

ABSTRACT
The peptide drug enfuvirtide (T20) is the only HIV-1 fusion inhibitor in clinical use, but it easily induces drug resistance, calling for new strategies for developing effective drugs. On the basis of the M-T hook structure, we recently developed highly potent short-peptide HIV-1 fusion inhibitors (MTSC22 and HP23), which mainly target the conserved gp41 pocket and possess high genetic barriers to resistance. Here, we focused on the selection and characterization of HIV-1 escape mutants of MTSC22, which revealed new resistance pathways and mechanisms. Two mutations (E49K and L57R) located at the inhibitor-binding site and two mutations (N126K and E136G) located at the C-terminal heptad repeat region of gp41 were identified as conferring high resistance either singly or in combination. While E49K reduced the C-terminal binding of inhibitors via an electrostatic repulsion, L57R dramatically disrupted the N-terminal binding of M-T hook structure and pocket-binding domain. Unlike E49K and N126K, which enhanced the stability of the endogenous viral six-helical bundle core (6-HB), L57R and E136G conversely destabilized the 6-HB structure. We also demonstrated that both primary and secondary mutations caused the structural changes in 6-HB and severely impaired the capability for HIV-1 entry. Collectively, our data provide novel insights into the mechanisms of short-peptide fusion inhibitors targeting the gp41 pocket site and help increase our understanding of the structure and function of gp41 and HIV-1 evolution.
IMPORTANCE The deep pocket on the N-trimer of HIV-1 gp41 has been considered an ideal drug target because of its high degree of conservation and essential role in viral entry. Short-peptide fusion inhibitors, which contain an M-T hook structure and mainly target the pocket site, show extremely high binding and inhibitory activities as well as high genetic barriers to resistance. In this study, the HIV-1 mutants resistant to MTSC22 were selected and characterized, which revealed that the E49K and L57R substitutions at the inhibitor-binding site and the N126K and E136G substitutions at the C-terminal heptad repeat region of gp41 critically determine the resistance phenotype. The data provide novel insights into the mechanisms of action of the M-T hook structure-based fusion inhibitors which will help further our understanding of the structure-function relationship of gp41 and molecular pathways of HIV-1 evolution and eventually facilitate the development of new anti-HIV drugs.
INTRODUCTION
The trimeric envelope (Env) glycoprotein of HIV-1 initiates infection by engaging cellular receptors and facilitating fusion of viral and target-cell membranes. In a generally accepted model, binding of the surface subunit gp120 to the host primary receptor CD4 and then to a chemokine receptor (CCR5 or CXCR4) triggers a cascade of conformational changes within the Env complex that include reduced interaction of gp120 with the transmembrane subunit gp41 (Fig. 1). The loss of constraint on gp41 activates its transition to an extended, membrane-bridging prehairpin conformation, with its N-terminal fusion peptide inserted into the target cell membrane and the transmembrane (TM) segment in the viral membrane. Subsequently, folding back of three C-terminal heptad repeats (CHR) onto the trimeric N-terminal repeats (NHR) creates a stable six-helix bundle (6-HB) that places the fusion peptide and TM segment at the same end of the molecule and thus pulls the viral and cellular membranes together, leading to bilayer fusion and viral entry (1–3).
FIG 1.
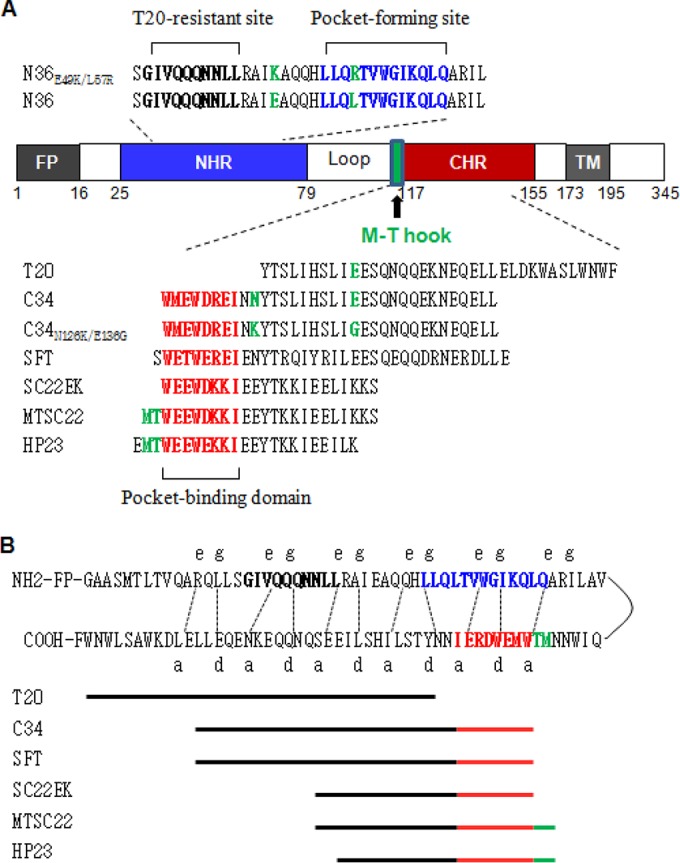
Schematic illustration of HIV-1 gp41 and peptide fusion inhibitors. (A) Functional domains of gp41 and peptide sequences. The gp41 numbering of HIV-1HXB2 is used. FP, fusion peptide; NHR, N-terminal heptad repeat; CHR, C-terminal heptad repeat; TM, transmembrane domain. The sequences corresponding to the T20-resistant site are in bold, the sequences corresponding to the NHR pocket region are in blue, and the sequences corresponding to the pocket-binding domain (PBD) are in red. The position and sequence of the M-T hook structure are shown in green. The peptide inhibitors and their sequences are listed, while the E49K and L57R mutations in N36 and the N126K and E136G mutations in C34 are in green. (B) The interaction between the NHR and CHR of gp41. In the current fusion model, the CHR region of gp41 folds back to the NHR region to form a hairpin structure. Three molecules of the hairpins associate with each other to form a 6-HB. The dashed lines between the NHR and CHR regions indicate the interaction between the residues located at the e and g positions and the a and d positions in the NHR and CHR, respectively. The peptide inhibitors are depicted as lines to express their sequences and binding sites.
Peptides derived from the NHR (N peptide) and CHR (C peptide) of gp41 can block 6-HB formation during the prehairpin intermediate state, thus preventing membrane fusion (4, 5). The drug enfuvirtide (T20), a 36-residue peptide with a native CHR sequence, is the only clinically approved HIV-1 fusion inhibitor; however, it has low antiviral activity and a genetic barrier to developing drug resistance (6–13). Almost all T20 resistance mutations appear within the inhibitor-binding sites on NHR, with the substitutions G36D/V/S, V38A/E/M, Q40H, N43D, and L45M predominating. The clinical development of peptide fusion inhibitor T1249 (39 residue) has been terminated due to its drug formulation problem and a similar resistance site (14, 15). Several next-generation peptides have been developed with significantly improved stability and potency, such as sifuvirtide (SFT; 36 residues) (16), T2635 (38 residues) (17), and SC34EK (34 residues) (18). Of them, SFT has been advanced to clinical phase III trials in China and will hopefully become the second clinically available HIV-1 fusion inhibitor; however, SFT has a similar low genetic barrier to resistance, with the mutations largely overlapping with T20 and T1249 resistance sites (19). The resistance profiles of T2635 and SC34EK have also been characterized by selecting escape viruses in vitro (14, 20, 21), which provide new insights into the molecular pathway and mechanism of HIV-1 resistance (22).
The hydrophobic pocket at the basis of the NHR grooves is nearly 16 Å long, 7 Å wide, and 5 to 6 Å deep. Three hydrophobic residues (Trp117, Trp120, and Ile124) from the pocket-binding domain of the CHR helix penetrate into the pocket to cause extensive hydrophobic interactions that stabilize the 6-HB structure. Since its discovery, the deep pocket has been considered an ideal drug target because of its high degree of conservation and essential role in viral entry (23, 24); however, inhibitors that specifically target the pocket site often lack a high antiviral activity, most likely due to their weak binding affinity (25–30). We recently found that two N-terminal residues (Met115 and Thr116) of a C peptide adopt a unique M-T hook structure, in which Thr116 redirects the peptide chain to position Met115 above the left side of the pocket so that its side chain caps the pocket to stabilize the inhibitor binding (31, 32). Indeed, the M-T hook structure-modified inhibitors exhibit greatly enhanced binding and inhibitory activities (31–37). On the basis of the M-T hook structure, we have developed two highly potent short-peptide-based inhibitors, MTSC22 and HP23, which mainly target the deep pocket rather than the T20- or SFT-resistant sites (34, 37). Promisingly, MTSC22 and HP23 also possess dramatically increased activities on the inhibition of diverse drug-resistant HIV-1 mutants and high genetic barriers to resistance; in sharp contrast, their template peptide SC22EK, which lacks the M-T hook structure, can select high resistance rather easily (35). The characterization of SC22EK-selected mutants revealed that substitution of Glu49 by lysine (E49K), located near the pocket site, conferred cross-resistance to diverse peptide fusion inhibitors, while the N126K substitution in CHR helix served as a secondary mutation that markedly boosted E49K-mediated resistance (38). To gain further insights into the genetic pathways and mechanisms of HIV-1 resistance to short-peptide fusion inhibitors targeting the gp41 pocket, we continued our efforts to select and characterize HIV-1 mutants resistant to MTSC22, which contains the M-T hook structure and has a dramatically improved target-binding affinity, virus-inhibitory activity, and genetic resistance barrier. Importantly, we found that besides the E49K and N126K mutations, the L57R mutation within the pocket and the E136G mutation in the CHR helix determine an extremely high resistance phenotype. The impact of MTSC22 resistance mutations on the binding affinity of inhibitors, the conformation and stability of 6-HB, and the functionality of viral Env function was investigated.
MATERIALS AND METHODS
Peptide synthesis.
N peptides, including N36, N36E49K, N36L57R, and N36E49K/L57R, and C peptides, including SC22EK, MTSC22, HP23, C34, C34N126K, C34E136G, C34N126K/E136G, T20, and SFT (Fig. 1), were synthesized using a standard solid-phase 9-fluorenylmethoxy carbonyl (FMOC) method as described previously (32). All peptides were protected by N-terminal acetylation and C-terminal amidation. They were purified by reversed-phase high-performance liquid chromatography (purity > 95%) and characterized for correct amino acid composition by mass spectrometry.
Selection of MTSC22-resistant viruses.
The in vitro selection of HIV-1 resistance to the peptide inhibitor MTSC22 was performed as described previously (19, 38). Briefly, viral stocks of HIV-1NL4-3 were generated by transfecting 293T cells with an encoding plasmid. MT-4 cells were seeded at 1 × 104 in RPMI 1640 medium containing 10% fetal bovine serum (FBS) on 12-well plates. The virus was used to infect the cells in the presence or absence of diluted MTSC22. Cells were incubated at 37°C with 5% CO2 until an extensive cytopathic effect was observed. Culture supernatants were harvested and used for the next passage on fresh MT-4 cells with a 1.5- to 2-fold increase in peptide concentrations. Cells and supernatant were collected at regular time points and stored at −80°C.
Site-directed mutagenesis.
HIV-1NL4-3 Env mutants were generated using double-stranded DNA templates and selection of mutants with a restriction enzyme (DpnI) as described previously (38). Briefly, two primers contained the desired mutation and occupied the same starting and ending positions on opposite strands of plasmid. DNA synthesis was performed by PCR in a 50-μl reaction volume using 1 ng of denatured plasmid template, a 50 pM concentration of upper and lower primers, and 5 U of the high-fidelity polymerase PrimeStar (TaKaRa, Dalian, China). PCR amplification was conducted for one cycle of denaturation at 98°C for 5 min, followed by 18 cycles of 98°C for 15 s and 68°C for 15 min, with a final extension at 72°C for 10 min. The amplicons were treated with DpnI for 1 h at 37°C, and DpnI-resistant molecules were recovered by transforming Escherichia coli strain DH5α to antibiotic resistance. The mutations were confirmed by DNA sequencing.
Single-cycle infection assay.
HIV-1 entry and its inhibition were measured by single-cycle infection assay as described previously (39). Briefly, HIV-1 pseudovirus was generated by cotransfecting 293T cells with an Env-expressing plasmid and a backbone plasmid, pSG3Δenv, that contains an Env-defective, luciferase-expressing HIV-1 genome. Supernatants were harvested 48 h after transfection, and 50% tissue culture infectious doses (TCID50) were determined in TZM-bl cells. Peptides were prepared in 3-fold dilutions, mixed with 100 TCID50 of viruses, and then incubated 1 h at room temperature. The mixture was added to TZM-bl cells (104/well) and incubated 48 h at 37°C. The luciferase activity was measured using luciferase assay reagents and a luminescence counter (Promega, Madison, WI, USA).
Cell-cell fusion assay.
A dual split-protein (DSP)-based fusion cell-cell assay (40, 41) was adapted to determine the effects of the introduced mutations on the fusion activity of NL4-3 Env. Briefly, a total of 1.5 × 104 293T cells (effector cells) were seeded on a 96-well plate and a total of 8 × 104 U87-CXCR4 cells (target cells) were seeded on a 24-well plate. On the following day, effector cells were transfected with a mixture of an Env-expressing plasmid and a DSP1-7 plasmid, and target cells were transfected with a DSP8-11 plasmid. Twenty-four hours posttransfection, the target cells were resuspended in 300 μl prewarmed culture medium, and 0.15 μl EnduRen live-cell substrate (Promega) was added to each well. Then, 75 μl of the target cell suspension was transferred to each well of the effector cells, and the cells were spun down to maximize cell-cell contact. Luciferase activity was measured by a luminescence counter (Promega).
ELISA.
A capture enzyme-linked immunosorbent assay (ELISA) was performed to determine the effects of introduced mutations on the Env expression. Briefly, the wells of an ELISA plate were coated with a sheep anti-gp120 antibody (D7324) at 10 μg/ml and blocked by 3% bovine serum albumin (BSA). Cell lysates or culture supernatants (50 μl) of Env-transfected cells were added to the wells and incubated at 37°C for 1 h. After four washes, 50 μl of human anti-gp120 monoclonal antibody (MAb) VRC01 or anti-gp41 MAb 10E8 diluted 10 μg/ml was added and incubated at 37°C for 1 h. The bound antibodies were detected by horseradish peroxidase (HRP)-conjugated goat anti-human IgG. The reaction was visualized by addition of 3,3,5,5-tetramethylbenzidine, and the A450 was measured.
To detect the 6-HBs, three 6-HB conformation-specific MAbs (NC-1, 17C8, and 2G8), which react with the N36 and C34 complex but not the isolated peptides, were described previously (42, 43). Briefly, the ELISA wells were coated with the isolated or mixed peptides at 10 μg/ml and blocked by 3% BSA. The anti-6-HB MAb at 5 μg/ml was added to the wells and incubated at 37°C for 1 h. After three washes, the bound antibodies were detected by HRP-conjugated anti-mouse IgG. Similarly, the reaction was visualized by addition of 3,3,5,5-tetramethylbenzidine, and the A450 was measured.
CD spectroscopy.
Circular dichroism (CD) spectroscopy was conducted according to our protocols described previously (44). Briefly, a C peptide was incubated with an equal molar concentration of N36 or its mutant at 37°C for 30 min in phosphate-buffered saline (PBS; pH 7.2). CD spectra were acquired on a Jasco spectropolarimeter (model J-815) using a 1-nm bandwidth with a 1-nm step resolution from 195 to 260 nm at room temperature. Spectra were corrected by subtraction of a solvent blank. The α-helical content was calculated from the CD signal by dividing the mean residue ellipticity ([θ]) at 222 nm by the value expected for 100% helix formation (−33,000 degrees cm−2 dmol−1). Thermal denaturation was performed by monitoring the ellipticity change at 222 nm from 20°C to 98°C at a rate of 1.2°C/min.
RESULTS
Selection of HIV-1 mutants highly resistant to MTSC22.
We previously conducted a parallel in vitro selection of escape HIV-1 mutants for SC22EK and MTSC22 peptides (35). While the viruses resistant to SC22EK were rather easily acquired, the selection of resistance against MTSC22 was much more difficult (35). We continued the selection culture for an additional 5 months, and the concentration of MTSC22 was finally escalated to 9,600 nM after 45 generations of virus passage over 9 months. To characterize the genetic pathways of MTSC22-induced resistance, the full Env genes of resistant viruses were amplified by PCR and cloned for sequencing. As shown in Fig. 2, all 10 Env clones (A to J) carry three consistent substitutions, including E49K in NHR, N126K in CHR, and N163D in the membrane-proximal external region (MPER) of gp41. Notably, nine of the Envs (A to I) possess a L57R substitution within the highly conserved pocket site. Furthermore, the clone A Env has an additional I37V substitution in the upstream of NHR, and the clones I and J carry an E136G substitution in the middle region of CHR. No obvious mutations were observed in the other regions of gp41 or in gp120. Notably, the E49K, N126K, and E136G substitutions were selected by both SC22EK and MTSC22 peptides, while the L57R substitution emerged only during MTSC22 selection, implying its association with the M-T hook structure that targets the gp41 pocket.
FIG 2.

MTSC22- and SC22EK-induced mutations in the NHR and CHR sites of HIV-1NL4-3 gp41. The amino acid sequences of wild-type (WT) and selected mutants are aligned. The positions of selected mutations are in bold, and numbering is according to that of HIV-1HXB2 gp41. The pocket-forming sequence in NHR and the pocket-binding domain in CHR with the M-T hook residues are underlined.
Resistance profiles of short-peptide fusion inhibitors targeting the pocket.
Apparently, four types of mutant viruses emerged during drug selection. A predominating virus had four amino acid mutations, E49K/L57R/N126K/N163D (designated KRGD virus); the second one also carried four mutations (E49K/N126K/E136G/N163D; designated KKGD virus). Two viruses had five mutations each, E49K/L57R/N126K/E136G/N163D (KRKGD virus) and I37V/E49K/L57R/N126K/N163D (VKRKD virus). To determine the mutations responsible for MTSC22-induced resistance, we generated a panel of HIV-1NL4-3 Env mutants carrying the amino acid substitutions either singly or in combination (Table 1). While the experimental design was focused on the newly identified mutations (L57R, E136G, and N163D), the previously characterized mutations (E49K and N126K) involved in SC22EK-induced resistance were also included for direct comparisons. After the introduced mutations were verified by DNA sequencing and the expression of Env glycoprotein was confirmed by Western blotting, the corresponding HIV-1 pseudoviruses were generated, and their susceptibility to three short-peptide inhibitors (MTSC22, HP23, and SC22EK) was determined. As shown in Table 1, the single mutations of E49K and N126K conferred resistance with the changes (n-fold) similar to the previous results. Strikingly, the L57R mutation caused extremely high resistance to the M-T hook structure-modified inhibitors MTSC22 and HP23 as well as their template SC22EK, with the changes reaching 402.08-, 567.95-, and 38.86-fold, respectively. It was also highly surprised that a single E136G mutation also conferred a moderate resistance to three inhibitors, with the changes being significantly higher than N126K-mediated resistance. As expected, the N163D mutation had no obvious effects on the anti-HIV activity of inhibitors.
TABLE 1.
Resistance profiles of HIV-1 mutants to short-peptide fusion inhibitorsa
| HIV-1NL4-3 mutation | MTSC22 |
HP23 |
SC22EK |
|||
|---|---|---|---|---|---|---|
| IC50 (nM) | Fold change | IC50 (nM) | Fold change | IC50 (nM) | Fold change | |
| Wild type (WT) | 1.86 ± 0.13 | 1.00 | 0.41 ± 0.09 | 1.00 | 44.13 ± 5 | 1.00 |
| E49K | 73.75 ± 13.53 | 39.74 | 5.34 ± 0.18 | 13.19 | 1,723.5 ± 184.13 | 39.06 |
| L57R | 746.12 ± 57 | 402.08 | 230.05 ± 39.19 | 567.95 | 1,714.5 ± 385.06 | 38.86 |
| N126K | 4.27 ± 0.98 | 2.3 | 1.37 ± 0.17 | 3.39 | 151.93 ± 12.15 | 3.44 |
| E136G | 8.78 ± 1.03 | 4.73 | 2.89 ± 0.52 | 7.14 | 477.05 ± 42.53 | 10.81 |
| N163D | 2.03 ± 0.3 | 1.09 | 0.54 ± 0.05 | 1.32 | 46.85 ± 7.84 | 1.06 |
| E49K/L57R | 4,527.86 ± 585.46 | 2,440.06 | 1,371.3 ± 561.56 | 3,385.51 | 6,694.96 ± 1,036.29 | 151.73 |
| L57R/N126K | 345.2 ± 34.06 | 186.03 | 98.64 ± 5.92 | 243.53 | 644.12 ± 244.72 | 14.60 |
| L57R/E136G | 1,553.08 ± 535.22 | 836.96 | 679.55 ± 191.15 | 1,677.69 | 5,322.97 ± 1,529.45 | 120.63 |
| L57R/N163D | 608.95 ± 147.94 | 328.16 | 244.95 ± 55.18 | 604.74 | 4,014.67 ± 652.8 | 90.98 |
| E49K/L57R/N126K | 1,148.68 ± 145.24 | 619.02 | 542.17 ± 141.94 | 1,338.52 | 3,226.73 ± 955.88 | 73.13 |
| E49K/L57R/E136G | >7,500 | >4041.75 | >1,666.67 | >4114.73 | >7,500 | >169.97 |
| E49K/N126K/E136G/N163D (KKGD) | 359.22 ± 86.44 | 193.58 | 44.26 ± 3.08 | 109.26 | >7,500 | >169.97 |
| E49K/L57R/N126K/E136G (KRKG) | 2,249.37 ± 655.77 | 1,212.18 | 1,389.6 ± 250.15 | 3,430.69 | 7,054 ± 617.91 | 159.86 |
| E49K/L57R/N126K/N163D (KRKD) | 5,932.8 ± 331.89 | 3,197.18 | 983.12 ± 184.14 | 2,427.15 | 5,959.18 ± 447.45 | 135.05 |
| I37V/E49K/L57R/N126K/N163D (VKRKD) | 3,081.67 ± 212.92 | 1,660.71 | 1,096.38 ± 444.81 | 2,706.79 | 5,649.09 ± 686.21 | 128.02 |
| E49K/L57R/N126K/E136G/N163D (KRKGD) | 3,854.04 ± 533.69 | 2,076.94 | >1,666.67 | >4114.73 | 5,797.24 ± 596.92 | 131.38 |
Assay was performed in triplicate and repeated three times. Data are expressed as means ± standard deviations. IC50, 50% inhibitory concentration. The change in the IC50 was determined relative to the wild-type level.
We next observed the effects of the combined mutations on the resistance profiles, which revealed multiple resistance phenotypes. First, the E49K/L57R combination resulted in markedly increased changes, indicating a synergistic effect. Second, the E136G significantly enhanced the resistance degree caused by the L57R single mutation and the E49K/L57R double mutation. Third, while N126K was previously shown to boost E49K-mediated resistance, it exhibited a reverse effect on L57R-mediated resistance, as shown by its counterbalance role in the virus carrying L57R/N126K or E49K/L57R/N126K. Fourth, the N126K/E136G combination synergistically boosted the E49K phenotype, but it did not act on the L57R phenotype, as shown by the KKGD and KRKGD viruses or the virus with E49K/L57R/N126K/E136G (KRKG virus). Fifth, the N163D mutation was not involved in the resistance even in the context of different combinations. Specifically, four corresponding mutant viruses (KRGD, KKGD, KRKGD, and VKRKD viruses) displayed extremely high resistance to MTSC22, HP23, and SC22EK.
Effects of MTSC22-induced mutations on the activity of long-peptide fusion inhibitors.
We previously showed that E49K rendered a moderate cross-resistance to the first-generation inhibitors T20 and C34 and the next-generation inhibitor SFT, and the resulting changes could be markedly enhanced by N126K (38). We were interested in determining whether L57R or its combinations mediated cross-resistance to three large inhibitors. Surprisingly, it was found that L57R did not cause the resistance instead of slightly increasing the sensitivity of T20, C34, and SFT (Table 2). Obviously, the viruses carrying L57R together with other resistant mutations (E49K, N126K, and E136G) could result in markedly increased activity of T20. For instance, the KKGD virus exhibited a resistance change of 5.84-fold, but the KRKG virus had a change of 0.13-fold, meaning a 7.64-fold increased potency for T20. Similarly, L57R also increased the sensitivity of C34 and SFT, especially in the context of other resistant mutations. In comparison, the mutant virus without L57R (KKGD) conferred high cross-resistance to C34 and SFT, but addition of L57R to the combination could abolish or significantly decrease the resistance.
TABLE 2.
Effect of MTSC22-induced mutations on the sensitivity of first- and next-generation fusion inhibitorsa
| HIV-1NL4-3 mutation | T20 |
C34 |
SFT |
|||
|---|---|---|---|---|---|---|
| IC50 (nM) | Fold change | IC50 (nM) | Fold change | IC50 (nM) | Fold change | |
| Wild-type (WT) | 81.77 ± 3.99 | 1.00 | 1.49 ± 0.25 | 1.00 | 1.87 ± 0.13 | 1.00 |
| E49K | 222.84 ± 13.24 | 2.73 | 4.76 ± 1.73 | 3.20 | 4.84 ± 0.46 | 2.59 |
| L57R | 42.85 ± 4.4 | 0.52 | 1.13 ± 0.15 | 0.76 | 1.04 ± 0.18 | 0.56 |
| N126K | 184.63 ± 18.32 | 2.26 | 3.52 ± 0.2 | 2.37 | 5.36 ± 2.87 | 2.87 |
| E136G | 175.68 ± 51.11 | 2.15 | 7.8 ± 1.3 | 5.25 | 9.62 ± 3.77 | 5.15 |
| N163D | 104.86 ± 50.14 | 1.28 | 1.97 ± 0.3 | 1.32 | 2.15 ± 0.17 | 1.15 |
| E49K/L57R | 18.75 ± 6.29 | 0.23 | 1.98 ± 0.31 | 1.33 | 1.6 ± 0.48 | 0.86 |
| L57R/N126K | 17.83 ± 4.24 | 0.22 | 1 ± 0.15 | 0.67 | 0.74 ± 0.1 | 0.40 |
| L57R/E136G | 34.18 ± 4.08 | 0.42 | 4.78 ± 0.61 | 3.21 | 4.02 ± 1.02 | 2.15 |
| L57R/N163D | 51.76 ± 7.11 | 0.63 | 2.54 ± 0.2 | 1.71 | 1.84 ± 0.63 | 0.99 |
| E49K/L57R/N126K | 10.59 ± 0.47 | 0.13 | 2.34 ± 0.25 | 1.57 | 0.85 ± 0.25 | 0.46 |
| E49K/L57R/E136G | 13.64 ± 2 | 0.17 | 6.2 ± 2.19 | 4.17 | 4.76 ± 1.43 | 2.55 |
| E49K/N126K/E136G/N163D (KKGD) | 477.43 ± 89.13 | 5.84 | 32.99 ± 1.99 | 22.18 | 32.96 ± 2.91 | 17.67 |
| E49K/L57R/N126K/E136G (KRKG) | 10.71 ± 1.1 | 0.13 | 3.87 ± 0.55 | 2.61 | 1.55 ± 0.14 | 0.83 |
| E49K/L57R/N126K/N163D (KRKD) | 12.35 ± 3.05 | 0.15 | 3.14 ± 0.65 | 2.11 | 0.93 ± 0.09 | 0.50 |
| I37V/E49K/L57R/N126K/N163D (VKRKD) | 29.38 ± 1.28 | 0.36 | 3.7 ± 0.67 | 2.49 | 1.41 ± 0.07 | 0.75 |
| E49K/L57R/N126K/E136G/N163D (KRKGD) | 12.22 ± 2.52 | 0.15 | 14.39 ± 2.6 | 9.68 | 3.05 ± 0.07 | 1.63 |
The assay was performed in triplicate and repeated three times. Data are expressed as means ± standard deviations. IC50, 50% inhibitory concentration. The change in the IC50 was determined relative to the wild-type level.
While the amino acid substitutions on the inhibitor-binding site of gp41 are considered primary mutations responsible for resistance, the CHR mutations have been recognized as secondary or compensatory mutations that can improve the kinetics of viral fusion (21, 45, 46). Previous studies demonstrate that N126K not only enhances the interaction between the NHR and CHR helices but also mediates a mild resistance to diverse fusion inhibitor peptides (20, 21, 38). Here, we found that a single E136G mutation rendered the virus resistant to three short-peptide inhibitors that specifically target the pocket site and its combinations with L57R or E49K/L57R dramatically boosted the resistance. We were interested in determining whether E136G conferred cross-resistance to T20, C34, and SFT. As shown in Table 2, the virus carrying E136G constantly displayed the resistance to three large inhibitors, with changes of 2.15-, 5.25-, and 5.15-fold, respectively. Compared with N126K, the E136G mutation caused resistance at a significantly higher level.
MTSC22-induced mutations severely impair the functionality of HIV-1 Env.
It is recognized that peptide-based HIV-1 fusion inhibitor-induced resistance mutations often result in a partial or substantial loss of viral Env's function (19-21, 38). Consistently, we demonstrated that E49K or its combination with N126K dramatically decreased HIV-1 Env-mediated cell entry. To elucidate whether the acquisition of HIV-1 resistance to MTSC22 was accompanied by a loss of gp41 function, we determined the entry efficiency of HIV-1NL4-3 pseudovirus that carry single or combined mutations (Fig. 3A). The infectivity of wild-type virus was set at 100%, and the relative infectivity of other mutants was calculated. As expected, L57R resulted in a dramatic decrease in virus infectivity, and its combinations with E49K or two secondary mutations reduced the infectivity further. Interestingly, a single N163D mutation facilitated the virus entry, and it was able to partially restore the infectivity damaged by other mutations, suggesting its compensatory role. Also, the virus with E136G largely retained its infectivity, similar to the virus having N126K. The functionality of the wild-type and mutant Envs was further evaluated by a dual split-protein-based cell-cell fusion assay, which displayed a profile similar to the entry data (Fig. 3B). Obviously, the introduced mutations did not affect the expression and secretion of Envs in transfected cells, as detected by anti-gp120 MAb VRC01 and anti-gp41 MAb 10E8 (Fig. 4). Combined, these data confirmed that the amino acids critical for HIV-1 resistance also play important roles for viral fusion and entry.
FIG 3.

Relative infectivity of HIV-1NL4-3 and its mutant viruses. (A) Entry efficiency of pseudoviruses. The wild-type HIV-1NL4-3 and mutant pseudoviruses were normalized to a fixed amount by p24 antigen, and viral infectivity was tested in TZM-bl cells using a single-cycle infection assay. The luciferase activity was measured and corrected for background. (B) HIV-1 Env-mediated cell-cell fusion determined by a dual split-protein assay (DSP). For both entry and fusion data, the luciferase activity of wild-type HIV-1NL4-3 (WT) was treated as 100% and the relative activities of other mutant viruses were calculated accordingly. Data were derived from the results of three (entry) or two (fusion) independent experiments and are expressed as means and standard deviations.
FIG 4.

Determination of HIV-1 Env expression by capture ELISA. 293T cells were transfected by plasmids expressing wild-type (WT) or mutant Envs. The proteins (gp120/gp41) in the lysates of transfected cells (A) or culture supernatants (B) were captured by sheep anti-gp120 antibody D7324 and detected by human MAbs VRC01 and 10E8.
The L57R mutation dramatically reduces the binding affinity of short-peptide inhibitors.
On the basis of SC22EK-induced resistance, we showed that E49K greatly reduced the binding stability of SC22EK, MTSC22, and HP23, while it enhanced the stability of the 6-HB structure formed by C34 and SFT (38). Here, we focused on investigating the effects of L57R and its combination with E49K on the binding of diverse inhibitors. Thus, the N-peptide N36 with a single mutation (N36E49K and N36L57R) or double mutations (N36E49K/L57R) were used as target surrogates and incubated with an inhibitor at equal molar concentrations, and their α-helicity and thermostability were then measured by CD spectroscopy. As shown in Fig. 5 and 6 and Table 3, the CD spectra of all five peptide pairs displayed double minima at 208 and 222 nm, which indicated the formation of α-helical secondary structures. Noticeably, the 6-HBs formed by N36E49K/L57R and short-peptide inhibitors showed a significantly decreased α-helicity, while no significant changes were observed for the 6-HBs between N36E49K/L57R and C34 or SFT. The thermostability of each 6-HB, defined as the midpoint of the thermal unfolding transition (Tm) value, was determined. Consistent with our previous data, the single E49K mutation resulted in a Tm value with an approximately 3 to 5°C decrease for SC22EK, MTSC22, and HP23, but it caused a 6°C increase for both C34 and SFT (Table 3). Strikingly, the single L57R mutation resulted in the Tm values for the three short-peptide inhibitors at 37, 40, and 49°C, respectively, which implied a 27, 37, or 37°C decrease relative to the wild-type N36. The combination of E49K and L57R further reduced the Tm values by approximately 4 to 5°C. Sharply differing from the E49K phenotype, the single L57R mutation also resulted in markedly decreased Tm values for C34 and SFT. Therefore, these data indicated that the L57R mutation can dramatically reduce the binding affinity of diverse peptide inhibitors.
FIG 5.

Binding stability of short peptide inhibitors determined by CD spectroscopy. The α-helicity and thermostability of 6-HBs formed by SC22EK (A and B), MTSC22 (C and D), or HP23 (E and F) with N36 or its mutants (N36E49K, N36L57R, and N36E49K/L57R) were measured. The Tm value was defined as the midpoint of the thermal unfolding transition. The final concentration of each peptide in PBS is 10 μM. The experiments were repeated at least twice, and representative data are shown.
FIG 6.

Binding stability of C34, SFT, and C34 mutants determined by CD spectroscopy. The α-helicity and thermostability of 6-HBs formed by C34 (A and B) or SFT (C and D) with N36 or its mutants (N36E49K, N36L57R, and N36E49K/L57R) were measured. The Tm value was defined as the midpoint of the thermal unfolding transition. The final concentration of each peptide in PBS is 10 μM. The binding stability of C34 or its mutants (C34N126K, C34E136G, and C34N126K/E136G) with N36 was determined by CD spectroscopy similarly (E and F). The experiments were repeated at least twice, and representative data are shown.
TABLE 3.
Effects of MTSC22 resistance mutations on the α-helicity and thermostability of 6-HB structurea
| NHR mutation type and peptide complex | [θ]222 | Helix content (%) | Tm (°C) |
|---|---|---|---|
| Primary | |||
| SC22+N36 | −28,470 | 86 | 64 |
| SC22+N36E49K | −31,360 | 95 | 61 |
| SC22+N36L57R | −28,586 | 86 | 37 |
| SC22+N36E49k/L57R | −20,052 | 61 | 32 |
| MTSC22+N36 | −31,170 | 94 | 77 |
| MTSC22+N36E49K | −28,570 | 87 | 72 |
| MTSC22+N36L57R | −28,150 | 85 | 40 |
| MTSC22+N36E49k/L57R | −21,683 | 66 | 35 |
| HP23+N36 | −31,877 | 97 | 86 |
| HP23+N36E49K | −30,246 | 92 | 83 |
| HP23+N36L57R | −30,441 | 92 | 49 |
| HP23+N36E49k/L57R | −28,102 | 85 | 45 |
| C34+N36 | −33,190 | 101 | 64 |
| C34+N36E49K | −31,240 | 95 | 70 |
| C34+N36L57R | −35,014 | 106 | 55 |
| C34+N36E49k/L57R | −33,757 | 102 | 60 |
| SFT+N36 | −31,389 | 95 | 71 |
| SFT+N36E49K | −30,292 | 92 | 77 |
| SFT+N36L57R | −32,819 | 100 | 62 |
| SFT+N36E49k/L57R | −34,625 | 105 | 69 |
| Secondary | |||
| N36+C34N126K | −27,708 | 84 | 70 |
| N36+C34E136G | −27,700 | 84 | 62 |
| N36+C34N126K/E136G | −26,229 | 80 | 65 |
| N36E49K+C34N126K | −34,893 | 105 | 74 |
| N36E49K+C34E136G | −33,386 | 101 | 66 |
| N36L57R+C34N126K | −33,557 | 102 | 57 |
| N36L57R+C34E136G | −36,014 | 109 | 50 |
| N36E49K+C34N126K/E136G | −34,900 | 106 | 67 |
| N36L57R+C34N126K/E136G | −35,929 | 108 | 51 |
| N36E49K/L57R+C34N126K | −33,086 | 100 | 62 |
| N36E49K/L57R+C34E136G | −35,414 | 107 | 54 |
| N36E49K/L57R+C34N126K/E136G | −32757 | 99 | 55 |
The α-helicity and thermostability of 6-HBs were measured by CD spectroscopy, and the experiments were repeated at least twice to verify the results.
The E136G mutation decreases the helical stability of 6-HB structure.
The secondary mutation N126K can significantly increase the thermal stability of 6-HB structure modeled by N36/C34 peptides, and the combination of E49K and N126K has a synergistic effect to stabilize the 6-HB (38). Here, we found that E136G, which is also considered a secondary CHR mutation, conferred cross-resistance to the diverse HIV-1 fusion inhibitor peptides tested. It was intriguing to determine the impact of E136G on the 6-HB. Thus, the C peptide C34 carrying N126K, E136G, or N126K/E136G was synthesized, and the α-helicity and thermostability of mutant peptide-based 6-HBs were determined by CD spectroscopy. Consistently, the peptide pair N36 and C34N126K created a 6-HB with an increased Tm value (70°C); however, the peptides N36 and C34E136G created a 6-HB with a decreased Tm value (62°C) (Fig. 6E and F and Table 3), indicating that N126K and E136G play different roles in 6-HB stability. The combination of N126K and E136G resulted in a 6-HB with a Tm value (65°C) which was close to that of the wild-type 6-HB (64°C). We also analyzed the 6-HBs formed by diverse N36 mutants and C34 mutants (Table 3), which provided additional information on the compensatory effects of NHR and CHR mutations.
MTSC22-induced mutations affect the conformation of the 6-HB core.
The 6-HB formed by the peptides N36 and C34 has been considered a core structure of gp41, which critically determines the fusion activity of gp41 (1). Here, we sought to determine the effects of MTSC22 resistance mutations on the conformation of 6-HB structure, which provided clues to elucidate how the mutations dictated the resistance, 6-HB stability, and virus entry. Three conformation-specific monoclonal antibodies (NC-1, 17C8, and 2G8) were applied in ELISA to detect 6-HBs formed by the wild-type or mutant N36 and C34 peptides. As shown in Fig. 7, two NHR mutations (E49K, L57R) resulted in significantly decreased reactivity with all three antibodies, while two CHR mutations (N126K and E136G) significantly increased the reactivity of NC-1 and 17C8 but sharply decreased the reactivity of 2G8. It is worth noting that the 6-HB with a L57R mutation had much lower 17C8 reactivity than the 6-HB with an E49K mutation, and the 6-HB with an E136G mutation exhibited much lower 2G8 reactivity than the 6-HB with a N126K mutation. Taken together, the results indicating decreased or increased exposure of antibody-recognized epitopes suggested that MTSC22-induced mutations can directly or indirectly cause a structural rearrangement of 6-HBs, which might correlate with the functionality of gp41 and the binding of inhibitors. While this may be one explanation, it is possible that the mutated residues are involved in the epitopes of three antibodies and that the changed stability of the mutated 6-HBs can determine the binding signals.
FIG 7.
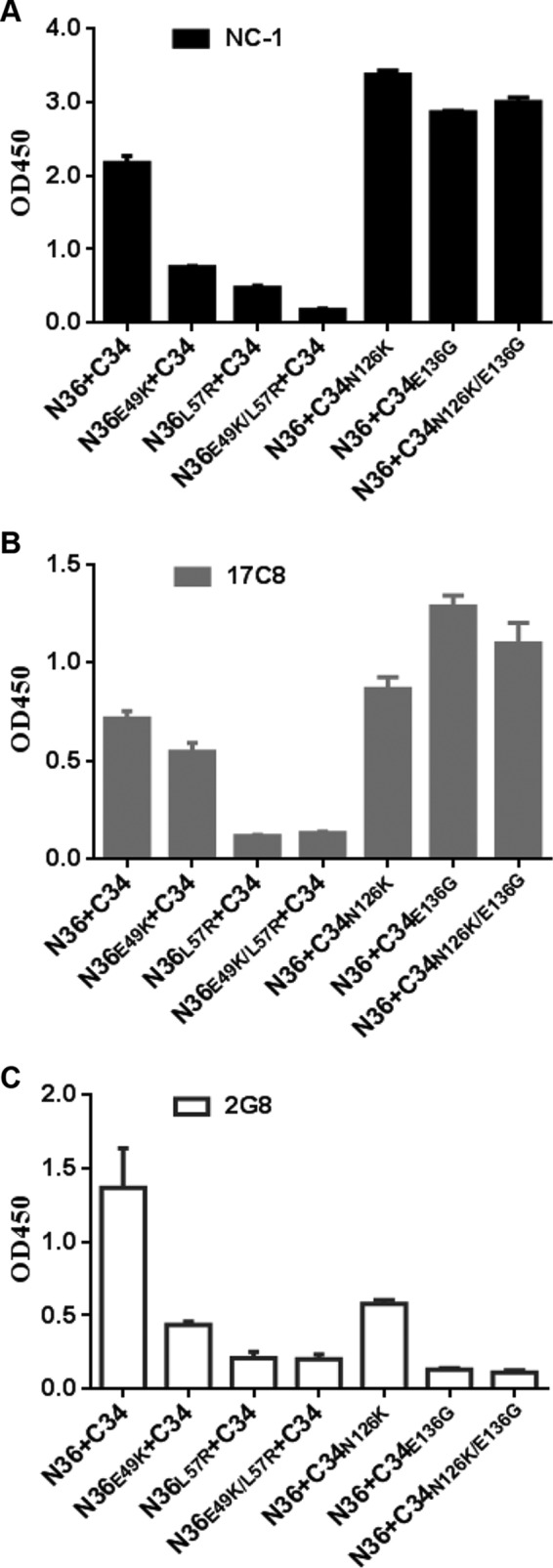
Effects of MTSC22-induced mutations on the conformation of 6-HB structure. The reactivity of 6-HBs formed by N36 and C34 or their mutants with conformation-dependent MAbs NC-1 (A), 17C8 (B), and 2G8 (C) was tested by ELISA. The peptide mixture was used to coat the plate wells at 10 μg/ml, and the final concentration of a tested MAb was 5 μg/ml. Data were derived from three independent experiments and are expressed as means and standard deviations.
Effects of MTSC22-induced mutations on the antiviral activity of N and C peptides.
Both N and C peptides have potent anti-HIV activity via blocking NHR-CHR association. Generally, a prototype N peptide has a much lower activity than a prototype C peptide (micromolar versus nanomolar), but N36 and C34 are widely used as templates to design novel HIV-1 fusion inhibitors with different specificity. To gain further insights into the impacts of MTSC22-induced mutations on the structure and function of gp41 and gp41-based inhibitors, we measured the anti-HIV activity of wild-type and mutant N36 and C34 peptides. As shown in Fig. 8, the peptides N36E49K and C34N126K displayed somewhat improved activities relative to the wild-type N36 in the inhibition of HIV-1 entry; however, the peptides N36L57R and C34E136G exhibited significantly decreased inhibitory activities. Here, we speculated that the effects of these mutations on the antiviral potency of peptides might be associated with the stability of 6-HB structures.
FIG 8.
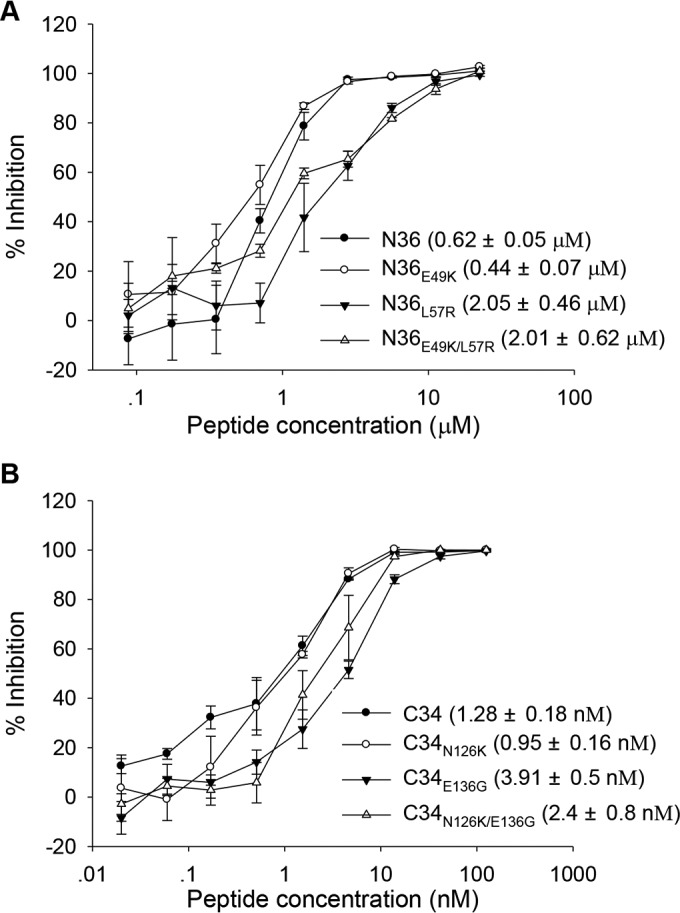
Inhibitory activity of wild-type and mutant N36 and C34 peptides determined by single-cycle infection assay. (A) Inhibition by N36 and its mutants of HIV-1NL4-3 pseudoviruses. (B) Inhibition by C34 and its mutants of HIV-1NL4-3 pseudoviruses. Data were derived from three independent experiments. The numbers in parentheses are IC50s (means and standard deviations).
Structural basis of MTSC22-induced resistance.
We previously analyzed E49K-mediated resistance by mapping the mutations on the crystal structure of SC22EK-based 6-HB, which revealed that E49K would disrupt a salt bridge between the negatively charged Glu49 and the positively charged lysine at position 20 (Lys20) of inhibitors; instead, an electrostatic repulsion is generated (38). On the other hand, E49K introduces a new salt bridge during the binding of C34 and SFT, which carry a natural glutamic acid corresponding to position 20 of short-peptide inhibitors. Thus, E49K would favor the interaction of endogenous NHR and CHR helices while disfavoring the inhibitor binding. When mapping L57R on the crystal structure of MTSC22-based 6-HB (Fig. 9A), the residue Leu57 is located on the left wall of the hydrophobic pocket and it makes an extensive hydrophobic interaction with the pocket-binding residues of inhibitors. More importantly, Leu57 is also targeted by the M-T hook structure of MTSC22, in which the N-terminal hydrophobic residue methionine physically interacts with the side chain of Leu57. It is conceivable that L57R can disrupt its interactions in the pocket site, thus contributing the resistance. Clearly, while E49K impairs the C-terminal binding of short-peptide inhibitors via an introduced electrostatic repulsion, L57R disrupts the binding of N-terminal M-T hook structure and pocket-binding domain; therefore, the E49K and L57R mutations can synergistically reduce the binding affinity of inhibitors.
FIG 9.
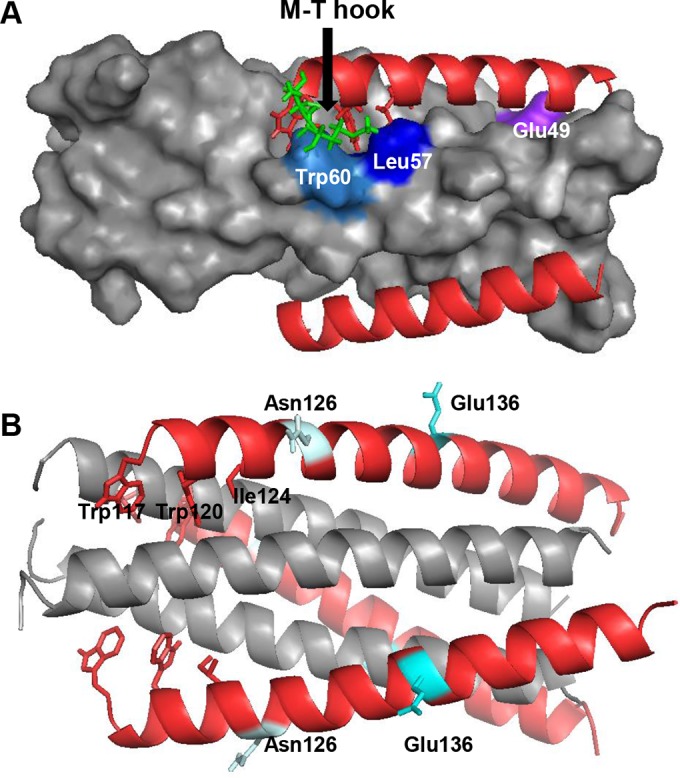
Modeling of MTSC22 resistance mutations on the 6-HB by the program PyMOL. (A) Analysis of the E49K and L57R mutations on the 6-HB formed by MTSC22 and an NHR-derived peptide (Protein Data Base ID code 3VU6). The pocket on the NHR helices is inserted by three hydrophobic residues (Trp117, Trp120, and Ile124) from the N-terminal pocket-binding domain of MTSC22. The M-T hook structure (green) of MTSC22 interacts with both Leu57 (blue) and Trp60 (marine) located at the left wall of the deep pocket. Therefore, Leu57 has extensive hydrophobic interactions with both the M-T hook residues and the pocket-binding residues to stabilize the binding of MTSC22. The residue Glu49 (purple) is located closely upstream of the pocket, and it can mediate a salt-bridge interaction with the positively charged lysine at position 20 (Lys20) of MTSC22. (B) Analysis of the N126K and E136G mutations on the 6-HB structure formed by N36 and C34 (Protein Data Base ID code 1AIK). The residues Asn126 and Glu136 are located, respectively, at the c and f positions of a CHR helix, corresponding to the outside of the binding surface of a CHR helix.
Next, we analyzed the N126K and E136G mutations on the 6-HB structure (Fig. 9B). These two residues are located at the c and f positions, respectively, in the CHR helix, corresponding to the outside of the binding surface of C peptides. Although N126K and E136G might enable the formation or disruption of intrahelical interactions of the isolated peptides, which can stabilize or destabilize the α-helicity and stability of 6-HB structure either, how the mutations at these locations affect the interhelical interactions and confer resistance remains a puzzle.
DISCUSSION
Six classes of HIV-1 drugs in clinical use are directed against specific steps of the viral life cycle, including cell entry, reverse transcription, integration, and maturation. Unlike other classes of anti-HIV drugs, which act after infection occurs, HIV-1 entry inhibitors block the virus before it invades the target cells (4, 47). However, there are only two entry inhibitors currently being used in combination therapy for HIV-1 infection. While maraviroc targets the cell coreceptor CCR5, T20 remains the only inhibitor that arrests the viral fusion step. Disappointingly, the clinical use of T20 has been limited due to its low antiviral activity and drug resistance. Due to the lack of pocket-binding residues by T20, the 34-residue peptide C34 has been widely used as a template to develop novel HIV-1 fusion inhibitors with improved pharmaceutical profiles. Unsatisfactorily, the resulting peptides usually inherit a longer sequence that also targets the T20-resistant sites. The selection and characterization of HIV-1 escaping mutants have revealed the resistance pathways shared by the first and new fusion inhibitors, particularly with the amino acid Gly36-Leu45 stretch, or GIV motif, being a hot spot (14, 19–21). Therefore, a potent HIV-1 fusion inhibitor possessing a high genetic barrier to resistance is required to meet the challenge.
Discovery of the M-T hook structure provides a powerful strategy to develop short-peptide inhibitors that specifically target the gp41 pocket rather than the T20-resistant site. As demonstrated, the electronically constrained short-peptide SC22EK mainly targets the pocket site, but it has low binding and inhibitory activities; however, the M-T hook structure-modified MTSC22 and HP23 display strikingly improved pharmaceutical profiles, including their helical structure, binding affinity, antiviral activity, and genetic resistance barrier (34, 37). The genetic pathways of SC22EK-induced resistance were recently characterized, which revealed the E49K and N126K mutations (38). Indeed, the resistance mutations in the pocket-forming residues were rarely observed in previous studies, except that the Q66R substitution was occasionally selected by T2635 (21). Here, it was very surprising to find the L57R mutation at the left wall of pocket, which conferred high resistance to MTSC22 and cross-resistance to HP23 and SC22EK. Consistent with its high degree of conservation and poor tolerance to substitution, the L57R mutation severely impaired the ability of virus for cell entry. The combination of E49K and L57R resulted in dramatically increased resistance changes and almost abolished HIV-1 infectivity.
Besides the E49K and L57R mutations, MTSC22 also selected N126K and E136G mutations in the CHR of gp41. It is generally accepted that substitutions in the inhibitor-binding site of NHR helix are primary mutations that dominate resistance, whereas substitutions in the CHR helix serve as secondary mutations that may compensate the delayed fusion kinetics of virus caused by primary resistance substitutions (21, 38, 45, 46, 48). The N126K mutation, located near the pocket-binding domain (PBD) of CHR helix, has been frequently observed during escape selection against diverse fusion inhibitors (8, 14, 19-21, 49) and in prolonged T20-containing clinical therapy (9–12, 45, 50, 51). Interestingly, while N126K was previously shown to enhance many primary mutations, it offsets the resistance level by L57R. Another interesting finding was the E136G mutation at the middle site of the CHR helix, which mediated a broad cross-resistance with significantly greater changes than N126K did.
Previous characterization of SC22EK-induced mutations revealed multiple molecular mechanisms of resistance (38). First, a reduced inhibitor-binding affinity can play a central role in resistance. Indeed, the E49K and L57R mutations dramatically reduced the binding affinity of short-peptide inhibitors either singly or in combination. As analyzed in crystal 6-HB structure, the L57R mutation in the pocket can disrupt the hydrophobic interactions of N-terminal two M-T hook residues and three pocket-binding residues of inhibitors, whereas the E49K mutation on another NHR helix would introduce an electrostatic repulsion. Second, an increased stability of endogenous 6-HB would provide more energy to retard the binding of inhibitors. In the cases of SC22EK- and MTSC22-induced mutations, the E49K and N126K mutations can synergistically enhance the interaction of viral NHR and CHR helices, thus outcompeting inhibitors. Third, the resistance to peptide fusion inhibitors often results in a partial or dramatic loss of viral Env's function. Here, the acquisition of HIV-1 resistance to MTSC22 was accompanied by a severe impairment of virus entry and a substantial change of the 6-HB conformation. It should be mentioned that a compensatory mutation, N163D, might significantly improve the functionality of viral Env glycoprotein to mediate the fusion. We think that the binding affinity, the structural changes, and the viral fusion kinetics are highly coordinated for resistance. From an evolutionary perspective, the virus would like to find a balance between the resistance and viral fitness. Additionally, the underlying mechanisms for several observed phenomena need to be addressed. First, how does the L57R mutation confer an extremely high resistance to short-peptide inhibitor targeting the pocket while increasing the sensitivity of T20, C34, and SFT, which lacks or contain the pocket-binding sequence? Second, how do the N126K and E136G mutations, which are located outside the binding surface, mediate cross-resistance to diverse peptides? Third, how does the N126K mutation boost the E49K-mediated resistance while counteracting L57R-mediated resistance? Fourth, while the E49K and N126K mutations enhance the stability of viral 6-HB, why do the L57R and E136G mutations act conversely? Fifth, what is the molecular pathway of N163D-mediated enhancement for HIV-1 entry? Therefore, it is critical to explore the interactions and compensatory effects between these primary and secondary mutations.
In summary, we have successfully selected and characterized HIV-1 variants that are highly resistant to MTSC22, a novel short-peptide fusion inhibitor containing the M-T hook structure. Two primary mutations, E49K and L57R, located at the inhibitor-binding site of gp41 NHR, and two secondary mutations, N126K and E136G, located at the gp41 CHR, were identified as conferring resistance either singly or in combination. We have demonstrated the effects of MTSC22-induced mutations on the binding affinity of inhibitors that specifically target the gp41 pocket, the Env-mediated virus entry, the conformation and stability of 6-HB structure, as well as on the antiviral activity of NHR- and CHR-derived model peptides. The present study provides new insights into the molecular pathways and mechanisms of HIV-1 resistance to diverse viral fusion inhibitors and will help further our understanding of the structure-function relationship of gp41 and the structure-activity relationship (SAR) of novel HIV-1 entry inhibitor peptides targeting gp41.
ACKNOWLEDGMENTS
We thank Zene Matsuda at the Institute of Medical Science of the University of Tokyo (Tokyo, Japan) for providing the DSP plasmids, Yinghua Chen at the College of Life Science of Tsinghua University (Beijing, China) for providing the 6-HB-specific MAbs 17C8 and 2G8, and Shibo Jiang at the Lindsley F. Kimball Research Institute of New York Blood Center (New York, NY, USA) for providing the 6-HB-specific MAb NC-1.
This work was supported by grants from the Natural Science Foundation of China (81473255, 81271830), National Science and Technology Major Project of China (2014ZX10001001, 2013ZX10004-601), and National 973 program of China (2014CB542502).
REFERENCES
- 1.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/S0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 2.Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 4.Eggink D, Berkhout B, Sanders RW. 2010. Inhibition of HIV-1 by fusion inhibitors. Curr Pharm Des 16:3716–3728. doi: 10.2174/138161210794079218. [DOI] [PubMed] [Google Scholar]
- 5.He Y. 2013. Synthesized peptide inhibitors of HIV-1 gp41-dependent membrane fusion. Curr Pharm Des 19:1800–1809. doi: 10.2174/1381612811319100004. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin CE, Sanders RW, Deng Y, Jurriaans S, Lange JM, Lu M, Berkhout B. 2004. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the T20 fusion inhibitor. J Virol 78:12428–12437. doi: 10.1128/JVI.78.22.12428-12437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg ML, Cammack N. 2004. Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother 54:333–340. doi: 10.1093/jac/dkh330. [DOI] [PubMed] [Google Scholar]
- 8.Nameki D, Kodama E, Ikeuchi M, Mabuchi N, Otaka A, Tamamura H, Ohno M, Fujii N, Matsuoka M. 2005. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and Rev-responsive element functions. J Virol 79:764–770. doi: 10.1128/JVI.79.2.764-770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu L, Pozniak A, Wildfire A, Stanfield-Oakley SA, Mosier SM, Ratcliffe D, Workman J, Joall A, Myers R, Smit E, Cane PA, Greenberg ML, Pillay D. 2005. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob Agents Chemother 49:1113–1119. doi: 10.1128/AAC.49.3.1113-1119.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cabrera C, Marfil S, Garcia E, Martinez-Picado J, Bonjoch A, Bofill M, Moreno S, Ribera E, Domingo P, Clotet B, Ruiz L. 2006. Genetic evolution of gp41 reveals a highly exclusive relationship between codons 36, 38 and 43 in gp41 under long-term enfuvirtide-containing salvage regimen. AIDS 20:2075–2080. doi: 10.1097/QAD.0b013e3280102377. [DOI] [PubMed] [Google Scholar]
- 11.Ray N, Harrison JE, Blackburn LA, Martin JN, Deeks SG, Doms RW. 2007. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J Virol 81:3240–3250. doi: 10.1128/JVI.02413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svicher V, Aquaro S, D'Arrigo R, Artese A, Dimonte S, Alcaro S, Santoro MM, Di Perri G, Caputo SL, Bellagamba R, Zaccarelli M, Visco-Comandini U, Antinori A, Narciso P, Ceccherini-Silberstein F, Perno CF. 2008. Specific enfuvirtide-associated mutational pathways in HIV-1 Gp41 are significantly correlated with an increase in CD4(+) cell count, despite virological failure. J Infect Dis 197:1408–1418. doi: 10.1086/587693. [DOI] [PubMed] [Google Scholar]
- 13.Ashkenazi A, Wexler-Cohen Y, Shai Y. 2011. Multifaceted action of Fuzeon as virus-cell membrane fusion inhibitor. Biochim Biophys Acta 1808:2352–2358. doi: 10.1016/j.bbamem.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 14.Eggink D, Baldwin CE, Deng Y, Langedijk JP, Lu M, Sanders RW, Berkhout B. 2008. Selection of T1249-resistant human immunodeficiency virus type 1 variants. J Virol 82:6678–6688. doi: 10.1128/JVI.00352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chinnadurai R, Rajan D, Munch J, Kirchhoff F. 2007. Human immunodeficiency virus type 1 variants resistant to first- and second-version fusion inhibitors and cytopathic in ex vivo human lymphoid tissue. J Virol 81:6563–6572. doi: 10.1128/JVI.02546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y, Xiao Y, Song H, Liang Q, Ju D, Chen X, Lu H, Jing W, Jiang S, Zhang L. 2008. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 283:11126–11134. doi: 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- 17.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. 2007. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci U S A 104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otaka A, Nakamura M, Nameki D, Kodama E, Uchiyama S, Nakamura S, Nakano H, Tamamura H, Kobayashi Y, Matsuoka M, Fujii N. 2002. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew Chem Int Ed Engl 41:2937–2940. doi:. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Shan M, Li L, Lu L, Meng S, Chen C, He Y, Jiang S, Zhang L. 2011. In vitro selection and characterization of HIV-1 variants with increased resistance to sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 286:3277–3287. doi: 10.1074/jbc.M110.199323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimura K, Nameki D, Kajiwara K, Watanabe K, Sakagami Y, Oishi S, Fujii N, Matsuoka M, Sarafianos SG, Kodama EN. 2010. Resistance profiles of novel electrostatically constrained HIV-1 fusion inhibitors. J Biol Chem 285:39471–39480. doi: 10.1074/jbc.M110.145789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eggink D, Bontjer I, Langedijk JP, Berkhout B, Sanders RW. 2011. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J Virol 85:10785–10797. doi: 10.1128/JVI.05331-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eggink D, Langedijk JP, Bonvin AM, Deng Y, Lu M, Berkhout B, Sanders RW. 2009. Detailed mechanistic insights into HIV-1 sensitivity to three generations of fusion inhibitors. J Biol Chem 284:26941–26950. doi: 10.1074/jbc.M109.004416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan DC, Chutkowski CT, Kim PS. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A 95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan DC, Kim PS. 1998. HIV entry and its inhibition. Cell 93:681–684. doi: 10.1016/S0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 25.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. 1999. Inhibiting HIV-1 entry: discovery of d-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103–115. doi: 10.1016/S0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 26.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. 2007. Potent d-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci U S A 104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steffen I, Pohlmann S. 2010. Peptide-based inhibitors of the HIV envelope protein and other class I viral fusion proteins. Curr Pharm Des 16:1143–1158. doi: 10.2174/138161210790963751. [DOI] [PubMed] [Google Scholar]
- 28.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. 2010. Design of a potent d-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol 84:11235–11244. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu F, Lu L, Liu Q, Yu X, Wang L, He E, Zou P, Du L, Sanders RW, Liu S, Jiang S. 2014. ADS-J1 inhibits HIV-1 infection and membrane fusion by targeting the highly conserved pocket in the gp41 NHR-trimer. Biochim Biophys Acta 1838:1296–1305. doi: 10.1016/j.bbamem.2013.12.022. [DOI] [PubMed] [Google Scholar]
- 30.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. 2004. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob Agents Chemother 48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chong H, Yao X, Qiu Z, Qin B, Han R, Waltersperger S, Wang M, Cui S, He Y. 2012. Discovery of critical residues for viral entry and inhibition through structural insight of HIV-1 fusion inhibitor CP621-652. J Biol Chem 287:20281–20289. doi: 10.1074/jbc.M112.354126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chong H, Yao X, Sun J, Qiu Z, Zhang M, Waltersperger S, Wang M, Cui S, He Y. 2012. The M-T hook structure is critical for design of HIV-1 fusion inhibitors. J Biol Chem 287:34558–34568. doi: 10.1074/jbc.M112.390393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Y, Cheng J, Li J, Qi Z, Lu H, Dong M, Jiang S, Dai Q. 2008. Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure: implications for designing novel anti-HIV fusion inhibitors. J Virol 82:6349–6358. doi: 10.1128/JVI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chong H, Yao X, Qiu Z, Sun J, Zhang M, Waltersperger S, Wang M, Liu SL, Cui S, He Y. 2013. Short-peptide fusion inhibitors with high potency against wild-type and enfuvirtide-resistant HIV-1. FASEB J 27:1203–1213. doi: 10.1096/fj.12-222547. [DOI] [PubMed] [Google Scholar]
- 35.Chong H, Qiu Z, Sun J, Qiao Y, Li X, He Y. 2014. Two M-T hook residues greatly improve the antiviral activity and resistance profile of the HIV-1 fusion inhibitor SC29EK. Retrovirology 11:40. doi: 10.1186/1742-4690-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chong H, Yao X, Qiu Z, Sun J, Qiao Y, Zhang M, Wang M, Cui S, He Y. 2014. The M-T hook structure increases the potency of HIV-1 fusion inhibitor sifuvirtide and overcomes drug resistance. J Antimicrob Chemother 69:6759. [DOI] [PubMed] [Google Scholar]
- 37.Chong H, Qiu Z, Su Y, Yang L, He Y. 2015. Design of a highly potent HIV-1 fusion inhibitor targeting the gp41 pocket. AIDS 29:13–21. doi: 10.1097/QAD.0000000000000498. [DOI] [PubMed] [Google Scholar]
- 38.Su Y, Chong H, Qiu Z, Xiong S, He Y. 2015. Mechanism of HIV-1 resistance to short-peptide fusion inhibitors targeting the Gp41 pocket. J Virol 89:5801–5811. doi: 10.1128/JVI.00373-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He Y, Liu S, Li J, Lu H, Qi Z, Liu Z, Debnath AK, Jiang S. 2008. Conserved salt bridge between the N- and C-terminal heptad repeat regions of the human immunodeficiency virus type 1 gp41 core structure is critical for virus entry and inhibition. J Virol 82:11129–11139. doi: 10.1128/JVI.01060-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondo N, Miyauchi K, Meng F, Iwamoto A, Matsuda Z. 2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem 285:14681–14688. doi: 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 42.Jiang S, Lin K, Lu M. 1998. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J Virol 72:10213–10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Chen X, Huang J, Jiang S, Chen YH. 2009. Identification of critical antibody-binding sites in the HIV-1 gp41 six-helix bundle core as potential targets for HIV-1 fusion inhibitors. Immunobiology 214:51–60. doi: 10.1016/j.imbio.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 44.He Y, Cheng J, Lu H, Li J, Hu J, Qi Z, Liu Z, Jiang S, Dai Q. 2008. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc Natl Acad Sci U S A 105:16332–16337. doi: 10.1073/pnas.0807335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ray N, Blackburn LA, Doms RW. 2009. HR-2 mutations in human immunodeficiency virus type 1 gp41 restore fusion kinetics delayed by HR-1 mutations that cause clinical resistance to enfuvirtide. J Virol 83:2989–2995. doi: 10.1128/JVI.02496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sivaraman V, Zhang L, Meissner EG, Jeffrey JL, Su L. 2009. The heptad repeat 2 domain is a major determinant for enhanced human immunodeficiency virus type 1 (HIV-1) fusion and pathogenicity of a highly pathogenic HIV-1 Env. J Virol 83:11715–11725. doi: 10.1128/JVI.00649-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lobritz MA, Ratcliff AN, Arts EJ. 2010. HIV-1 entry, inhibitors, and resistance. Viruses 2:1069–1105. doi: 10.3390/v2051069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Feo CJ, Wang W, Hsieh ML, Zhuang M, Vassell R, Weiss CD. 2014. Resistance to N-peptide fusion inhibitors correlates with thermodynamic stability of the gp41 six-helix bundle but not HIV entry kinetics. Retrovirology 11:86. doi: 10.1186/s12977-014-0086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lohrengel S, Hermann F, Hagmann I, Oberwinkler H, Scrivano L, Hoffmann C, von Laer D, Dittmar MT. 2005. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J Virol 79:10237–10246. doi: 10.1128/JVI.79.16.10237-10246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loutfy MR, Raboud JM, Montaner JS, Antoniou T, Wynhoven B, Smaill F, Rouleau D, Gill J, Schlech W, Brumme ZL, Mo T, Gough K, Rachlis A, Harrigan PR, Walmsley SL. 2007. Assay of HIV gp41 amino acid sequence to identify baseline variation and mutation development in patients with virologic failure on enfuvirtide. Antiviral Res 75:58–63. doi: 10.1016/j.antiviral.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 51.Poveda E, Rodes B, Labernardiere JL, Benito JM, Toro C, Gonzalez-Lahoz J, Faudon JL, Clavel F, Schapiro J, Soriano V. 2004. Evolution of genotypic and phenotypic resistance to Enfuvirtide in HIV-infected patients experiencing prolonged virologic failure. J Med Virol 74:21–28. doi: 10.1002/jmv.20141. [DOI] [PubMed] [Google Scholar]