

ABSTRACT
Herpesvirus entry into cells is mediated by the viral fusogen gB, which is thought to refold from the prefusion to the postfusion form in a series of large conformational changes that energetically couple refolding to membrane fusion. In contrast to most viral fusogens, gB requires a conserved heterodimer, gH/gL, as well as other nonconserved proteins. In a further mechanistic twist, gB-mediated cell-cell fusion appears restricted by its intraviral or cytoplasmic domain (cytodomain) because mutations within it result in a hyperfusogenic phenotype. Here, we characterized a panel of hyperfusogenic HSV-1 gB cytodomain mutants and show that they are fully functional in cell-cell fusion at shorter coincubation times and at lower temperatures than those for wild-type (WT) gB, which suggests that these mutations reduce the kinetic energy barrier to fusion. Despite this, the mutants require both gH/gL and gD. We confirm previous observations that the gH cytotail is an essential component of the cell-cell fusion mechanism and show that the N-terminal portion of the gH cytotail is critical for this process. Moreover, the fusion levels achieved by all gB constructs, WT and mutant, were proportionate to the length of the gH cytotail. Putting these results together, we propose that the gH cytotail, in addition to the gH/gL ectodomain, plays an essential role in gB activation, potentially acting as a “wedge” to release the gB cytodomain “clamp” and enable gB activation.
IMPORTANCE Herpesviruses infect their hosts for life and cause a substantial disease burden. Herpes simplex viruses cause oral and genital sores as well as rare yet severe encephalitis and a panoply of ocular ailments. Infection initiates when the viral envelope fuses with the host cell membrane in a process orchestrated by the viral fusogen gB, assisted by the viral glycoproteins gH, gL, and gD and a cellular gD receptor. This process is more complicated than that of most other viruses and is subject to multiple regulatory inputs. Antiviral and vaccine development would benefit from a detailed mechanistic knowledge of this process and how it is regulated.
INTRODUCTION
Herpesviruses, large, enveloped, double-stranded DNA (dsDNA) viruses, enter cells by the merger of the viral envelope and a host cell membrane, catalyzed by the conserved viral glycoprotein gB. As for other viral fusogens, gB is thought to refold from the prefusion to the postfusion form in a series of large conformational changes that provides the energy necessary to overcome the kinetic barrier associated with membrane fusion (1). However, unlike most viral fusogens, gB does not mediate fusion on its own and requires a conserved heterodimer, gH/gL (2), as well as other nonconserved proteins. For example, herpes simplex virus 1 (HSV-1) and HSV-2, members of the alphaherpesvirus subfamily, require the receptor-binding glycoprotein gD and a cellular gD receptor such as nectin-1 in addition to gB and gH/gL (3). These five proteins also mediate the fusion of transfected cells in the absence of any other viral proteins. It is unclear why HSV-mediated fusion requires so many proteins, nor is the mechanism known. According to the current model (4), based on the work of several laboratories (5–11), fusion is initiated when gD binds one of its cellular receptors and undergoes a conformational change (12, 13). The subsequent events are less well understood, but it is generally thought that this activated gD triggers gH/gL (5, 7, 10), which, in turn, activates gB (9, 11, 14), although neither mechanism has been elucidated. Activation of gB by gH/gL is presumed to involve direct interactions between their respective ectodomains, and both gB-gH/gL interactions and cell fusion can be inhibited by neutralizing antibodies against either participant (11, 14).
The requirement of the ectodomain of gH bound to gL (gH/gL) for fusion is well documented (10, 14–16). Less is known about the roles of the intraviral, or cytoplasmic, portions of gB and gH. The 109-amino-acid cytoplasmic domain (cytodomain) of gB appears to restrict the fusion activity of gB. Although cell-cell fusion is not normally associated with HSV infection in tissue culture (17), certain clinical isolates induce extensive cell-cell fusion manifested as multinucleated cells, or syncytia (18, 19). The syncytial (syn) phenotype of these isolates can result from mutations in several glycoproteins but is often due to a single point mutation in the gB cytodomain (19–26). In a virus-free cell-cell fusion system, such syn gB mutations result in increased fusion (25, 27, 28), termed hyperfusion (27). A number of engineered mutations within the gB cytodomain likewise result in hyperfusion (24, 25, 27, 29, 30), which suggests that the cytodomain has an inhibitory role. Deletion of the entire cytoplasmic domain results in gB that is unable to complement a gB-null virus and is misfolded, as judged by its glycosylation pattern (31).
The soluble gB cytodomain expressed in Escherichia coli is a trimer with 5 putative helices, predicted on the basis of bioinformatic analysis and proteolytic sensitivity (Fig. 1B). The soluble gB cytodomain binds anionic liposomes, and at least two of these helices are fully formed only in the presence of membranes, suggesting that membrane association stabilizes the cytodomain fold. C-terminally truncated gB variants with reduced membrane binding are hyperfusogenic, which supports the idea that membrane interactions by the gB cytodomain are necessary for its function in fusion regulation (32). A model for the gB cytodomain was previously proposed by our laboratory based on biochemical evidence (27) (Fig. 1B).
FIG 1.

The HSV-1 gB cytodomain. (A) Known syn point mutations, truncations, and insertion in HSV-1 and HSV-2 gB within the primary amino acid sequence of the HSV-1 gB cytodomain. Locations of putative helices proposed in previous work (27) are shown below the sequence. Mutations generated in this work are marked below the sequence in green. MPR, membrane-proximal region; TM, transmembrane region. (B) Previously proposed model of the gB cytoplasmic domain fully folded in the presence of the membrane. (Panel B reprinted from reference 27.)
Like the gB cytodomain, the 14-amino-acid cytoplasmic tail (cytotail) of gH is necessary for fusion but may have an activating, rather than an inhibitory, role because truncations, insertions, or point mutations reduce cell-cell fusion of infected or transfected cells mediated by wild-type (WT) gB (33–36). Additionally, truncations within the gH cytotail could suppress syncytium formation in cells infected with a syn HSV-1 strain (37) and showed delayed virus penetration (34). Previously, we showed that a gHSAP mutant, truncated at residue 832 and containing the point mutation V831A, reduced cell-cell fusion regardless of whether WT gB or a syn allele containing the A855V mutation was present (36). These results established the importance of the gH cytotail in regulating fusion and suggested that it participates at a rate-limiting step, yet its contribution to fusion has not yet been systematically investigated.
Here, we generated a panel of gB cytodomain mutants and characterized their fusion activity to further refine our model of the cytodomain. Four hyperfusogenic mutants were characterized further. We found that these mutants were fully functional at short coincubation times and at low temperatures, conditions that reduce the fusion activity of WT gB to ∼35%. This suggests that hyperfusogenic gB mutations reduce the kinetic energy barrier to fusion. However, none of the hyperfusogenic mutants were capable of bypassing the requirement for gH/gL and gD. We confirm previous observations that the gH cytotail is an essential component of the cell-cell fusion mechanism and show that the N-terminal portion of the gH cytotail is critical for this process. Moreover, the fusion levels achieved by all gB constructs, WT and mutant, were proportionate to the length of the gH cytotail. Putting these results together, we propose that the gH cytotail, in addition to the gH/gL ectodomain, may be essential for gB activation.
MATERIALS AND METHODS
Cells and plasmids.
CHO cells were a gift from J. M. Coffin and were grown in Ham's F-12 medium supplemented with 10% fetal bovine serum (FBS) at 37°C in the presence of 5% CO2, except where noted otherwise. Plasmids pPEP98, pPEP99, pPEP100, and pPEP101 carry the full-length HSV-1 (strain KOS) gB, gD, gH, and gL genes, respectively, in a pCAGGS vector and were gifts from P. G. Spear (3). Plasmids pCAGT7 (carrying the T7 polymerase gene) and pT7EMCLuc (carrying the firefly luciferase gene) (38) were also gifts from P. G. Spear. Plasmid pSC386 carrying the herpesvirus entry mediator (HVEM) gene (39) and the pCAGGS vector were gifts from G. H. Cohen and R. J. Eisenberg. Plasmid pmCherry-C1, a Clontech pAcGFP1-C1 vector carrying mCherry in place of enhanced green fluorescent protein (GFP), was a gift from R. R. Isberg. Plasmid pJLS8 (gB with an R858H mutation) was generated previously (27).
gB cytodomain mutants.
The R858H point mutant (pJLS8) in gB expression plasmid pPEP98 was described previously (27). Point mutations in the cytodomain of the full-length gB gene were generated in pPEP98 by using “splicing by overlap extension” PCR (SOE PCR) (40). The forward primer 5′-TCCCACGTGCGTGCCGGTCGC-3′ (the PmlI site is underlined) and the reverse primer 5′-CAGAGGGAAAAAGATCTGCTAGAC-3′ (the BglII site is underlined) were used in each SOE PCR. Primer pairs used for each mutant are as follows: 5′-CCACCAAGGAGCACAAGAACCCCA-3′ and 5′-TGGGGTTCTTGTGCTCCTTGGTGG-3′ for the L817H mutant, 5′-GAGGGCAGTTCGGGCGGTGACTTTGAC-3′ and 5′-GTCAAAGTCACCGCCCGAACTGCCCTC-3′ for the E830S/E831S mutant, 5′-CTAGCGAGTGCGAGGGAGATGATACGG-3′ and 5′-TATCATCTCCCTCGCACTCGCTAGCT-3′ for the E842S mutant, 5′-GCCGAGGCAAGGTCTATGATACGGTAC-3′ and 5′-GTACCGTATCATAGACCTTGCCTCGGC-3′ for the E845S mutant, and 5′-AAGGCCAGCAGTTCAGGCACGAGC-3′ and 5′-GCTCGTGCCTGAACTGCTGGCCTT-3′ for the K864S/K865S/K866S mutant. The resulting plasmids were pJLS19 (L817H), pJLS21 (E830S/E831S), pJLS23 (E842S), pJLS25 (E845S), and pJLS29 (K864S/K865S/K866S). These initial plasmids contained cloning errors at the 5′ PmlI site and were subcloned back into pPEP98 using PvuI and BglII sites to generate plasmids pJLS19F, pJLS21F, pJLS23F, pJLS25F, and pJLS29F, which were used in all experiments reported here. The L817H/R858H double mutant (pHR21) was generated by digesting pJLS8 and pJLS19F with MluI, ligating the 2 smaller fragments from pJLS19F with the large fragment from pJLS8, and screening for the correct orientation by restriction digestion.
gH cytodomain mutants.
The gH truncation gH832 was generated as described previously (36). The gH truncation gH828 was generated by SOE PCR using the primer pair 5′-AGCCTTCTGATAGCCTCGGCCCTGTGTACGT-3 and 5′-CGGGACTTATTACCGGAGAACCTTTAGGA-3′ and the primer pair 5′-TCCTAAAGGTTCTCCGGTAATAAGTCCCGTTTTTTTG-3′ and 5′-GTCCCCATAATTTTTGGCAGAGGGAAAAA-3′ (double stop codons are in boldface type). The PCR product was subcloned into pPEP100 by using Bsu36I and BglII. Truncations in the cytodomain of gH were generated by PCR from pPEP100 and cloning the fragment back into pPEP100, except for gH828, which was generated by using SOE PCR. The forward primer 5′-AGCCTTCTGATAGCCTCGGCCCTGTGTACGT-3′ was used in each case, which is before the Bsu36I restriction site. The reverse primer 5′-CCAAAAAGATCTTTATTATGTCCGGAGAACCTTTAGGA-3′ (the BglII site is underlined, and the double stop codon is in boldface type) was used to generate gH829. The reverse primer 5′-AAAACGGAGATCTTTATTAGAGAACCTTTAGGATGCCAGCCAGG-3′ (the BglII site is underlined, and the double stop codon is in boldface type) was used to generate gH827. The reverse primer 5′-GGACACTAGATCTTTAAACCTTTAGGATGCCAGCCA-3′ (the BglII site is underlined, and the stop codon is in boldface type) was used to generate gH826. The reverse primer 5′-CACTTGTAGATCTTTACTTTAGGATGCCAGCCAGGGC-3′ (the BglII site is underlined, and the double stop codon is in boldface type) was used to generate gH825. The reverse primer 5′-TGTCCGAGATCTTTATAGGATGCCAGCCAGGGCGG-3′ (the BglII site is underlined, and the stop codon is in boldface type) was used to generate gH824. All constructs were verified by sequencing.
Cell fusion assay.
Cell fusion was measured by using the well-established luciferase gene reporter system (3, 27, 41, 42). CHO cells were seeded into 6-well and 24-well plates and transfected the following day at 70 to 90% confluence by using Lipofectamine 2000. Target cells in 6-well plates were transfected with 1.6 μg pT7EMCLuc and 0.4 μg pSC386 per well in 1 ml Optimem with 5 μl Lipofectamine 2000. Effector cells were transfected in triplicate with 80 ng each of gB (pPEP98), gB cytoplasmic domain mutants, or an empty vector (pCAGGS), along with gH (pPEP100 or the gH mutant), gL (pPEP101), gD (pPEP99), and pT7 (pCAGT7), in 200 μl Optimem with 1 μl Lipofectamine 2000 per well. For the initial experiments shown in Fig. 2A, target cells at 3 h posttransfection were washed with phosphate-buffered saline (PBS), treated with trypsin, and cocultured with effector cells washed with PBS at a 1:1 ratio for 20 h. For later experiments, cells were cocultured at 4 h posttransfection for 18 h or the indicated times. Cells were washed with 1 ml PBS per well, lysed with 200 μl of 1× lysis buffer (Promega), and assayed or frozen at −80°C until later use. Luciferase production was assayed by adding 100 μl of the substrate (Promega) to the mixture and measuring luminescence by using a BioTek plate reader. In each experiment, light output from the vector was subtracted from that from each sample and expressed as a percentage of the output under WT conditions. Values reported here represent averages of the results from at least three experiments unless otherwise noted. To test fusion without gH/gL or gD, the expression plasmids were replaced with pCAGGS during transfection.
FIG 2.
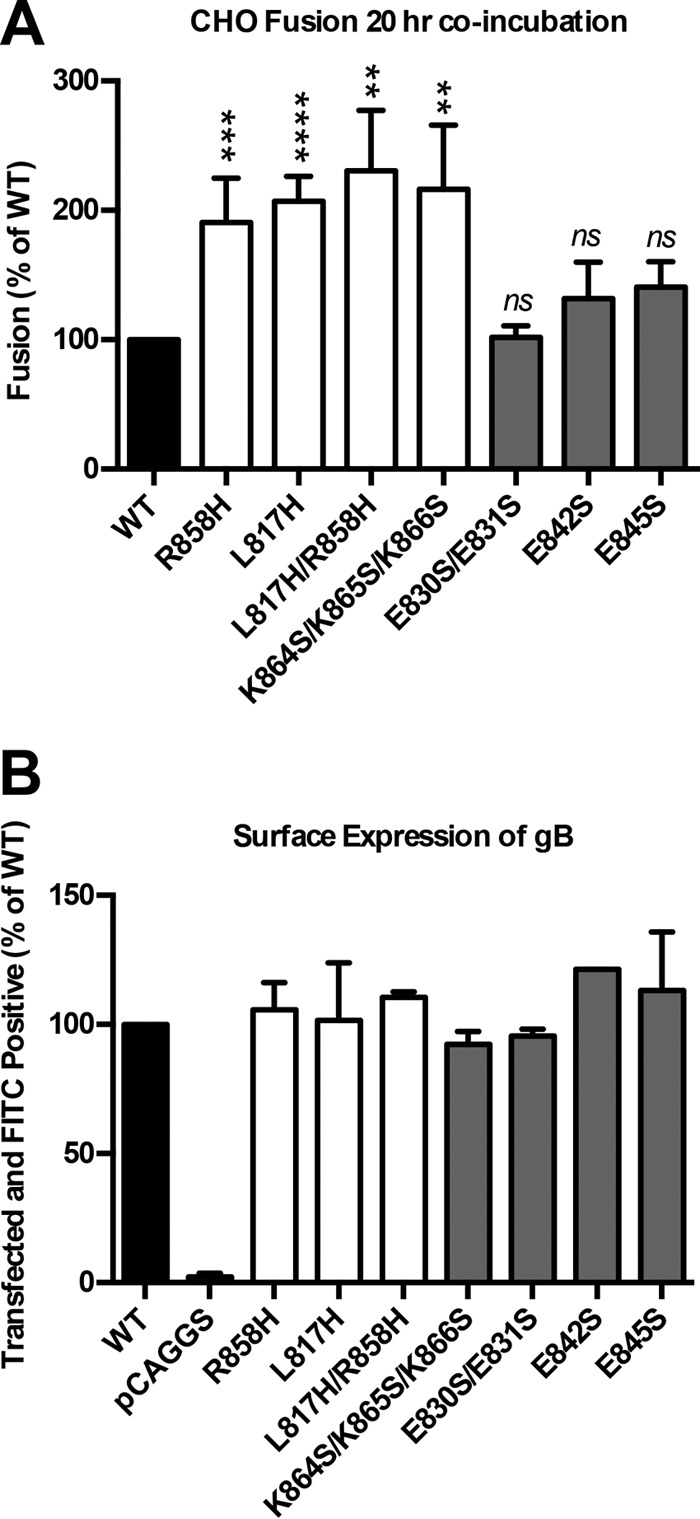
Fusion activity and surface expression of gB cytodomain mutants. (A) Luciferase production was used to quantify the fusion of CHO cells mediated by WT gB, the vector, or the gB mutant indicated below each bar. Glycoproteins gH, gL, and gD as well as pT7 were transfected with each gB mutant or the control. Target cells were transfected with pHVEM and pLuciferase, which is under the control of the T7 promoter. Cell populations were coincubated in a 1:1 ratio for 20 h at 37°C. In each experiment, the value for the vector control was subtracted from the value for each sample (performed in triplicate), and the values are expressed as a percentage of the value for WT gB. Reported values represent averages of data from at least three experiments. Error bars represent standard deviations. Statistical analysis was performed by using a paired Student t test comparing each mutant to the WT control (****, P < 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; ns, not significant). (B) Expression of WT gB and each mutant determined by FACS analysis using anti-gB polyclonal antibody R68 and fluorescein isothiocyanate (FITC)-conjugated anti-rabbit secondary antibody. mCherry was included as a transfection control. The percentages of cells double positive for mCherry and fluorescein isothiocyanate were normalized to WT expression levels. Reported values represent averages of data from two experiments. Error bars represent standard deviations.
FACS analysis.
Surface expression of gB and gB mutants was assessed by fluorescence-activated cell sorter (FACS) analysis as previously described (27), with modifications. CHO cells seeded previously into 6-well plates were transfected with 1.6 μg gB (pPEP98), the indicated mutant, or the empty vector (pCAGGS) plus 0.4 μg pmCherry-C1 with 5 μl Lipofectamine 2000 in 1 ml Optimem. The transfection solution was replaced after 4 h and incubated for 18 to 20 h in 2 ml growth medium. Cells were treated with trypsin and resuspended in PBS with 3% FBS (FACS medium). A total of 1 × 106 cells were washed with FACS medium and resuspended in 50 μl primary antibody (1:1,000 in FACS medium). To test the expression of gB and the gH truncation mutants together, CHO cells were transfected with 0.4 μg each of gB, gH or the indicated mutant, gL, gD, and pmCherry-C1 with 5 μl Lipofectamine 2000 in 1 ml Optimem as described above, except the cells were incubated for 12 h after replacement of the transfection solution.
Dye transfer assay.
CHO cells were transfected as described above, with the following modifications. Effector cell transfections were performed by using pmCherry instead of pT7, and target cell transfections were performed by using pCAGGs instead of pT7EMCLUC. The transfection solution was replaced with medium at 4 h posttransfection. Target cells were incubated with 3 μM DiO [DiOC18(3) (3,3′-dioctadecyloxacarbocyanine perchlorate)] (Molecular Probes) in Ham's F-12 medium containing 10% FBS for 20 min at 37°C. Medium was replaced twice over 20 min to remove unincorporated dye. The target and effector cells were detached by using Versene solution at 12 h posttransfection, and 1.5 × 105 cells each were coincubated on glass coverslips in a 24-well plate at 37°C. Four hours later, the cells were washed with PBS, fixed by using 4% paraformaldehyde (PFA) in PBS, and stained by using DAPI (4′,6-diamidino-2-phenylindole) at 1 μg/ml for 20 min at room temperature. The coverslips were mounted by using Prolong Gold (Invitrogen). Coverslips were imaged with a Spot RT2 color digital camera mounted on a Nikon E800 microscope. mCherry-positive cells were counted in random visual fields at a ×40 magnification, and the numbers of mCherry- and DiO-positive cells were quantified. Nuclei in each double-positive group were also examined.
Statistics.
Statistical analysis was performed for each experiment on the raw luminescence units following log10 transformation by using GraphPad PRISM 6 software. Repeated-measures analysis of variance (ANOVA) was used when all conditions/mutants were tested together, followed by either Dunnett's posttest for comparisons against the WT control or Bonferroni's posttest for specific comparisons.
RESULTS
Choice of gB cytodomain mutations.
Previously, we proposed a model for the secondary structure and the tertiary fold of the cytoplasmic domain of HSV-1 gB based on bioinformatic sequence analysis and biochemical characterization of the isolated cytodomain, which included membrane binding assays and protection from proteolysis in the presence of membranes (Fig. 1) (27). To refine this model and to gain further mechanistic insights into the gB cytodomain function, we generated a panel of the following mutants: L817H, L817H/R858H, K864S/K865S/K866S, E830S/E831S, E842S, and E845S.
Within the HSV gB cytodomain, clinical syn mutations cluster into two “hot spots,” residue 817 in putative helix h1b (hot spot 1) (21, 23) and putative helix h2b (hot spot 2) (19, 21, 28, 43–45) (Fig. 1). We previously showed that the A855V (21, 43) and R858H (19) syn mutants, both in hot spot 2, have hyperfusogenic phenotypes in cell-cell fusion (27). Here, we wanted to determine whether the L817H syn mutant (21) also had a hyperfusogenic phenotype in cell-cell fusion and whether combining syn mutations from two hot spots would have an additive effect on fusion.
We previously showed that the removal of the putative helix h3 reduced membrane binding by the cytodomain, which we attributed to a cluster of basic residues at the C terminus of helix h3 (32). Removal of h3 also resulted in hyperfusion, consistent with the idea that membrane binding by the gB cytodomain plays an important role in fusion restriction. The simultaneous removal of putative helices h3 and h2b eliminated membrane binding, which suggested that residues within helix h2b contact the membrane (32). Moreover, h2b residues 857 to 867 are protected from proteolysis when the gB cytodomain is bound to anionic membranes (27). We hypothesized that a conserved cluster of basic residues at the C terminus of helix h2b may be important for membrane binding and, thus, fusion regulation. The K864S/K865S/K866S triple mutant was generated to test this hypothesis.
Several known syncytial mutations target basic residues R876, R800, K839, and R858. The gB cytodomain also contains a large number of acidic residues, which could form salt bridges with the basic residues. We hypothesized that their side chains may participate in salt bridges, and to identify potential counterparts, we mutated several glutamates conserved in HSV to generate the E830S/E831S, E842S, and E845S mutants. Residues E830 and E831 are located in the proteolytically resistant region between putative helices h1b and h2a, while residues E842 and E845 map to putative helix h2a.
The L817H and K864S/K865S/K866S mutants are hyperfusogenic in the cell-cell fusion assay.
All gB cytodomain mutants were first tested by using a quantitative virus-free luciferase cell-cell fusion assay (3). The previously characterized R858H mutant (27) served as a positive control for the hyperfusogenic phenotype. The E830S/E831S mutant had WT fusogenicity, while the E842S and E845S mutants had fusogenicity that was only 30% above that of the WT (Fig. 2A), which suggested that the four glutamates played a minor role, if any, in either fusion or its regulation. The R858H, L817H, L817H/R858H, and K864S/K865S/K866S mutants mediated fusion at ∼2-fold-higher levels than those of the WT following a 20-h incubation (Fig. 2A). These results show that syn mutation L817H from hot spot 1 increases cell-cell fusion to a similar extent as syn mutation R858H from hot spot 2 and that combining these two mutations does not cause a further increase in fusion, which implies that ∼2-fold fusion over that of the WT represents the upper fusion limit under these experimental conditions. The hyperfusogenic phenotype of the new K864S/K865S/K866S mutant supports the idea that the basic cluster at the C terminus of putative helix h2b plays a role in fusion regulation. The hyperfusogenic phenotype of the four gB mutants was not due to overexpression because all mutants were expressed on the cell surface at WT levels (Fig. 2B). These mutants were characterized further.
Hyperfusogenic mutants require gH/gL and gD for fusion.
Certain truncation mutants of Epstein-Barr virus (EBV) gB are fusogenic on their own (46, 47). The increased activity of the hyperfusogenic mutants raised the question of whether gH/gL and gD were still required for fusion. In the absence of either gH/gL or gD, the fusion of all four hyperfusogenic mutants was at background (no-gB) levels (Fig. 3). Therefore, all hyperfusogenic gB mutants depend on gD and gH/gL for fusion activity.
FIG 3.
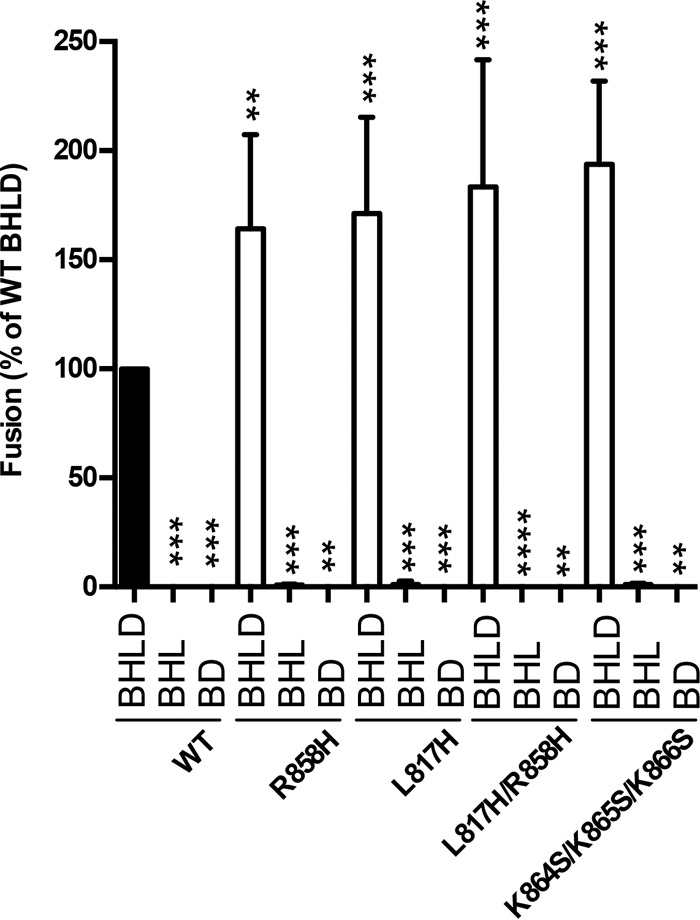
Hyperfusogenic gB mutants require gH/gL and gD for fusion. Fusion experiments were carried out as described in the legend of Fig. 2 except that cell populations were coincubated in a 1:1 ratio for 18 h at 37°C. The vector was transfected in place of gD or gH/gL, as indicated. Transfected glycoproteins (B, D, H, and L) are indicated below each bar, and the gB construct used is indicated below each set of bars. In each experiment, the value for the vector control was subtracted from the value for each sample (performed in triplicate), and the values are expressed as a percentage of the WT value. Reported values represent averages of data from at least three experiments. Error bars represent standard deviations. Comparison to the WT for each set (BHLD, BHL, and BD) was done by repeated-measures ANOVA (P < 0.001 for all sets) with Dunnett's post hoc test (****, P < 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01).
Unlike WT gB, the mutants are fully hyperfusogenic at lower temperatures or shorter coincubation times.
In all previous experiments, the cell-cell fusion activity of HSV-1 gB was assayed at 37°C after an 18- to 20-h coincubation of target and effector cells. To characterize the temporal and thermal dependencies of gB-mediated fusion, we measured the fusion activity of WT and mutant gB proteins after 6, 12, or 18 h of coincubation at 37°C (Fig. 4) or after 18 h of coincubation at 28°C. The fusion activity of WT gB decreased proportionately to the coincubation time and at 6 h was only at 37% of its 18-h level. In contrast, all hyperfusogenic mutants reached their full fusion activity by 6 h. Similarly, at 28°C, WT gB mediated only 36% fusion relative to its 37°C level, while all the mutants were just as active at 28°C as they were at 37°C (Fig. 5A). Cell surface expression levels of the mutant and WT gB proteins were similar at 28°C and could not account for the large differences in fusion activity at low temperatures between WT gB and the mutants (Fig. 5B). Thus, unlike WT gB, all four mutants were fully fusogenic at lower temperatures and shorter coincubation times, implying that each mutation reduced the kinetic energy barrier to fusion.
FIG 4.
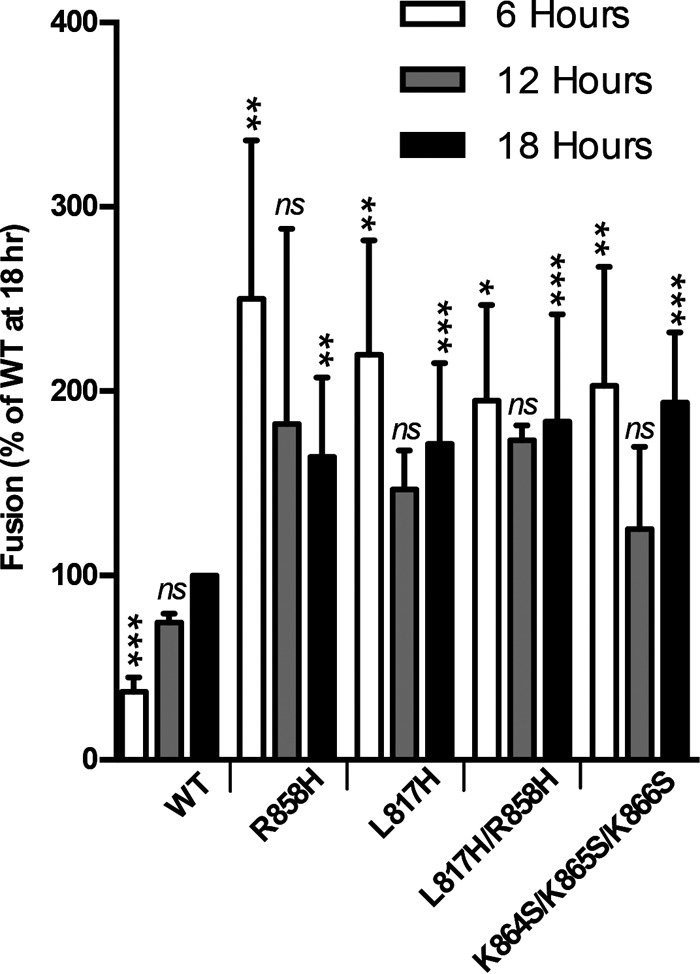
Hyperfusogenic mutants complete fusion faster than the WT. Fusion experiments were carried out as described in the legend of Fig. 2 except that the two populations were coincubated for the indicated times at 37°C before the luciferase assay. Reported values represent averages of data from three experiments. Error bars represent standard deviations. Comparison to the WT for each set (6 h, 12 h, and 18 h) was done by repeated-measures ANOVA (18 h, P = 0.0002; 12 h, P = 0.017; 6 h, P < 0.0001) with Dunnett's post hoc test (***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05; ns, not significant).
FIG 5.
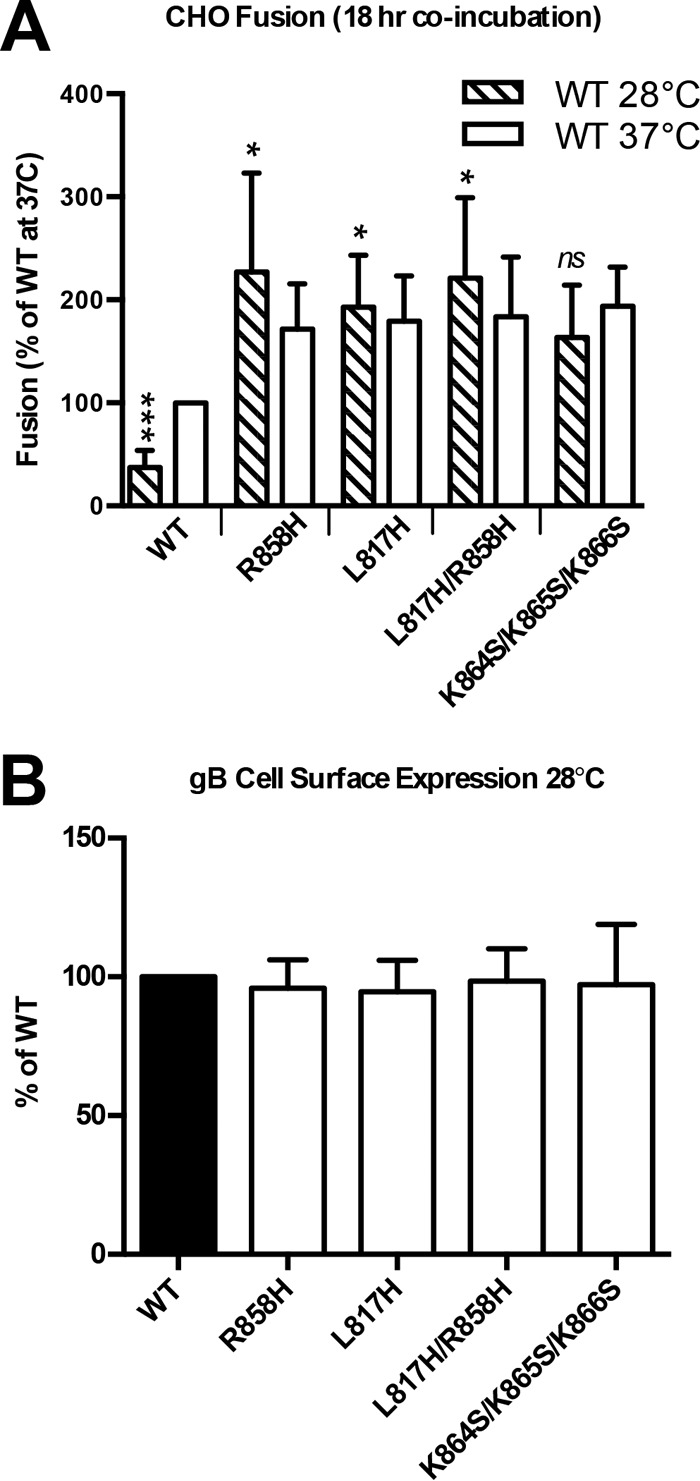
Hyperfusogenic mutants fuse at lower temperatures. (A) Fusion experiments were carried out as described in the legend of Fig. 2 except that the two populations were coincubated for 18 h at 28°C before the luciferase assay. Reported values represent averages of data from three experiments. Error bars represent standard deviations. Data from 18 h at 37°C (Fig. 4) are shown for comparison. Comparison to the WT was done by repeated-measures ANOVA (P < 0.0001) with Dunnett's post hoc test (***, P ≤ 0.001; *, P ≤ 0.05; ns, not significant). (B) Expression levels of WT gB and each mutant were determined by FACS analysis as described in the legend of Fig. 2 except that the cells were moved to 28°C following transfection. Reported values represent averages of data from two experiments. Error bars represent standard deviations.
Hyperfusogenic mutants require less gH/gL to achieve WT gB fusion levels.
A hyperfusogenic truncation mutant of EBV gB has been shown to achieve the same fusion levels as the WT with less gH/gL (48). We tested whether hyperfusogenic mutants of HSV-1 gB had the same ability by measuring cell-cell fusion in the presence of different amounts of transfected WT gH. To keep the total amount of transfected gH constant, we complemented WT gH with a gH truncation mutant lacking the cytotail (gH824), which is expressed on the cell surface at WT gH levels (see Fig. 7B) but is unable to support fusion (see Fig. 7A). For any given amount of gH, the gB mutants achieved higher fusion levels than those of WT gB. For example, in the presence of only 1/16 of the WT gH level, WT gB fusion levels were reduced to 20%, while those of the mutants were reduced to ∼37 to 70%, depending on the mutant (Fig. 6A). However, while higher fusion levels were achieved overall, the proportional response of mutant versus WT gB to gH amounts appeared similar. For example, with one-eighth of the gH level, fusion levels of WT gB and the R858H/L817H and K864S/K865S/K866S gB mutants were reduced 2-fold from their maximum levels (Fig. 6B). Thus, while the gB mutants achieved higher fusion levels than those of WT gB, their dependence upon gH/gL was similar.
FIG 7.

Hyperfusogenic mutants do not rescue gH truncations that reduce fusion. (A) gH truncations generated in this work. (B) Expression of gB and gH/gL was determined by FACS analysis using either anti-gB monoclonal antibody SS10 or anti-gH polyclonal antibody R137 and Alexa 488-conjugated anti-mouse secondary antibody or fluorescein isothiocyanate-conjugated anti-rabbit secondary antibody, respectively. mCherry was included as a transfection control. Percentages of cells double positive for mCherry and fluorescein isothiocyanate were normalized to WT expression levels. (C) Surface expression of gH/gL or the indicated gH truncation with gL determined by FACS analysis using anti-gH/gL monoclonal antibody LP11 and Alexa 488-conjugated anti-mouse secondary antibody. Percentages of cells double positive for mCherry and fluorescein isothiocyanate were normalized to WT expression values. Reported values represent averages of data from two experiments. Error bars represent standard deviations. (D) Fusion experiments were carried out as described in the legend of Fig. 2 except that the two populations were coincubated for 12 h at 37°C. In each experiment, the value for the vector control was subtracted from the value for each sample (performed in triplicate), and the values are expressed as a percentage of the WT value. Reported values represent averages of data from three experiments. Error bars represent standard deviations. Linear regression was performed with WT and gB mutants against gH cytotail length, omitting control WT gB and WT gH (R2 = 0.96 for WT gB, R2 = 0.74 for the R858H mutant, R2 = 0.75 for the L817H mutant, R2 = 0.75 for the L817H/R858H mutant, and R2 = 0.72 for the K864S/K865S/K866S mutant; P < 0.001 for each condition). (E) Fusion results from panel D reported as a ratio of each gH truncation to full-length gH.
FIG 6.
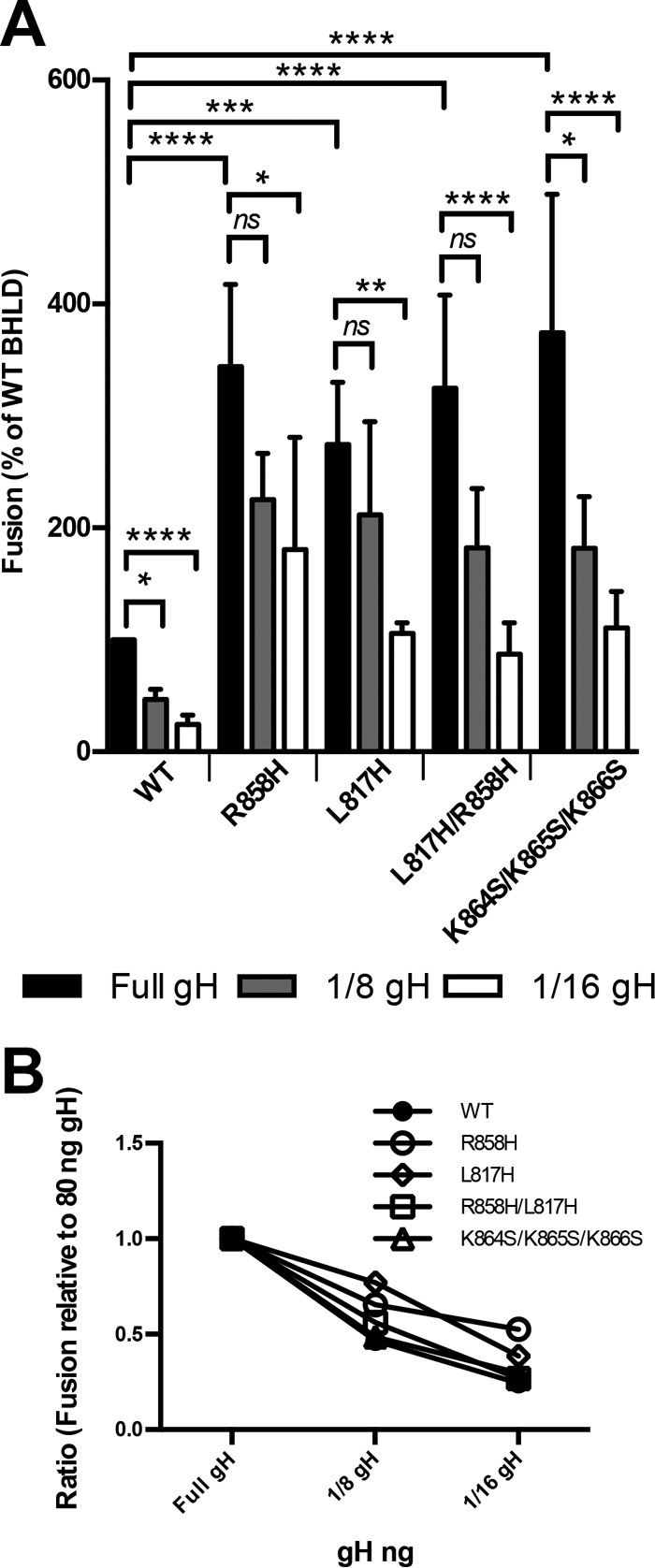
Hyperfusogenic mutants require less gH/gL for fusion activation. (A) Fusion experiments were carried out as described in the legend of Fig. 2 except that the two populations were coincubated for 12 h at 37°C. In each experiment, the value for the vector control was subtracted from the value for each sample (performed in triplicate), and the values are expressed as a percentage of the WT value. gH was transfected in smaller amounts, with the gH824 mutant making up the difference. Reported values represent averages of data from 3 experiments. Error bars represent standard deviations. Comparison to the WT was done by repeated-measures ANOVA (P < 0.0001) with Bonferroni's multiple-comparison test (****, P < 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; *, P ≤ 0.05; ns, not significant). (B) Fusion results from panel A reported as ratios of reduced gH transfection to full gH transfection.
Fusion of WT gB and hyperfusogenic mutants is proportionate to the length of the gH cytotail.
To systematically assess the dependence of cell-cell fusion on the length of the gH cytotail, we generated a panel of gH truncation mutants (Fig. 7A). A coincubation time of 12 h was selected based on previous experiments (Fig. 4) to maximize fusion levels while permitting the detection of small amounts of fusion. Truncations of the gH cytotail did not change the surface expression levels of either gH/gL or gB (Fig. 7B). Moreover, truncations of the gH cytotail did not affect the conformation of the extracellular portion of the gH/gL ectodomain because they did not alter the reactivity of several truncated gH/gL constructs with a conformational monoclonal antibody, LP11 (Fig. 7C), which is considered the “gold standard” in the field (49). gH832 did not reduce cell-cell fusion, whereas the removal of the entire gH cytotail abolished fusion with all gB constructs (Fig. 7D), consistent with data from previous reports (33, 36). The rest of the truncations progressively reduced the fusion activity of both WT gB and the hyperfusogenic mutants (Fig. 7D and Table 1) proportionately to the length of the truncation. With any given gH mutant, hyperfusogenic gB mutants mediated higher fusion levels than those of WT gB. For example, in the presence of gH828, the mutants mediated between 85 and 161% fusion (relative to WT gB/WT gH), whereas WT gB mediated only 27% fusion. In the presence of gH826, the mutants mediated between 29 and 63% fusion (relative to WT gB/WT gH), whereas WT gB mediated only 7% fusion. These results show that although the fusion of all gB variants requires the gH cytotail, the gB mutants can achieve similar fusion levels with a shorter gH cytotail, e.g., gH827 versus gH829 and gH826 versus gH828 (Fig. 7D and Table 1). We also found that the gB mutants require at least one residue within the gH cytotail to mediate fusion above the background level, whereas WT gB requires at least two residues. Despite this, gH truncations reduced fusion to a similar extent for gB constructs, be they WT or hyperfusogenic (Fig. 7E). For example, the gH829 truncation reduced fusion by ∼2-fold (relative to WT gH levels), whereas the gH827 truncation reduced fusion by ∼5-fold (Fig. 7E). Thus, the fusion activity of all gB variants, mutant or WT, is dependent upon the length of the gH cytotail to similar extents.
TABLE 1.
Fusion in the presence of different gB and gH variantsa
| Protein | Mean % fusion ± SD |
||||
|---|---|---|---|---|---|
| WT gB | R858H | L817H | L817H/R858H | K864S/K865S/K866S | |
| WT gH | 100 | 322 ± 137 | 309 ± 108 | 271 ± 73 | 272 ± 85 |
| gH832 | 94 ± 13 | 415 ± 107 | 388 ± 102 | 343 ± 112 | 365 ± 117 |
| gH829 | 54 ± 23 | 170 ± 69 | 159 ± 64 | 156 ± 41 | 155 ± 50 |
| gH828 | 27 ± 18 | 161 ± 20 | 140 ± 16 | 85 ± 41 | 121 ± 33 |
| gH827 | 19 ± 6 | 61 ± 19 | 59 ± 10 | 62 ± 24 | 50 ± 29 |
| gH826 | 7 ± 1 | 57 ± 11 | 33 ± 6 | 27 ± 8 | 27 ± 5 |
| gH825 | 2 ± 3 | 19 ± 4 | 7 ± 2 | 4 ± 1 | 6 ± 2 |
| gH824 | −3 ± 1 | −2 ± 2 | −3 ± 0 | −4 ± 1 | −4 ± 1 |
Glycoproteins gL and gD as well as pT7 were transfected with each gB (or the empty vector) and gH variant. Target cells were transfected with pHVEM and pLuciferase, which is under the control of the T7 promoter. In each experiment, the value of the vector control with WT gH was subtracted from the value for each sample, and fusion values (percent) were normalized to the value obtained in the presence of WT gH and WT gB, set to 100%. Reported values represent averages of data from 3 experiments.
Hyperfusogenic mutants and the gH cytotail exert their effect prior to lipid mixing.
To narrow down the step(s) in fusion targeted by the hyperfusogenic mutations in gB and by the gH cytotail, we tested for transfer of the lipophilic dye DiO, as a marker of lipid transfer, from target CHO cells to effector CHO cells expressing mCherry, gL, gD, WT gB or gB mutants, and WT gH or gH824 (Fig. 8). In the absence of gB, nonspecific transfer of DiO to mCherry-positive cells was limited to <1% (Fig. 8A). In the presence of WT gB and WT gH/gL, we observed a number of DiO/mCherry-positive cells containing 2 or more nuclei (Fig. 8B). In the presence of the R858H mutant and WT gH/gL, we observed a higher number of DiO/mCherry-positive cells containing 2 or more nuclei (Fig. 8C). If the hyperfusogenic mutants increased the rate of fusion at a step after hemifusion, we would have expected to find some mCherry/DiO-positive cells with single nuclei for WT gB with WT gH/gL. Instead, all DiO/mCherry-positive cells contained 2 or more nuclei, which implies that the hyperfusogenic mutations exert their effect at a step prior to lipid mixing. Additionally, we tested the R858H mutant with the gH824 mutant and found that DiO transfer to mCherry-positive cells was at a background level (Fig. 8D). The cytoplasmic tail of gH is thus required not only for full fusion (Fig. 7) but also for DiO transfer to effector cells, which is a marker for hemifusion. This result extends previous findings with a fusion-null gH tail insertion mutant (35) to CHO cell-cell fusion.
FIG 8.
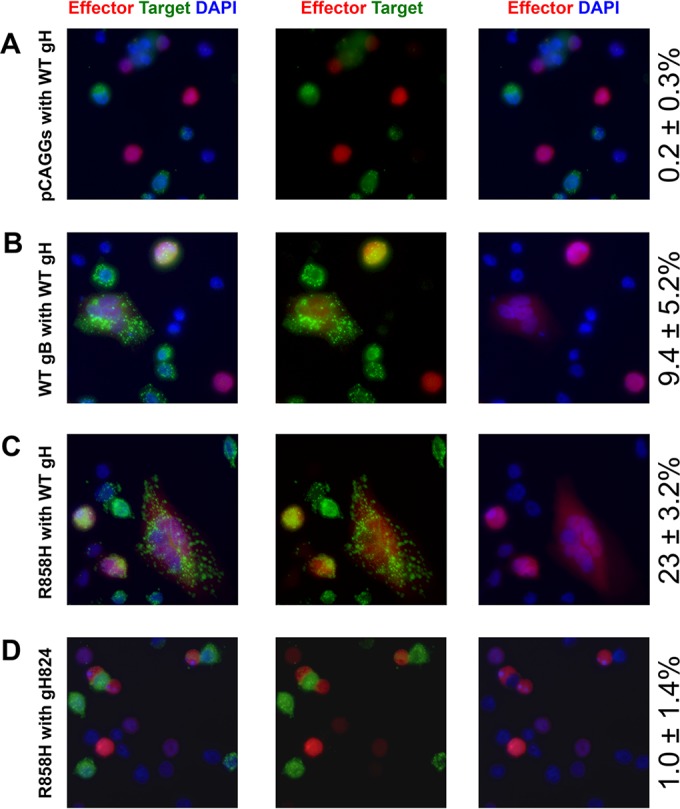
Lipid transfer requires gB and the gH cytoplasmic tail. CHO cells were transfected with pHVEM for target cells and with gL, gD, pmCherry, and the indicated gB and gH plasmids for effector cells. Following transfection, target cells were labeled with the lipophilic dye DiO. Target and effector cells were coincubated at a 1:1 ratio on a glass coverslip for 4 h at 37°C before fixing with PFA and staining of nuclei with DAPI. mCherry-positive cells were counted in random visual fields at a ×40 magnification, and the numbers of mCherry- and DiO-positive cells were quantified. Values on the right indicate averages ± standard deviations for the percentages of mCherry cells that were DiO positive. The experiment was performed twice, and representative images are shown.
DISCUSSION
Updated gB cytodomain model.
The cytodomain of gB plays an important regulatory role in membrane fusion that is yet unclear, at least in part, due to the lack of any direct structural information on the gB cytodomain. Previously, we proposed a model for the secondary and tertiary structures of the gB cytodomain on the basis of bioinformatic analysis and biochemical data (Fig. 1) (27). Here, we generated and characterized a panel of mutants, E830S/E831S, E842S, E845S, and K864S/K865S/K866S, with the goal of refining this model. The K864S/K865S/K866S triple mutant was generated to test the hypothesis that a conserved cluster of basic residues at the C terminus of helix h2b is important for fusion regulation by binding the membrane. The K864S/K865S/K866S mutant was hyperfusogenic, which supports the role of this region in fusion repression. Although the involvement of these residues in membrane interactions has not yet been tested directly, for now, we place the C terminus of helix h2b in proximity to the membrane in our revised model (Fig. 9A).
FIG 9.
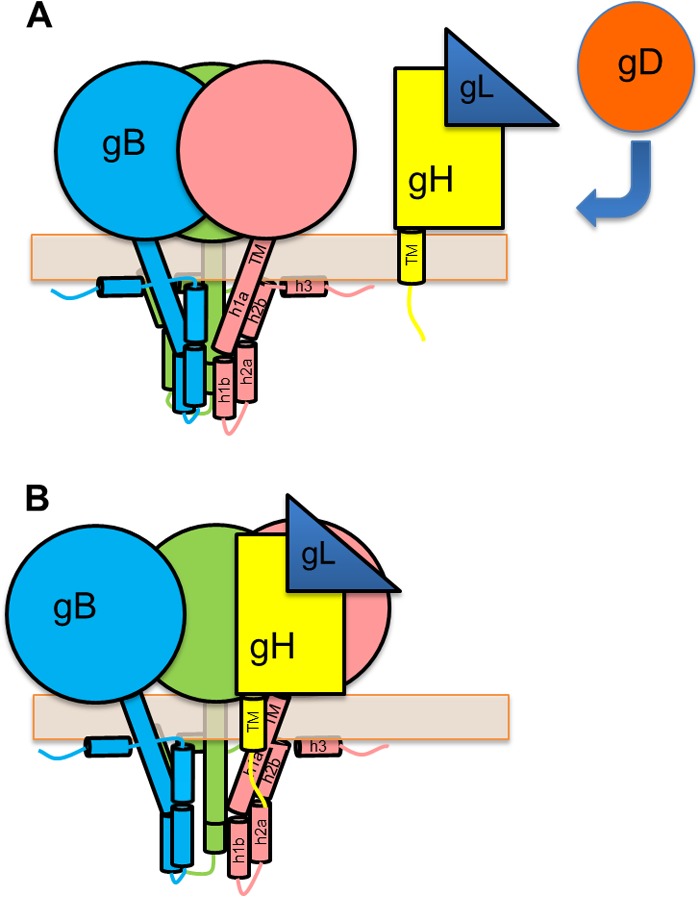
Working model for gB activation by the gH cytotail. (A) gB cytoplasmic domain model updated to show the end of h2b and h3 interacting with the membrane, supporting the idea that the cytodomain functions as a clamp that stabilizes prefusion gB. The region between helices h1b and h2a, which is protected from proteolysis even in the absence of the membrane, is placed inside the “cage” formed by the interacting protomers. Receptor-bound gD activates gH/gL, which causes the ectodomains of gB and gH/gL to interact. (B) The gB cytodomain and gH cytotail transiently interact, which disrupts the interprotomer interactions within the gB cytodomain and releases the clamp. This allows gB to undergo a conformational change.
The E842S and E845S mutants were used to test the hypothesis that one of these residues in helix h2a may form a salt bridge with R858 in helix h2b. Previously, we found that the soluble gB cytodomain containing the R858H mutation had an additional proteolytic cleavage site after residue R844 in helix h2a not found in the soluble WT gB cytodomain (27). We speculated that helices h2a and h2b interacted, possibly, through a salt bridge formed by R858 with either E842 or E845 and that the disruption of the salt bridge by the R858H mutation could explain why R844 became more accessible to cleavage (27). If either E842 or E845 participated in a salt bridge with the R858H mutation, the E842S or E845S mutant would have been expected to be hyperfusogenic like R858H. However, neither mutant was hyperfusogenic, so R858 probably does not form a salt bridge with either of these residues. To reflect this, helices h2a and h2b are no longer shown as interacting in the new model (Fig. 9A).
The E830S/E831S double mutant was designed to probe the functional role of these conserved residues located in the proteolytically resistant region between putative helices h1b and h2a. The double mutation did not affect the fusion activity of gB, which is notable because relatively few such mutations within the cytodomain have been described (29). Why these two glutamates are conserved remains unclear.
Hyperfusogenic gB mutations reduce the kinetic barrier to fusion.
The four hyperfusogenic mutants of HSV-1 gB investigated here achieved the same levels of fusion sooner than did WT gB. The differences in fusion between WT gB and the mutants became smaller with longer incubation times, which means that the WT was catching up to the mutants. Thus, the effect of hyperfusogenic mutants is kinetic. These observations highlight the importance of monitoring fusion at early time points because the total number of possible fusion events is finite.
For the first time, we tested whether combining syn mutations from two “hot spots” could have an additive effect on fusion. The L817H/R858H double mutant mediates fusion at the same level as the single mutants, suggesting that these mutations target the same step in fusion. Alternatively, these mutations could cause the hyperfusogenic phenotype by disrupting the same structural element.
The hyperfusogenic mutants were fully functional even at reduced incubation times and temperatures, suggesting that mutations reduced the overall kinetic barrier to fusion (Fig. 9B). This is in agreement with data from a recent report in which a hyperfusogenic truncation mutant of EBV gB mediated fusion at levels well above those of WT gB even with reduced incubation times and temperatures (48). We (27) and others (48) have proposed that hyperfusogenic mutants either exist in an activated conformation or require less energy to achieve such a conformation. If so, they could potentially mediate fusion in the absence of gD or gH/gL. We found, however, that in the absence of either gH/gL or gD, fusion was at background levels (no gB) for all tested gB mutants. Although we previously observed that some hyperfusogenic mutants, including the R858H mutant used here, could mediate low-level fusion in the absence of either gH/gL or gD (27), this was probably due to a higher background level and lower signal-to-noise ratio of the previous experimental setup. Despite having a lower overall activation energy barrier, the hyperfusogenic mutants require gH/gL, yet they can achieve the same fusion levels with less gH/gL. We propose that the hyperfusogenic mutations in gB increase the probability that any particular gB-gH/gL interaction will activate gB. In other words, these gB mutants are hypersensitive to gH: they show a similar response to gH truncation as that of WT gB but are more likely to drive fusion at any given gH level. In this scenario, the subsequent rate of fusion remains unchanged. Current data do not exclude the alternative model, however, in which both WT gB and the hyperfusogenic mutants require gH/gL to be activated, but once triggered, the mutants drive fusion at a higher rate than does WT gB. Data from lipid transfer experiments suggest that the hyperfusogenic mutants accelerate a step prior to lipid mixing, but further experiments are needed to identify this step and to distinguish the two models.
The gH cytotail is essential for fusion.
We selected the CHO cell-cell fusion assay to systematically investigate the activity of the hyperfusogenic gB mutants and their dependence on the length of the gH cytotail. This system provides a sensitive assay for the effect of gH truncations while permitting comparisons between our work and those of others. Our results confirm previously reported observations that the gH cytotail is an essential component of the cell-cell fusion mechanism and show that the N-terminal portion of the gH cytotail is critical for this process. Eight residues of the 14-residue gH cytotail were sufficient to mediate fusion at WT gH levels, whereas the complete removal of the cytotail abrogated fusion. Fusion levels achieved by all gB constructs, WT or mutant, in the presence of gH with truncated cytotails were reduced and proportionate to the remaining cytotail length, and at least a 1-residue-long gH cytotail was necessary to produce measurable fusion. Based on the results presented here, we propose that, in addition to the gH/gL ectodomain, the gH cytotail is involved in gB activation.
How could the gH cytotail activate gB? We ruled out the possibility that the gH cytotail truncations could play an indirect role by altering the conformation of the ectodomain. Previously, we hypothesized that the gB cytodomain restricts fusion by stabilizing the prefusion form, like a clamp, and preventing its premature refolding into the postfusion form (50) and that a fully folded cytodomain is necessary for this effect (27). The hyperfusogenic gB mutations could destabilize the cytodomain “clamp” to allow gB to be triggered more readily. Here, we further hypothesize that the gH cytotail also destabilizes the cytodomain clamp by disrupting the interprotomer contacts like a wedge (Fig. 9B). Shorter gH cytotails, being smaller wedges, are less efficient at disrupting the gB cytodomain fold. Indeed, truncations resulted in a gradual reduction in fusion levels rather than an abrupt loss of activity at a specific length. Although no interaction between the recombinant gB cytodomain expressed in E. coli and a synthetic peptide encompassing the gH cytotail was detected in in vitro pulldown experiments (36), the interaction may be transient, or it may require the transmembrane regions (TMs) of gB and gH. Although the roles of the TMs in fusion have not yet been delineated, mutations in either TM reduce fusion (33, 51).
To summarize, hyperfusogenic mutations within the gB cytodomain can either render it more sensitive to triggering by gH/gL or increase the rate of fusion at a posttriggering step prior to hemifusion. Recent studies monitoring the kinetics of fusion of single influenza A virus virions or West Nile virus-like particles with a membrane bilayer (52–54) distinguished two steps prior to hemifusion: activation of the fusogen characterized by the exposure and outward extension of the fusion peptide (or loop) followed by the clustering of activated fusogens in a number sufficient to overcome the energy barrier to membrane deformation. Future single-particle membrane fusion studies will help delineate the steps in HSV fusion and the respective contributions of various regulatory elements.
ACKNOWLEDGMENTS
We thank Jessica Silverman for her early contributions to this project and for generating the initial versions of the gB mutant plasmids. We thank Joan Mecsas for the gift of DAPI stain, James Schwob and the members of his laboratory for help with the microscopy experiments and the gift of DiO dye, Roselyn Eisenberg and Gary Cohen for the gift of antibodies, and Helena Browne for the gift of monoclonal antibody LP11. We also thank Stephen Kwok and Allen Parmelee at the Tufts Laser Cytometry Core facility for their help with FACS experiments.
This work was funded by NIH grant 1R21AI107171.
REFERENCES
- 1.Harrison SC. 2015. Viral membrane fusion. Virology 479:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mocarski ES., Jr 2007. Comparative analysis of herpesvirus-common proteins, p 8 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 3.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 4.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuller AO, Lee WC. 1992. Herpes simplex virus type 1 entry through a cascade of virus-cell interactions requires different roles of gD and gH in penetration. J Virol 66:5002–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J Virol 81:11532–11537. doi: 10.1128/JVI.01343-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A 104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4:e00046–13. doi: 10.1128/mBio.00046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazear E, Carfi A, Whitbeck JC, Cairns TM, Krummenacher C, Cohen GH, Eisenberg RJ. 2008. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J Virol 82:700–709. doi: 10.1128/JVI.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J 24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns TM, Friedman LS, Lou H, Whitbeck JC, Shaner MS, Cohen GH, Eisenberg RJ. 2007. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J Virol 81:5102–5111. doi: 10.1128/JVI.00097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol 67:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 18.Weise K, Kaerner HC, Glorioso J, Schroder CH. 1987. Replacement of glycoprotein B gene sequences in herpes simplex virus type 1 strain ANG by corresponding sequences of the strain KOS causes changes of plaque morphology and neuropathogenicity. J Gen Virol 68(Part 7):1909–1919. [DOI] [PubMed] [Google Scholar]
- 19.Bzik DJ, Fox BA, DeLuca NA, Person S. 1984. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology 137:185–190. doi: 10.1016/0042-6822(84)90022-9. [DOI] [PubMed] [Google Scholar]
- 20.Cai WH, Gu B, Person S. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol 62:2596–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel JP, Boyer EP, Goodman JL. 1993. Two novel single amino acid syncytial mutations in the carboxy terminus of glycoprotein B of herpes simplex virus type 1 confer a unique pathogenic phenotype. Virology 192:112–120. doi: 10.1006/viro.1993.1013. [DOI] [PubMed] [Google Scholar]
- 22.Baghian A, Huang L, Newman S, Jayachandra S, Kousoulas KG. 1993. Truncation of the carboxy-terminal 28 amino acids of glycoprotein B specified by herpes simplex virus type 1 mutant amb1511-7 causes extensive cell fusion. J Virol 67:2396–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diakidi-Kosta A, Michailidou G, Kontogounis G, Sivropoulou A, Arsenakis M. 2003. A single amino acid substitution in the cytoplasmic tail of the glycoprotein B of herpes simplex virus 1 affects both syncytium formation and binding to intracellular heparan sulfate. Virus Res 93:99–108. doi: 10.1016/S0168-1702(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 24.Gage PJ, Levine M, Glorioso JC. 1993. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J Virol 67:2191–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foster TP, Melancon JM, Kousoulas KG. 2001. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18–29. doi: 10.1006/viro.2001.1004. [DOI] [PubMed] [Google Scholar]
- 26.Lin E, Spear PG. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc Natl Acad Sci U S A 104:13140–13145. doi: 10.1073/pnas.0705926104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J Virol 86:8171–8184. doi: 10.1128/JVI.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muggeridge MI. 2000. Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J Gen Virol 81:2017–2027. doi: 10.1099/0022-1317-81-8-2017. [DOI] [PubMed] [Google Scholar]
- 29.Ruel N, Zago A, Spear PG. 2006. Alanine substitution of conserved residues in the cytoplasmic tail of herpes simplex virus gB can enhance or abolish cell fusion activity and viral entry. Virology 346:229–237. doi: 10.1016/j.virol.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Fan Z, Grantham ML, Smith MS, Anderson ES, Cardelli JA, Muggeridge MI. 2002. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell-cell fusion, but these properties are unrelated. J Virol 76:9271–9283. doi: 10.1128/JVI.76.18.9271-9283.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai WZ, Person S, Debroy C, Gu BH. 1988. Functional regions and structural features of the gB glycoprotein of herpes-simplex virus type-1—an analysis of linker insertion mutants. J Mol Biol 201:575–588. doi: 10.1016/0022-2836(88)90639-0. [DOI] [PubMed] [Google Scholar]
- 32.Chowdary TK, Heldwein EE. 2010. Syncytial phenotype of C-terminally truncated herpes simplex virus type 1 gB is associated with diminished membrane interactions. J Virol 84:4923–4935. doi: 10.1128/JVI.00206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harman A, Browne H, Minson T. 2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J Virol 76:10708–10716. doi: 10.1128/JVI.76.21.10708-10716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Browne HM, Bruun BC, Minson AC. 1996. Characterization of herpes simplex virus type 1 recombinants with mutations in the cytoplasmic tail of glycoprotein H. J Gen Virol 77(Part 10):2569–2573. [DOI] [PubMed] [Google Scholar]
- 35.Jackson JO, Lin E, Spear PG, Longnecker R. 2010. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J Virol 84:2038–2046. doi: 10.1128/JVI.02215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silverman JL, Heldwein EE. 2013. Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fusogenicity of gB in transfected cells. J Virol 87:10139–10147. doi: 10.1128/JVI.01760-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson DW, Davis-Poynter N, Minson AC. 1994. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J Virol 68:6985–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okuma K, Nakamura M, Nakano S, Niho Y, Matsuura Y. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235–244. doi: 10.1006/viro.1998.9530. [DOI] [PubMed] [Google Scholar]
- 39.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Eisenberg RJ, Cohen GH. 2002. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J Virol 76:10894–10904. doi: 10.1128/JVI.76.21.10894-10904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heckman KL, Pease LR. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 41.McShane MP, Longnecker R. 2005. Analysis of fusion using a virus-free cell fusion assay. Methods Mol Biol 292:187–196. [DOI] [PubMed] [Google Scholar]
- 42.Connolly SA, Longnecker R. 2012. Residues within the C-terminal arm of the herpes simplex virus 1 glycoprotein B ectodomain contribute to its refolding during the fusion step of virus entry. J Virol 86:6386–6393. doi: 10.1128/JVI.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haanes EJ, Nelson CM, Soule CL, Goodman JL. 1994. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J Virol 68:5825–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walev I, Lingen M, Lazzaro M, Weise K, Falke D. 1994. Cyclosporin A resistance of herpes simplex virus-induced “fusion from within” as a phenotypical marker of mutations in the Syn 3 locus of the glycoprotein B gene. Virus Genes 8:83–86. doi: 10.1007/BF01703606. [DOI] [PubMed] [Google Scholar]
- 45.Muggeridge MI, Grantham ML, Johnson FB. 2004. Identification of syncytial mutations in a clinical isolate of herpes simplex virus 2. Virology 328:244–253. doi: 10.1016/j.virol.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 46.McShane MP, Longnecker R. 2004. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc Natl Acad Sci U S A 101:17474–17479. doi: 10.1073/pnas.0404535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia NJ, Chen J, Longnecker R. 2013. Modulation of Epstein-Barr virus glycoprotein B (gB) fusion activity by the gB cytoplasmic tail domain. mBio 4:e00571–12. doi: 10.1128/mBio.00571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J, Zhang X, Jardetzky TS, Longnecker R. 2014. The Epstein-Barr virus (EBV) glycoprotein B cytoplasmic C-terminal tail domain regulates the energy requirement for EBV-induced membrane fusion. J Virol 88:11686–11695. doi: 10.1128/JVI.01349-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng T, Ponce de Leon M, Novotny MJ, Jiang H, Lambris JD, Dubin G, Spear PG, Cohen GH, Eisenberg RJ. 1998. Structural and antigenic analysis of a truncated form of the herpes simplex virus glycoprotein gH-gL complex. J Virol 72:6092–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vitu E, Sharma S, Stampfer SD, Heldwein EE. 2013. Extensive mutagenesis of the HSV-1 gB ectodomain reveals remarkable stability of its postfusion form. J Mol Biol 425:2056–2071. doi: 10.1016/j.jmb.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wanas E, Efler S, Ghosh K, Ghosh HP. 1999. Mutations in the conserved carboxy-terminal hydrophobic region of glycoprotein gB affect infectivity of herpes simplex virus. J Gen Virol 80(Part 12):3189–3198. [DOI] [PubMed] [Google Scholar]
- 52.Chao LH, Klein DE, Schmidt AG, Pena JM, Harrison SC. 2014. Sequential conformational rearrangements in flavivirus membrane fusion. eLife 3:e04389. doi: 10.7554/eLife.04389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC. 2013. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2:e00333. doi: 10.7554/eLife.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Floyd DL, Ragains JR, Skehel JJ, Harrison SC, van Oijen AM. 2008. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A 105:15382–15387. doi: 10.1073/pnas.0807771105. [DOI] [PMC free article] [PubMed] [Google Scholar]