

ABSTRACT
Our understanding of adenovirus (Ad) biology is largely extrapolated from human species C Ad5. Most humans are immune to Ad5, so lower-seroprevalence viruses like human Ad6 and Ad26 are being tested as therapeutic vectors. Ad6 and Ad26 differ at the DNA level by 34%. To better understand how this might impact their biology, we examined the life cycle of the two viruses in human lung cells in vitro. Both viruses infected A549 cells with similar efficiencies, executed DNA replication with identical kinetics within 12 h, and began killing cells within 72 h. While Ad6-infected cells remained adherent until death, Ad26-infected cells detached within 12 h of infection but remained viable. Next-generation sequencing (NGS) of mRNA from infected cells demonstrated that viral transcripts constituted 1% of cellular mRNAs within 6 h and 8 to 16% within 12 h. Quantitative PCR and NGS revealed the activation of key early genes at 6 h and transition to late gene activation by 12 h by both viruses. There were marked differences in the balance of E1A and E1B activation by the two viruses and in the expression of E3 immune evasion mRNAs. Ad6 was markedly more effective at suppressing major histocompatibility complex class I (MHC I) display on the cell surface and in evading TRAIL-mediated apoptosis than was Ad26. These data demonstrate shared as well as divergent life cycles in these genetically distant human adenoviruses. An understanding of these differences expands the knowledge of alternative Ad species and may inform the selection of related Ads for therapeutic development.
IMPORTANCE A burgeoning number of adenoviruses (Ads) are being harnessed as therapeutics, yet the biology of these viruses is generally extrapolated from Ad2 and Ad5. Here, we are the first to compare the transcriptional programs of two genetically distant Ads by mRNA next-generation sequencing (NGS). Species C Ad6 and Ad26 are being pursued as lower-seroprevalence Ad vectors but differ at the DNA level by 34%. Head-to-head comparison in human lung cells by NGS revealed that the two viruses generally conform to our general understanding of the Ad transcriptional program. However, fine mapping revealed subtle and strong differences in how these two viruses execute these programs, including differences in the balance of E1A and E1B mRNAs and in E3 immune evasion genes. This suggests that not all adenoviruses behave like Ad2 and Ad5 and that they may have unique strategies to infect cells and evade the immune system.
INTRODUCTION
There are currently nearly 60 genotypes of adenoviruses (Ads) that infect humans, causing an array of ocular, respiratory, digestive, and systemic infections (reviewed in reference 1). Human mastadenoviruses (HAdVs) fall into genetically grouped species A (HAdV-A) through species G (HAdV-G), with the level of sequence diversity across the virome being as high as 40% (2). Despite this drastic genetic diversity, the biology of most Ads is often extrapolated from the study of just two respiratory serotypes of HAdV-C, Ad2 and Ad5.
Transcriptional activation of adenovirus early (E) genes and late (L) genes is pivotal to the viral life cycle (Fig. 1A). All subsequent effects on the infected cell and host are defined by these early and late transcriptional events (reviewed in reference 3). Briefly, the viral life cycle is initiated by the activation of E1A and E1B transcription, followed by a cascade of activation of E2 and E4 genes. Ad E3 immune evasion genes are activated early but can extend into late stages. Finally, the major late promoter (MLP) is activated to coordinate the expression of capsid and accessory proteins involved largely in genome encapsidation. While these processes are well understood, particularly for species C adenoviruses Ad2 and Ad5, there is less extensive study of other human and nonhuman Ad species.
FIG 1.

Schematic of genome organizations and comparison of E3 genes of Ad6 and Ad26. Shown are a basic schematic of all adenoviruses, including early (gray arrows) (E1A, E1B, E2A, E2B, E3, and E4), intermediate (open arrows) (pIX and Iva2), and late (black arrows) (L1, L2, L3, L4, and L5) transcription units (top) and a diagram of the unique E3 regions of Ad6 and Ad26 (bottom). Species C Ads (Ad6) have two distinct E3-encoded products (6.7K and ADP) (gray), while species D Ads (Ad26) have three species-specific protein-encoding regions (23K, 31K, and 49K) (white). The five conserved protein-encoding regions (12.5K, E3-19K, RID-alpha, RID-beta, and 14.7K) are shown in black. The direction of arrows indicates coding strand and transcription direction.
Recently, high-throughput genetic analyses using microarrays and mRNA next-generation sequencing (NGS) have been applied to better understand transcriptional events after infection with human species C Ad2 (4–7) and after infection by bat adenovirus (8). These studies demonstrate that these individual viruses execute programs that generally conform to our knowledge of the adenovirus transcriptional cascade and host cell responses to the virus but in markedly more depth and with scrutiny of many more viral and host mRNAs than was feasible with earlier technologies. While these analyses provide useful insights into the biology of each virus, it is unclear in these separate studies how these two viruses or any two adenoviruses may execute their transcriptional programs in similar or different ways.
Human Ad6 falls into species C with Ad2 and Ad5 and shares 98 and 94% identities at the DNA level, respectively (2). Ad6 was originally isolated from the tonsils of a child and is generally thought to be a respiratory virus like other species C viruses (9). Human Ad26 falls into species D, the largest group of human adenoviruses (1). While Ad26 falls into this species, which includes viruses that infect ocular surfaces, it is unclear what natural site Ad26 infects. Both Ad6 and Ad26 are thought to infect different initial sites, but both viruses are shed from the digestive track for months after infection (1). Ad6 and Ad26 differ at the DNA genome level by 34% and represent two ends of the viral phylogenetic tree (2). Like other HAdV-C serotypes, Ad6 uses the coxsackievirus and adenovirus receptor (CAR) and αv integrins as well as binds vitamin K-dependent clotting factors that modulate its pharmacology (reviewed in reference 10). In contrast, Ad26 is thought to bind CD46 as its primary receptor (11) and to use αv integrins (12) but does not appear to bind clotting factors (13). Given their lower seroprevalence and unique biological properties, both Ad6 and Ad26 are being developed as vaccine and oncolytic vectors (12, 14–20).
In this work, we compare the postentry effects of two genetically distinct human adenoviruses, including transcriptional mRNA expression by NGS, as well as provide insights into their differing expression by looking at specific functional effects on host cell responses. We compare human species C Ad6, which is a lower-seroprevalence family member of Ad2 and Ad5, with human species D Ad26.
MATERIALS AND METHODS
Cell lines.
Human 293 cells were purchased from Microbix (Mississauga, Ontario, Canada). A549 lung cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in HyClone Dulbecco's high-glucose modified Eagle's medium (Thermo Scientific, Waltham, MA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Adenoviruses.
The human Ad6 “Tonsil 99” and Ad26 “BP-2” strains were purchased from the ATCC. Viruses were grown in 293 cells and purified by double CsCl banding, and viral particle concentrations were calculated from measurements of the optical density determined by absorbance at 260 nm (OD260) using a Nanodrop ND-1000 spectrophotometer (Labtech International, Ringmer, United Kingdom). The bioactivity of the two viruses was measured by serial dilution on A549 cells in replicates of 8. Infectivity was calculated 2 weeks after infection from wells suffering cytopathic effect (CPE) to derive a 50% tissue culture infectious dose (TCID50) based on the Reed-Muench method (66). Ad6 and Ad26 E3-deleted viruses were generated by red recombination in bacteria, as described previously (19, 21).
Cell infections.
A549 cell monolayers at 50% confluence were infected with Ad6 or Ad26 at the indicated multiplicity of infection (MOI), in terms of viral particles per cell. To synchronize infection, cells were first incubated for 1 h at 37°C to permit particle entry. Following this, cells were treated with trypsin and washed 3 times with phosphate-buffered saline (PBS) to remove bound and unbound particles. Cells were collected after washing (0-h time point) or returned to culture conditions for an additional 6, 12, 24, or 48 h after entry. For each treatment condition (mock, Ad6, or Ad26 infection), cells were infected en masse in a T225 culture flask to ensure homogeneous infection, and 106 cells were then divided into separate T25 culture flasks after washing for later collection. At the indicated time points, the medium from each T25 flask was saved, adherent cells were detached with trypsin, and the two were combined. Total cells were pelleted by centrifugation and processed to collect DNA or RNA for subsequent analyses.
Quantitative PCR for viral genome replication.
Viral genome replication was quantified by quantitative PCR (qPCR) as described previously (16). DNA was purified from ∼106 cells by using DNeasy blood and tissue kits (Qiagen, Valencia, CA). The total DNA concentration was determined by using a Nanodrop ND-1000 spectrophotometer (Labtech International, Ringmer, United Kingdom) as described above. Ad genomes were quantified by using species-specific primers against the Ad6 (species C) or Ad26 (species D) hexon, as previously described (16). Twenty nanograms of DNA template was analyzed in a 20-μl reaction mixture containing 300 nM F primer, 300 nM R primer, and SYBR green by using an AB7900HT instrument (Applied Biosystems). Genome quantification was achieved by comparison to a standard curve for each virus by using plasmid DNA at 10-fold dilutions from 109 to 103 viral genomes (vg). Samples were run in triplicate.
RNA purification.
Total RNA was purified from ∼106 cells by using an RNeasy minikit (Qiagen, Valencia, CA) according to the manufacturer's protocol, including on-column DNase treatment. RNA was quantified by using a Nanodrop Abs260 ND-1000 spectrophotometer (Labtech International, Ringmer, United Kingdom). A total of 1,500 ng of RNA was submitted to the Genome Expression Core in the Medical Genome Facility, Mayo Clinic, for RNA quality testing, library preparation, and subsequent sequencing. RNA quality was determined by using a 2100 Bioanalyzer (Agilent, Santa Clara, CA). All RNA samples were rated with an RNA integrity number (RIN) of 7.7 or greater and deemed of acceptable quality for subsequent analyses.
mRNA library construction.
TruSeq mRNA libraries (Illumina, San Diego, CA) were generated for three treatments (mock, Ad6, and Ad26) at two time points (6 and 12 h). RNA libraries were prepared according to the manufacturer's instructions for the TruSeq RNA Sample Prep v2 kit by using an Eppendorf EpMotion 5075 robot (Eppendorf, Hamburg, Germany). Reverse transcription and adaptor ligation steps were performed manually. Briefly, poly(A) mRNA was purified from total RNA by using oligo(dT) magnetic beads. The purified mRNA was fragmented at 95°C for 8 min, eluted from the beads, and primed for first-strand cDNA synthesis. RNA fragments were reverse transcribed into cDNA by using SuperScript III reverse transcriptase with random primers (Invitrogen, Carlsbad, CA). Second-strand cDNA synthesis was performed by using DNA polymerase I and RNase H. Double-stranded cDNA was purified by using a single AMPure XP bead (Agencourt, Danvers, MA) cleanup step. cDNA ends were repaired and phosphorylated by using Klenow fragment, T4 polymerase, and T4 polynucleotide kinase, followed by a single AMPure XP bead cleanup. A single 3′ adenosine was added to these blunt-ended cDNAs with Klenow exo- (Illumina). Paired-end DNA adaptors (Illumina) with a single “T” base overhang at the 3′ end were immediately ligated to the “A-tailed” cDNA population. Unique indexes included in the standard TruSeq kits (12 set A and 12 set B) were incorporated at the adaptor ligation step for multiplex sample loading onto the flow cells. The adaptor-modified DNA fragments were purified by two rounds of AMPure XP bead cleanup steps and were enriched by 12 cycles of PCR using primers included in the Illumina Sample Prep kit. The concentration and size distribution of the libraries were determined on an Agilent Bioanalyzer DNA 1000 chip (Agilent, Santa Clara, CA). A final quantification, using Qubit fluorometry (Invitrogen, Carlsbad, CA), was done to confirm the sample concentration.
mRNA library sequencing.
Libraries were loaded onto paired-end flow cells at concentrations of 8 to 10 pM to generate cluster densities of 700,000 cells/mm2 according to Illumina's standard protocol, using Illumina cBot and the cBot paired-end cluster kit version 3. Libraries were indexed on the flow cell, accommodating 3 or 4 libraries per lane. The flow cells were sequenced as 101-by-2 paired-end reads on an Illumina HiSeq 2000 instrument using TruSeq SBS sequencing kit version 3 and HCS v2.0.12 data collection software. Base calling was performed by using Illumina RTA version 1.17.21.3. An initial sequencing run was performed on single mock-, Ad6-, and Ad26-infected samples followed by a second batch of samples, for a final “n” of 3 samples per test group. The triplicate data set was used for downstream analyses.
NGS data analysis.
Sequenced reads from the instrument were aligned, using Bowtie2 aligner (22), to a reference consisting of the Ad6 genome (GenBank accession number HQ413315.1) and the Ad26 genome (GenBank accession number EF153474.1). In order to quantify the number of reads mapped to adenovirus genes, the Bowtie2-aligned SAM (sequence alignment map) files were converted to BAM (binary alignment map) file format. BEDTools (23) was used on the BAM files and GenBank annotation files to report the number of mapped reads intersecting with the adenovirus gene features. Custom shell and perl scripts were used for parsing the data. Total numbers of mRNA reads as well as reads per kilobase per million mapped viral reads (RPKM) or reads per kilobase per million total reads (RPKMtotal) were quantified for each Ad region of interest (see Table 2). Undefined Ad regions in the main serotype-specific references were predicted through sequence alignment of Ad6 and Ad26 sequences to other species C (GenBank accession number FJ349096.1 for Ad6 and accession number NC_001405.1 for AdC) and species D (accession number NC_010956.1 for Ad54 and AdD) GenBank reference sequences, respectively. All definitions of mapped coding regions used are given in Table 2. Raw reads mapping across Ad genomes were visualized by using Integrative Genomics Viewer (IGV) (24). The image detail and complexity generated with IGV favored the use of 500-bp-read mapping, as shown in Fig. 3B.
TABLE 2.
Summary of adenovial sequence definitionsb
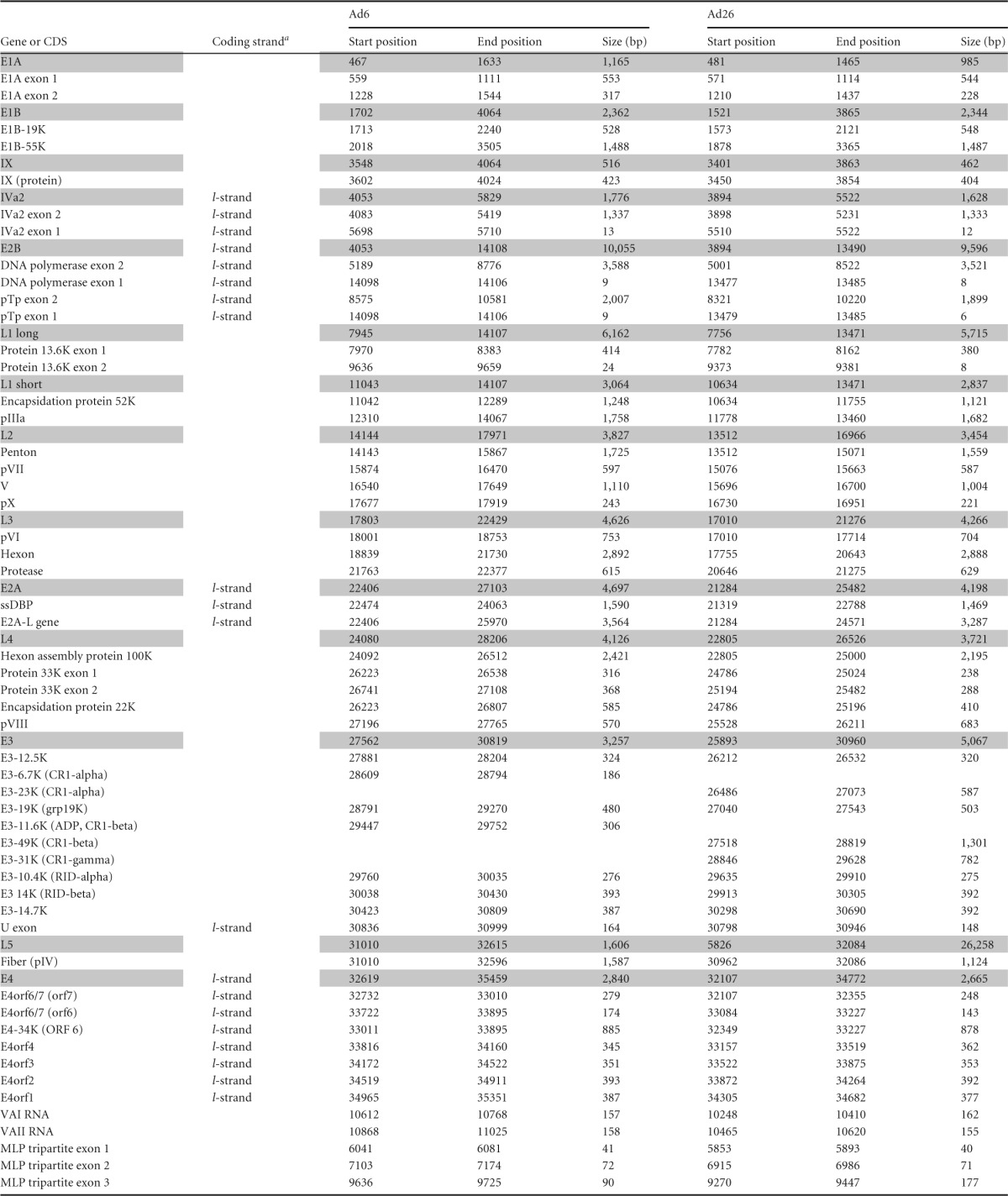
a The coding strand is typical unless otherwise indicated. The l-strand designation indicates “leftward transcription.”
CDS, coding sequence; ssDBP, single-stranded DNA binding protein. Shading indicates the major adenoviral gene regions; those not shaded are the smaller coding regions or ORFs belonging to the shaded coding region above.
FIG 3.

Influence of physical and genetic characteristics of adenoviruses on morphology of infected A549 cells. (A) Cells infected with 10,000 vp/cell of virus after no treatment (untreated) or virus that was treated with UV for 15 min, heat inactivated at 55°C for 15 min, or both. Images were taken after 17 h of infection. Magnification, ×100. (B) Cells were infected with 10,000 vp/cell of replication-competent E3-deleted Ad26 (Ad26ΔE3-GL) or replication-defective E1-deleted Ad26 (Ad26ΔE1-GL) or uninfected. Images were taken after 16 h of infection. Phase-contrast and fluorescence microscopy images are shown. Magnification, ×100.
Reverse transcription-quantitative PCR (RT-qPCR) for viral mRNAs.
Total RNA was isolated from infected cells as described above under “RNA purification.” cDNA was prepared from purified RNA samples by using the First Strand Synthesis SuperMix kit (Invitrogen/Life Technologies), according to the manufacturer's protocol, with oligo(dT) primers. Ad mRNA expression from cDNA samples was evaluated by using species-specific primers for E1A (5′-TGAAGAGGGTGAGGAGTTTG-3′ and 5′-GGTGATAATGACAAGACCTG-3′ for Ad6 and 5′-ACCGACTCTGTCACCCATAC-3′ and 5′-AAACCTTCCTCATAACACCG-3′ for Ad26), E1B-55K (5′-ACAACATACTGACCCGCTG-3′ and 5′-GCATTGGTAAGGTAGGAACAC-3′ for Ad6 and 5′-GCAACTTTAGCCAGACCAAG-3′ and 5′-AGATGCCGTTCAGGTTCAC-3′ for Ad26), E3-19K (5′-CCCCCACAAAAGTGTTTAGAG-3′ and 5′-TGTAATAAGCAGAGCGGTG-3′ for Ad6 and 5′-TGGGAAATGTATGGGTGGG-3′ and 5′-GACAGTGACCGTGTAGTTC-3′ for Ad26), E3-10.4K (5′-TGCGTGCTCTACATTGGCTG-3′ and 5′-ACTGTGAAAGGTGGGATGC-3′ for Ad6 and 5′-TTCCCCACCTACCTCCTCTTTG-3′ and 5′-ATGACCAGGCAGACAATGC-3′ for Ad26), hexon (5′-CAACCTGAACCACAAGTAGG-3′ and 5′-TAAGAACCCTTCCACCAGC-3′ for Ad6 and 5′-TTCCAGCCAGAACCTCAAG-3′ and 5′-AAGAGCCCTTCCTCCATAG-3′ for Ad26), and fiber (5′-TGCTTTTGACAACACCGC-3′ and 5′-ATTTCCGTTTGAGGAGTCTG-3′ for Ad6 and 5′-AAGGGGAATAGGAACGGAG-3′ and 5′-AAGTCTTACACGCAAGCC-3′ for Ad26). Ten nanograms of the cDNA template was analyzed in a 20-μl reaction mixture containing 300 nM forward (F) primer, 300 nM reverse (R) primer, and SYBR green on an ABI ViiA 7 system (Applied Biosystems). Absolute quantification was achieved by comparison to a standard curve for each virus by using purified viral DNA at 10-fold dilutions from 108 to 103 viral genomes. Samples were run in triplicate.
Flow cytometry for major histocompatibility complex class I in infected cells.
Cells were infected with Ads or mock infected for the indicated times, and major histocompatibility complex class I (MHC I) was detected by flow cytometry after staining for 1 h with anti-HLA-A, -B, and -C (W6/32; Abcam, Cambridge MA) or the isotype control in fluorescence-activated cell sorter (FACS) buffer (PBS, 1% bovine serum albumin [BSA], 0.1% sodium azide), washed twice with FACS buffer, and fixed in a 4% paraformaldehyde solution. Cells were evaluated on a FACScan flow cytometer (Becton-Dickinson, Franklin Lakes, NJ). The geometric mean fluorescence intensities (gMFI) were compared between samples. E3-19K does not affect preexisting MHC I on the cell surface, so these MHC I molecules were removed from the cells prior to measurement of the effects of E3-19K. Trypsin failed to remove cell surface MHC I (data not shown), so MHC I was instead removed by dissociating the MHC I-β2 microglobulin-peptide complexes by transient exposure to low-pH citric acid buffer, as described previously (25). For strip-and-recovery MHC I experiments, cells were infected at 1,000 virus particles (vp)/cell. After 6 h of infection, cells were disassociated from flasks and collected. Cell pellets were resuspended in a solution containing 133 mM citric acid and 66 mM sodium phosphate (pH 3.1) with 1% BSA for 2 min. The cells were washed twice in culture medium and returned to culture flasks. Following a 3-h “recovery” incubation, surface staining of MHC I for flow cytometry was carried out as stated above.
Evaluation of resistance to TRAIL-induced apoptosis.
A549 cells were infected with 1,000 vp/cell of the specified Ads in 96-well plates. After 6 h of infection, cells were sensitized with cycloheximide (CHX) and treated with 0 to 50 ng/ml of soluble recombinant tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) (catalog number cyt-443; Prospec Bio, Ness Ziona, Israel). Samples were run in triplicate. Cell viability and apoptosis were determined after 24 h by using the Tali apoptosis kit (catalog number A10788; Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Briefly, culture media and detached cells were collected from wells, and the remaining adherent cells were disassociated by using trypsin and added to the collection. Cells were stained with the Tali apoptotic marker (annexin V-Alexa Fluor 488) and viability dye (propidium iodide). By using the Tali system, dead cells are defined by propidium iodide inclusion independent of annexin V staining, apoptotic cells are defined by annexin V positivity, and live nonapoptotic cells are defined as annexin V- and propidium iodide-double-negative cells.
Differential expression and statistical analyses.
Total reads were calculated and converted to RPKM values for each region of interest. Since we are assessing Ad mRNA, these RPKM values are normalized to reads that map to the viral genome and ignore reads assigned to the host genome. Unpaired, 2-tailed t tests of log2 RPKM values were performed on triplicate values for each comparison between Ads at a given time point. Significance is noted in the figure legends as P values of <0.05, <0.01, <0.001, and <0.0001. Similarly, regions of interest were compared for their change over time (ΔT12 h–6 h) by first calculating the ΔT12 h–6 h value for each replicate, as paired observations, and subjecting the triplicate ΔT values for each viral treatment to a t test as described above (unpaired and 2 tailed). Thus, paired log2 RPKM values from 6 h and 12 h from a single replicate were subtracted, giving rise to the log2 fold change in RPKM values from 6 to 12 h: T12 h–6 h = log2 RPKM12 h − log2 RPKM6 h and ΔT12 h–6 h = log2(RPKM12 h/RPKM6 h).
Accession numbers.
Details and raw sequencing reads can be found in NCBI databases through the following accession numbers: PRJNA296866 (BioProject), SAMN04168604 to SAMN04168615 (BioSample), and SRP064863 (Sequence Read Archive).
RESULTS
Infection of human A549 lung cells by Ad6 and Ad26.
The kinetics of infection for the two viruses in A549 human lung carcinoma cells were compared. A549 cells have often been used to study adenoviral infection and are highly permissive to many species and serotypes, including Ad6 and Ad26. A549 cells were infected at various multiplicities of infection, and cell morphology and viability were assessed by microscopy and propidium iodide exclusion (Fig. 2A and C). Infections with 100, 1,000, 3,000, 10,000, 30,000, and 100,000 vp per cell produced a cell rounding phenotype 12 h after infection by Ad26 (Fig. 2A and data not shown). Ad6 cells remained adherent for 30 h after infection and detached only by 48 h. In contrast, a large fraction of Ad26-infected cells detached from the plate within 12 h and remained detached over 48 h (Fig. 2A). At all of these time points, the cells appeared viable, with no indications of apoptosis or necrosis as assessed by propidium iodide exclusion and annexin V staining, respectively (Fig. 2C and data not shown). Only at late time points after 48 h was a loss of membrane integrity observed.
FIG 2.

Ad6 and Ad26 infection of A549 cells. (A) A549 cells were infected with 1,000 vp/cell of Ad6 or Ad26 and imaged over time. (B) Time course of detection of Ad genomes in A549 cells 0, 6, 12, 24, 36, and 48 h after infection with 10,000 vp per cell. Twenty nanograms of DNA was evaluated by qPCR at each time. Averages and standard errors for three independent experiments are shown. (C) Viability of infected A549 cells (10,000 vp per cell) evaluated by a propidium iodide exclusion assay. Averages and standard errors for three replicates are shown (*, P < 0.05; ****, P < 0.001; ns, not significant).
This detachment-and-rounding phenotype induced by Ad26 required functional infectious viruses, since UV and heat inactivation of Ad26 ablated this effect (Fig. 3A). This effect also depended on E1 expression, since E1-deleted, replication-defective Ad26 (Ad26ΔE1) did not induce the same degree of rounding (Fig. 3B). Early cell rounding was observed for other cell types (data not shown) and was not specific to Ad26 E1 genes, since Ad26ΔE1 induced the phenotype in 293 cells that express Ad2 E1A in trans (data not shown). This rounding phenotype was also not mediated by E3 proteins, since E3-deleted Ad26ΔE3 still mediated the phenotypes (Fig. 3B).
Viral genome replication by Ad6 and Ad26.
The kinetics of viral entry and DNA replication of the two viruses in A549 human lung carcinoma cells were next compared by quantitative PCR (qPCR) (Fig. 2B). A549 cells were infected at various MOIs for 1 h, and the cells were washed with trypsin to remove viruses that had not yet entered the cells to synchronize infections. At the indicated times, total DNA was extracted, and viral DNA was quantified by qPCR. Infections with 100, 1,000, and 10,000 vp per cell produced increasing numbers of viral genomes in the cells, which were proportionate to the MOI after a 1-h exposure (data not shown). One hour of exposure with 10,000 vp/cell yielded ∼2 × 105 viral genomes (vg) per 20 ng of genomic DNA for both viruses (Fig. 2B). This translates into ∼60 to 90 viral genomes per A549 cell for both Ad6 and Ad26 (A549 cells are hypotriploid, so they may have 2 or 3 copies of each human genome). This indicated that the two viruses internalized their DNAs into the cells to similar degrees after 1 h of synchronous infection.
qPCR analysis of Ad6 and Ad26 DNAs after 1 h of infection revealed that genome copy numbers remained constant for 6 h, but these copy numbers were amplified drastically from 6 to 12 h (Fig. 2B). Genome copy numbers increased through 36 to 48 h and then plateaued. The maximum level of viral genome production by this time was ∼109 viral genomes per 20 ng of genomic DNA. This means that the initial infections that began with 60 to 90 viral genomes were amplified ∼10,000-fold to a final level of 105 genomes per cell for both viruses.
Quantitative comparison of E1, E3, and late gene transcripts by RT-qPCR.
We performed reverse transcription-quantitative PCR (RT-qPCR) on mRNA from A549 cells 6, 12, and 24 h after infection with Ad6 or Ad26, to compare mRNA output from a subset of viral genes: E1A, E1B-55K, E3-19K, E3-10.4K, hexon, and fiber (Fig. 4). E1A was scrutinized because of its importance in representing the immediate early response and master regulator. E1B-55K regions were scrutinized as a representative of early gene transcription downstream of E1A activation. Two E3 open reading frames (ORFs) shared between Ad6 and Ad26, E3-19K and E3-10.4K, were evaluated, since E3 is uniquely described as being expressed both early and late in the viral life cycle. Finally, the mRNAs for hexon and fiber capsomer proteins were also analyzed as benchmarks for late gene activation.
FIG 4.

Detection of viral expression in A549 cells by RT-qPCR. Expression of E1A and E1B-55K (A), E3-19K and E3-10.4K (RID-α) (B), and fiber and hexon late gene (C) cDNAs detected in Ad6- and Ad26-infected samples 6, 12, and 24 h after infection. Averages and standard errors for triplicate experimental samples are plotted. Statistical significance was determined by one-way analysis of variance for all genes at 6, 12, or 24 h. Significance and P values are noted in Results.
RT-qPCR revealed that rates of E1A mRNA expression differed between Ad6 and Ad26. Notably, of the time points examined, levels of E1A were the highest, at 3 × 105 mRNA copies per 10 ng 6 h after Ad6 infection, while E1A levels peaked at 12 h for Ad26 (Fig. 4A). At 12 h, quantities were similar, as Ad6 E1A levels steadily decreased from 6 h to 12 h, while Ad26 levels rose from 6 h to 12 h. Finally, after 24 h, E1A expression declined for both viruses but at a much higher rate for Ad6. These differences resulted in 2-fold-higher Ad6 E1A expression levels at 6 h (P < 0.0001), a minimal difference at 12 h, and >3-fold-higher Ad26 E1A expression levels at 24 h.
High levels of E1B-55K were found for both viruses at 6 h (Fig. 4A). However, E1B-55K levels were higher than those of E1A in Ad26 samples after 6 h (P < 0.0001), while Ad6 had higher levels of E1A expression than of E1B-55K expression at this time (P < 0.0001). E1B-55K levels increased from 6 to 12 h and were higher than E1A levels for both treatments after 12 h. However, this difference was far greater for Ad26 than for Ad6, resulting in differences in expression levels of 4-fold (P < 0.001) and <2-fold (P < 0.001) between the two early genes in Ad26 and Ad6, respectively. E1B-55K levels diminished from 12 h to 24 h, with levels in Ad26 remaining higher than those in Ad6.
The level of transcription of E3 mRNAs was relatively high at 6 h but, unlike E1A and E1B-55K, steadily increased out to 24 h after infection (Fig. 4B). At 6 h, Ad6 E3-19K mRNA levels were 10-fold higher than Ad26 E3-19K levels (P < 0.0001). E3-10.4K mRNA levels were more similar between viruses, but Ad26 had slightly higher levels than did Ad6 (P < 0.01). Overall, 10.4K levels were intermediate between the Ad6 E3-19K and Ad26 E3-19K mRNA levels. Therefore, Ad6 expressed E3-19K more strongly than it expressed E3-10.4K, and Ad26 expressed E3-10.4K more strongly than it expressed E3-19K.
mRNA levels for both hexon and fiber 6 h after infection were not significantly above background levels but increased dramatically 12 h after infection and increased steadily out to 24 h (Fig. 4C). This observation is consistent with the dogma of adenovirus late gene synthesis occurring after the onset of viral gene replication, which was absent at 6 h but initiated by 12 h (Fig. 3C). Interestingly, Ad26 had higher levels of hexon than did Ad6 at both 12 and 24 h (P < 0.0001). Fiber levels were also moderately higher for Ad26 at 12 h and significantly higher than those for Ad6 at 24 h (P < 0.05).
Viral mRNA expression profiles for Ad6 and Ad26 6 and 12 h after infection.
These data suggested that Ad6 and Ad26 internalize and replicate their DNA similarly after a synchronous 1-h infection. However, the two viruses provoke markedly different downstream events, including cell rounding. RT-qPCR evaluation of Ad genes revealed some unique differences in Ad6 and Ad26, implying transcriptional differences. To examine if the viruses activate their total transcriptional programs differently, next-generation sequencing (NGS) of viral mRNAs was performed. We chose the time point of 6 h after infection to perform NGS mRNA sequencing, since this corresponds to a time just before the initiation of viral genome replication (Fig. 2B). We also chose the time point of 12 h after infection as a time to examine viral mRNAs by NGS since it represents a time after the onset of viral DNA replication and expression of late genes.
Cells were infected with 10,000 vp/cell of Ad6 or Ad26 for 1 h, as shown in Fig. 2B and C. In addition, uninfected control or “mock-infected” cells were treated in parallel. Total cellular RNA was extracted from cells 6 h or 12 h after infection. Triplicate samples were prepared for each condition, and 18 mRNA sequencing (mRNA-seq) libraries were prepared and subjected to mRNA-seq. mRNA-seq resulted in upwards of 100 million sequencing reads per sample (range, 96 million to 234 million) (Table 1). Over 99% of reads were of acceptable quality and were used for downstream analysis.
TABLE 1.
Summary of sequencing reads from mRNA-seq sample sets
| Parameter | Value for sample set |
|||||
|---|---|---|---|---|---|---|
| 1 |
2 |
3 |
||||
| 6 h | 12 h | 6 h | 12 h | 6 | 12 | |
| Ad6 infection | ||||||
| Total no. of reads | 180,782,044 | 210,282,090 | 135,071,614 | 108,274,812 | 118,058,558 | 121,554,014 |
| No. of reads used | 180,463,643 | 209,914,762 | 134,893,355 | 108,130,934 | 117,900,862 | 121,404,039 |
| Cellular results | ||||||
| Total no. of mapped reads | 165,041,538 | 159,043,960 | 126,368,625 | 86,774,447 | 110,615,057 | 97,662,010 |
| % of mapped reads | 91.45 | 75.77 | 93.68 | 80.25 | 93.82 | 80.44 |
| Viral results | ||||||
| Total no. of mapped reads | 2,942,687 | 36,619,873 | 1,150,964 | 15,251,237 | 1,130,752 | 16,931,217 |
| No. of properly paired mapped reads | 2,755,224 | 34,702,806 | 1,070,978 | 14,227,066 | 1,051,572 | 15,817,326 |
| % of mapped reads | 1.63 | 17.45 | 0.85 | 14.10 | 0.96 | 13.95 |
| Ad26 infection | ||||||
| Total no. of reads | 196,950,760 | 198,932,178 | 135,228,756 | 116,166,350 | 110,402,754 | 131,713,602 |
| No. of reads used | 196,600,536 | 198,582,602 | 135,061,729 | 116,020,082 | 110,267,078 | 131,545,913 |
| Cellular results | ||||||
| No. of mapped reads | 182,415,868 | 160,373,156 | 127,298,181 | 105,704,250 | 103,757,608 | 120,185,933 |
| % cellular mapped reads | 92.79 | 80.76 | 94.25 | 91.11 | 94.10 | 91.36 |
| Viral results | ||||||
| No. of mapped reads | 2,226,651 | 25,852,093 | 179,986 | 3,955,512 | 143,315 | 4,005,895 |
| No. of properly paired mapped reads | 2,122,808 | 24,463,322 | 168,616 | 3,728,562 | 134,076 | 3,781,340 |
| % of virus mapped | 1.13 | 13.02 | 0.13 | 3.41 | 0.13 | 3.05 |
| Mock infection | ||||||
| Total no. of reads | 170,005,708 | 234,859,512 | 115,569,388 | 127,419,472 | 96,004,636 | 121,989,820 |
| No. of reads used | 169,674,316 | 234,389,298 | 115,423,082 | 127,271,387 | 95,892,810 | 121,848,550 |
| Cellular results | ||||||
| No. of mapped reads | 159,035,022 | 219,390,337 | 108,857,737 | 120,348,547 | 90,486,964 | 114,675,762 |
| % cellular mapped reads | 93.73 | 93.60 | 94.31 | 94.56 | 94.36 | 94.11 |
| Viral results | ||||||
| No. of mapped reads | 22 | 351 | 1,962 | 10 | 22 | 76 |
| No. of properly paired mapped reads | 22 | 322 | 686 | 10 | 22 | 72 |
| % of virus mapped | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
For mock-treated cells, 93 to 94% of these reads mapped to the human genome at both time points (Table 1). For Ad6, 91 to 93% of reads mapped to the host at 6 h, and this proportion fell to 75 to 80% by 12 h. In parallel, 0.85 to 1.63% of mRNA-seq reads mapped to the Ad6 genome at 6 h, and this proportion increased to 14 to 17% by 12 h. For Ad26, 91 to 92% of reads mapped to the host at 6 h, and this proportion fell to 80 to 91% by 12 h. At 6 h, 0.13 to 1.13% of mRNA-seq reads mapped to the Ad26 genome. This proportion increased to 3 to 13% of Ad26 viral reads by 12 h, reflecting greater variation in reads from Ad26 infections. These data indicate substantial conversion of transcription toward viral mRNAs from 6 to 12 h after synchronous infection.
Global adenoviral transcription across the Ad6 and Ad26 genomes.
To correct for various read outputs between samples and for differences in sequencing depth due to various transcript lengths, RNA-seq data are often normalized to reads per million mapped reads (RPM) and reads per kilobase per million mapped reads (RPKM), respectively. Since we are specifically assessing Ad mRNA, RPM and RPKM values are normalized to only total viral reads and not host reads (Table 1).
To assess where the RNA-seq reads were expressed along the Ad6 and Ad26 genomes, we plotted these reads across the viral genome in 500-bp increments (Fig. 5A and B). When aligned to Ad early and late gene regions (Fig. 1A and 5A), a clear shift in this mRNA “landscape” is evident for both Ad6 and Ad26 from 6 to 12 h after infection, which was similar to alignments performed by using Integrated Genomics Viewer (data not shown). At 6 h, peaks were evident at multiple positions across the genome, and largely corresponded to early (E) gene regions, as expected (Fig. 5B). At 12 h, the proportions of reads mapping to most early region reads were reduced, and late gene peaks increased across the genome. While most mapped reads were similar between the two viruses, there were notable differences between Ad6 and Ad26 in the distribution of reads from 500 to 4,000 bp as well as from 26,000 to 31,000 bp corresponding to the E1A and E1B regions and the E3 region, respectively (Fig. 5B).
FIG 5.

mRNA sequencing reads across Ad genomes at 6 and 12 h. (A) Ad genome organization schematic from Fig. 1 for ease of comparison. Early (gray arrows) (E1A, E1B, E2A, E2B, E3, and E4), intermediate (open arrows) (pIX and Iva2), and late (black arrows) (L1, L2, L3, L4, and L5) transcription units are shown. The direction of arrows indicates the coding strand and transcription direction. (B) mRNA RPM values. RPM values were calculated and are displayed in 500-bp increments across the Ad6 (left) and Ad26 (right) genomes 6 h (top) and 12 h (bottom) after infection. Average values and standard errors for three data sets are shown. (C) Quantification of RPKM values for the specific Ad6 (black) and Ad26 (gray) regions indicated after 6 h (top) and 12 h (bottom) (see Materials and Methods and Table 2 for quantification methods and sequence coordinates for defined regions). Averages and standard errors for three data sets are shown. Significance between Ad6 and Ad26 values was determined by an unpaired 2-tailed t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Following visualization of the transcriptional landscape, reads were mapped specifically to entire early and late transcription units for quantitative and statistical comparisons (Fig. 5C). Reads were similar between the two viruses in many regions but notably different in others. At 6 h, all early genes were expressed, but only a few late genes were transcribed. Both viruses expressed the late L4 cassette, but only Ad26 had reads in L5 at 6 h. Ad6 transcription of the pivotal E1 regions was stronger from E1A than from E1B at 6 h, whereas Ad26 transcript levels were higher for E1B than for E1A. Expression of E2A was strong at 6 h, but that of E2B was minimal. Overall, E3 transcription was stronger than those of other early regions with Ad6 infection, while Ad26 E3 was transcribed less on average than both E4 and E1B at this early time point.
By 12 h, both viruses devoted a substantial proportion of their total transcriptional activity to late genes, as evidenced by increases in late mRNA levels along with reductions in RPKM values for E1A, E3, and E4 (Fig. 5C). Interestingly, the relative E1B expression level remained high at 6 and 12 h, whereas E2B expression increased from 6 to 12 h. All late genes were activated by 12 h to similar degrees in both viruses. Ad26 expression levels of L2, L3, and L4 were slightly higher than those for Ad6 at 12 h.
Early gene expression. (i) E1.
E1 encodes a number of master regulators of the adenoviral life cycle that interact with the tumor suppressor proteins pRB and p53 and many other cellular targets (26, 27). Overall, E1A and E1B reads differed between the two viruses at 6 h (Fig. 6A). Ad6 drove E1A more strongly than it drove E1B, and Ad26 drove E1B more strongly than it drove E1A. The difference in E1B expression (Fig. 5C) mapped to the E1B-19K and -55K ORFs (Fig. 6A). Interestingly, E1B-19K reads were quite similar for Ad6 and Ad26 at both time points, whereas E1B-55K RPKM values were significantly higher for Ad26 at both time points (P = 0.0458 at 6 h, and P = 0.0229 at 12 h). Interestingly, the relative expression of E1 products shifted from 6 to 12 h when E1B-19K became dominant over E1A and E1B-55K for both viruses.
FIG 6.

mRNA expression across E1, E2, and E4 coding regions. Quantification of RPKM values for ORFs in E1 (A), E2 (B), and E4 (C) is shown. RPKM values were calculated for 6-h (top) and 12-h (bottom) time points. Ad6 is shown in black, and Ad26 is shown in gray (see Materials and Methods and Table 2 for quantification methods and base pair coordinates for defined regions). Averages and standard errors for three data sets are shown. Significance between Ad6 and Ad26 values was determined by an unpaired 2-tailed t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
(ii) E4.
E4 encodes pivotal early proteins that work in cooperation with E1 to activate the viral transcription-and-replication program (28, 29). Six hours after infection, Ad6 and Ad26 had similar RPKM values for E4 (Fig. 5C). While overall RPKM values were similar, Ad6 and Ad26 had distinctly different distributions of reads that mapped to different E4 ORFs (Fig. 6C). Ad6 reads mapped similarly to E4-ORF7 (of E4-ORF6/7), E4-ORF6 (34K), and E4-ORF4. However, mapping to these regions in Ad26 was somewhat different than that for Ad6. E4-ORF3, -ORF2, and -ORF1 mapped reads were similar between the two Ads, and lower than those for the other E4 ORFs. Distributions of reads across E4 remained similar for each virus from 6 to 12 h after infection (Fig. 6C). However, the RPKM value was higher overall for the E4 region in Ad26 than in Ad6 (Fig. 5C).
(iii) E2.
E2 region genes encode a number of proteins that are critical to viral DNA replication. Surprisingly, the transcriptional dynamics of the two E2 regions (E2A and E2B) differed markedly. E2A encoding the single-stranded DNA binding protein (DBP) was transcribed at levels comparable to the levels for the E1 products (∼100,000 RPKM) (Fig. 6A and B). In contrast, transcription of E2B encoding the viral DNA polymerase (DNA Pol) and preterminal protein (pTp) was barely detectable in comparison to E1 and E2A, with values of <5,000 RPKM (Fig. 6B). These differences translate into 85- and 41-fold-higher DBP expression levels than DNA Pol expression levels for Ad6 and Ad26, respectively, and 142- and 115-fold-higher DBP expression levels than pTp expression levels in Ad6 and Ad26, respectively. By 12 h, the relative expression levels of DBP remained similar, with average values of ∼105,000 and 96,000 RPKM for Ad6 and Ad26, respectively. In contrast, expression levels of DNA Pol and pTp increased from 6 to 12 h after infection. The DNA Pol transcription level increased 8-fold for Ad6 and 4-fold for Ad26. Similarly, pTp expression levels increased to a greater extent, resulting in 27-fold and 14-fold increases from 6 to 12 h for Ad6 and Ad26, respectively.
Intermediate gene expression.
Ad pIX and Iva2 genes make up the “intermediate” genes, as their expression from species C viruses peaks at times between those for E and L genes (30, 31). pIX functions as a minor capsid structural protein and may also act as a transcriptional activator via nuclear reorganization (32). pIX and IVa2 expression requires the synthesis of a new DNA template and begins immediately after the onset of viral DNA replication (30, 31). IVa2 is critical for Ad genome recognition and packaging through its interaction with the viral packaging signal and subsequent recruitment and interaction with critical packaging proteins, including L4-22K, L4-33K, L1-52/55K, and E2 DBP (33, 34). IVa2 has also been implicated as a transcriptional activator of the major late promoter (MLP) (35). For Ad6 and Ad26, pIX expression was observed at 6 h, with a 2-fold increase in expression at 12 h (Fig. 7A). IVa2 expression was also seen at both time points; however, RPKM was markedly lower than those of early or late genes. Similar to pIX, IVa2 relative reads increased from 6 to 12 h.
FIG 7.

mRNA expression across intermediate and late regions at 6 h and 12 h. Sequence reads (RPKM) were mapped to protein-coding regions of intermediate gene regions (A) or late gene regions L1 to L5 (B to F) for Ad6 (black) and Ad26 (gray). RPKM values were calculated for the 6-h (top) and 12-h (bottom) time points, as indicated in Materials and Methods, using the sequence definitions given in Table 2. Averages and standard errors for three data sets are shown. Significance between Ad6 and Ad26 values was determined by an unpaired 2-tailed t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Late gene expression.
Expression of adenoviral late genes arises almost exclusively from the MLP (reviewed in reference 36). Extensive splicing occurs within five primary transcripts (L1, L2, L3, L4, and L5) arising from the MLP and constitutes the major late transcription unit (MLTU) (37).
In this comparison of species C Ad6 and species D Ad26, late gene expression largely recapitulated previous observations (Fig. 4 and 5). Most late gene transcripts were “off” and nearly undetectable 6 h after infection (Fig. 4C and 7). By 12 h, late gene expression was “on” and increased 80-, 83-, 63-, and 21-fold for L1, L2, L3, and L5, respectively, in Ad6. Similarly, Ad26 late gene expression increased 69-, 71-, 39-, 3-, and 1.5-fold for L1, L2, L3, L4, and L5, respectively, by 12 h. RPKM values for L1 to L4 were similar between the two viruses (Fig. 4C and 7).
While most late genes were not expressed at 6 h, two were: L4 and L5. L4 transcripts had higher RPKM values than those of other late regions at both time points (Fig. 4C and 7). This is consistent with previous observations of species C Ads where moderate expression of L4 precedes MLP activation through an independent promoter, presumably since L4 products are critical for the shift from early to late phase (38–40). The L5 transcript encodes only one protein, fiber. The fiber protein functions as the major in vitro attachment protein for adenovirus binding to primary receptors on cells (reviewed in references 10, 41, and 42). Fiber proteins for Ad6 and Ad26 differ significantly both structurally and functionally. The Ad6 fiber is ∼35 nm long and contains 22 β turn repeats, whereas the Ad26 fiber is much shorter, with only 8 repeats. Species C Ads, including Ad6, utilize coxsackievirus and adenovirus receptor (CAR) as their primary receptor, whereas species D Ad26 may use sialic acid and CD46. Ad6 and Ad26 regulated fiber expression differently (Fig. 7F). While both viruses drove fiber expression by 12 h, only Ad26 expressed fiber mRNA early at 6 h.
(i) U.
The U exon protein (UXP) is expressed from the complementary l-strand of the viral genome from an exon (U) located within E3, with two subsequent exons located in the 100K and DBP coding regions (Table 2) (43). Activation of the U promoter element located in fiber has been shown to occur after, and to be dependent on, Ad genome replication (44). In concordance with these observations, transcripts from U are found at later time points, consistent with late gene transcripts.
When mRNA levels for Ad6 and Ad26 were assessed, reads across the U exon were observed at 12 h for both viruses (Fig. 7F). Interestingly, Ad6 also had detectable U exon expression at 6 h. Ad6 had ∼5,000 RPKM at 6 h, which increased to ∼8,000 RPKM at 12 h. In contrast, Ad26 U reads at 6 h were <500 RPKM but increased substantially at 12 h (∼10,000 RPKM), resulting in a 24-fold increase.
(ii) E3.
Unlike E1, E2, and E4, the E3 region is dispensable for the viral life cycle and is instead devoted to proteins involved in immune evasion, avoidance of apoptosis, and, for species C viruses, accelerated cell death (Fig. 1) (45, 46). As such, most adenoviral vectors have the E3 region deleted to make space for the insertion of exogenous transgenes in the viral genome. The E3 region is known to be one of the most genetically variable regions among different Ad species (45). Indeed, the E3 region encodes two distinct products for Ad6 (E3-6.7K and E3-11.6K) that are not present in Ad26 (Fig. 1). Conversely, Ad26 encodes three unique products (E3-23K, E3-31K, and E3-49K).
Since E3 is dispensable in vitro, it was interesting to observe that Ad6 E3 actually had more transcript reads per kilobase pair that mapped to this region at 6 h than to any other region in the viral genome (Fig. 5). In addition, E3 provided some of the greatest differences in mapped reads between Ad6 and Ad26 (Fig. 5C). Specifically, Ad6 had more RPKM that mapped to the first few coding regions, including E3-6.7K, E3-19K, and E3-11.6K than to later ORFs, including E3-10.4K, E3-14K, and E3-14.7K (Fig. 8). In contrast, Ad26 had three to six times as many RPKM mapping to later E3-10.4K, -14K, and -14.7K ORFs as those mapping to earlier E3-23K, -19K, -49K, and -31K ORFs (Fig. 8). These E3 expression dynamics were largely maintained through the 12-h time point although with less overall transcriptional activity dedicated to E3 than at 6 h.
FIG 8.

mRNA expression across Ad6 and Ad26 E3 genes at 6 h and 12 h. (Top) RPKM values calculated for the 6-h (top) and 12-h (bottom) time points, as indicated in Materials and Methods, by using the sequence definitions given in Table 2. (Bottom) RPKMtotal values calculated based on total reads as opposed to viral reads and represented as described above. Averages and standard errors for three data sets are shown. Significance between Ad6 and Ad26 values was determined by an unpaired 2-tailed t test.
NGS provides relative quantitation of transcripts. To correct for various read outputs among samples and for differences in sequencing depth, the NGS data in Fig. 3 through 7 are normalized to RPKM values (Table 1). This method allows comparisons of mRNA distributions of reads across the genome of each adenovirus and is used to evaluate the relative investment of that virus in its own transcription. For example, for E3 ORFs, RPKM evaluation emphasizes that Ad6 invests more of its viral transcription in 6.7K, 19K, and its unique ADP ORF than later E3 ORFs at 6 h. In contrast, RPKM analysis shows that Ad26 invests more in later 10.4K, 14K, and 14.7K ORFs than in 23K, 19K, 49K, and 31K ORFs (Ad26 lacks ADP but has unique 49K and 31K ORFs) (Fig. 8). However, normalization of reads to total viral reads by RPKM analysis ignores differences in the levels of mRNAs between the two viruses. When viral reads are instead normalized to all mRNA reads from the cell (RPKMtotal), the same balance in transcription is observed, but differences in mRNA levels between the viruses are revealed (Fig. 8). Consistent with the qPCR data (Fig. 4B), RPKMtotal values show that E3-19K is more highly expressed by Ad6 than by Ad26, while E3-10.4K expression levels are nearly equal between the two Ads by this analysis (Fig. 8).
Effects of Ad6 and Ad26 on cellular functions. (i) Effects of E3-19K on MHC I.
The qPCR and NGS data described above suggest select differences in mRNA expression by Ad6 and Ad26 primarily in E1 and E3 regions. To evaluate if these differences may impact the interactions of the viruses with host cells, we compared select cellular functions after infection in A549 cells. E3-19K (also known as E3grp19K) enhances viral immune evasion in part by preventing newly synthesized MHC I proteins from reaching the cell surface (reviewed in references 45 and 46). By all measures, Ad6 expressed E3-19K more strongly than did Ad26, particularly 6 h after infection (Fig. 4B and 8). To test if the two viruses affect the display of MHC I on cells differently, A549 cells were infected with Ad6 and Ad26, and the display of newly synthesized MHC I was measured by flow cytometry (Fig. 9A). A549 cells were infected for 6 h to allow the expression of E3-19K to impact MHC I loading. Preexisting cell surface MHC I was removed at this time point by a brief low-pH wash, and the cells were allowed to load MHC I onto their surfaces for 3 h before MHC I staining by flow cytometry (Fig. 9A). Under these conditions, Ad26 did not reduce MHC I levels significantly compared to the levels in uninfected cells. In contrast, Ad6 reduced cell surface MHC I levels by 40% compared to those in uninfected cells (P < 0.001). Ad6 downregulation of MHC I was significantly stronger than that for Ad26 (P < 0.01). These effects of Ad6 were mediated by the E3 region, since Ad6 reduced MHC I levels significantly compared to E3-deleted Ad6 (P < 0.001). This difference in the inhibition of MHC I display between the viruses was transient, since both viruses were able to reduce these levels after 24 h of infection (Fig. 9B). Both Ad6 and Ad26 downregulated MHC I to levels comparable to those of the benchmark Ad5 at this time point (data not shown).
FIG 9.

Phenotype of immune evasion functions of Ad6 and Ad26. (A) Surface MHC I (pan-HLA-A, -B, and -C) detection by flow cytometry on A549 cells after mock infection or infection with Ad6, Ad26, or E3-deleted viruses after 6 h of infection and 3 h of MHC I recovery (see Materials and Methods for details). Averages and standard errors for five independent experiments are shown. (B) Surface MHC I (pan-HLA-A, -B, and -C) detection by flow cytometry on A549 cells after 24 h of infection. Averages and standard errors for three experiments are shown. (C) TRAIL-induced cell death and resistance after mock infection or infection with Ad6, Ad26, or E3-deleted virus. Percent viable (propidium iodide-negative [PI−] and annexin V-negative) cells plotted are averages of data from three experimental samples. Significance was determined by two-way analysis of variance with a Tukey posttest. Significance at a specific concentration is denoted with a symbol or number next to the asterisk(s), where ****# indicates significance of Ad6 infection compared to mock infection at 0.5 mg/liter and **∞ indicates significance of Ad26 and Ad6ΔE3 infections compared to mock infection at 0.5 mg/liter (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.001).
(ii) Effects of Ad6 and Ad26 on TRAIL-mediated cell death.
Ad2 and Ad5 evade apoptosis by expressing their E3 receptor internalization and degradation (RID) (10.4K and 14K) proteins and E3-14.7K proteins, which reduce the display of TNF family death receptors, including Fas, TRAIL receptor 1 (TRAIL-R1), and TRAIL-R2, and inhibit DISC complex formation, respectively (47, 48). Ad6 expressed these regions to similar levels as Ad26 (Fig. 4 and 8). To test how these differences in expression impact the ability to evade apoptosis, Ad6- and Ad26-infected cells were treated with TRAIL at increasing concentrations (0 to 50 mg/liter), and levels of apoptosis induction were compared (Fig. 9C). In uninfected cells, increasing concentrations of TRAIL induced decreasing numbers of viable cells. In contrast, Ad6 and Ad26 significantly increased the number of viable, nonapoptotic cells (P < 0.05 to P < 0.01). However, Ad6-infected cells were more resistant to TRAIL than were Ad26-infected cells (P < 0.001). E3 deletion reduced protection against TRAIL by Ad6 slightly but only at the highest concentration of TRAIL (50 mg/liter) (P < 0.05). Otherwise, E3 deletion in either virus had weak effects on protection against TRAIL, suggesting that other viral proteins mediated most of this effect.
DISCUSSION
Human species C mastadenoviruses Ad2 and Ad5 are the foundation for most human adenovirus therapies. While they are potent for many uses, rampant preexisting immunity to these viruses in most humans makes them likely to be ineffective in patients. This has led to the evaluation of alternate adenoviral serotypes to avoid immune neutralization (14, 49). In the course of our work on oncolytic virotherapy, we identified species C Ad6 and species D Ad26 as two promising lower-seroprevalence viruses (2, 12, 16, 18, 50–52). Ad6 appears promising as an oncolytic agent against prostate cancer, whereas Ad26 holds promise against B cell malignancies. However, both viruses can infect and kill cells like A549 cells with similar kinetics but with differing effects on cell phenotypes. In this work, we explored the innate biology of these clinically relevant viruses to determine if they activate similar or different life cycles in cells that are permissive for both Ads.
We first established the biological boundaries within which the two viruses act: genome delivery through cell death. Between these two boundaries, we examined the kinetics of how the two viruses replicate their DNA and found them to be nearly identical.
We first measured the number of Ad6 and Ad26 genomes that enter A549 cells after a 1-h exposure by qPCR. Ad6 and Ad26 virions were allowed 1 h at 37°C to bind and then be internalized, followed by trypsinization to remove virions that are on the surface. This somewhat synchronizes the infection, so subsequent events occur with similar timelines. If these bound virions were allowed to persist, they might asynchronously slowly enter and smear the kinetics of subsequent events. Under these conditions, wherein each cell was exposed to 104 viral particles/vg for 1 h, we observed ∼2 × 105 vg in 20 ng of total cellular DNA after 1 h at 37°C and after a trypsin wash. A549 cells are hypotriploid, with ∼40% being triploid and 60% being diploid, so each diploid cell would have ∼6 pg of genomic DNA and each triploid cell would have ∼9 pg. Based on this estimate, 2 × 105 vg in 20 ng of total cellular DNA would translate into ∼60 to 90 vg per cell after a 1-h exposure and a trypsin wash.
This number of viruses entering in an hour may differ to some degree from those reported in other studies. For example, in one study, adenoviruses were permitted to interact with cells at 10,000 vp/cell (53). In this case, the cells were exposed to virus for 1 h at 4°C, such that the virus binds but does not enter; the medium was then removed; and any bound virus was allowed to enter at 37°C. Under these conditions, slightly less than 100 vg of adenovirus were observed per cell. In another study (54), cells were exposed to 5,000 vg/cell, and ∼200 vg/cell were observed. This is about twice our result at a lower multiplicity of infection. While there may be differences in the numbers of virions per cell reported by different studies, these differences are unlikely to affect the interpretation of the downstream transcriptional events that we report here.
We determined that the two boundaries of viral entry and cell death were nearly identical for these two evolutionarily divergent adenoviruses. While many aspects were similar, we observed that Ad26 induced a strong rounding phenotype but did not induce apoptosis or a loss of membrane integrity. This effect did not appear related to differences in cellular receptor binding, since heat- or UV-inactivated viruses did not produce the effect. This was therefore a postentry effect likely independent of receptor engagement. E1 deletion inactivated the ability of Ad26 to induce this phenotype, suggesting again that events after early activation of transcription were involved and were different between the two viruses.
Given this, we tested whether the two viruses activated pivotal E1A and E1B mRNAs differentially and found that there were substantial differences in their balance. Both viruses drove similar levels of E1A at 6 h; however, Ad26 drove its E1B mRNA to levels as high as 5 times above Ad6 E1B mRNA levels and E1A mRNA levels of both viruses. RT-qPCR also demonstrated that Ad26 continued driving E1B-55K expression strongly over 24 h, while other E1 transcript levels waned over this same time. E3 transcript levels were also markedly different between the two viruses, as determined by RT-qPCR, where Ad6 appeared to drive the strongest expression of its E3-19K mRNA compared to its E3-10.4K mRNA, whereas Ad26 drove these mRNAs in the reverse balance. While E1 and E3 mRNA balances were struck quite differently, both viruses drove the late hexon and fiber mRNAs similarly over 24 h, consistent with their similar end-of-life programs terminating in a loss of viability beginning after 48 h.
These observations of differential regulation in four of six mRNAs justified a more global study of Ad6 and Ad26 mRNA regulation by NGS of viral mRNAs. NGS mRNA sequencing provided a tremendous depth of scrutiny of viral mRNAs by analysis of >100 million separate mRNA sequence reads for each virus and each control. Importantly, this NGS analysis was performed in triplicate for each sample and time point to ensure that the observed mRNA changes were not due simply to biological variation within one sample. Here, we compared the biologies of two genetically divergent adenoviruses using ∼1.5 billion mRNA reads, allowing similarities and differences in adenoviral transcriptional control and persistence to be evaluated in one study.
Six hours after infection, ∼1% of each of their half billion mRNA reads for Ad6 and Ad26 mapped to their viral genomes. At 12 h, ∼15% of reads mapped to the Ad6 genome, and 3 to 13% mapped to Ad26, emphasizing the degree to which biological variation can occur within replicate infections and the need for these replicates despite the high cost of NGS. When these viral mRNA reads were mapped to their full genomes, Ad6 and Ad26 activated distinct viral mRNA landscapes at 6 and 12 h. This provided a global comparison of viral transcription to complement the granular analysis provided by RT-qPCR. Looking at transcriptional distributions across the viral genomes, the relationships between E1A and E1B expression and E3 expression that was observed by qPCR were magnified and reinforced by NGS.
Consistent with the results of previous studies examining a single Ad (5, 8), a large number of mRNA reads mapped to the early gene region E1 in Ad6 and Ad26 6 h after infection. However, unlike previous work examining one virus, in this case, where two adenoviruses were compared, significant differences in E1 activation between the viruses were also observed by qPCR and by NGS. E1B-19K RPKM values for Ad6 and Ad26 were quite similar. Interestingly, the relative expression levels of E1A and E1B-55K products for both viruses decreased significantly from 6 to 12 h, while E1B-19K reads persisted and predominated over E1A and E1B-55K reads. This is perhaps unsurprising given the role of E1B-19K in preventing apoptosis, a function potentially needed longer into the adenoviral life cycle than E1A and E1B-55K functions. In addition to activating adenoviral genes, E1A also interacts with a number of cell cycle and transcriptional regulators, including pRB, p53, and p300, to enable viral replication (26, 27).
Considering the level of amino acid divergence in E1 proteins between Ad6 and Ad26 (Fig. 10), it is also not surprising that the two viruses drive the expression of interacting viral and host proteins in different ways. Ad6 E1A has the known Ad2 and Ad5 motifs to bind the transcription factors pRB and p300, but these are not conserved in Ad26. The Ad26 E1A protein also has a natural deletion protein that ablates the BS69 binding site that inactivates transactivation by species C E1A 289R proteins (55). These differences are interesting given that E1A proteins by themselves induce cell rounding phenotypes (56) similar to those observed after Ad26 but not Ad6 infection. The activation of E1A within 6 h of infection before cell rounding by Ad26 at 12 h suggests that its E1A proteins could be involved in this different cell phenotype. However, the fact that this phenotype can be observed in Ad26ΔE1-infected 293 cells, where there is no Ad26 E1 but there is Ad2 E1, suggests that this phenotype is not due directly to Ad26 E1 proteins. This finding suggests that this is an event downstream of E1.
FIG 10.

Amino acid alignments of Ad6 and Ad26 E1 proteins. (A) E1A. Boxes highlight residues for interaction with the transcription p300 (FXD/EXXXL), pRB (DLXCXE), and BS69. (B) E1B-19K. (C) E1B-55K.
The most profound difference in E1 expression between the two viruses was the substantially stronger and more persistent induction of E1B-55K by Ad26. E1B-55K binds to p53, releasing its interactions with E2F transcription factors (23, 24, 26, 27, 54, 57). The Ad26 E1B-55K expression level peaked 12 h after infection and was as much as 5 times higher than the transcription levels of E1B of Ad6 and E1A of both viruses. The N terminus of Ad26 E1B-55K diverges substantially from the species C conserved region in Ad6 (Fig. 10C). In contrast, the C terminus of Ad26 E1B-55K is more conserved and still appears to bear p53 binding and zinc finger motifs (58). It is interesting that mutations in species C E1B-55K perturb cell morphology and increase cell rounding during species C Ad infection (59). It is therefore also possible that the stronger induction of E1B-55K on downstream events by Ad26 may be associated with the distinct cell rounding phenotype observed in Ad26- but not Ad6-infected cells.
The most notable differences in viral activation between the two viruses were observed in E3 immune evasion ORFs. This is emphasized particularly in the degree to which the two viruses “invest” in their E3 ORFs relative to other early and late genes. At 6 h, both viruses generate E3 mRNAs at levels that are comparable to the expression levels of the pivotal E1 and E4 genes. The E3 expression level actually remained higher than those of E1 through 24 h and E4 through 12 h, supporting that this gene region is important for virus biology. The fact that Ad6 preferentially drives E3-19K and early ORFs over later ORFs while Ad26 drives the inverse balance also suggests that the two viruses have evolved different immune evasion strategies.
The reality check for these speculations based on E3 mRNAs was revealed in the ability of the viruses to downregulate MHC I and avoid apoptosis. Ad6 more rapidly downregulated the cell surface display of MHC I than did Ad26 at 6 h. At the same time, Ad26 had little effect on hiding this immunologic “red flag.” However, over time, Ad26 was able to catch up to Ad6 by 24 h. Considering that the T cell response take days to weeks to evolve, it may well be that this difference in kinetics may have little consequence for Ad26 in vivo, at least during a first infection. However, faster downregulation may favor a virus during a second infection, where memory responses may impact infected cells quickly.
Previous studies comparing the sequence homologies and functions of E3-19K molecules across many Ad species and serotypes showed that E3-19K from HAdV-D, such as Ad37, can have a drastically lower binding affinity for MHC I molecules than E3-19K from HAdV-B, -C, and -E (60). Furthermore, E3-19K from species B, C, and E had a higher affinity for HLA-A than for HLA-B, and interactions were allele specific with these molecules. Given the early low number of transcript reads across E3-19K for our species D representative Ad26, the limited effect on surface MHC I, and the low affinity of other species D Ads for HLA molecules, it is possible that species D Ads may have evolved to invest more transcriptional and translational activity in other E3 proteins.
The balance of E3 expression favoring later ORFs over early ORFs by Ad26 is in line with its slower use of E3-19K functions. Ad26 had significantly higher mRNA expression levels of the E3B genes E3-10.4K, -14K, and -14.7K than of E3A. However, the absolute expression level of E3-10.4K (RID-alpha) determined by RNA-seq or RT-qPCR was only moderately higher for Ad26 than for Ad6. RID-alpha and RID-beta combine to form the RID complex, which reduces the display of Fas, TRAIL-R1, and TRAIL-R2 on the cell surface (45–47). Since the total expression level of RID-alpha as determined by RNA-seq or RT-qPCR was moderately higher for Ad26, with RNA-seq data suggesting similar levels for RID-beta and 14.7K, we hypothesized that Ad26-infected cells may be more resistant to the extrinsic activation of apoptosis than cells that infected with Ad6. However, this hypothesis was not supported by in vitro test results; both viruses induced significant resistance to TRAIL compared to infection of mock-infected cells. Indeed, Ad6 was even more resistant than Ad26. This suggests that the Ad6 proteins may be more effective at evading TRAIL. While we did not find evidence of increased TRAIL resistance by Ad26, this is only one potential effect of E3B proteins and is also influenced by multiple proteins, including RID, 14.7K, and E1B-19K (61). Our results also point to the functionality of the Ad26 E1B-19K protein since E3-deleted Ad26 was also resistant to TRAIL apoptosis. More comprehensive evaluation of immune evasion differences between Ad6 and Ad26 would necessarily include permissive animal models for the two viruses.
Most data in the literature were obtained from replication-defective E1- and E3-deleted vectors to make space for transgenes and to avoid potentially dangerous adenovirus infections. It should therefore be noted that immune evasion functions for Ad6 or Ad26 will be relevant in humans only if (i) the viruses are replication competent and (ii) their E3 region has not been deleted. Applications where this may be relevant include natural Ad infections, replication-competent oncolytic Ads, replication-competent Ad vaccines, and single-cycle replicating Ad vectors (19, 20).
Beyond subtle differences, most other early and late genes were activated in a similar fashion in Ad6 and Ad26 before and after the initiation of DNA synthesis. However, this global transcription evaluation of the two viruses still provides confirmation of prior data and new insights into the key biology of human adenoviruses. E2A, E2B, and E4 expressions had subtle differences in the two viruses, but by and large, their levels of activation 6 and 12 h after infection were relatively similar. This is consistent with the conserved need to express these workhorse genes for DNA replication and host cell takeover. The L1, L2, and L3 regions were barely expressed by both viruses at 6 h and were markedly upregulated by 12 h. In contrast, the L4 region was expressed at 6 h, with 2- to 3-fold increases by 12 h. At 6 h, Ad6 expressed the late proteins L4-22K and L4-33K more strongly than did Ad26. Ad6 also induced higher levels of IVa2 and E4orf3 than did Ad26 at this early time point. Interestingly all four proteins play a role in the shift from early to late transcription (35, 39, 40, 62, 63). This suggests that the transition from early to late transcription occurs earlier in Ad6 infection than in Ad26 infection. This is also supported by the kinetics of UXP expression that occurs at significantly higher levels in Ad6 samples than in Ad26 samples at 6 h, even though UXP has traditionally been characterized as a late gene. These results are consistent with those reported previously by Wu et al. (8) for bat adenovirus, who also found the U exon, 22K, and 33K proteins to have a more “intermediate” level of expression than traditionally seen.
L5 encodes the fiber protein involved in receptor binding. L5 was not expressed early by Ad6 but was surprisingly expressed by Ad26 at 6 h. This is interesting, since fiber proteins are expressed in a 100-fold excess of the amount need for encapsidation. For Ad5, this excess fiber has been shown to bind the coxsackievirus and adenovirus receptor after cell lysis and opening of tight junctions (64, 65). While it is possible that early expression of Ad26 fiber could be associated with the rounding phenotype at 12 h, the membranes of the cells remain intact, suggesting that this early fiber expression is not related to this effect.
In summary, this was the first head-to-head deep mRNA sequencing comparison of two genetically divergent adenoviruses after infection of cells permissive for both viruses. We found that much of the temporal expression of viral mRNAs for Ad6 and Ad26 in A549 cell infection was similar for the two viruses. Many of these events were comfortingly familiar and consistent with the literature on Ad biology. At the same time, this comparison revealed key differences in the activation of E1 and E3 proteins that are pivotal for the in vivo biology of wild versions of these viruses and for their replication-competent vectors. An understanding of the differences between these two viruses in a shared permissive cell model lays the foundation for comparisons of their biologies in cells where their efficacy is diametrically opposite and examination of in vivo immune evasion functions.
ACKNOWLEDGMENTS
This work was supported by NIH/NIAID grants R01 AI096967 and R01 CA136945 to M.A.B. This work was also supported by the Division of Infectious Diseases in the Department of Medicine and the Department of Molecular Medicine at Mayo Clinic.
This work was performed in part by the Medical Genome Facility and the Division of Biomedical Statistics and Informatics in the Department of Health Sciences Research at Mayo Clinic. We thank Mary Barry for excellent technical assistance as well as Eric Weaver, Asha Nair, Yuji Zhang, and Diane Grill for their helpful consultations.
REFERENCES
- 1.Davison AJ, Benko M, Harrach B. 2003. Genetic content and evolution of adenoviruses. J Gen Virol 84:2895–2908. doi: 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- 2.Weaver EA, Hillestad ML, Khare R, Palmer D, Ng P, Barry MA. 2011. Characterization of species C human adenovirus serotype 6 (Ad6). Virology 412:19–27. doi: 10.1016/j.virol.2010.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berk AJ. 2007. Adenoviridae: the viruses and their replication, p 2355–2394. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Zhao HX, Granberg F, Pettersson U. 2007. How adenovirus strives to control cellular gene expression. Virology 363:357–375. doi: 10.1016/j.virol.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Zhao H, Dahlo M, Isaksson A, Syvanen AC, Pettersson U. 2012. The transcriptome of the adenovirus infected cell. Virology 424:115–128. doi: 10.1016/j.virol.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Zhao H, Chen M, Pettersson U. 2013. Identification of adenovirus-encoded small RNAs by deep RNA sequencing. Virology 442:148–155. doi: 10.1016/j.virol.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Zhao HX, Chen MS, Pettersson U. 2014. A new look at adenovirus splicing. Virology 456–457:329–341. doi: 10.1016/j.virol.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Wu L, Zhou P, Ge X, Wang LF, Baker ML, Shi Z. 2013. Deep RNA sequencing reveals complex transcriptional landscape of a bat adenovirus. J Virol 87:503–511. doi: 10.1128/JVI.02332-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rowe WP, Huebner RJ, Gilmore LK, Parrott RH, Ward TG. 1953. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Proc Soc Exp Biol Med 84:570–573. doi: 10.3181/00379727-84-20714. [DOI] [PubMed] [Google Scholar]
- 10.Khare R, Chen CY, Weaver EA, Barry MA. 2011. Advances and future challenges in adenoviral vector pharmacology and targeting. Curr Gene Ther 11:241–258. doi: 10.2174/156652311796150363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li HL, Rhee EG, Masek-Hammerman K, Teigler JE, Abbink P, Barouch DH. 2012. Adenovirus serotype 26 utilizes CD46 as a primary cellular receptor and only transiently activates T lymphocytes following vaccination of rhesus monkeys. J Virol 86:10862–10865. doi: 10.1128/JVI.00928-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CY, Senac JS, Weaver EA, May SM, Jelinek DF, Greipp P, Witzig T, Barry MA. 2011. Species D adenoviruses as oncolytics against B-cell cancers. Clin Cancer Res 17:6712–6722. doi: 10.1158/1078-0432.CCR-11-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, Pink R, Buckley SM, Greig JA, Denby L, Custers J, Morita T, Francischetti IM, Monteiro RQ, Barouch DH, van Rooijen N, Napoli C, Havenga MJ, Nicklin SA, Baker AH. 2008. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 132:397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Abbink P, Lemckert AAC, Ewald BA, Lynch DM, Denholtz M, Smits S, Holterman L, Damen I, Vogels R, Thorner AR, O'Brien KL, Carville A, Mansfield KG, Goudsmit J, Havenga MJE, Barouch DH. 2007. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J Virol 81:4654–4663. doi: 10.1128/JVI.02696-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, O'Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, Landucci G, Forthal DN, Montefiori DC, Carville A, Mansfield KG, Havenga MJ, Pau MG, Goudsmit J, Barouch DH. 2009. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 457:87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senac JS, Doronin K, Russell SJ, Jelinek DF, Greipp PR, Barry MA. 2010. Infection and killing of multiple myeloma by adenoviruses. Hum Gene Ther 21:179–190. doi: 10.1089/hum.2009.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weaver EA, Chen CY, May SM, Barry ME, Barry MA. 2011. Comparison of adenoviruses as oncolytics and cancer vaccines in an immunocompetent B cell lymphoma model. Hum Gene Ther 22:1095–1100. doi: 10.1089/hum.2011.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shashkova EV, May SM, Barry MA. 2009. Characterization of human adenovirus serotypes 5, 6, 11, and 35 as anticancer agents. Virology 394:311–320. doi: 10.1016/j.virol.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crosby CM, Barry MA. 2014. IIIa deleted adenovirus as a single-cycle genome replicating vector. Virology 462–463:158–165. doi: 10.1016/j.virol.2014.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crosby CM, Nehete P, Sastry KJ, Barry MA. 2015. Amplified and persistent immune responses generated by single-cycle replicating adenovirus vaccines. J Virol 89:669–675. doi: 10.1128/JVI.02184-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camacho ZT, Turner MA, Barry MA, Weaver EA. 2014. CD46-mediated transduction of a species D adenovirus vaccine improves mucosal vaccine efficacy. Hum Gene Ther 25:364–374. doi: 10.1089/hum.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugawara S, Abo T, Kumagai K. 1987. A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J Immunol Methods 100:83–90. doi: 10.1016/0022-1759(87)90175-X. [DOI] [PubMed] [Google Scholar]
- 26.Barker DD, Berk AJ. 1987. Adenovirus proteins from both E1b reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology 156:107–121. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- 27.Kao CC, Yew PR, Berk AJ. 1990. Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 179:806–814. doi: 10.1016/0042-6822(90)90148-K. [DOI] [PubMed] [Google Scholar]
- 28.Leppard KN. 1997. E4 gene function in adenovirus, adenovirus vector and adeno-associated virus infections. J Gen Virol 78(Part 9):2131–2138. [DOI] [PubMed] [Google Scholar]
- 29.Weitzman MD. 2005. Functions of the adenovirus E4 proteins and their impact on viral vectors. Front Biosci 10:1106–1117. doi: 10.2741/1604. [DOI] [PubMed] [Google Scholar]
- 30.Matsui T, Murayama M, Mita T. 1986. Adenovirus 2 peptide IX gene is expressed only on replicated DNA molecules. Mol Cell Biol 6:4149–4154. doi: 10.1128/MCB.6.12.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crossland LD, Raskas HJ. 1983. Identification of adenovirus genes that require template replication for expression. J Virol 46:737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parks RJ. 2005. Adenovirus protein IX: a new look at an old protein. Mol Ther 11:19–25. doi: 10.1016/j.ymthe.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 33.Ahi YS, Vemula SV, Mittal SK. 2013. Adenoviral E2 IVa2 protein interacts with L4 33K protein and E2 DNA-binding protein. J Gen Virol 94:1325–1334. doi: 10.1099/vir.0.049346-0. [DOI] [PubMed] [Google Scholar]
- 34.Christensen JB, Ewing SG, Imperiale MJ. 2012. Identification and characterization of a DNA binding domain on the adenovirus IVa2 protein. Virology 433:124–130. doi: 10.1016/j.virol.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutz P, Kedinger C. 1996. Properties of the adenovirus IVa2 gene product, an effector of late-phase-dependent activation of the major late promoter. J Virol 70:1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akusjarvi G. 2008. Temporal regulation of adenovirus major late alternative RNA splicing. Front Biosci 13:5006–5015. [DOI] [PubMed] [Google Scholar]
- 37.Shaw AR, Ziff EB. 1980. Transcripts from the adenovirus-2 major late promoter yield a single early family of 3′ coterminal mRNAs and five late families. Cell 22:905–916. doi: 10.1016/0092-8674(80)90568-1. [DOI] [PubMed] [Google Scholar]
- 38.Hayes BW, Telling GC, Myat MM, Williams JF, Flint SJ. 1990. The adenovirus L4 100-kilodalton protein is necessary for efficient translation of viral late mRNA species. J Virol 64:2732–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farley DC, Brown JL, Leppard KN. 2004. Activation of the early-late switch in adenovirus type 5 major late transcription unit expression by L4 gene products. J Virol 78:1782–1791. doi: 10.1128/JVI.78.4.1782-1791.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris SJ, Scott GE, Leppard KN. 2010. Adenovirus late-phase infection is controlled by a novel L4 promoter. J Virol 84:7096–7104. doi: 10.1128/JVI.00107-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 42.Campos SK, Barry MA. 2006. Comparison of adenovirus fiber, protein IX, and hexon capsomeres as scaffolds for vector purification and cell targeting. Virology 349:453–462. doi: 10.1016/j.virol.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 43.Tollefson AE, Ying B, Doronin K, Sidor PD, Wold WS. 2007. Identification of a new human adenovirus protein encoded by a novel late l-strand transcription unit. J Virol 81:12918–12926. doi: 10.1128/JVI.01531-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ying BL, Tollefson AE, Wold WSM. 2010. Identification of a previously unrecognized promoter that drives expression of the UXP transcription unit in the human adenovirus type 5 genome. J Virol 84:11470–11478. doi: 10.1128/JVI.01338-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horwitz MS. 2004. Function of adenovirus E3 proteins and their interactions with immunoregulatory cell proteins. J Gene Med 6(Suppl 1):S172–S183. doi: 10.1002/jgm.495. [DOI] [PubMed] [Google Scholar]
- 46.Lichtenstein DL, Toth K, Doronin K, Tollefson AE, Wold WS. 2004. Functions and mechanisms of action of the adenovirus E3 proteins. Int Rev Immunol 23:75–111. doi: 10.1080/08830180490265556. [DOI] [PubMed] [Google Scholar]
- 47.Hilgendorf A, Lindberg J, Ruzsics Z, Honing S, Elsing A, Lofqvist M, Engelmann H, Burgert HG. 2003. Two distinct transport motifs in the adenovirus E3/10.4-14.5 proteins act in concert to down-modulate apoptosis receptors and the epidermal growth factor receptor. J Biol Chem 278:51872–51884. doi: 10.1074/jbc.M310038200. [DOI] [PubMed] [Google Scholar]
- 48.Schneider-Brachert W, Tchikov V, Merkel O, Jakob M, Hallas C, Kruse ML, Groitl P, Lehn A, Hildt E, Held-Feindt J, Dobner T, Kabelitz D, Kronke M, Schutze S. 2006. Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J Clin Invest 116:2901–2913. doi: 10.1172/JCI23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tatsis N, Blejer A, Lasaro MO, Hensley SE, Cun A, Tesema L, Li Y, Gao GP, Xiang ZQ, Zhou DM, Wilson JM, Ertl HCJ. 2007. A CD46-binding chimpanzee adenovirus vector as a vaccine carrier. Mol Ther 15:608–617. doi: 10.1038/sj.mt.6300078. [DOI] [PubMed] [Google Scholar]
- 50.Weaver EA, Nehete PN, Buchl SS, Senac JS, Palmer D, Ng P, Sastry KJ, Barry MA. 2009. Comparison of replication-competent, first generation, and helper-dependent adenoviral vaccines. PLoS One 4:e5059. doi: 10.1371/journal.pone.0005059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weaver EA, Nehete PN, Nehete BP, Buchl SJ, Palmer D, Montefiori DC, Ng P, Sastry KJ, Barry MA. 2009. Protection against mucosal SHIV challenge by peptide and helper-dependent adenovirus vaccines. Viruses 1:920. doi: 10.3390/v1030920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weaver EA, Nehete PN, Nehete BP, Yang G, Buchl SJ, Hanley PW, Palmer D, Montefiori DC, Ferrari G, Ng P, Sastry KJ, Barry MA. 2013. Comparison of systemic and mucosal immunization with helper-dependent adenoviruses for vaccination against mucosal challenge with SHIV. PLoS One 8:e67574. doi: 10.1371/journal.pone.0067574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puig-Saus C, Rojas LA, Laborda E, Figueras A, Alba R, Fillat C, Alemany R. 2014. iRGD tumor-penetrating peptide-modified oncolytic adenovirus shows enhanced tumor transduction, intratumoral dissemination and antitumor efficacy. Gene Ther 21:767–774. doi: 10.1038/gt.2014.52. [DOI] [PubMed] [Google Scholar]
- 54.Le LP, Everts M, Dmitriev IP, Davydova JG, Yamamoto M, Curiel DT. 2004. Fluorescently labeled adenovirus with pIX-EGFP for vector detection. Mol Imaging 3:105–116. doi: 10.1162/1535350041464874. [DOI] [PubMed] [Google Scholar]
- 55.Hateboer G, Gennissen A, Ramos YFM, Kerkhoven RM, Sonntagbuck V, Stunnenberg HG, Bernards R. 1995. Bs69, a novel adenovirus E1a-associated protein that inhibits E1a transactivation. EMBO J 14:3159–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roberts BE, Miller JS, Kimelman D, Cepko CL, Lemischka IR, Mulligan RC. 1985. Individual adenovirus type 5 early region 1A gene products elicit distinct alterations of cellular morphology and gene expression. J Virol 56:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berk AJ. 1986. Functions of adenovirus E1A. Cancer Surv 5:367–387. [PubMed] [Google Scholar]
- 58.Blackford AN, Grand RJ. 2009. Adenovirus E1B 55-kilodalton protein: multiple roles in viral infection and cell transformation. J Virol 83:4000–4012. doi: 10.1128/JVI.02417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turner RL, Wilkinson JC, Ornelles DA. 2014. E1B and E4 oncoproteins of adenovirus antagonize the effect of apoptosis inducing factor. Virology 456–457:205–219. doi: 10.1016/j.virol.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu J, Li LN, Bouvier M. 2011. Adenovirus E3-19K proteins of different serotypes and subgroups have similar, yet distinct, immunomodulatory functions toward major histocompatibility class I molecules. J Biol Chem 286:17631–17639. doi: 10.1074/jbc.M110.212050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tollefson AE, Toth K, Doronin K, Kuppuswamy M, Doronina OA, Lichtenstein DL, Hermiston TW, Smith CA, Wold WS. 2001. Inhibition of TRAIL-induced apoptosis and forced internalization of TRAIL receptor 1 by adenovirus proteins. J Virol 75:8875–8887. doi: 10.1128/JVI.75.19.8875-8887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ali H, LeRoy G, Bridge G, Flint SJ. 2007. The adenovirus L4 33-kilodalton protein binds to intragenic sequences of the major late promoter required for late phase-specific stimulation of transcription. J Virol 81:1327–1338. doi: 10.1128/JVI.01584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Backström E, Kaufmann KB, Lan X, Akusjärvi G. 2010. Adenovirus L4-22K stimulates major late transcription by a mechanism requiring the intragenic late-specific transcription factor-binding site. Virus Res 151:220–228. doi: 10.1016/j.virusres.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 64.Goosney DL, Nemerow GR. 2003. Adenovirus infection: taking the back roads to viral entry. Curr Biol 13:R99–R100. doi: 10.1016/S0960-9822(03)00037-X. [DOI] [PubMed] [Google Scholar]
- 65.Wang HJ, Li ZY, Yumul R, Lara S, Hemminki A, Fender P, Lieber A. 2011. Multimerization of adenovirus serotype 3 fiber knob domains is required for efficient binding of virus to desmoglein 2 and subsequent opening of epithelial junctions. J Virol 85:6390–6402. doi: 10.1128/JVI.00514-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reed LJ, Muench M. 1938. A simple method for estimating fifty per cent endpoints. Am J Hyg 27:493–497. [Google Scholar]