

ABSTRACT
In lethal prion neurodegenerative diseases, misfolded prion proteins (PrPSc) replicate by redirecting the folding of the cellular prion glycoprotein (PrPC). Infections of different durations can have a subclinical phase with constant levels of infectious particles, but the mechanisms underlying this plateau and a subsequent exit to overt clinical disease are unknown. Using tandem biophysical techniques, we show that attenuated accumulation of infectious particles in presymptomatic disease is preceded by a progressive fall in PrPC level, which constricts replication rate and thereby causes the plateau effect. Furthermore, disease symptoms occurred at the threshold associated with increasing levels of small, relatively less protease-resistant oligomeric prion particles (oPrPSc). Although a hypothetical lethal isoform of PrP cannot be excluded, our data argue that diminishing residual PrPC levels and continuously increasing levels of oPrPSc are crucial determinants in the transition from presymptomatic to symptomatic prion disease.
IMPORTANCE Prions are infectious agents that cause lethal brain diseases; they arise from misfolding of a cell surface protein, PrPC to a form called PrPSc. Prion infections can have long latencies even though there is no protective immune response. Accumulation of infectious prion particles has been suggested to always reach the same plateau in the brain during latent periods, with clinical disease only occurring when hypothetical toxic forms (called PrPL or TPrP) begin to accumulate. We show here that infectivity plateaus arise because PrPC precursor levels become downregulated and that the duration of latent periods can be accounted for by the level of residual PrPC, which transduces a toxic effect, along with the amount of oligomeric forms of PrPSc.
INTRODUCTION
Prions are proteinaceous, infectious particles responsible for a group of incurable neurodegenerative diseases in humans and animals. A posttranslationally misfolded version of the cellular prion protein (PrPC), known as PrPSc, is the primary component of a prion and propagates by acting as a template for the conformational conversion of PrPC substrate (1). Analysis of brain material by prion bioassays has shown that infectivity plateaus can exist early during disease, suggesting that infections can be divided into an infectivity phase and a toxicity phase (2–4). The accumulation of a hypothetical toxic PrP form (PrPL, “L” for lethal), distinct from PrPSc, has been proposed to explain the transition from a subclinical phase to the appearance of clinical signs and progression to end-stage disease at the time when the prion levels plateau. It has been further suggested that prions are infectious, but nontoxic, entities that act as a catalyst for the generation of toxic PrPL at a rate with direct proportionality to PrPC expression levels in the animal models used for these experiments (2). However, this hypothetical protein has yet to be isolated.
In other studies, based on falling levels of the PrP-like Shadoo (Sho) protein, we recently demonstrated that the extended asymptomatic stage encompasses a different chemical parameter, namely, the posttranslational downregulation of PrPC levels (5). We surmised that a functional relationship might exist between the PrPC downregulation seen at disease endpoint (“ΔPrPC,” as measured against mock-infected control mice) in different prion diseases and the formation of PrPSc measured by conformation-dependent immunoassay (CDI) (5). We explored this relationship for RML, 139A, Sc237, and CJD prion isolates using an in vitro protein misfolding cyclic amplification system (6, 7). In titrated amplification reactions using low rPrPSc concentrations (to model an early stage in disease), a 30% decrease in PrPC substrate produced a 54% drop in replication rate (from 118- to 54-fold) of protease-resistant PrPSc (5). However, these earlier studies neither addressed the full time course of disease nor the formation of infectious prion particles. Here, we used standard scrapie cell assay (SSCA) data to better understand the relationship between the infectivity plateau and substrate reduction in mice of three genotypes.
MATERIALS AND METHODS
Mouse lines, prion bioassays, and brain homogenates.
TgPrnp mice (abbreviated to Tg20 [8]), a low-expresser Tg line (Tg.Prnp-AL), wild-type (wt) mice (Prnp+/+), and hemizygous Prnp-null mice (Prnp0/+) were used as described previously (5) in accordance with the Canadian Council on Animal Care and were approved by the Institutional Animal Care and Use Committees at the University of Alberta. Mice were inoculated at ∼6 weeks of age with 30 to 50 μl of 1% (wt/vol) brain homogenate containing RML mouse-adapted scrapie prions. The mice were sacrificed in a time course regimen designed to monitor changes in PrP and infectivity over the complete duration of the disease. These animals were monitored over this period for neurological signs associated with RML prion disease, including ataxia, a rigid tail, kyphosis, a bobbing head, and a rough coat. Prnp0/+ mice were sacrificed on days 1 (n = 4), 60 (n = 4), 100 (n = 4), 140 (n = 4), 180 (n = 4; 0/4 were exhibiting prion disease symptoms), and 363 (n = 2) postinoculation when all animals reached terminal end-stage disease, as judged by symptomatic progression. Prnp+/+ mice were sacrificed on days 1 (n = 3), 30 (n = 3), 60 (n = 3), 100 (n = 3), 140 (n = 3; first symptomatic time point for 2/3), and 162.67 ± 7.84 (n = 3) postinoculation when all animals reached terminal end-stage disease, as judged by symptomatic progression. Tg20 mice were sacrificed on days 1 (n = 3), 15 (n = 3), 30 (n = 3), 45 (n = 3), 60 (n = 3; first symptomatic time point for 2/3), and 65 (n = 3) postinoculation when all animals reached terminal end-stage disease, as judged by symptomatic progression. Brain homogenate was made by serial passage through needles at 10% (wt/vol) in phosphate-buffered saline (PBS; pH 7.4).
Sucrose gradient evaluation.
Velocity gradient centrifugation was used to separate monomeric, oligomeric, and high molecular assemblies of PrP as previously described (5). Briefly, the 400-μl aliquots of 10% brain homogenate containing 2% Sarkosyl were clarified and applied to a 10 to 45% sucrose gradient prepared in PBS (pH 7.4) containing 1% Sarkosyl. Ultracentrifugation was performed at 237,000 × g for 73 min at 5°C. Gradient fractions were bottom harvested.
CDI evaluation of PrPC and PrPSc levels, replication rate, and specific infectivity.
The validation of CDI has been reported by the authors and other laboratories (9–18). Here, we utilized the recently modified protocol that adapts CDI to murine samples (5). First, Lumitrac 600 High-Binding 96-well plates (E&K Scientific) were coated with monoclonal antibody (MAb) 8H4 (epitope 175-185) in 200 mM NaH2PO4 containing 0.03% (wt/vol) NaN3 (pH 7.5) (19). Next, 20-μl aliquots from each fraction containing 0.007% (vol/vol) of Patent Blue V (Sigma-Aldrich) were directly loaded into wells prefilled with 200 μl of assay buffer (Perkin-Elmer). Then, the captured PrP was detected by europium-conjugated MAb 12B2 (epitope 88-92) (20). The time-resolved fluorescence (TRF) signals were measured by the multimode microplate reader PHERAstar Plus (BMG LabTech).
A calibration curve was prepared with either recombinant PrP(23-231, 129M) for samples containing full-length PrPSc or recombinant PrP(90-231, 129M) for samples containing truncated rPrPSc [PrP(27-30)] after proteinase K (PK) treatment. The PK-untreated sample containing PrP was divided into a native and a denatured aliquot, where the latter was denatured with 4 M guanidinium HCl for 5 min at 80°C. Using europium-labeled 12B2 for detection, the TRF signal of native samples corresponds to the epitope 88-92 that is exposed in α-helical PrPC and hidden in PrPSc, thus allowing a direct measurement of the PrPC concentration. The signal of the denatured aliquot corresponds to the total PrP in a sample. The concentration of PrPSc was calculated according to the following formula: [PrPSc] = [PrPD] – [PrPN], where D stands for the denatured aliquot, and N stands for the native aliquot. Next, the concentration of rPrPSc was calculated in samples subjected to PK treatment, followed by complete denaturation using the PrP(90-231, 129M) calibration curve. The separate calibration for the PK-treated and untreated sample is critical for correct results due to the lower affinity of MAb 12B2 to denatured full-length human PrP(23-231, 129M) compared to PrP(90-231, 129M). Furthermore, the concentration of sPrPSc was calculated according the following formula: [sPrPSc] = [PrPSc] – [rPrPSc]. Replication was defined as the increase in the value of rPrPSc between two time points, thus allowing the replication rate to be deduced by determining the rate of change in the slope of the rPrPSc values as a dy/dx function. Lastly, specific infectivity was defined as the amount of infectious PrPSc, and determined by measuring the units of (SSCA-estimated) infectivity per mass of (CDI-estimated) PrPSc in the sample.
SSCA.
SSCA was performed by exposing L929 cells to various concentrations of brain homogenates (0.1 to 0.0001% [wt/vol]) for 5 days in 96-well culture plates (5, 21). The cells were passaged three times (1:4 and 1:7), with 20,000 cells collected at the third passage and loaded on to MultiscreenHTS IP 96-well, 0.45-μm-pore-size filter plates (Millipore). Cells were subjected to PK digestion (5 μg/ml) and subsequent denaturation with 3 M guanidine thiocyanate. The enzyme-linked immunospot (ELISpot) reaction was performed using a mouse anti-PrP antibody (SAF83; 1:1,000) and a goat anti-mouse alkaline phosphatase-conjugated secondary antibody (1:5,000). The plates were developed using BCIP/NBT and analyzed using an Autoimmun Diagnostika GmbH ELISpot plate reader (ELR07). The plate reader was set at intensity 4, size 1, and gradient 0, using algorithm B; these settings produced a background noise level in control samples of fewer than 30 spots. Time course infectivity data are presented with a best-line fit by nonlinear regression plotted with GraphPad Prism software (sigmoidal function). Low-power views of ELISpot data from the three mouse genotypes were adjusted with the same parameters (−20% brightness, +40% contrast, and +50% sharpen).
RESULTS
Effect of PrPC expression levels on the prion infectivity plateaus.
Using brain homogenates derived from mice at end-stage with the Rocky Mountain Laboratory (RML) isolate of scrapie prions, we performed intracerebral inoculations on groups of mice expressing different levels of PrPC: Tg20 mice (also known as tga20) overexpressing PrPC by ∼6-fold (8), wild-type mice (wt; Prnp+/+), and hemizygous Prnp-null mice (Prnp+/0). Animals were sacrificed at designated time points throughout infection, including the terminal stage of disease. PrPC levels from these time point studies (Fig. 1A) were determined as described previously (5), with the SSCA with L929 cells also being used to determine the prion infectivity of unfractionated brain homogenate (0.1% [wt/vol]) from animals at each time point (Fig. 1B). As expected, all three groups of animals exhibited a rapid exponential increase in infectivity early in the incubation period. However, while wt animals tended to plateau and a plateau could be clearly discerned in hemizygous mice, infectivity continued to rise until the Tg20 overexpresser mice were scored as being in the terminal phase of disease. To assess whether the perceived decrease in replication rate for infectious titers in wt and hemizygous mice was merely derived from a “ceiling effect” of the assays performed in L929 cells, SSCA was repeated using a further 10-fold dilution of the brain homogenate samples (0.01% [wt/vol]). Here, the profiles of infectivity from the diluted samples resembled their more concentrated counterparts, arguing against such a ceiling effect (gray traces, Fig. 1B). In contrast to the concept of a single unifying plateau value for a given prion isolate measured in PK1 N2a cells (2, 3), the maximum values for titers measured in L929 cells differed significantly between wt and hemizygous mice with the same PrP allelic type (Prnpa), where infectivity in wt mice was 1 log greater than hemizygous mice and 2 logs greater than Tg20 mice (P < 0.0001 for both 0.1 and 0.01% homogenates; Table 1). Low-power views of representative ELISpot filters used for the time course analyses are presented in Fig. 1C to complement the numerical data of Fig. 1B and illustrate the plateau effect especially apparent in infected hemizygous mice.
FIG 1.

Presence or absence of plateau effects for prion infectivity in three mouse genotypes. (A) PrPC levels during the time course of RML infection in Tg20 (Tg.Prnp) mice overexpressing the a allele of PrPC, wild-type mice (Prnp+/+), and mice hemizygous for the PrP gene (Prnp0/+). Note the different scales for the y axes. dpi, days postinoculation. Some data points originate from Mays et al. (5). (B) Prion titer in the same time course studies expressed as spot counts per 20,000 cells. SSCA was performed in L929 cells to determine infectivity for brain material in these longitudinal studies of infection (0.1% [wt/vol] homogenate plotted in black; 0.01% homogenate plotted in gray). A red trace depicts the first derivative to represent replication rate for prion titer (spot counts) for the 0.1% (wt/vol) homogenate, with a diminished replication rate being correlated with diminished PrPC. The data represent averages ± the SEM. Black arrows on the y axis indicate maximum of infectivity values by SSCA using 0.1% (wt/vol) homogenate (n = 8 to 16). (C) Representative low-power images of the ELISpot wells are presented for each host genotype.
TABLE 1.
Plateau effects and biophysical parameters of PrP isoforms in prion-infected micea
| Parameter | Genotype |
||
|---|---|---|---|
| Tg20 mice (Tg.Prnp) | wt mice (Prnp+/+) | Hemizygote mice (Prnp0/+) | |
| Plateau effect for infectious particles | Equivocalb | Equivocal | Yes |
| Initial PrPC level (ng/ml)c | 3,104 ± 190 | 1,133 ± 15 | 449 ± 12 |
| Endpoint PrPC level (ng/ml) | 2,760 ± 311 | 737 ± 5 | 218 ± 5 |
| Mean percentage of PrPC ± the SD at the disease end stage (downregulation at disease endpoint) | 89% ± 12% | 65% ± 3.9% | 48% ± 3.3% |
| Mean infectivity ± the SD at last time point brain material (spots per 20,000 L929 cells) | 1,347 ± 157d | 4,285 ± 254 | 3,581 ± 270e |
| Specific infectivity of unfractionated brain material (spots per ng of PrPSc) | 7.7 × 105 | 7.7 × 105 | 7.2 × 105 |
| Gradient fractions with the highest specific infectivity | Low Mr fractions 5 to 9 | Low Mr fractions 5 to 9 | Low Mr fractions 5 to 9 |
Relationship between PrPC downregulation and the prion infectivity plateau.
Infectivity continued to rise to disease endpoint in time course studies of inoculated Tg20 mice (Fig. 1B), although the replication rates at the last two time points [(75 ± 18)-fold and (133 ± 29)-fold, expressed as means ± the standard errors of the mean (SEM), at 60 and 65 days postinoculation (dpi), respectively] were not significantly different (P = 0.1). An infectivity plateau was apparent in the time course for RML infection in Tg20 mice with sampling every single day between 60 and 65 dpi (2, 3), compared to our sampling on days 60 and 65 dpi. Clear attenuations for the rate of prion replication in wt and hemizygous mice (with an unambiguous plateau in hemizygous mice) were preceded by the occurrence of PrPC downregulation (Fig. 1A and B and Table 1). Although a linear increase in infectivity was recognized between 60 and 140 dpi for wt and hemizygous mice alike (Fig. 1B), continual reductions in PrPC levels apparently prevent substantial increases in infectivity from 140 dpi until terminal disease is reached. For RML-infected wt mice, replication rate fell 58% between 90 and 120 dpi by (69 ± 4)-fold to (29 ± 6)-fold (P = 0.0001), whereas the PrPC levels in the same interval dropped by ∼20% from 1,124 ± 2 ng/ml to 899 ± 9 ng/ml (P < 0.01). For RML-infected hemizygous mice, the changes were greater, such that the replication rate fell 67% between 100 and 140 dpi by (46 ± 1.5)-fold to (15 ± 3.2)-fold (P = 0.0001), while the PrPC levels dropped by ∼30% from 408 ± 8 ng/ml to 344 ± 21 ng/ml (P < 0.001).
Progression of oligomer formation during prion infection.
We wanted to determine whether a specific form of PrP, perhaps compatible with the hypothetical species called “PrPL,” is responsible for triggering PrPC downregulation and/or a surge of neurotoxicity causing the transition from subclinical disease to neurological end-stage disease. For biophysical analysis, we used velocity gradient centrifugation coupled with CDI to track different PrP species in mice of the aforementioned genotypes at different stages of disease (Fig. 2 and 3). In terms of isoform concentration, higher-molecular-weight species in fraction 2 predominated and increased in a steady manner through the disease course irrespective of the Prnp genotype; furthermore, the overall shape of the gradient profiles of PrPSc and rPrPSc species did not differ notably as the dpi values increased (Fig. 2). The only possible exception was the emergence of lower-molecular-weight (Mr) species centered on fraction 8 at later time points (most obvious for PrPSc in hemizygous mice at 363 dpi; Fig. 2, yellow trace, top right-hand panel). To assess this area of the gradient more closely, PrPSc and rPrPSc values for fraction 8 (containing oligomeric species) were plotted versus dpi (Fig. 3A). These expanded analyses did not reveal a singular performance for hemizygous mice; rather, they illustrated a spectrum of responses wherein similar levels (∼20 ng/ml) of oligomeric PrPSc accrued at later time points in all three genotypes, but albeit at tempos that varied between the Tg20, wt, and hemizygous animals (Fig. 3A). In these analyses, smaller oligomeric forms of PrP assemblies in fraction 8 were more PK-sensitive than large aggregates in fraction 2 (Fig. 3A versus 3B). Lastly, potentially relevant to the triggering of downregulation for wt and hemizygous mice, rPrPSc species were evident in fraction 2 at time points when PrPC downregulation was already present (at 120 and 140 dpi), respectively (compare Fig. 2A and 3A with Fig. 1A).
FIG 2.

Time course for the appearance of misfolded PrP isoforms in three mouse genotypes. CDI analyses of gradient fractions from brains of mice of three genotypes (Tg20, Prnp+/+, and Prnp0/+) plotted versus different times after inoculation with the RML prion isolate to represent the gradient profiles for total PrPSc (A) and rPrPSc (B) after PK treatment. Time points (dpi) of the source material are indicated within each panel by color codes. Each data point is an average ± the SEM. of triplicate independent CDI measurements in three to four mice brains. CDI values for 30 and 45 dpi for Tg20 mice were below assay threshold and are not presented.
FIG 3.

Progressive accumulation of misfolded PrP isoforms in gradient fractions 8 and 2 for three mouse Prnp genotypes. (A) Sequential appearance of total PrPSc and rPrPSc in fraction 8 containing low-Mr species plotted versus dpi. (B) Sequential appearance of total PrPSc and rPrPSc in fraction 2 containing high-Mr species plotted versus dpi. Note the different y axes for panels A and B. Each data point is an average ± the SEM of triplicate independent CDI measurements in three to four mice brains. As in Fig. 2, the CDI values for PrPSc in Tg20 mice at 30 and 45 days lay below the assay threshold.
Gradient profiles of infectivity during prion infection.
As a prelude to extending these types of biophysical analyses to include infectivity measures, measurements of unfractionated brain samples normalized by volume determined that prion infectivity was significantly lower in unfractionated homogenates of Tg20 mice versus the other two genotypes (Table 1; P = 0.0001); this finding is in accord with replication still being in an exponential phase and thus failing to attain maximum possible values. However, specific infectivity expressed in spots per 20,000 cells per ng of PrPSc was similar in all three genotypes, indicating that PrPC expression level in the different models did not influence the overall characteristics of the RML prion isolate. Next, we assessed profiles of gradient-fractionated brain lysates for their infectious content in the context of disease time course. Since unfractionated 30 and 45 dpi samples from Tg20 mice were at the assay baseline, their gradient-fractionated counterparts were not assayed, but we addressed fractionated samples from the last two time points (60 and 65 dpi). For wt mice, we tested the last three time points (120, 140, and 162.67 ± 7.84 dpi) and for hemizygous mice we tested the last four time points (100, 140, 180, and 363 dpi) (Fig. 4A). The data in each fraction was plotted as a specific infectivity value to produce corrected gradient profiles (Fig. 4B). In these corrected analyses, the gradient fractions from terminal Tg20, wt, and hemizygous mice closely resembled one another. In each case, the peak of infectivity occurred in either fraction 7 or 8, thus indicating higher infectivity for relatively more PK sensitive, oligomeric PrPSc in comparison to PK-resistant PrPSc (Fig. 4B and Table 1). Moreover, within each given genotype, the gradient profile at the last time point did not differ notably in shape from prior time points in the subclinical phase of disease, and the differences in magnitude were an inevitable consequence of alterations in titer over the disease course. A tendency for lower net titers at endpoint in Prnp0/+ mice (Fig. 4B) reached significance in 3 of 10 fractions (P = 0.049, 0.001, and 0.049, respectively, for fractions 10, 9, and 4). Taken together with the gradient profiles of the PrP isoforms (Fig. 2), our data did not provide evidence of an emergent peak in the infectivity profile or PrP isoform profile that might provide a physiochemical signature for the hypothetical PrPL species. Instead, the data for each genotype revealed gradual alterations in the sedimentation profile favoring the accumulation of small oligomeric forms of PrP (Fig. 4B).
FIG 4.

Sequential appearance of infectivity in gradient fractionated material from three Prnp genotypes. (A) Uncorrected values for infectivity (spot counts) expressed per unit volume. Note the linear scale for the y axis. The infectivity data are averages ± the SEM obtained with 8× to 1× SSCA titrations for each fraction and time point. (B) Specific infectivity of gradient fractions from brains from different time points in disease and with genotypes as indicated.
Ratiometric relationship between PrPC downregulation and disease phase.
To explore the possible role of oligomers further, we undertook regression analyses. These indicated a statistically significant correlation between incubation time and PrPSc levels in the low-Mr fraction 8 containing oligomeric species (R = 0.834) but not in the high-Mr fraction 2 (Fig. 5). For these analyses, we also included data from a low-copy number Tg.Prnpa-AL mouse line (5, 22) infected with the same RML prion isolate. Since PrPC is likely a crucial factor in disease by mediating toxic signaling concentration (23), we next plotted the ratios of residual PrPC to total PrPSc at the disease endpoint versus the corresponding incubation times using PrPSc values from either fraction 2 (high Mr) or fraction 8 (low Mr, oligomeric; Fig. 6). Considering these four genotypes infected with the RML prion isolate, the dynamic range of PrPC to PrPSc ratios extended only from 0.6 to 1.0 for high Mr fractions. However, the dynamic range of ratios expanded ∼16-fold (extending from 4.5 to 122) when measuring PrPSc from oligomeric fractions. Furthermore, this relationship reliably predicted the incubation time and symptomatic stage of disease with a regression correlation value of 0.998 (P = 0.004).
FIG 5.
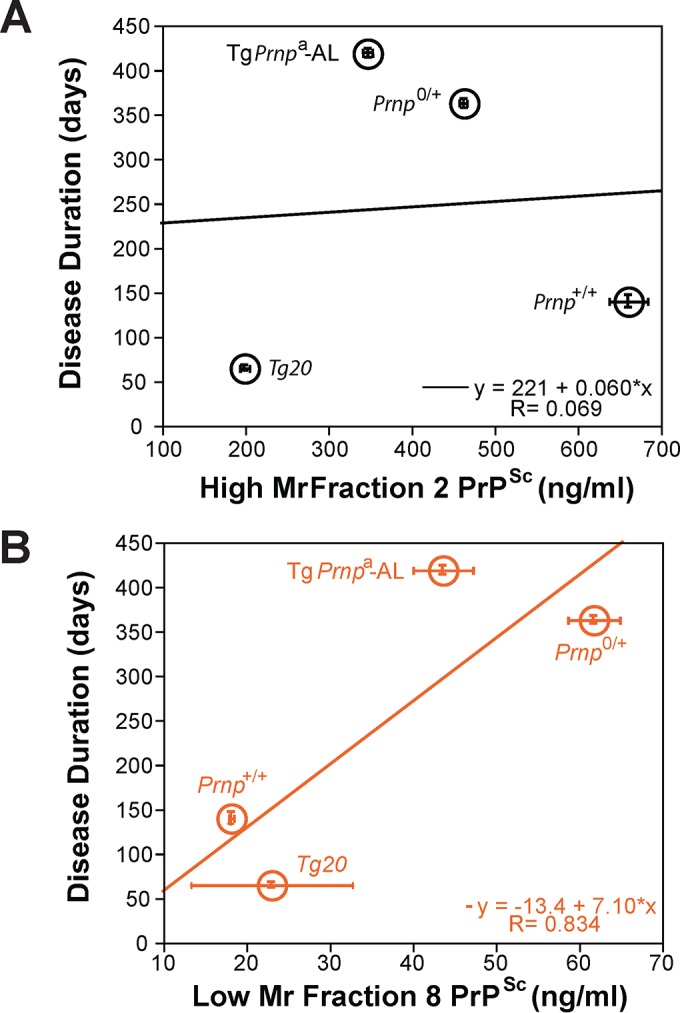
Regression analysis of PrPSc species in gradient fractions 2 and 8 versus disease incubation time. (A) Regression analysis of high-Mr PrPSc from gradient fraction 2. (B) Regression analysis of low-Mr oligomeric PrPSc species from gradient fraction 8.
FIG 6.
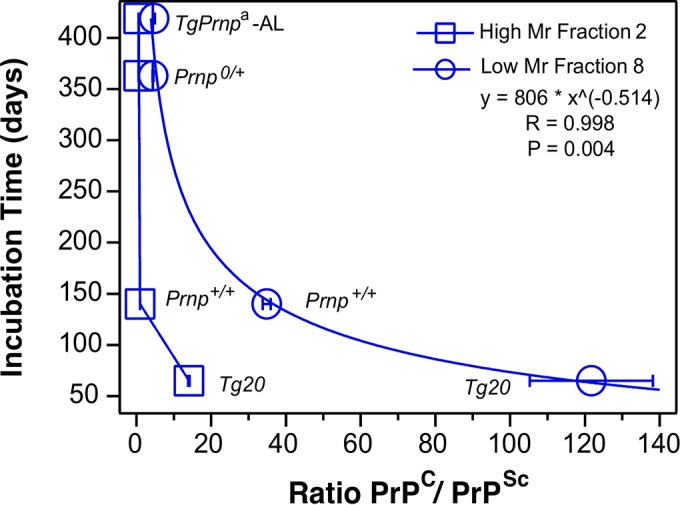
Ratiometric relationship between residual PrPC, oPrPSc, and duration of prion disease. Regression analysis of disease course versus a ratio of residual PrPC concentration at endpoint expressed as a ratio versus concentration of low-Mr (oligomeric) or high-Mr PrPSc. The infectivity data are averages ± the SEM obtained with 8× to 1× SSCA titrations for each fraction and time point.
DISCUSSION
In prior theoretical studies, it has been posited that the exponential rate of prion replication eventually inhibits and overtakes prion degradation, although reduced substrate availability simultaneously causes prion replication to decline and prion accumulation to plateau (24). Our study method of velocity gradient fractionation in the presence of Sarkosyl, followed by SSCA and CDI, offers a means to sample diverse PrP species during infection. The data obtained by these methods are summarized in Table 1. Strong effect sizes for PrPC downregulation in infected wt and hemizygous mice, as well as the synchronicity in reductions in replication rate (Fig. 1), lead us to infer that a relationship exists between PrPC downregulation and infectivity dropping toward a plateau level during the subclinical phase of prion disease (2, 3). As discussed below, we would assert that this is a simple cause-and-effect relationship. Given that plateau effects can exist, does a hypothetical PrPL species necessarily follow as a unifying determinant of toxicity and entry into the clinical disease phase? In the experiments here, the PrP allelic type (denoted Prnpa) and the prion strain (RML) were held constant and our analyses defined similar profiles after gradient fractionation (Fig. 2A), paralleling the unchanging overall specific infectivity of the samples prior to fractionation (Fig. 4B). The peak for specific infectivity in each case agrees with analyses that the “most infectious” particles (here in fractions 7 and 8) correspond to oligomers rather than high-molecular-weight fibrillar assemblies. These data are in agreement with other velocity gradient analyses (10, 25) and compatible with an analysis where PrP aggregates were dissociated and then fractionated by a native method (26). Importantly, in preclinical samples, the gradient profiles of PrPSc isoforms detected in a highly sensitive assay and the specific infectivity (Fig. 2 and 4 and Table 1) differed from endpoint samples only in quantity and not quality. A toxic monomeric alpha-helical form of PrP (“TPrP”) has been deduced from refolding unglycosylated recombinant PrP and assay upon the PK-1 subline of N2a neuroblastoma cells (27). Toxicity mediated by TPrP was observed in the range of 0.5 to 5 μg/ml (27, 28), which is in the same general range as PrPC levels measured here in brain homogenates of Prnp+/+ and Prnp0/+ mice (i.e., ∼1.1 and 0.4 μg/ml, respectively; Fig. 1). Although immunological reagents to distinguish “TPrP” in brain homogenates from PrPC would facilitate study of this putative toxic alpha-helical form, we already know that the PrPC levels detected with the 12B2 antibody in Prnp+/+ and Prnp0/+ mice are falling—not rising—as animals progress through preclinical disease (to levels of 0.7 and 0.2 μg/ml, respectively, Fig. 1) (5), making a simultaneous, superimposed rise in TPrP levels unlikely. Instead, our data are notable for failing to define a specific form of PrP emerging at the last time points in infection, when the animals exhibit clinical disease. Although our studies did not assay directly for neurotoxicity in cell cultures, the responses in these types of experiments will be dependent upon the particular cells selected. Indeed, it is possible that a protracted search for a hypothetical PrPL molecule might ultimately prove fruitless and that ratios of tangible PrP species are, instead, the critical determinants for disease manifestation. From prior analyses of eight experimental prion diseases at endpoint, we suggested (5) that the entry into and duration of the clinical phase of disease depends on the ratio of PrPSc to residual PrPC at endpoint. In addition to its role as a substrate, PrPC's importance here fits with the prior hypothesis that PrPC serves as a PrPSc receptor to mediate toxic signaling (23, 24). By extending our analyses to include fractionation by velocity gradients, we have identified the low-Mr oligomeric forms of PrPSc (oPrPSc), rather than high-Mr species, as being particularly important in this ratiometric relationship at the end stage (Fig. 4 to 6). Based on our previous calibrations (10), we estimate the prions in these fractions are assembled from 20 to 78 monomers of PrPSc.
Returning to consider preclinical events leads to the “whys and wherefores” of downregulation, a process that sculpts PrPC/oPrPSc ratios. Although cell-based mechanisms to attenuate the accumulation of rPrPSc or infectivity may exist (for example, clearance [29] and phagocytosis [30]), it is unlikely they could have the same magnitude of impact as the downregulation of PrPC, which is closely linked in time to the drop in replication rate for infectivity and PrPSc isoforms. This is because (i) PrPC is an obligatory precursor for the formation of PrPSc and infectivity (shown by the use of PrP transgenic mice [31] and Prnp0/0 knockout mice [32]) and, conversely, (ii) there is no alternative pathway of cross-seeding to create mouse-adapted prions by misfolding of Sho protein (33–35). Although it remains possible that a particular subpool of PrPC is eligible for conversion to PrPSc (for example, partially unfolded or particularly glycosylated molecules), the size of this hypothetical pool would have to expand and contract in direct proportion with the net starting concentration of PrPC because constitutive and inducible transgenes that alter PrPC levels have a profound effect upon the generation of prion infectivity and disease incubation times (31, 36, 37). Indeed, altered expression levels of PrPC eclipse the effect of allelic variation that was once thought to be the preeminent determinant of scrapie incubation time in mice (38–40). On a practical level, bulk PrP exists in di-, mono-, and unglycosylated forms, but PrP alleles with amino acid substitutions affecting the attachment of N-glycans are not the equivalent of wt PrP in conversion to PrPSc after adjusting for expression levels (41–43). During the preclinical downregulation effects that impact PrPC and Sho, the mature glycosylated forms of these proteins are preferentially depleted (5, 44). Furthermore, for Sho, the proportionality of downregulation of mature glycoprotein at endpoint is unaffected by transgene expression levels (44, 45), calling to mind the proportional scaling effect for PrPC expression mentioned above. Overall, we deduce that the supply of mature diglysosylated PrPC, reduced by downregulation versus partial sparing of unglycosylated and monoglycosylated isoforms (5), is a bottleneck for prion replication. Since PrPC is a precursor, this controlling variable inevitably lies “upstream” of all subsequent events. Extending analyses to deal with infectivity (Fig. 1 and Table 1), this concept builds on previous studies that used calibrated in vitro reactions to determine the impact of decreasing PrPC concentration upon decreasing replication rate to make rPrPSc (5). While we infer that the downregulation of PrPC and Sho (both glycosylphosphatidylinositol-anchored proteins) triggered by the accumulation of protease resistant PrP (44, 45) might represent a partially effective host response to infection, interventions to further depress PrPC may have the benefit of reducing plateau values for infectivity, extending the length of subclinical disease and the amount of residual cell surface molecules available to transduce the toxic effects of misfolded PrPs.
Cumulatively, our measurements in three mouse genotypes infected with the RML prion strain failed to define a consistent plateau phenomenon, but rather data that existed on a continuum with dropping levels of PrPC being crucial in the asymptomatic phase of disease. Rising levels of mobile small oligomeric prions can then transform this phase of prion disease into a symptomatic stage at the threshold ratio between available PrPC and oPrPSc (Fig. 6). These aspects argue for dynamic interactions, where fewer small oligomeric prions are able to trigger symptoms at high surface cell concentrations of PrPC, receptors for toxicity, and vice versa.
ACKNOWLEDGMENTS
We thank Witold Surewicz for recombinant PrP used for calibration in CDI, Earl Poptic from Cleveland Clinic Hybridoma Core Facility for the production of 8H4 MAb, and J. Langeveld for providing 12B2 MAb (epitope 88-92). We thank F. Jirik (University of Calgary, Calgary, Alberta, Canada) and V. Sim (University of Alberta) for Tg20 mice and J. Yang, J. Grams, and K. Bergen for maintenance of the mouse colony.
This study was supported by grants from the National Institute for Neurological Disorders and Stroke (NS074317), the Centers for Disease Control and Prevention (UR8/CCU515004), the Charles S. Britton Fund, the Canada Foundation for Innovation, the Canadian Institutes of Health Research (MOP36377), the Alberta Prion Research Institute, and Alberta Innovates-Health Solutions (through a postdoctoral fellowship supporting C.E.M.).
Experiments were designed by C.E.M., J.V.D.M., and D.W. Experiments were performed and analyzed by C.E.M., J.V.D.M., C.K., T.H., D.M., J.G.S., and D.W. The manuscript was written by C.E.M., J.G.S., and D.W.
REFERENCES
- 1.Prusiner SB. 1998. Prions. Proc Natl Acad Sci U S A 95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. 2011. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 470:540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- 3.Sandberg MK, Al-Doujaily H, Sharps B, De Oliveira MW, Schmidt C, Richard-Londt A, Lyall S, Linehan JM, Brandner S, Wadsworth JDF, Clarke AR, Collinge J. 2014. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat Commun 5:4347. doi: 10.1038/ncomms5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. 1994. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1:19–30. [PMC free article] [PubMed] [Google Scholar]
- 5.Mays CE, Kim C, Haldiman T, van der Merwe J, Lau A, Yang J, Grams J, Di Bari MA, Nonno R, Telling GC, Kong Q, Langeveld J, McKenzie D, Westaway D, Safar JG. 2014. Prion disease tempo determined by host-dependent substrate reduction. J Clin Invest 124:847–858. doi: 10.1172/JCI72241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saborio GP, Permanne B, Soto C. 2001. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 7.Castilla J, Morales R, Saá P, Barria M, Gambetti P, Soto C. 2008. Cell-free propagation of prion strains. EMBO J 27:2557–2566. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 9.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. 1998. Eight prion strains have PrPSc molecules with different conformations. Nat Med 4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 10.Kim C, Haldiman T, Surewicz K, Cohen Y, Chen W, Blevins J, Sy M-S, Cohen M, Kong Q, Telling GC, Surewicz WK, Safar JG. 2012. Small protease sensitive oligomers of PrPSc in distinct human prions determine conversion rate of PrPC. PLoS Pathog 8:e1002835. doi: 10.1371/journal.ppat.1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, Miller BL, DeArmond SJ, Prusiner SB. 2005. Diagnosis of human prion disease. Proc Natl Acad Sci U S A 102:3501–3506. doi: 10.1073/pnas.0409651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim C, Haldiman T, Cohen Y, Chen W, Blevins J, Sy M-S, Cohen M, Safar JG. 2011. Protease-sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt-Jakob disease are indicator of progression rate. PLoS Pathog 7:e1002242. doi: 10.1371/journal.ppat.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Safar JG, Lessard P, Tamgüney G, Freyman Y, Deering C, Letessier F, DeArmond SJ, Prusiner SB. 2008. Transmission and detection of prions in feces. J Infect Dis 198:81–89. doi: 10.1086/588193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, Serban H, Groth D, Burton DR, Prusiner SB, Williamson RA. 2002. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol 20:1147–1150. doi: 10.1038/nbt748. [DOI] [PubMed] [Google Scholar]
- 15.Thackray AM, Hopkins L, Bujdoso R. 2007. Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochem J 401:475–483. doi: 10.1042/BJ20061264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellon A, Seyfert-Brandt W, Lang W, Baron H, Gröner A, Vey M. 2003. Improved conformation-dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J Gen Virol 84:1921–1925. doi: 10.1099/vir.0.18996-0. [DOI] [PubMed] [Google Scholar]
- 17.Choi YP, Gröner A, Ironside JW, Head MW. 2011. Comparison of the level, distribution and form of disease-associated prion protein in variant and sporadic Creutzfeldt-Jakob diseased brain using conformation-dependent immunoassay and Western blot. J Gen Virol 92:727–732. doi: 10.1099/vir.0.026948-0. [DOI] [PubMed] [Google Scholar]
- 18.McCutcheon S, Hunter N, Houston F. 2005. Use of a new immunoassay to measure PrPSc levels in scrapie-infected sheep brains reveals PrP genotype-specific differences. J Immunol Methods 298:119–128. doi: 10.1016/j.jim.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 19.Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS. 1998. Prion protein expression in different species: analysis with a panel of new MAbs. Proc Natl Acad Sci U S A 95:8812–8816. doi: 10.1073/pnas.95.15.8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langeveld JPM, Jacobs JG, Erkens JHF, Bossers A, Zijderveld FG, van Keulen LJM. 2006. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet Res 9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Merwe J, Aiken J, Westaway D, McKenzie D. 2015. The standard scrapie cell assay: development, utility and prospects. Viruses 7:180–198. doi: 10.3390/v7010180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau A, McDonald A, Daude N, Mays CE, Walter ED, Aglietti R, Mercer RCC, Wohlgemuth S, van der Merwe J, Yang J, Gapeshina H, Kim C, Grams J, Shi B, Wille H, Balachandran A, Schmitt-Ulms G, Safar JG, Millhauser GL, Westaway D. 2015. Octarepeat region flexibility impacts prion function, endoproteolysis, and disease manifestation. EMBO Mol Med 7:339–356. doi: 10.15252/emmm.201404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weismann C, Aguzzi A. 1996. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 24.Aguzzi A, Falsig J. 2012. Prion propagation, toxicity and degradation. Nat Neurosci 15:936–939. doi: 10.1038/nn.3120. [DOI] [PubMed] [Google Scholar]
- 25.Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, Laude H, Béringue V. 2010. The physical relationship between infectivity and prion protein aggregates is strain dependent. PLoS Pathog 6:e1000859. doi: 10.1371/journal.ppat.1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. 2005. The most infectious prion protein particles. Nature 437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou M, Ottenberg G, Sferrazza GF, Lasmézas CI. 2012. Highly neurotoxic monomeric α-helical prion protein. Proc Natl Acad Sci U S A 109:3113–3118. doi: 10.1073/pnas.1118090109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou M, Ottenberg G, Sferrazza GF, Hubbs C, Fallahi M, Rumbaugh G, Brantley AF, Lasmézas CI. 2015. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 138:992–1008. doi: 10.1093/brain/awv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, Prusiner SB, Tremblay P. 2005. Prion clearance in bigenic mice. J Gen Virol 86:2913–2923. doi: 10.1099/vir.0.80947-0. [DOI] [PubMed] [Google Scholar]
- 30.Brown GC, Neher JJ. 2014. Microglial phagocytosis of live neurons. Nat Rev Neurosci 15:209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 31.Prusiner SB, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, Torchia M, Yang S-L, Serban D, Carlson GA, Hoppe PC, Westaway D, DeArmond SJ. 1990. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63:673–686. doi: 10.1016/0092-8674(90)90134-Z. [DOI] [PubMed] [Google Scholar]
- 32.Büeler H, Aguzzi A, Sailer A, Greiner R-A, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 33.Premzl M, Sangiorgio L, Strumbo B, Marshall Graves JA, Simonic T, Gready JE. 2003. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 314:89–102. doi: 10.1016/S0378-1119(03)00707-8. [DOI] [PubMed] [Google Scholar]
- 34.Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, Sy MS, Yoong L, Young R, Mastrangelo P, Bergeron C, Fraser PE, Carlson GA, Mount HT, Schmitt-Ulms G, Westaway D. 2007. The CNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reduced levels in prion infections. EMBO J 26:4038–4050. doi: 10.1038/sj.emboj.7601830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daude N, Wohlgemuth S, Brown R, Pitstick R, Gapeshina H, Yang J, Carlson GA, Westaway D. 2012. Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrPC deficiency. Proc Natl Acad Sci U S A 109:9035–9040. doi: 10.1073/pnas.1202130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlson GA, Ebeling C, Yang S-L, Telling G, Torchia M, Groth D, Westaway D, DeArmond SJ, Prusiner SB. 1994. Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci U S A 91:5690–5694. doi: 10.1073/pnas.91.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mallucci G, Dickinson A, Linehan J, Klöhn P-C, Brandner S, Collinge J. 2003. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 38.Dickinson AG, Fraser H. 1979. An assessment of the genetics of scrapie in sheep and mice, p 367–386. In Prusiner SB, Hadlow WJ (ed), Slow transmissible diseases of the nervous system, vol 1 Academic Press, Inc, New York, NY. [Google Scholar]
- 39.Westaway D, Mirenda CA, Foster D, Zebarjadian Y, Scott M, Torchia M, Yang S-L, Serban H, DeArmond SJ, Ebeling C, Prusiner SB, Carlson GA. 1991. Paradoxical shortening of scrapie incubation times by expression of prion protein transgenes derived from long incubation period mice. Neuron 7:59–68. doi: 10.1016/0896-6273(91)90074-A. [DOI] [PubMed] [Google Scholar]
- 40.Moore RC, Hope J, McBride PA, McConnell I, Selfridge J, Melton DW, Manson JC. 1998. Mice with gene targetted prion protein alterations show that Prnp, Sinc, and Prni are congruent. Nat Genet 18:118–125. doi: 10.1038/ng0298-118. [DOI] [PubMed] [Google Scholar]
- 41.Rogers M, Taraboulos A, Scott M, Borchelt D, Serban D, Gyuris T, Prusiner SB. 1992. Modification and expression of prion proteins in cultured cells, p 457–469. In Prusiner SB, Collinge J, Powell J, Anderton B (ed), Prion diseases of humans and animals. Ellis Horwood, London, United Kingdom. [Google Scholar]
- 42.Neuendorf E, Weber A, Saalmueller A, Schatzl H, Reifenberg K, Pfaff E, Groschup MH. 2004. Glycosylation deficiency at either one of the two glycan attachment sites of cellular prion protein preserves susceptibility to bovine spongiform encephalopathy and scrapie infections. J Biol Chem 279:53306–53316. doi: 10.1074/jbc.M410796200. [DOI] [PubMed] [Google Scholar]
- 43.Tuzi NL, Cancellotti E, Baybutt H, Blackford L, Bradford B, Plinston C, Coghill A, Hart P, Piccardo P, Barron RM, Manson JC. 2008. Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biol 6:e100. doi: 10.1371/journal.pbio.0060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Westaway D, Genovesi S, Daude N, Brown R, Lau A, Lee I, Mays CE, Coomaraswamy J, Canine B, Pitstick R, Herbst A, Yang J, Ko KWS, Schmitt-Ulms G, DeArmond SJ, McKenzie D, Hood L, Carlson GA. 2011. Downregulation of Shadoo in prion infections traces a preclinical event inversely related to PrPSc accumulation. PLoS Pathog 7:e1002391. doi: 10.1371/journal.ppat.1002391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watts JC, Stöhr J, Bhardwaj S, Wille H, Oehler A, DeArmond SJ, Giles K, Prusiner SB. 2011. Protease-resistant prions selectively decrease Shadoo protein. PLoS Pathog 7:e1002382. doi: 10.1371/journal.ppat.1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]