

Abstract
The von Willebrand factor (vWF) is a plasma glycoprotein that plays an essential role in hemostasis by supporting platelet adhesion and thrombus formation in response to vascular injury. Plasma levels of vWF are an independent risk factor for patients with acute myocardial infarction (AMI); however, clinical data have demonstrated a marked variation of vWF levels in patients with AMI, the reason for which has not yet been identified. In the present study, a rat model of ST-segment elevation AMI was established, and cardiac and peripheral blood was collected for a time-course examination of the plasma levels of vWF and tumor necrosis factor-α (TNF-α). The level of vWF in the blood plasma increased, peaked at 1 h and decreased to normal levels by day 7 following AMI, while the level of TNF-α peaked at 24 h and remained elevated until day 7. The effects of TNF-α on vWF secretion and expression were examined in cultured human umbilical vascular endothelial cells (HUVECs). TNF-α treatment increased vWF secretion from the HUVECs but inhibited the mRNA and protein expression of vWF in the HUVECs. These results indicate that vWF secretion from endothelial cells is transiently elevated following AMI, and then decreases as the expression of vWF is inhibited by TNF-α. The present study increases the understanding of the pathophysiology of vWF and indicates that the determination of vWF levels may be useful in the clinical evaluation of AMI.
Keywords: von Willebrand factor, acute myocardial infarction, secretion, cardiac blood, TNF-α
Introduction
The von Willebrand factor (vWF) is a large multimeric plasma glycoprotein produced by endothelial cells and megakaryocytes (1). vWF can be secreted through a constitutive pathway following synthesis, or by a regulated pathway involving storage and release by secretagogues (2,3). Although platelets secrete vWF, plasma vWF levels have been shown to depend almost entirely on vWF from endothelial cells (4). A large amount of vWF is stored in Weibel-Palade bodies in endothelial cells and released towards the lumen of blood vessels in response to various stimuli (5,6). In blood circulation, vWF is well known for its role in homeostasis, where it binds to platelet receptor glycoprotein Ib and to the constituents of the sub-endothelial connective tissue (4,7). vWF also binds to blood coagulation factor VIII, another blood clotting protein, and acts as its carrier in the circulation (8). In addition to homeostasis, vWF has recently been recognized as a critical regulator in angiogenesis, inflammation and cell proliferation (9–11).
The vWF gene is located on chromosome 12p and comprises 52 exons spanning ~178 kb of genomic sequence (12). Mutations in the vWF gene are responsible for von Willebrand disease, a bleeding disorder that prolongs the blood clotting process (13). The vWF gene is also a marker of endothelial cell heterogeneity as demonstrated by the existence of regional variations in vWF protein and mRNA levels within the vascular tree (14). The vascular-bed specific expression of vWF is regulated by a group of distinct signaling pathways, each communicating with different regions of the promoter (15,16). In pathological conditions such as sepsis, diabetes and cancer development, vWF expression in endothelial cells is regulated by specific groups of transcription factors (17–21).
The plasma levels of vWF have been found to be associated with cardiovascular diseases, particularly myocardial infarction (MI) (22,23). MI is a common cause of mortality; as of 2008, >3,000,000 individuals exhibited ST elevation MIs and 4,000,000 non-ST elevation MIs every year worldwide (24). As a result of inadequate coronary artery blood flow, MI triggers ischemic responses, which include myocyte death, endothelial cell dysfunction and abnormal tissue repair with fibrosis (25). To date, no clinical marker is available for the accurate assessment of this process (26). Plasma vWF concentration has been shown to be increased in patients with acute MI (AMI) (27,28). However, other studies have reported that no difference in the vWF levels was identified between patients with MI and the controls (29,30). It is therefore necessary to accurately assess the plasma vWF level during MI.
In the present study, a rat model of ST-elevation AMI was established, and the plasma vWF level in the cardiac and peripheral blood was examined at different time-points. The plasma level of TNF-α, an inflammatory cytokine, was also examined and the effects of TNF-α on vWF secretion and expression in cultured endothelial cells were investigated. The data obtained may be important for evaluating the association between plasma vWF and AMI progression.
Materials and methods
Animal model
Fifty-seven male Wistar rats (age, 8 weeks; weight, 280–300 g) were obtained from the Animal Center of Shandong University (Jinan, China) and randomized into four groups: Sham-operated and MI 1-h, 24-h and 7-day groups. Each group contained 12–15 rats. Following anesthesia with 3% sodium pentobarbital (30 mg/kg intraperitoneally; Sigma-Aldrich, St. Louis, MO, USA), the rats were intubated via tracheotomy and ventilated with a small-animal ventilator (HX-300S; Chengdu TME Technology Co., Chengdu, China). The heart was exposed through left fourth intercostal lateral thoracotomy, and the AMI model was induced by permanently ligating the left anterior descending artery ~2 mm from its origin as previously described (31,32). At each time-point, ~5-µl blood samples were collected from the coronary sinus and inferior vena cava using a 30-gauge needle. Electrodes were attached to the four limbs for electrocardiography, and successful creation of AMI was verified by ST-segment elevation shown by electrocardiography. The rats of the sham-operated group underwent thoracotomy and pericardiotomy without coronary artery ligation. The study was approved by the Animal Care and Use Committee of Shandong University and all procedures involving animals were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (publication no. 85–23, revised 1996).
Masson's trichrome staining
Masson's trichrome staining was performed as previously described (33). Briefly, the rats of the MI 7-day group were decapitated following blood collection. Subsequently, their hearts were removed, washed and fixed in Bouin's solution [Tiangen Biotech (Beijing) Co., Ltd.,China] for 2 h. They were then embedded in optimum cutting temperature compound (Tissue-Tek; Miles Laboratories, Inc., Elkhart, IN, USA) by dry ice-ethanol bath. The frozen-embedded rat hearts were transversely sectioned (6 µm) and stained with Weigert's iron hematoxylin (Shanghai Jinpan Biotech Co., Ltd., Shanghai, China) for 10 min, then washed and stained in 1% ponceau-acetic acid solution (mixture of equal volumes of 0.5% ponceau 2R in 1% acetic acid and 0.5% acid fuchsin in 1% acetic acid) for 5 min. Following washing with phosphate-buffered saline (PBS), the sections were incubated with 1% phosphomolybdic acid for 5 min and counterstained light green. The images were captured under an Olympus FSX200 microscope (Olympus Corporation, Tokyo, Japan).
ELISA
The rat blood samples were centrifuged at 1,075 × g for 2 min at 4°C, and the supernatant (blood plasma) was collected. Then, the plasma samples were diluted 1:20 with PBS for the detection of vWF and TNF-α protein levels using a vWF ELISA kit (Ramco Laboratories Inc., Stafford, TX, USA) and a TNF-α Rat ELISA kit (Abcam, Cambridge, MA, USA), respectively. For cell culture, HUVECs were grown to confluence and serum/growth factor-free media containing 0, 10 or 50 ng/ml TNF-α (Miltenyi Biotec, Bergisch Gladbach, Germany) was applied. At each time-point, the media were collected for detection of the vWF protein level using the vWF ELISA kit. The measurements were performed 4 times (n=4).
Cell culture
Human umbilical vascular endothelial cells (HUVECs) were purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained in endothelial growth media (EGM-2) supplemented with EGM™-2-MV bullet kit (Lonza, Basel, Switzerland) and containing antibiotics (100 IU/ml penicillin and 100 µg/ml streptomycin), in humidified air at 37°C with 5% CO2.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from HUVECs treated with 10 ng/ml TNF-α at 37°C at each time-point (0, 0.5, 1, 2, 6, 12 and 24 h) using the RNeasy Mini kit (Qiagen, Hilden, Germany). cDNA was synthesized using the High Capacity RNA-to-cDNA Master Mix (Applied Biosystems-Life Technologies, Carlsbad, CA, USA). RT-qPCR was performed using SYBR Green master mixes (Life Technologies) with a ViiA7 Real-Time PCR system (Life Technologies). All PCR reactions were repeated in triplicate. The relative expression of vWF was calculated using GAPDH as an endogenous internal control. The primer sequences are listed in Table I.
Table I.
Reverse transcription-quantitative polymerase chain reaction primer sequences.
| Gene | Sense/antisense | Primer sequence | Size (bp) | Tm (°C) |
|---|---|---|---|---|
| vWF | Sense | CGGCTTGCACCATTCAGCTA | 90 | 61.5 |
| Antisense | TGCAGAAGTGAGTATCACAGCCATC | |||
| GAPDH | Sense | TGATGACATCAAGAAGGTGGTGAAG | 240 | 57.9 |
| Antisense | TCCTTGGAGGCCATGTGGGCCAT |
All sequences are written in the 5′ to 3′ orientation. Tm, melting temperature.
Immunofluorescence
HUVEC monolayers grown on fibronectin-coated glass chamber slides were exposed to 0 or 10 ng/ml TNF-α. After 24 h, the media was aspirated and the monolayers were washed with PBS containing 100 mM L-glycine, fixed with 4% paraformaldehyde, and re-washed with PBS. Immunofluorescence assay was performed using a primary polyclonal rabbit anti-human vWF antibody (cat. no., A008229; dilution, 1:200; Dako, Glostrup, Denmark) and an Alexa Fluor® 546 polyclonal anti-rabbit secondary antibody (cat. no., A-11035; dilution,1:200; Life Technologies). The cells were incubated with the primary antibody overnight at 4°C, washed with PBS and then incubated with the secondary antibody for 1 h at room temperature. Photographic images of the slides were captured using an Olympus FSX200 microscope (Olympus Corporation) with an excitation wavelength of 546 nm.
Statistical analysis
Data are expressed as the mean ± standard error. An unpaired, two-tailed Student's t-test was used for the comparison between means. The SPSS 17.0 software for Windows (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
Model establishment
The establishment of the AMI model was confirmed by the significant ST-segment elevation shown on electrocardiography (Fig. 1A). Following blood sample collection at each time-point, the rats were sacrificed and the hearts were extracted to verify the effects of AMI. The Masson's trichrome staining results of the sections of the cardiac tissues from the rats of the AMI model after 7 days are shown in Fig. 1. In these sections, the normal myocardium was stained red, while the region of fibrosis that caused infarction damage was stained blue. In the sham-operated control group, only the perivascular area was stained light blue (Fig. 1B). By contrast, the infarcted zone of the AMI group contained a large quantity of blue fibrotic tissues (Fig. 1B), suggesting that the AMI model has been successfully developed in the Wistar rats.
Figure 1.

Establishment of an AMI model in male Wistar rats. (A) Representative electrocardiography of the rats from the sham-operated and MI groups. Rats with MI showed a clear ST-segment elevation when compared with the sham-operated group. (B) Representative image of sections of rat hearts from the sham-operated and MI (7-day) groups stained with Massion's trichrome. AMI, acute myocardial infarction; MI, myocardial infarction.
vWF levels in blood plasma
The concentrations of vWF in blood plasma were detected using ELISA. As shown in Fig. 2, the level of vWF in the cardiac blood collected from the coronary sinus underwent a 1.31-fold increase (P<0.01) at 1 h and a 0.88-fold increase (P<0.05) at 24 h following AMI, but decreased to normal levels by day 7. The level of vWF in the peripheral blood collected from the inferior vena cava underwent a 0.37-fold increase at 1 h (P<0.05) and a 0.18-fold increase at 24 h (P<0.05), but decreased to normal levels at day 7. Thus, following AMI, the concentrations of vWF in the cardiac and peripheral blood increased, peaked at 1 h and then gradually decreased to normal levels. At the peak level, the increase of vWF in the peripheral blood (0.37-fold) was considerably smaller than that in the cardiac blood (1.31-fold), suggesting that vWF was secreted from the heart following MI and diluted through the circulatory system.
Figure 2.
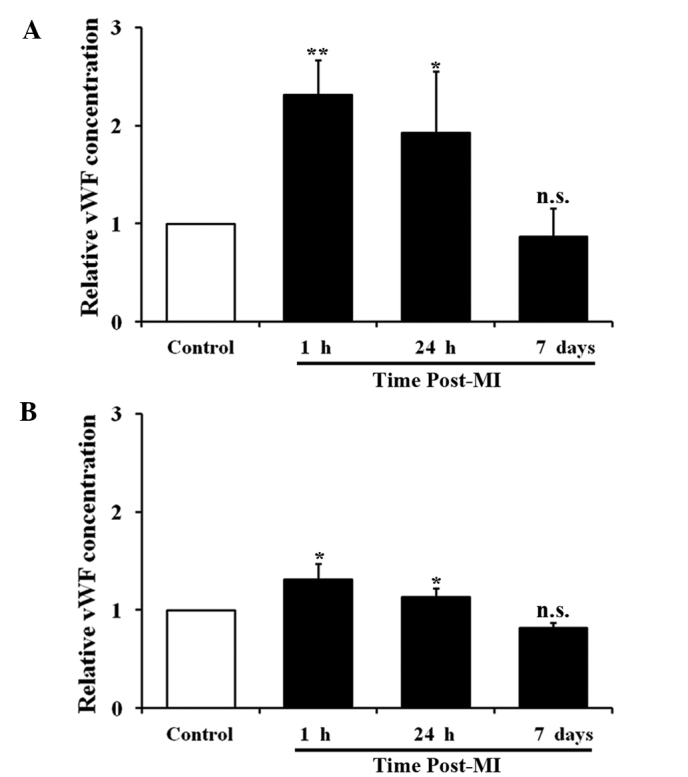
vWF concentration in cardiac and peripheral blood from male Wistar rats with AMI. Time-course measurement of vWF concentration in (A) cardiac blood and (B) peripheral blood of rats with AMI (n=12 for each assay; *P<0.05 and **P<0.01 compared with the control group). vWF, von Willebrand factor; AMI, acute myocardial infarction; MI, myocardial infarction; n.s., not significant.
TNF-α levels in blood plasma
TNF-α, a pro-inflammatory cytokine, regulates vWF expression in the vasculature (34). The concentration of TNF-α in the blood plasma collected from the rat model of AMI was examined. The cardiac plasma level of TNF-α was increased at 1 h (0.89-fold; P<0.05), 24 h (1.32-fold; P<0.05) and 7 days (1.23-fold; P<0.05) after AMI. The peripheral plasma level of TNF-α was also increased at all time-points as follows: 0.53-fold increase at 1 h (P<0.05), 0.71-fold increase at 24 h (P<0.05) and 0.72-fold increase at 7 days (P<0.01). During AMI, TNF-α was also produced in the heart and diluted through the circulatory system. Unlike vWF, the level of TNF-α peaked at 24 h, and a high level was maintained until day 7 after AMI (Fig. 3).
Figure 3.
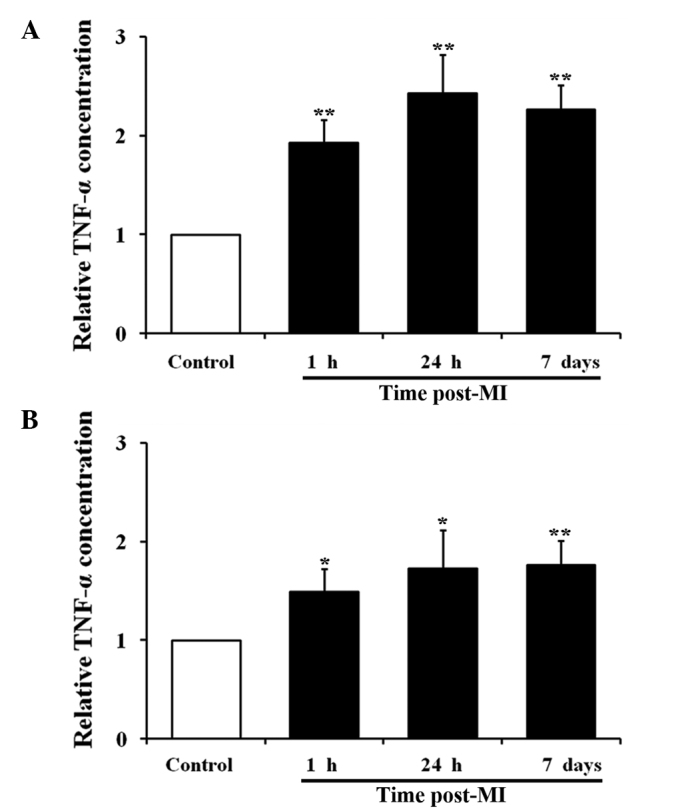
TNF-α concentration in cardiac and peripheral blood from male Wistar rats with AMI. Time-course measurement of TNF-α concentration in (A) cardiac blood and (B) peripheral blood of rats with AMI (n=12 for each assay; *P<0.05 and **P<0.01 compared with the control group). TNF-α, tumor necrosis factor-α; AMI, acute myocardial infarction; MI, myocardial infarction.
Effect of TNF-α on HUVECs
TNF-α was added to the medium of cultured HUVECs and the vWF secretion in vitro was examined. Following treatment with 10 or 50 ng/ml TNF-α, the level of vWF secreted into the medium was significantly increased at all time-points (Fig. 4), suggesting that TNF-α stimulates vWF secretion. In addition, vWF expression in the HUVECs treated with 10 ng/ml TNF-α for 24 h was examined. RT-qPCR analysis indicated that the mRNA level of vWF was significantly decreased at 2 h and continued to decrease until 24 h after treatment (Fig. 5A). An immunofluorescence assay using a rabbit anti-human vWF antibody was performed on the HUVEC monolayer. As shown on Fig. 5B, the vWF protein was abundant on the membranes of the untreated cells but its expression on the cells following a 24-h treatment with 10 ng/ml TNF-α was considerably reduced. Therefore, TNF-α treatment increased the secretion of vWF by HUVECs, but decreased the expression of vWF in the HUVECs.
Figure 4.
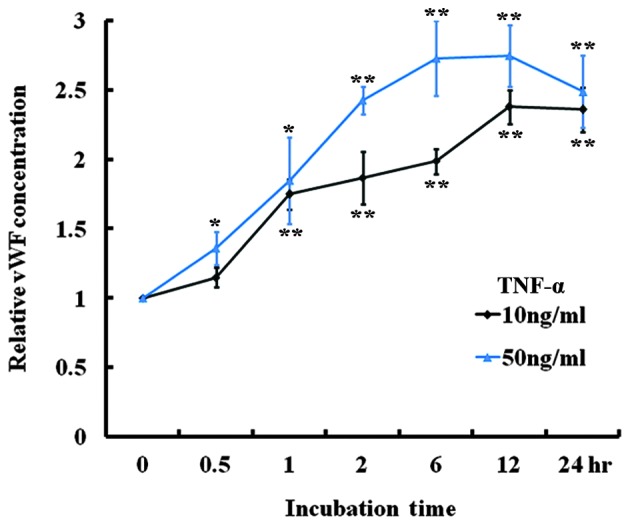
TNF-α induced vWF secretion in HUVECs. vWF ELISA of the culture media of HUVECs treated with 10 and 50 ng/ml TNF-α (n=4; *P<0.05 and **P<0.01 compared with baseline). TNF-α, tumor necrosis factor-α; vWF, von Willebrand factor; HUVEC, human umbilical vascular endothelial cell.
Figure 5.

TNF-α induced downregulation of vWF expression in HUVECs. (A) Reverse transcription-quantitative polymerase chain reaction analysis of vWF mRNA expression in HUVECs treated with 10 ng/ml TNF-α (n=4; *P<0.05 and **P<0.01 compared with baseline). (B) Immunofluorescence of vWF in HUVECs treated with 0 and 10 ng/ml TNF-α for 24 h (magnification, ×100). vWF, von Willebrand factor; HUVECs, human umbilical vascular endothelial cells; TNF-α, tumor necrosis factor-α.
Discussion
In the present study, a rat model of AMI was successfully developed, which was validated by the ST-segment elevation shown on electrocardiography and the results of Masson's twrichrome staining on the cardiac tissues. The plasma level of vWF, as determined by ELISA, was transiently increased following AMI, and then decreased in the cardiac and peripheral blood, while the level of TNF-α continuously increased until it peaked at 24 h and remained elevated for 7 days. The in vitro experiments indicated that TNF-α stimulated vWF secretion but inhibited the expression of vWF in cultured endothelial cells. The present study, therefore, partially elucidated the cause of the variation in human plasma vWF levels during AMI and the underlying mechanisms.
The plasma level of vWF is a potential clinical marker for AMI and may be a risk factor for recurrent myocardial infarction (35). A number of studies have investigated the associations between vWF plasma levels and AMI events (22,23); however the results are inconsistent and the conclusions remain controversial. The present study determined the plasma level of vWF in a rat AMI model, and indicated that vWF is transiently increased following AMI in the cardiac and peripheral blood. This time-course is consistent with the results of a clinical study that recorded the progression of AMI precisely, and reported that the vWF concentration was significantly increased at the onset stage of AMI and normalized 14 days following AMI (36). The present findings suggest that a higher plasma level of vWF is associated with the early stage but not the later stage of AMI. In addition, the discrepancies between this and previous studies are, at least partially, due to the different time-points of blood collection from patients.
Inflammation plays an important role in AMI, and TNF-α is the central regulator of inflammation (37,38). At low levels, TNF-α exhibits a cardioprotective effect, whereas high levels of TNF-α induce myocardial damage (39,40). TNF-α is produced primarily by activated macrophages and expressed by multiple cell types in the heart (41,42). During AMI, the expression of TNF-α is increased in cardiac tissues (43). In the present study, it was found that the plasma level of TNF-α continuously increased during the first 24 h and was maintained at a high level for the 7-day period following AMI, suggesting that circulating TNF-α could be associated with AMI. These results are consistent with the findings of clinical studies, which support that observation that the TNF-α level is higher in patients with AMI (25,44). The increase in the level of TNF-α may be caused by both the ischemia-induced damage of myocardial tissues and the late consequences of tissue repair (40,45).
TNF-α in blood plasma interacts with its receptors on the membrane of endothelial cells, triggers a collection of signaling pathways and regulates various cellular processes (46,47). In healthy humans, the intravenous injection of recombinant TNF-α has been found to stimulate the release of vWF from storage into the circulation, which is one of the consequences of TNF-α-induced endothelial cell activation (48). Through in vitro experiments in the present study, it was also observed that TNF-α-induced an increase in vWF secretion; however, TNF-α was shown to downregulate the mRNA and protein expression of vWF (Fig. 6). It has previously been discovered that transcription factor GATA3 and ETS transcription factor ERG are positive regulators of vWF expression (12,34). TNF-α inhibits the expression of these transcription factors in endothelial cells (18,49), thus repressing the expression of vWF. As the expression of vWF is decreased, the TNF-α-induced secretion of vWF reaches a peak shortly afterwards and then decreases.
Figure 6.

Schematic illustration of TNF-α regulated vWF secretion and expression. TNF-α, tumor necrosis factor-α; TNFR1, tumor necrosis factor receptor 1; von Willebrand factor.
In conclusion, the present study found that the plasma level of vWF is transiently elevated in a rat model of AMI, which may result from a TNF-α-induced increase in vWF secretion and reduction in vWF expression. The present results suggest that plasma level of vWF may be a clinical marker of the early stage of AMI. Future studies are required in order to elucidate the role of vWF in the progression of AMI and the underlying mechanism.
Acknowledgements
This study was supported by a grant from the National Natural Science Foundation of China (grant no. 81370269). The authors are grateful for the funding received from the Shandong Taishan Scholarship.
References
- 1.Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. 2003;1:1335–1342. doi: 10.1046/j.1538-7836.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 2.Lenting PJ, Christophe OD, Denis CV. von Willebrand factor biosynthesis, secretion and clearance: Connecting the far ends. Blood. 2015;125:2019–2028. doi: 10.1182/blood-2014-06-528406. [DOI] [PubMed] [Google Scholar]
- 3.Sadler JE. von Willebrand factor assembly and secretion. J Thromb Haemost. 2009;7(Suppl 1):S24–S27. doi: 10.1111/j.1538-7836.2009.03375.x. [DOI] [PubMed] [Google Scholar]
- 4.Kanaji S, Fahs SA, Shi Q, Haberichter SL, Montgomery RR. Contribution of platelet vs. endothelial VWF to platelet adhesion and hemostasis. J Thromb Haemost. 2012;10:1646–1652. doi: 10.1111/j.1538-7836.2012.04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metcalf DJ, Nightingale TD, Zenner HL, Lui-Roberts WW, Cutler DF. Formation and function of Weibel-Palade bodies. J Cell Sci. 2008;121:19–27. doi: 10.1242/jcs.03494. [DOI] [PubMed] [Google Scholar]
- 6.Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J Thromb Haemost. 2013;11(Suppl 1):S192–S201. doi: 10.1111/jth.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yee A, Kretz CA. Von Willebrand factor: Form for function. Semin Thromb Hemost. 2014;40:17–27. doi: 10.1055/s-0033-1363155. [DOI] [PubMed] [Google Scholar]
- 8.Lenting PJ, Pegon JN, Groot E, de Groot PG. Regulation of von Willebrand factor-platelet interactions. Thromb Haemost. 2010;104:449–455. doi: 10.1160/TH09-11-0777. [DOI] [PubMed] [Google Scholar]
- 9.Franchini M, Frattini F, Crestani S, Bonfanti C, Lippi G. von Willebrand factor and cancer: A renewed interest. Thromb Res. 2013;131:290–292. doi: 10.1016/j.thromres.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Lenting PJ, Casari C, Christophe OD, Denis CV. von Willebrand factor: The old, the new and the unknown. J Thromb Haemost. 2012;10:2428–2437. doi: 10.1111/jth.12008. [DOI] [PubMed] [Google Scholar]
- 11.Randi AM, Laffan MA, Starke RD. Von Willebrand factor, angiodysplasia and angiogenesis. Mediterr J Hematol Infect Dis. 2013;5:e2013060. doi: 10.4084/mjhid.2013.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jahroudi N, Lynch DC. Endothelial-cell-specific regulation of von Willebrand factor gene expression. Mol Cell Biol. 1994;14:999–1008. doi: 10.1128/mcb.14.2.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lillicrap D. von Willebrand disease: Advances in pathogenetic understanding, diagnosis and therapy. Blood. 2013;122:3735–3740. doi: 10.1182/blood-2013-06-498303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto K, de Waard V, Fearns C, Loskutoff DJ. Tissue distribution and regulation of murine von Willebrand factor gene expression in vivo. Blood. 1998;92:2791–2801. [PubMed] [Google Scholar]
- 15.Aird WC, Jahroudi N, Weiler-Guettler H, Rayburn HB, Rosenberg RD. Human von Willebrand factor gene sequences target expression to a subpopulation of endothelial cells in transgenic mice. Proc Natl Acad Sci USA. 1995;92:4567–4571. doi: 10.1073/pnas.92.10.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aird WC, Edelberg JM, Weiler-Guettler H, Simmons WW, Smith TW, Rosenberg RD. Vascular bed-specific expression of an endothelial cell gene is programmed by the tissue microenvironment. J Cell Biol. 1997;138:1117–1124. doi: 10.1083/jcb.138.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertagna A, Jahroudi N. The NFY transcription factor mediates induction of the von Willebrand factor promoter by irradiation. Thromb Haemost. 2001;85:837–844. [PubMed] [Google Scholar]
- 18.Liu J, Kanki Y, Okada Y, Jin E, Yano K, Shih SC, Minami T, Aird WC. A +220 GATA motif mediates basal but not endotoxin-repressible expression of the von Willebrand factor promoter in Hprt-targeted transgenic mice. J Thromb Haemost. 2009;7:1384–1392. doi: 10.1111/j.1538-7836.2009.03501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mojiri A, Nakhaii-Nejad M, Phan WL, Kulak S, Radziwon-Balicka A, Jurasz P, Michelakis E, Jahroudi N. Hypoxia results in upregulation and de novo activation of von Willebrand factor expression in lung endothelial cells. Arterioscler, Thromb Vasc Biol. 2013;33:1329–1338. doi: 10.1161/ATVBAHA.113.301359. [DOI] [PubMed] [Google Scholar]
- 20.Kleinschmidt AM, Nassiri M, Stitt MS, Wasserloos K, Watkins SC, Pitt BR, Jahroudi N. Sequences in intron 51 of the von Willebrand factor gene target promoter activation to a subset of lung endothelial cells in transgenic mice. J Biol Chem. 2008;283:2741–2750. doi: 10.1074/jbc.M705466200. [DOI] [PubMed] [Google Scholar]
- 21.Nassiri M, Liu J, Kulak S, Uwiera RR, Aird WC, Ballermann BJ, Jahroudi N. Repressors NFI and NFY participate in organ-specific regulation of von Willebrand factor promoter activity in transgenic mice. Arterioscler, Thromb Vasc Biol. 2010;30:1423–1429. doi: 10.1161/ATVBAHA.110.206680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulinska P, Spiel A, Jilma B. Role of von Willebrand factor in vascular disease. Hamostaseologie. 2009;29:32–38. [PubMed] [Google Scholar]
- 23.Spiel AO, Gilbert JC, Jilma B. von Willebrand factor in cardiovascular disease: Focus on acute coronary syndromes. Circulation. 2008;117:1449–1459. doi: 10.1161/CIRCULATIONAHA.107.722827. [DOI] [PubMed] [Google Scholar]
- 24.White HD, Chew DP. Acute myocardial infarction. Lancet. 2008;372:570–584. doi: 10.1016/S0140-6736(08)61237-4. [DOI] [PubMed] [Google Scholar]
- 25.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81:457–464. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 26.White HD, Thygesen K, Alpert JS, Jaffe AS. Clinical implications of the third universal definition of myocardial infarction. Heart. 2014;100:424–432. doi: 10.1136/heartjnl-2012-302976. [DOI] [PubMed] [Google Scholar]
- 27.Peyvandi F, Hollestelle MJ, Palla R, Merlini PA, Feys HB, Vanhoorelbeke K, Lenting PJ, Mannucci PM. Active platelet-binding conformation of plasma von Willebrand factor in young women with acute myocardial infarction. J Haemost. 2010;8:1653–1656. doi: 10.1111/j.1538-7836.2010.03896.x. [DOI] [PubMed] [Google Scholar]
- 28.Goto S, Sakai H, Ikeda Y, Handa S. Acute myocardial infarction plasma augments platelet thrombus growth in high shear rates. Lancet. 1997;349:543–544. doi: 10.1016/S0140-6736(97)80095-5. [DOI] [PubMed] [Google Scholar]
- 29.Jansson JH, Nilsson TK, Johnson O. von Willebrand factor in plasma: A novel risk factor for recurrent myocardial infarction and death. Br Heart J. 1991;66:351–355. doi: 10.1136/hrt.66.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chion CK, Doggen CJ, Crawley JT, Lane DA, Rosendaal FR. ADAMTS13 and von Willebrand factor and the risk of myocardial infarction in men. Blood. 2007;109:1998–2000. doi: 10.1182/blood-2006-07-038166. [DOI] [PubMed] [Google Scholar]
- 31.Hu H, Xuan Y, Wang Y, Xue M, Suo F, Li X, Cheng W, Li X, Yin J, Liu J, Yan S. Targeted NGF siRNA delivery attenuates sympathetic nerve sprouting and deteriorates cardiac dysfunction in rats with myocardial infarction. PloS One. 2014;9:e95106. doi: 10.1371/journal.pone.0095106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Liu J, Suo F, Hu HS, Xue M, Cheng WJ, Xuan YL, Yan SH. Metoprolol-mediated amelioration of sympathetic nerve sprouting after myocardial infarction. Cardiology. 2013;126:50–58. doi: 10.1159/000351074. [DOI] [PubMed] [Google Scholar]
- 33.Zhang WB, Du QJ, Li H, Sun AJ, Qiu ZH, Wu CN, Zhao G, Gong H, Hu K, Zou YZ, Ge JB. The therapeutic effect of rosuvastatin on cardiac remodelling from hypertrophy to fibrosis during the end-stage hypertension in rats. J Cell Mol Med. 2012;16:2227–2237. doi: 10.1111/j.1582-4934.2012.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Yuan L, Molema G, Regan E, Janes L, Beeler D, Spokes KC, Okada Y, Minami T, Oettgen P, Aird WC. Vascular bed-specific regulation of the von Willebrand factor promoter in the heart and skeletal muscle. Blood. 2011;117:342–351. doi: 10.1182/blood-2010-06-287987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutten B, Maseri A, Cianflone D, Laricchia A, Cristell NA, Durante A, Spartera M, Ancona F, Limite L, Hu D, et al. Plasma levels of active Von Willebrand factor are increased in patients with first ST-segment elevation myocardial infarction: A multicenter and multiethnic study. Eur Heart J Acute Cardiovasc Care. 2015;4:64–74. doi: 10.1177/2048872614534388. [DOI] [PubMed] [Google Scholar]
- 36.Sakai H, Goto S, Kim JY, Aoki N, Abe S, Ichikawa N, Yoshida M, Nagaoka Y, Handa S. Plasma concentration of von Willebrand factor in acute myocardial infarction. Thromb Haemost. 2000;84:204–209. [PubMed] [Google Scholar]
- 37.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/S0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 38.Dinarello CA, Pomerantz BJ. Proinflammatory cytokines in heart disease. Blood Purif. 2001;19:314–321. doi: 10.1159/000046960. [DOI] [PubMed] [Google Scholar]
- 39.Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell Cardiol. 2006;41:580–591. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Kleinbongard P, Schulz R, Heusch G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev. 2011;16:49–69. doi: 10.1007/s10741-010-9180-8. [DOI] [PubMed] [Google Scholar]
- 41.Mann DL. Tumor necrosis factor-induced signal transduction and left ventricular remodeling. J Card Fail. 2002;8(Suppl 6):S379–S386. doi: 10.1054/jcaf.2002.129253. [DOI] [PubMed] [Google Scholar]
- 42.Bradham WS, Bozkurt B, Gunasinghe H, Mann D, Spinale FG. Tumor necrosis factor-alpha and myocardial remodeling in progression of heart failure: A current perspective. Cardiovasc Res. 2002;53:822–830. doi: 10.1016/S0008-6363(01)00503-X. [DOI] [PubMed] [Google Scholar]
- 43.Jacobs M, Staufenberger S, Gergs U, Meuter K, Brandstätter K, Hafner M, Ertl G, Schorb W. Tumor necrosis factor-alpha at acute myocardial infarction in rats and effects on cardiac fibroblasts. J Mol Cell Cardiol. 1999;31:1949–1959. doi: 10.1006/jmcc.1999.1007. [DOI] [PubMed] [Google Scholar]
- 44.Blake GJ. Inflammatory biomarkers of the patient with myocardial insufficiency. Curr Opin Crit Care. 2003;9:369–374. doi: 10.1097/00075198-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 45.Schulz R, Aker S, Belosjorow S, Heusch G. TNFalpha in ischemia/reperfusion injury and heart failure. Basic Res Cardiol. 2004;99:8–11. doi: 10.1007/s00395-003-0431-x. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H, Park Y, Wu J, Chen Xp, Lee S, Yang J, Dellsperger KC, Zhang C. Role of TNF-alpha in vascular dysfunction. Clin Sci (Lond) 2009;116:219–230. doi: 10.1042/CS20080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 48.van der Poll T, van Deventer SJ, Pasterkamp G, van Mourik JA, Büller HR, ten Cate JW. Tumor necrosis factor induces von Willebrand factor release in healthy humans. Thromb Haemost. 1992;67:623–626. [PubMed] [Google Scholar]
- 49.Umetani M, Mataki C, Minegishi N, Yamamoto M, Hamakubo T, Kodama T. Function of GATA transcription factors in induction of endothelial vascular cell adhesion molecule-1 by tumor necrosis factor-alpha. Arterioscler Thromb Vasc Biol. 2001;21:917–922. doi: 10.1161/01.ATV.21.6.917. [DOI] [PubMed] [Google Scholar]