope II, and epitope III, respectively (Fig. 1A and B). The E2661 proteins were expressed in HEK293T cells and concentrated from the tissue culture fluid. The amount of E2661 used to coat ELISA plates was standardized by using anti-His antibody in ELISAs. The majority of mutations did not affect the binding of conformation-dependent antibody H53 or CBH-7, indicating that the overall global fold was maintained. The exceptions were mutations N540A and W549A, which reduced H53 and CBH-7 binding. However, mutations N540A and W549A did not affect the binding of the majority of antibodies within our panel, consistent with these amino acids being part of the H53 and CBH-7 epitopes, as described previously (35, 36). The results of E2661 mutant-MAb binding studies reveal that the binding ability of MAbs 6, 13, 14, 22, 23, 25, 39, and 44 was affected by mutations within the region spanning epitope III (Table 1). Two antibodies, MAb24 and MAb33, were affected by mutations in epitope I (Table 1). The binding of MAb26, whose epitope is contained in the region spanning residues 645 to 661, was reduced ∼50% by the G436A mutation in epitope II (Table 1). Six MAbs (MAbs 1, 7, 10, 12, and 23) could not be mapped by using this method due to poor reactivity with the relatively low concentrations of E2661 proteins used for this method (not shown). In addition, we examined the ability of the E2661 mutants to bind recombinant CD81 MBP-LEL113–201. Mutations that reduce binding to CD81 include those at W420, H421, S424, L427, N430, G436, W437, L438, A439, G440, L441, F442, Y443, G523, P525, Y527, W529A, G530, D535, N540, W549, Y613, and Y616 (Table 1).
TABLE 1.
Reactivity of antibodies to E2661 mutantsa
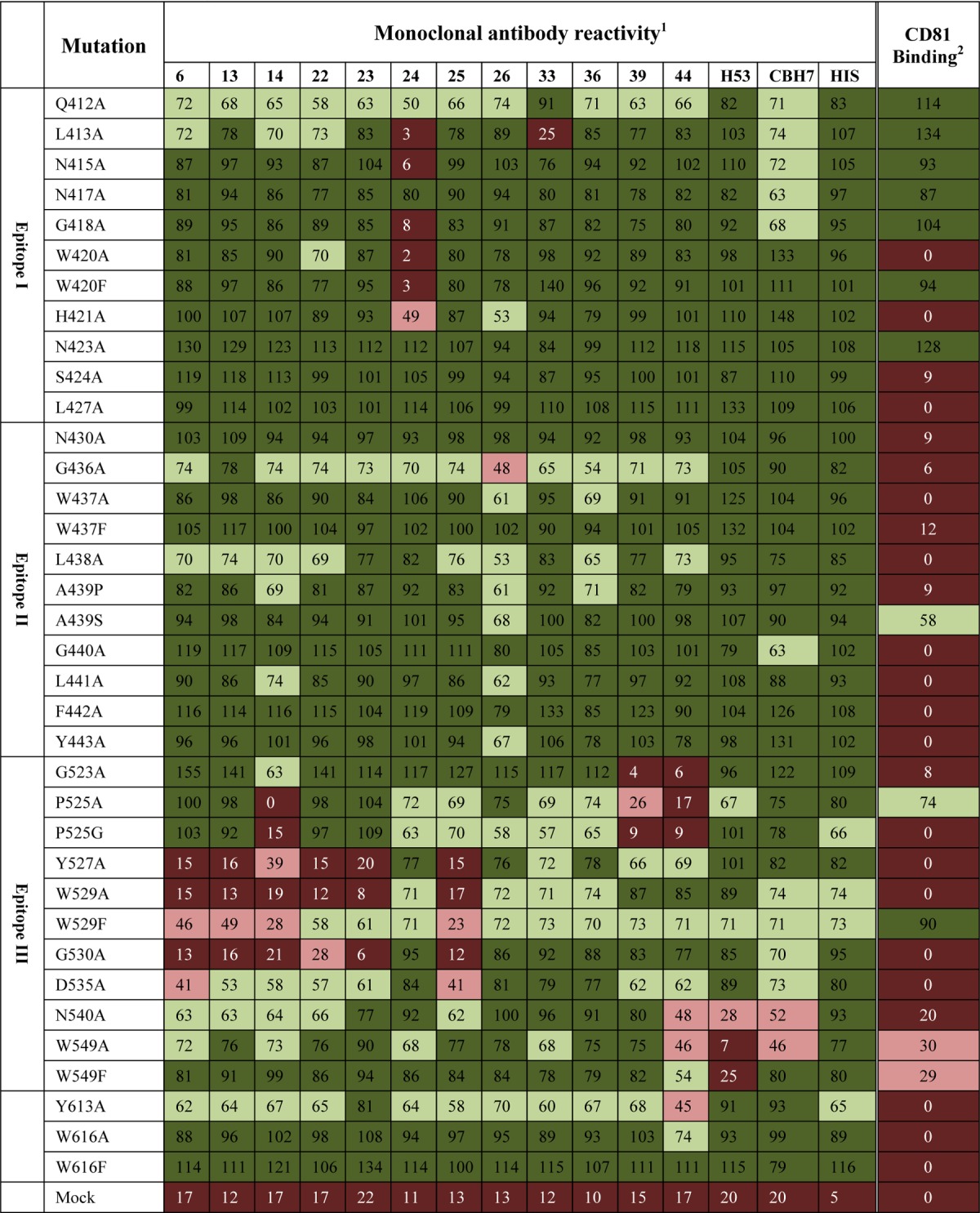
1, mean antibody binding to the mutant E2661 protein, where binding is expressed as a percentage relative to E2661 (n = 2); 2, mean mutant E2661 binding to MBP-LEL113–201, where binding is expressed as a percentage relative to E2661 (n = 3); dark red, 0 to 25% binding; light red, 26 to 49% binding; light green, 50 to 74% binding; dark green, 75 to 100% binding.
Reactivity of MAbs to intact E2661 and E2661 proteins containing variable region deletions.
We examined whether the reactivity of antibodies to E2661 was modulated by the presence of the variable regions. A solid-phase assay was used to assess the binding of MAbs to WT E2661 and E2661 containing deletions of HVR1 (Δ1), HVR2 (Δ2), the igVR (Δ3), or combinations thereof (Δ12, Δ13, Δ23, and Δ123) (12). First, equivalent coating levels for all antigens were confirmed by using an antibody to the C-terminal six-histidine tag (anti-His) (Fig. 3). MAbs 6, 13, 14, 22, 23, 24, 25, 26, 39, and 44 reacted equally to all antigens; binding curves for E2661 and Δ123 are shown in Fig. 3, and the apparent affinities of binding calculated for each MAb against each antigen are shown in Table 2. However, the reactivity of MAbs 1, 7, 12, 16, and 20, all raised to Δ123, was at least 2-fold lower for E2661 than for Δ123 (Fig. 3 and Table 2). In the case of MAbs 1, 7, 12, 16, and 20, improved binding was observed when any one of the variable regions was deleted (Table 2). In the case of HVR1-specific MAb36, deletion of HVR1, individually or in combination with other variable region deletions, ablated binding (Fig. 3). Deletion of HVR1 also blocked conformation-independent MAb33 binding to E2661, suggesting that its epitope includes residues in HVR1 as well as L413 in epitope I (Fig. 3 and Tables 1 and 2). Deletion of the igVR alone (Δ3) resulted in a complete or partial loss of reactivity for MAb33 or MAb36, respectively. However, deletion of the igVR together with HVR2 (Δ23) improved MAb33 and MAb36 binding, indicating that their epitopes do not directly include residues from the igVR but that their epitopes are affected by a conformation induced by the isolated deletion of the igVR from E2661.
FIG 3.

Direct binding of MAbs to E2661 with single and multiple variable region deletions. Data shown are the means ± standard deviations of data from at least two independent experiments. Percent binding was calculated relative to the binding observed for WT E2661. Nonlinear regression analysis was performed with Prism v 6.0f. Data from two independent analyses of MAb24 reactivity are shown for reproducibility. The amounts of E2661 containing single and multiple deletions of the variable regions added to plates were equivalent, as indicated by GNA-lectin capture of proteins followed by detection with an antibody to the 6×His epitope tag (anti-His) from two independent analyses performed three times (means ± standard deviations).
TABLE 2.
Apparent affinity of monoclonal antibodies for E2661 containing variable region deletionsa
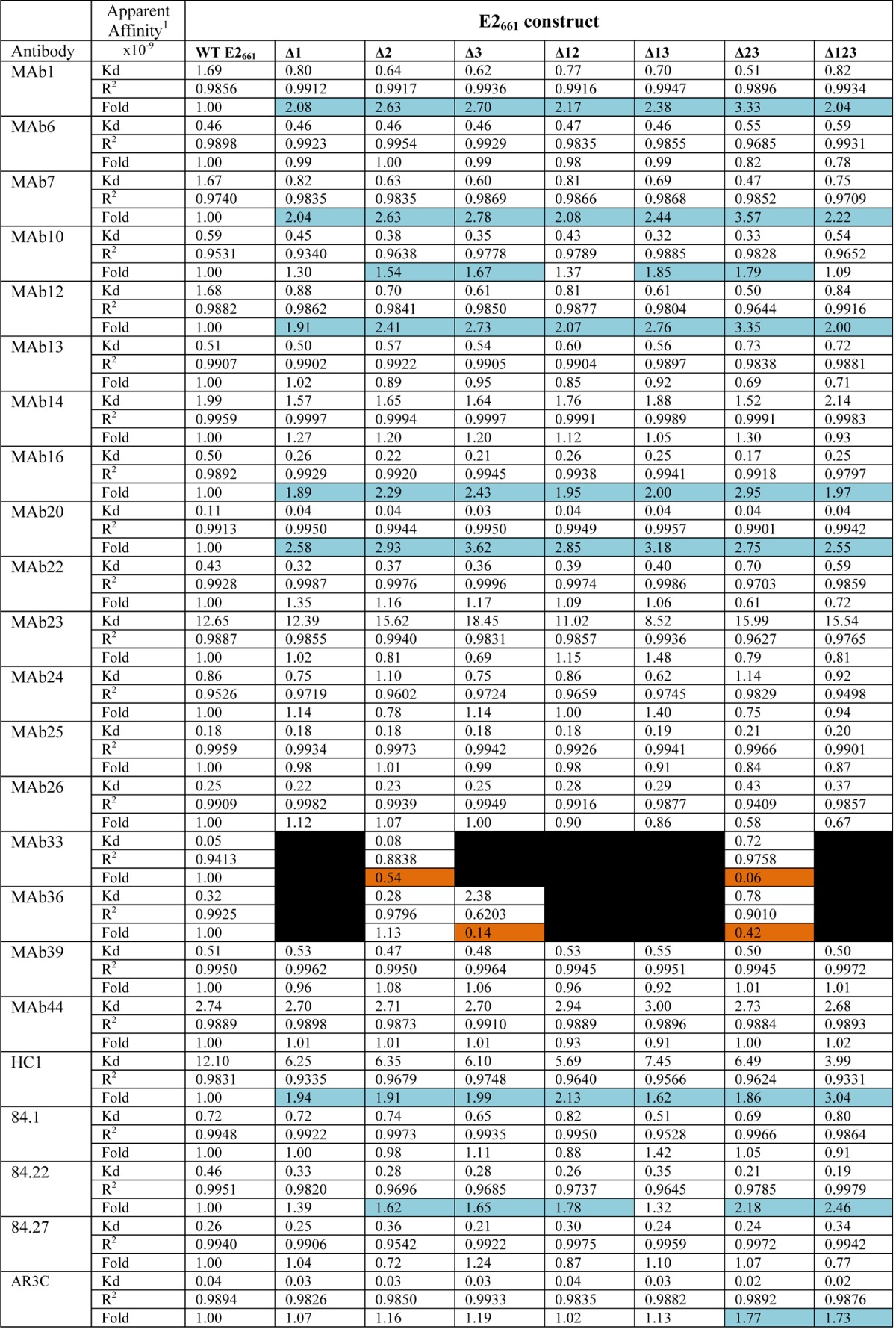
1, apparent affinity calculated from nonlinear regression analysis with Prism v 6.0f, which is the concentration of antibody required for half-maximal binding to the antigen. The fold difference is relative to the WT dissociation constant. Differences in binding to WT E2661 of ≥1.5-fold are shaded light blue, and differences in binding to WT E2661 of ≤0.5-fold are in orange. Black indicates no binding.
We also examined whether binding to E2661 was modulated by variable regions using previously described human MAbs HC1, HC84.1, HC84.22, HC84.27, and AR3C. The level of binding to Δ123 for HC1, HC84.22, and AR3C was >1.5-fold higher than the binding toward WT E2661 (Fig. 3 and Table 2). These results suggest that all three variable regions modulate the exposure of antibody epitopes on the E2 core domain.
Ability of MAbs to inhibit interaction with CD81.
Hepatitis C virus-mediated entry requires an interaction between glycoprotein E2 and the LEL of the surface tetraspanin CD81. Antibodies with the ability to prevent this interaction can also have the capacity to neutralize virus entry. We examined whether any of the antibodies were able to prevent the interaction between E2 and CD81 using a dimeric form of the LEL, MBP-LEL113–201, previously used to define the E2 binding site (19). A constant amount of E2 protein was incubated with serial dilutions of MAb and applied to ELISA plates coated with MBP-LEL113–201.
Ten of 18 MAbs (MAbs 1, 7, 10, 12, 16, 20, 24, 33, 39, and 44) were able to inhibit E2 binding to CD81 (Fig. 4 and Table 3). Three antibodies, MAbs 10, 39, and 44, showed similar abilities to inhibit E2661 and Δ123 binding to CD81. In contrast, MAbs 1, 7, 12, 16, 20, and 24 showed a stronger inhibitory capacity against Δ123, with the most striking example being MAb7 (raised to Δ123), which failed to block E2661-CD81 binding while completely inhibiting Δ123-CD81 binding (50% inhibitory concentration [IC50] of 2 μg/ml) (Fig. 4 and Table 3). Conversely, MAb33, whose epitope involves residues in HVR1 as well as L413, moderately inhibited the binding of E2661 to CD81 (IC50 of 16 μg/ml) but failed to block Δ123-CD81 binding, consistent with the deletion of HVR1 from the latter construct (Fig. 4A and Table 3). The ability of MAb24, specific to epitope I, to inhibit E2-CD81 binding was 10-fold greater for Δ123 than for E2661. Monoclonal antibodies isolated from HCV-infected humans, AR3C, HC1, HC84.1, HC84.22, and HC84.27, also demonstrated a greater capacity to inhibit Δ123-CD81 binding, suggesting that this property is not unique to the use of E2661 or Δ123 for MAb production and is a feature of antibodies isolated from infected humans also (Fig. 4A).
FIG 4.

Ability of MAbs to inhibit binding between E2661 or Δ123 and recombinant MBP-LEL113–201. (A) Serial dilutions of antibody were mixed with 50 ng E2 E2661 or Δ123 and applied to plates coated with purified dimeric MBP-LEL113–201. Bound E2 was detected with rabbit anti-His and horseradish peroxidase-labeled goat anti-rabbit IgG. Results shown are the means ± standard deviations of data from at least 2 independent experiments. Data were normalized to the percentage of E2 binding to CD81 in the absence of MAb. Curves were fitted with one-site-specific binding with the Hill slope equation in Prism v 6.0f. (B) Binding of E2661 proteins containing one or more variable region deletions to solid-phase MBP-LEL113–201. The inset graph shows the capture of E2 proteins with GNA-lectin and detection with anti-His antibody and confirms that similar amounts of E2 protein were present in every well. (C) Biosensor analysis of binding of E2661 and variants containing one or more variable region deletions to dimeric MBP-LEL113–201. Four concentrations of each E2661 protein were flowed over biosensor chips coated with MBP-LEL113–201, and the curves generated with 100 μg/ml E2 protein are shown.
TABLE 3.
Summary of characteristics of the 18 MAbs raised against E2661 and Δ123
| Antigen used to raise MAb | MAb | ELISA reactivity to E2 genotypea: |
IC50 (μg/ml) for inhibition of binding ofb: |
Reactivity to reduced E2661c | Epitope exposure on viral particlesd | IC50 (μg/ml) for neutralization of HCVppe | Amino acid(s) within epitopef | Isotype | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 2a | 3a | 4a | 5a | 6a | E2661 to CD81 | Δ123 to CD81 | |||||||
| Δ123 | 1 | + | − | − | − | − | − | − | 9 | 2.5 | − | − | > | NM | 2a |
| 6 | + | + | + | + | + | + | + | > | > | + | ++ | > | Y527, W529, G530, D535 | 2b | |
| 7 | + | − | − | − | + | − | − | > | 2 | − | − | > | NM | 2b | |
| 10 | + | − | − | + | − | + | − | 3 | 5 | − | − | 34 | NM | 1 | |
| 12 | + | − | − | − | − | − | − | 12 | 2 | − | − | > | NM | 2a | |
| 13 | + | + | + | + | + | + | + | > | > | + | ++ | > | Y527, W529, G530 | 2b | |
| 14 | + | + | + | + | + | + | + | > | > | ± | ++ | > | P525, Y527, W529, G530 | 2b | |
| 16 | + | − | − | − | − | − | − | 2.5 | 0.7 | − | − | > | NM | 1 | |
| 20 | + | − | − | − | − | − | − | 8 | 1.5 | − | − | > | NM | 2a | |
| 22 | + | + | + | + | + | + | + | > | > | + | ++ | > | Y527, W529, G530 | 2b | |
| 23 | + | + | + | + | + | + | + | > | > | − | + | > | Y527, W529, G530 | 2b | |
| 24 | + | + | + | + | + | + | + | 18 | 1.5 | + | +++ | 6 | L413, I414, N415, T416, G418, W420, H421 | 2b | |
| E2661 | 25 | + | + | + | + | + | − | − | > | > | − | +++ | > | Y527, W529, G530, D535 | 2a |
| 26 | + | + | + | + | − | + | − | > | > | + | +++ | > | Residues 645–661 | 2b | |
| 33 | + | − | − | − | + | − | + | 16 | > | + | +++ | 0.1 | HVR1 + L413 | 1 | |
| 36 | + | − | − | − | − | − | − | > | > | + | + | 53 | HVR1 | 1 | |
| 39 | + | − | − | − | − | − | − | 3 | 3 | + | +++ | > | G523, P525 | 1 | |
| 44 | + | − | − | − | − | − | − | 3.1 | 1.9 | +/− | + | 8 | G523, P525, N540, W549, Y613 | 1 | |
+, reactivity of at least 20% of that observed for genotype 1a binding. —, <20% binding observed relative to genotype 1a binding.
IC50s for E2-CD81 inhibition from Fig. 4. >, no inhibition observed.
+, strong binding; −, no binding; +/−, weak binding (Fig. 1B).
−, >1 μg/ml required to achieve an optical density of 0.1 units; +, 1 to 0.1 μg/ml required; ++, 0.1 to 0.01 μg/ml required; +++, <0.01 μg/ml required (Fig. 5).
For neutralization, the IC50 was calculated against HCVpp incorporating genotype 1a E1E2 glycoproteins in a 6-point dilution curve performed in triplicate. >, 50% neutralization not achieved.
We examined whether the data could be influenced by a difference in the binding of the E2661 proteins to CD81 using MBP-LEL113–201. Our previous study determined an affinity of ∼30 nM for the E2661–MBP-LEL113–201 interaction in a competition ELISA (19). Using a solid-phase binding assay, affinity-purified E2661 with one or two variable regions deleted displayed similar binding to MBP-LEL113–201 and was even slightly improved for Δ123, relative to WT E2661 binding (Fig. 4B). Biosensor analysis of the binding of E2661 proteins to MBP-LEL113–201 was also performed (Fig. 4C). Although the maximum response units (RU) varied between the deletions due to different concentrations of the functional monomer in individual protein preparations, they all displayed association and dissociation curves similar to those of the WT E2661 protein and deletants, indicating that they share similar binding kinetics (Fig. 4C). Global fitting of the concentration-independent dissociation rates (koff) also demonstrated that each of the deletion constructs retained high-affinity (koff = 10−3 s) binding to MBP-LEL113–201 (Table 4). These data suggest that deletion of the individual (or multiple) variable regions does not substantially alter the MBP-LEL113–201 binding affinity (or kinetics of binding), and so cannot account for the differences in the abilities of these MAbs to inhibit the E2-CD81 interaction. These results suggest that the variable regions of E2 modulate the accessibility of epitopes within the core domain of E2, thereby interfering with the ability of antibodies to inhibit the interaction between E2 and CD81.
TABLE 4.
Comparative analysis of the abilities of MAbs to inhibit E2 binding to CD81 and dissociation of E2 from CD81
| Construct | IC50 (fold change)a |
E2-CD81 koff (s−1[10−3]) (χ2 value)b | |
|---|---|---|---|
| MAb24 | MAb7 | ||
| WT E2661 | 17.5 (21.4) | > (>) | 1.43 (8.3) |
| Δ1 | 3.7 (4.5) | > (>) | 1.90 (6.6) |
| Δ2 | 3.0 (3.6) | 6.4 (5.4) | 2.71 (7.3) |
| Δ3 | 5.0 (6.1) | 7.5 (6.4) | 3.26 (6.8) |
| Δ12 | 2.1 (2.6) | 6.5 (5.5) | 2.75 (10) |
| Δ13 | 2.4 (2.9) | 2.2 (1.9) | 3.27 (5.3) |
| Δ23 | 1.0 (1.2) | 1.5 (1.3) | 3.29 (8.4) |
| Δ123 | 0.8 (1.0) | 1.8 (1) | 1.73 (18) |
The IC50 for E2-CD81 inhibition was calculated from the means of data from at least two independent experiments, and the fold increases in IC50 values relative to those for Δ123 are shown in parentheses. > denotes that 50% inhibition was not achieved. Data were derived from an analysis independent of those for Fig. 4A and Table 3.
Global fitting of the data was performed by using BiaEvaluation software to determine the off rate, and χ2 values are shown in parentheses.
Variable regions modulate the accessibility of MAb epitopes in the CD81 binding site on E2661.
We further examined the contribution of each variable region of E2 to modulating the accessibility of epitopes recognized by MAbs capable of blocking E2-CD81 binding using single (Δ1, Δ2, and Δ3) and double (Δ12, Δ13, and Δ23) variable region deletants in addition to E2661 and Δ123 (12). MAbs 24 and 7 were examined because they showed a >10-fold difference in their ability to inhibit binding of E2661 to CD81 versus binding of Δ123 to CD81.
Removal of HVR1 together with either HVR2 (Δ12) or the igVR (Δ13), removal of HVR2 and the igVR (Δ23), or removal of all three variable regions at once (Δ123) increased the ability of MAb24 to inhibit E2-CD81 binding at similar levels (Table 4). Removal of only one variable region (Δ1, Δ2, or Δ3) resulted in intermediate levels of inhibition. These differences are not explained by a difference in the binding preferences of MAb24 for different E2661 antigens (Table 2). In addition, the dissociation rates of E2661 and deletants with CD81 differed by <2-fold (Fig. 4B and Table 4) and cannot account for the ≥4-fold differences in the ability of MAb24 to inhibit Δ1, Δ2, Δ3, and WT E2661 binding to CD81.
MAb7 was unable to inhibit CD81 binding by either E2661 or E2661 containing HVR2 and the igVR (Δ1) (Table 4). However, deletion of all three variable regions (Δ123), HVR1 plus the igVR (Δ13), or HVR2 plus the igVR (Δ23) resulted in markedly increased inhibitory activity, whereas deletion of HVR1 plus HVR2 (Δ12), HVR2 (Δ2), or the igVR alone (Δ3) had an intermediate effect (Table 4). MAb7 shows a preference for binding E2661 when one or more variable regions are deleted, as we observed a 2-fold increase in apparent affinity, which may in part account for these differences in E2-CD81 inhibition (Table 2) but does not explain the failure of MAb7 to inhibit Δ1-CD81 interactions. Furthermore, this pattern of inhibition does not correlate with dissociation rates for E2 binding to CD81 and are distinct from the IC50s obtained by using MAb24. These data suggest that the epitope of MAb7 is occluded by HVR2 and the igVR and that deletion of both is necessary to reveal its ability to inhibit E2-CD81 binding. Overall, the variable regions modulate the accessibility of the antibody epitopes of MAbs 24 and 7 and can influence their ability to block the interaction between E2 and CD81.
Ability of antibodies to recognize epitopes on virus-like particles.
To examine whether the antibodies were able to bind their epitopes on E1E2 glycoproteins present on viral particles, a solid-phase ELISA was employed, where VLPs were bound to plates and all steps were performed in the absence of detergent. The pelleted VLPs contained E1 and E2 glycoproteins as well as the components of the HIV capsid (Fig. 5A). The ELISA results show that anticapsid antibody binding was enhanced 4-fold only if VLPs were permeabilized with Triton X-100, confirming that VLPs were largely intact in the absence of detergent (Fig. 5B). No binding was observed by using an irrelevant antibody or in the absence of primary antibody (Fig. 5B). A high level of binding of MAbs 6, 13, 14, 22, 24, 25, 26, 33, and 39 to intact VLPs was observed, suggesting that their epitopes are highly accessible in VLP-incorporated E1E2, while MAbs 23, 36, and 44 showed an intermediate level of reactivity (Fig. 5C and Table 3). The remaining MAbs, MAbs 1, 7, 10, 12, 16, and 20, reacted weakly or not at all to VLPs, suggesting that their epitopes are occluded in the structure of E1E2 on VLPs (Fig. 5C and Table 3) and correlating with a weaker ability to immunoprecipitate E1E2 complexes from cell lysates (Fig. 2). This subset of antibodies correlates with those that could not be mapped by using an E2661 mutant panel and with a binding preference for E2 containing at least one variable region deletion, suggesting that their epitopes are occluded in solubilized E1E2 and VLPs.
FIG 5.

Ability of MAbs to bind their epitopes on the surface of VLPs. (A) VLPs containing genotype 1a H77c E1E2 glycoproteins were pelleted through a sucrose cushion, subjected to reducing SDS-PAGE, and transferred onto nitrocellulose. Membranes were probed with a mixture of H52 (anti-E2) and A4 (anti-E1) or with IgG obtained from an HIV-positive individual. (B) Binding of anticapsid antibody to VLPs is enhanced by permeabilization with Triton X-100. Capsid protein (anti-CA) was detected with MAb183. No binding was observed by using an irrelevant MAb to a Myc epitope tag (anti-myc) or in the absence of primary antibody (No primary). (C) Ability of MAbs to bind VLPs in a direct binding ELISA. Data shown are the means ± standard deviations of data from two independent experiments. OD, optical density.
Ability of MAbs to neutralize HCV.
To date, two major specificity classes have been identified for HCV NAbs that recognize E2. The first class comprises those antibodies that are specific to HVR1, which usually mediate type-specific neutralization, with little or no cross-reactivity with heterologous genotypes or subtypes. The second class comprises NAbs that can prevent the interaction between E2 and CD81; such NAbs are sometimes able to neutralize more than one genotype of HCV. The cross-reactivity of MAbs toward the 6 major genotypes of HCV was examined in a direct binding ELISA using genotype 1 to 6 E2661 proteins secreted from transfected HEK293T cells. The results show that MAbs 1, 12, 16, 20, 36, 39, and 44 were entirely specific to genotype 1a E2661, while MAbs 6, 13, 14, 22, 23, and 24 were completely cross-reactive against all 6 genotypes. The remaining MAbs had variable cross-reactivity to heterologous genotypes (Table 3).
The neutralization abilities of MAbs were examined with retroviral luciferase reporter particles pseudotyped with genotype 1a H77c E1E2 (HCVpp). Five of the 18 MAbs, MAb10, MAb24, MAb33, MAb36, and MAb44, showed at least 50% neutralization against homologous H77c HCVpp (Table 3). The neutralizing abilities of these MAbs correlated with their ability to inhibit E2 binding to CD81 (MAbs 10, 24, 33, and 44) or HVR1 reactivity (MAb36). Six MAbs that inhibited the E2-CD81 interaction were unable to prevent >50% of homologous HCVpp entry (not shown).
Neutralizing MAb24 was able to recognize E2661 proteins of 6 HCV genotypes. We therefore examined the ability of MAb24 to mediate cross-genotype neutralization using either HCVpp or HCVcc. The IC50 of MAb24 against homologous genotype 1a HCVpp was found to be 6 μg/ml, with cross-neutralizing activity against genotype 2a, 3a, 4a, 5a, 6a, and 7a viruses being detected (Fig. 6A). However, 90% neutralizing activity was observed only against genotype 1a, 2a, 4a, and 5a viruses (Fig. 6A). MAb24, specific to epitope I, overlaps an epitope recognized by other broadly reactive antibodies, AP33, HCV1, and 3/11 (34, 37, 38). The IC50 of MAb24 against H77 HCVpp is similar to that of AP33 (1 μg/ml) (37) but higher than that of HCV1 (1 nM) (38). The use of E2661 mutants indicated that the epitope of MAb24 includes residues L413, N415, G418, W420, and H421 (Table 1). An alanine scan of the synthetic peptide spanning residues 411 to 428 confirmed these results and identified additional mutations, I414A and T416A, which substantially reduced MAb24 binding in the context of a peptide (Fig. 6B). A comparison of MAb24 with antibodies whose epitopes are also located within this region is shown in Fig. 6C. Common to all antibodies that bind the region spanning residues 411 to 428 is the recognition of N415 and W420. What distinguishes MAb24 from previously described MAbs to this region is its sensitivity to mutations at I414 and T416 as well as those at H421 (Fig. 6C). While this region is well conserved across each of the representative members of the 7 genotypes, variation at I414 and T416 is observed (Fig. 6D).
FIG 6.

Properties of MAb24. (A) Ability of MAb24 to mediate neutralization of different HCV genotypes. IC50s and IC90s (micrograms per milliliter) were derived from neutralization assays performed with HCVpp (genotype 1a) and HCVcc (genotypes 2a and 4a to 7a). X indicates that no neutralization was observed at the highest concentration of antibody tested. Data shown are the means ± standard errors of the means of data from at least three independent experiments. Genotype 3a neutralization was performed by incubating MAb24 with HCVcc and applying this mixture to Huh7.5 cells plated onto coverslips. Foci were visualized 3 days later after staining with anti-NS5A antibody and Alexa 488-labeled anti-mouse antibody. The mean percent reductions in foci on 2 coverslips relative to the no-antibody control and standard deviations are shown. Results are representative of data from two independent experiments. (B) Binding of MAb24 to the peptide spanning residues 411 to 428 with alanine substitutions at each position. Binding values are the means of data from duplicate samples ± standard deviations. (C) Comparison of the epitopes recognized by murine antibody AP33, rat antibody 3/11, and an antibody produced in transgenic mice containing human antibody genes, HCV1, for the region spanning residues 412 to 421. Mutations that abolish binding are shown in dark gray. Mutations that reduce binding are shown in light gray. (D) Alignment of the region spanning residues 412 to 421 in representative isolates of the 7 HCV genotypes.
MAb33 cross-reacted with E2 proteins of genotypes 4a and 6a. We therefore tested its ability to mediate cross-genotype neutralization and found that it weakly neutralized genotype 4a, 6a, and 7a viruses, with IC50s of 52 μg/ml, 78 μg/ml, and 78 μg/ml, respectively, but failed to reach 90% inhibition.
Comparison of the VH and VL sequences of MAb24 with those of analogous MAbs.
To examine whether the mechanism of MAb24 binding to E2 and its ability to cross-neutralize diverse strains of HCV resembled those of other MAbs recognizing this region of E2, the amino acid sequences of the variable light and variable heavy chain regions of MAb24 were aligned with those of AP33 (Fig. 7A and B). Sequences of MAb24 and murine AP33 are similar, with the following exceptions. In VL, CDR1 has 3 amino acid changes (G27SVL, N30KVL, and F36YVL), CDR2 contains one amino acid change (N55EVL), and CDR3 contains 2 changes (Q89HVL and V92EVL). In the VH sequence, CDR1 is identical to AP33. However, CDR2 contains 3 changes (S56DVH, L61PVH, and R64KVH), and CDR3 is elongated and differs at each position with respect to AP33. The most likely germ lines from which MAb24 has evolved are IGKV3-10*01 F (96% identical), IGKJ1*01 (100% identical), IGHV3-8*02 F (96.49% identical), IGHJ3*01 F or IGHJ3*02 P (79.17% identical), and IGHD3-2*01. Comparison of MAb24 with MAb AP33 reveals that the same germ line ancestors are used for both the VH and VL genes but that a different VH J gene is used for AP33, IGHJ4*01 F.
FIG 7.

Mapping of the major sites of structural differences between AP33 and MAb24. Shown are orthogonal ribbon representations of the structure of AP33 (16) (PDB accession number 4GAG), with side chains shown as sticks and the E2 peptide (residues 412 to 425 [ELINTNGSWHVN]) shown as spheres. The heavy and light chains are in red and blue, respectively, and the alignment is shown beneath the structure. CDR3 of the heavy chain is the most divergent region in MAb24 and is highlighted in magenta. Other sequence differences between MAb24 and AP33 are highlighted in green and labeled for residues mentioned in the text. The gray regions correspond to CDRs according to the Kabat numbering system (41, 42).
The structure of AP33 was used to map specific features of MAb24 (Fig. 7A and B). Most residues that delimit the peptide binding pocket are conserved, suggesting that they play a similar role in MAb24 (Tyr28VL, Phe32VL, Asn91/Asn92VL, Trp96VL, and Tyr33-50-53-58VH). The most significant differences are located in the heavy chain of CDR3, which is longer by 1 residue in MAb24 and has little sequence similarity with that of AP33. In particular, the three contact residues Ile95VH, Thr97VH, and Tyr100VH are not equivalent in MAb24. The corresponding Met95/Tyr97/Gln100VH residues have a similar pharmacophore, although the polar and aromatic residues are swapped in the sequence (Fig. 7A and B). High-resolution structural studies will be needed to establish whether this motif also plays a role in the docking of the peptide. Among the other four differences in MAb24 located around the paratope (S56DVH, R64KVH, G27SVL, and N30KVL), position 27 is notable because of its proximity to CD81 contact residue His421 within E2. In the absence of conformational rearrangements, the hydroxyl group of Ser27 would be only 5 Å away from His421 in the E2 protein, which may explain the increased importance of this residue compared to AP33.
DISCUSSION
Glycoprotein E2 contains three surface-exposed variable regions that can be deleted simultaneously from E2661 (Δ123) while retaining CD81 binding and NAb epitopes. In this study, we generated 18 new HCV E2-specific MAbs, raised to either Δ123 or E2661, that were used to examine whether HVR1, HVR2, or the igVR affected their ability to recognize antigen or inhibit E2-CD81 interactions. In addition, well-characterized MAbs isolated from HCV-infected humans were examined. These studies reveal that the variable regions affect the ability of both neutralizing and nonneutralizing antibodies to bind their epitopes and modulate their ability to inhibit E2-CD81 interactions.
Five of the new murine MAbs were able to neutralize virus, and neutralization correlated with either HVR1 reactivity or the ability to prevent E2 binding to CD81. Two neutralizing antibody epitopes (MAb33 and MAb36) involved HVR1. Interestingly, deletion of the igVR alone in an HVR1-containing construct (Δ3) substantially reduced binding by these MAbs, suggesting that the conformation and/or accessibility of HVR1 is altered when the igVR is deleted from E2. However, binding was at least partially restored when the igVR and HVR2 were deleted simultaneously (Δ23), suggesting that HVR1, HVR2, and the igVR are structurally linked in E2. Examination of the E2 core structure places the igVR in the vicinity of the HVR2 stem-loop, which is on the opposite face of E2 with respect to the likely location of HVR1 (13, 14) (Fig. 8A). It is possible that if E2, like other viral envelope glycoproteins, forms dimers or trimers on the surface of virions, intersubunit contacts may place HVR1, HVR2, and the igVR proximal to each other in the quaternary structure, thereby modulating the formation and/or structure of the MAb36 and MAb33 HVR1-dependent epitopes. In turn, sequence changes in HVR2 and the igVR could potentially impact the HVR1 structure and provide an additional mechanism of allosteric modulation of HVR1 neutralization epitopes.
FIG 8.

(A) Three-dimensional structure of the E2 core domain showing the location of CD81 contact residues (Table 1) as spheres. (B to E) Amino acids involved in binding of neutralizing MAb44 (B) and nonneutralizing MAb39 (C), MAb25 (D), and MAb14 (E) identified in this study, shown as spheres. Coloring is according to the coloring described in the legend of Fig. 1.
MAb24 recognized the epitope I region of E2, was completely cross-reactive against HCV genotypes 1 to 6 in solid-phase binding assays, and was able to neutralize HCV genotypes 1 to 7 but with a large range of IC50s. Epitope I is proximal to the neutralizing face of E2 and contains two CD81 contact residues, W420 and H421 (33, 35). Binding data obtained with VLPs suggest that it is a highly surface-accessible region in H77c genotype 1a E1E2 glycoproteins. Alanine scanning indicated that W420 and H421, as well L413, N415, and G418, are essential for MAb24 binding. Interestingly, relatively few mutations occur within epitope I in the HCV strains used for neutralization assays here: I414V occurs in genotypes 1b, 2a (J6), 3a, and 6a, while T416S is observed for G4a. However, epitope I sequence conservation did not correlate with neutralization. For example, MAb24 potently neutralized V414-containing G2a strain J6 but poorly neutralized the G6a strain with an identical epitope I sequence. Furthermore, a G7a strain was poorly neutralized by MAb24 despite having an epitope I sequence identical to that of G1a and G5a viruses. These data suggest that the ability of MAb24 to neutralize virus is dependent on sequences external to epitope 1 that may modulate its exposure and/or conformation. Consistent with this idea, removal of HVR1, HVR2, or the igVR improved the ability of MAb24 to inhibit E2-CD81 binding by 3- to 6-fold. We predict that the exposure of the MAb24 epitope and the ability to neutralize virus will therefore be affected by sequence changes in HVR1, HVR2, and the igVR and their ability to occlude epitope I on the surface of virus particles. In the three-dimensional structure of E2, HVR1 is proximal to epitope I (Fig. 1B), and previous studies demonstrated its ability to modulate CD81 binding (11). However, HVR2 and the igVR are on the opposite face of E2, and thus, why their presence occludes MAb binding on the neutralizing face remains unclear. Similarly to the proximal HVR1 region, it is possible that occlusion of epitope I by HVR2 and the igVR occurs in the context of E2 dimers or trimers present in virions, but this remains unknown.
In total, 8 MAbs recognized amino acids in epitope III: MAbs 6, 13, 22, 39, and 44 strongly recognized their epitope on denatured E2, while MAbs 14, 23, and 25 recognized a conformation-sensitive epitope and reacted weakly to denatured E2661 (Fig. 2B and Table 3). In the E2 core domain structure, epitope III overlaps the “CD81 binding loop” (residues 519 to 535) on the neutralizing face of E2 (13), and our panel of E2661 mutants (Table 1 and Fig. 8B) indicates that the residues involved in CD81 binding map to the neutralizing face of E2. Consistent with the predicted prominent exposure of this region on virions, all epitope III-specific MAbs recognized E1E2 on the surface of intact VLPs. Two epitope III-specific MAbs (MAbs 39 and 44) inhibited E2 binding to CD81 and were distinguished from noninhibitory MAbs by a requirement for G523 for E2661 binding. Of these, only MAb44 exhibited neutralizing activity, which appears to correlate with a more extensive epitope involving G523, P525, N540, and W549 in epitope III and Y613 (Fig. 8B). These data suggest that neutralizing activity is dependent on the engagement of an extensive antibody contact area (including G523 and W549) for effective E2-CD81 blockade (Fig. 8B). In comparison, the E2 region that was recognized by nonneutralizing/non-E2-CD81-inhibitory epitope III-specific MAbs (i.e., MAbs 6, 13, 14, 22, and 23) only partially overlaps the MAb44 epitope, resulting in an inability to prevent E2-CD81 interactions. Together, the data suggest that the angle of MAb engagement with E2 may be critical in determining its ability to both block E2-CD81 binding and prevent the entry of HCV. Further structural studies similar to those performed by using CD4 binding site antibodies and HIV are required to resolve this question (39, 40).
Differences in the ability to block E2-CD81 interactions were found to be dependent on the presence of variable regions and were detected for a subset of both the new murine antibodies characterized here and MAbs previously isolated from HCV-infected humans. These differences in inhibitory capacity could not be explained by differences in E2-CD81 binding or the antibody-E2 binding capacity alone. The results suggest that, in addition to HVR1, both HVR2 and the igVR contribute to the occlusion of antibody epitopes and their ability to prevent E2-CD81 interactions. As HVR2 and the igVR are essential components of E1E2 glycoprotein assembly, it was not possible to examine how their deletion affects neutralization. Structures of E2 containing HVR1, HVR2, and the igVR; knowledge of the organization of (E1)E2 on virions; and structures of intact E2 with CD81 are required to resolve how the CD81 site and underlying NAb epitopes are modulated by all three variable regions in recombinant and virion-incorporated forms of E2.
ACKNOWLEDGMENTS
We thank Charles Rice, Jens Bukh, Steven Foung, Mansun Law, and Jean Dubuisson for the kind provision of reagents. We thank Kirsten Vandenberg and Steve Rockman for expression of the MAbs.
This work was supported by NHMRC project grants 1020175, 543113, and 1009809 and the Australian Centre for HIV and Hepatitis Virology. H.E.D. is currently supported by NHMRC senior research fellowship 1041897 and was previously supported by R. D. Wright fellowship 433929. F.C. is a Future Fellow of the Australian Research Council. We gratefully acknowledge the contribution to this work of the Victorian Operational Infrastructure Support Program, received by the Burnet Institute.
REFERENCES
- 1.Morin TJ, Broering TJ, Leav BA, Blair BM, Rowley KJ, Boucher EN, Wang Y, Cheslock PS, Knauber M, Olsen DB, Ludmerer SW, Szabo G, Finberg RW, Purcell RH, Lanford RE, Ambrosino DM, Molrine DC, Babcock GJ. 2012. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog 8:e1002895. doi: 10.1371/journal.ppat.1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanwolleghem T, Bukh J, Meuleman P, Desombere I, Meunier JC, Alter H, Purcell RH, Leroux-Roels G. 2008. Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology 47:1846–1855. doi: 10.1002/hep.22244. [DOI] [PubMed] [Google Scholar]
- 3.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 4.Pestka JM, Zeisel MB, Blaser E, Schurmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, Rispeter K, Blum HE, Roggendorf M, Baumert TF. 2007. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 104:6025–6030. doi: 10.1073/pnas.0607026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowd KA, Netski DM, Wang XH, Cox AL, Ray SC. 2009. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 136:2377–2386. doi: 10.1053/j.gastro.2009.02.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lavillette D, Morice Y, Germanidis G, Donot P, Soulier A, Pagkalos E, Sakellariou G, Intrator L, Bartosch B, Pawlotsky JM, Cosset FL. 2005. Human serum facilitates hepatitis C virus infection, and neutralizing responses inversely correlate with viral replication kinetics at the acute phase of hepatitis C virus infection. J Virol 79:6023–6034. doi: 10.1128/JVI.79.10.6023-6034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 8.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michalak JP, Wychowski C, Choukhi A, Meunier JC, Ung S, Rice CM, Dubuisson J. 1997. Characterization of truncated forms of hepatitis C virus glycoproteins. J Gen Virol 78:2299–2306. doi: 10.1099/0022-1317-78-9-2299. [DOI] [PubMed] [Google Scholar]
- 10.Weiner AJ, Geysen HM, Christopherson C, Hall JE, Mason TJ, Saracco G, Bonino F, Crawford K, Marion CD, Crawford KA. 1992. Evidence for immune selection of hepatitis C virus (HCV) putative envelope glycoprotein variants: potential role in chronic HCV infections. Proc Natl Acad Sci U S A 89:3468–3472. doi: 10.1073/pnas.89.8.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M, Herrmann E, Zeisel MB, Baumert TF, Keck ZY, Foung SK, Pecheur EI, Pietschmann T. 2010. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol 84:5751–5763. doi: 10.1128/JVI.02200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCaffrey K, Boo I, Poumbourios P, Drummer HE. 2007. Expression and characterization of a minimal hepatitis C virus glycoprotein E2 core domain that retains CD81 binding. J Virol 81:9584–9590. doi: 10.1128/JVI.02782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, Wilson IA, Law M. 2013. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, Grakoui A, Marcotrigiano J. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509:381–384. doi: 10.1038/nature13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meola A, Tarr AW, England P, Meredith LW, McClure CP, Foung SK, McKeating JA, Ball JK, Rey FA, Krey T. 2015. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J Virol 89:2170–2181. doi: 10.1128/JVI.02190-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter JA, Owsianka AM, Jeffery N, Matthews DJ, Keck ZY, Lau P, Foung SK, Taylor GL, Patel AH. 2012. Toward a hepatitis C virus vaccine: the structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J Virol 86:12923–12932. doi: 10.1128/JVI.02052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong L, Giang E, Nieusma T, Robbins JB, Deller MC, Stanfield RL, Wilson IA, Law M. 2012. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J Virol 86:13085–13088. doi: 10.1128/JVI.01939-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong L, Giang E, Robbins JB, Stanfield RL, Burton DR, Wilson IA, Law M. 2012. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A 109:9499–9504. doi: 10.1073/pnas.1202924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummer HE, Wilson KA, Poumbourios P. 2002. Identification of the hepatitis C virus E2 glycoprotein binding site on the large extracellular loop of CD81. J Virol 76:11143–11147. doi: 10.1128/JVI.76.21.11143-11147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drummer HE, Boo I, Maerz AL, Poumbourios P. 2006. A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J Virol 80:7844–7853. doi: 10.1128/JVI.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drummer HE, Maerz A, Poumbourios P. 2003. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett 546:385–390. doi: 10.1016/S0014-5793(03)00635-5. [DOI] [PubMed] [Google Scholar]
- 22.Gottwein JM, Jensen TB, Mathiesen CK, Meuleman P, Serre SB, Lademann JB, Ghanem L, Scheel TK, Leroux-Roels G, Bukh J. 2011. Development and application of hepatitis C reporter viruses with genotype 1 to 7 core-nonstructural protein 2 (NS2) expressing fluorescent proteins or luciferase in modified JFH1 NS5A. J Virol 85:8913–8928. doi: 10.1128/JVI.00049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. 2008. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850. doi: 10.1002/hep.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deleersnyder V, Pillez A, Wychowski C, Blight K, Xu J, Hahn YS, Rice CM, Dubuisson J. 1997. Formation of native hepatitis C virus glycoprotein complexes. J Virol 71:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keck ZY, Li TK, Xia J, Gal-Tanamy M, Olson O, Li SH, Patel AH, Ball JK, Lemon SM, Foung SK. 2008. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J Virol 82:6061–6066. doi: 10.1128/JVI.02475-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, Bukh J, Rey FA, Foung SK. 2012. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8:e1002653. doi: 10.1371/journal.ppat.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chesebro B, Wehrly K, Nishio J, Perryman S. 1992. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol 66:6547–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krey T, Meola A, Keck ZY, Damier-Piolle L, Foung SK, Rey FA. 2013. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog 9:e1003364. doi: 10.1371/journal.ppat.1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 32.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 33.Boo I, Tewierek K, Douam F, Lavillette D, Poumbourios P, Drummer HE. 2012. Distinct roles in folding, CD81 receptor binding and viral entry for conserved histidines of HCV glycoprotein E1 and E2. Biochem J 443:85–94. doi: 10.1042/BJ20110868. [DOI] [PubMed] [Google Scholar]
- 34.Tarr AW, Owsianka AM, Timms JM, McClure CP, Brown RJ, Hickling TP, Pietschmann T, Bartenschlager R, Patel AH, Ball JK. 2006. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology 43:592–601. doi: 10.1002/hep.21088. [DOI] [PubMed] [Google Scholar]
- 35.Owsianka AM, Timms JM, Tarr AW, Brown RJ, Hickling TP, Szwejk A, Bienkowska-Szewczyk K, Thomson BJ, Patel AH, Ball JK. 2006. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J Virol 80:8695–8704. doi: 10.1128/JVI.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owsianka AM, Tarr AW, Keck ZY, Li TK, Witteveldt J, Adair R, Foung SK, Ball JK, Patel AH. 2008. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J Gen Virol 89:653–659. doi: 10.1099/vir.0.83386-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Owsianka A, Tarr AW, Juttla VS, Lavillette D, Bartosch B, Cosset FL, Ball JK, Patel AH. 2005. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J Virol 79:11095–11104. doi: 10.1128/JVI.79.17.11095-11104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Broering TJ, Garrity KA, Boatright NK, Sloan SE, Sandor F, Thomas WD Jr, Szabo G, Finberg RW, Ambrosino DM, Babcock GJ. 2009. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J Virol 83:12473–12482. doi: 10.1128/JVI.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou T, Lynch RM, Chen L, Acharya P, Wu X, Doria-Rose NA, Joyce MG, Lingwood D, Soto C, Bailer RT, Ernandes MJ, Kong R, Longo NS, Louder MK, McKee K, O'Dell S, Schmidt SD, Tran L, Yang Z, Druz A, Luongo TS, Moquin S, Srivatsan S, Yang Y, Zhang B, Zheng A, Pancera M, Kirys T, Georgiev IS, Gindin T, Peng HP, Yang AS, Program NCS, Mullikin JC, Gray MD, Stamatatos L, Burton DR, Koff WC, Cohen MS, Haynes BF, Casazza JP, Connors M, Corti D, Lanzavecchia A, Sattentau QJ, Weiss RA, West AP Jr, Bjorkman PJ, Scheid JF, Nussenzweig MC, et al. 2015. Structural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell 161:1280–1292. doi: 10.1016/j.cell.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran K, Poulsen C, Guenaga J, de Val N, Wilson R, Sundling C, Li Y, Stanfield RL, Wilson IA, Ward AB, Karlsson Hedestam GB, Wyatt RT. 2014. Vaccine-elicited primate antibodies use a distinct approach to the HIV-1 primary receptor binding site informing vaccine redesign. Proc Natl Acad Sci U S A 111:E738–E747. doi: 10.1073/pnas.1319512111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabat EA, Wu TT, Bilofsky H, Reid-Miller M, Perry H. 1983. Sequences of proteins of immunological interest. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, Bethesda, MD. [Google Scholar]
- 42.Wu TT, Kabat EA. 1970. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med 132:211–250. [DOI] [PMC free article] [PubMed] [Google Scholar]