

Abstract
Posttranslational modifications to the intracellular domain of the EGFR are known to regulate EGFR functions; however, modifications to the extracellular domain and their effects remain relatively unexplored. Here, we determined that methylation at R198 and R200 of the EGFR extracellular domain by protein arginine methyltransferase 1 (PRMT1) enhances binding to EGF and subsequent receptor dimerization and signaling activation. In a mouse orthotopic colorectal cancer xenograft model, expression of a methylation-defective EGFR reduced tumor growth. Moreover, increased EGFR methylation sustained signaling activation and cell proliferation in the presence of the therapeutic EGFR monoclonal antibody cetuximab. In colorectal cancer patients, EGFR methylation level also correlated with a higher recurrence rate after cetuximab treatment and reduced overall survival. Together, these data indicate that R198/R200 methylation of the EGFR plays an important role in regulating EGFR functionality and resistance to cetuximab treatment.
Introduction
The EGFR family is one of the most well characterized RTK systems. In addition to their function in normal development, aberrant expression of EGFR is involved in abnormal cell proliferation, reduced apoptosis, cell migration, metastasis, and angiogenesis in cancer patients (1, 2). Activation of EGFR converts extracellular stimulations into intracellular signals to regulate cellular responses through protein modifications. Crosstalk between methylation and phosphorylation on intracellular domain regulates downstream signaling activation (3), whereas ubiquitination mediates EGFR stability, trafficking, and signal sustainability (4). While intracellular domain modifications of transmembrane proteins have been well studied, only a few types of extracellular domain modifications have been identified (5, 6). There is evidence that extracellular modifications of transmembrane proteins have important physiological functions. For example, extracellular domain phosphorylation of cadherin protein by intracellular Golgi kinase or ectokinase regulates cell adhesion, cell growth, and cell polarity (6, 7). In addition, glycosylation on the extracellular domain of RTK is critical for protein stabilization and subcellular localization (5, 8). However, other types of extracellular domain protein modifications besides phosphorylation and glycosylation are not well explored. Interestingly, our MS analysis revealed several methylated arginines on EGFR extracellular domain. Since the discovery of protein arginine methyltransferases (PRMTs), arginine methylation has been implicated in many biological processes and human diseases (9). During arginine methylation, PRMTs transfer methyl groups from S-adenosylmethionine (SAM) to the guanidine nitrogen of specific arginine residues on their target proteins, which in turn alter the protein structure, protein-protein interaction, protein localization, and enzyme activity that are critical for various cellular functions (10). For example, signal transduction, RNA processing, DNA repair, and gene transcription are regulated by arginine methylation (3, 11, 12).
The observation of EGFR extracellular domain methylation prompted us to ask whether this modification affects EGFR functionality and the efficacy of extracellular domain–targeted therapeutic monoclonal antibody, cetuximab, in colorectal cancer treatment. Colorectal cancer is the third leading cause of cancer deaths in the United States. Optimization of dosing and scheduling of chemotherapeutic agents have improved the response and survival rate of colorectal patients. Currently, rational targeting of molecular signaling pathways that are involved in the etiology of malignancies is one of the most promising strategies in novel anticancer drug development (13). Owing to the role of EGFR in tumorigenesis, new classes of drugs that target EGFR are among the most clinically advanced molecular-targeted therapies. Although EGFR tyrosine kinase inhibitors combined with chemotherapy presented severe toxicity and limited effect (14), the combination of EGFR monoclonal antibody, such as cetuximab and panitumumab, with chemotherapy has shown efficacy in colorectal cancer treatment (15). Unfortunately, resistance to EGFR-targeted therapy has recently been observed. Many mechanisms have been proposed to explain the poor response to cetuximab, including activation of HER2 or MET signaling, mutation of PIK3CA and BRAF, or expression status of PTEN, but retrospective analyses revealed inconsistent and controversial findings (16). So far, the most accepted predictive marker for poor cetuximab response is mutant KRAS status, due to its association with poor survival rate under cetuximab treatment in colorectal cancer clinical trials (17–20). Therefore, American Society of Clinical Oncology recommended cetuximab treatment for only patients with WT KRAS (21). However, there is increasing evidence showing that WT KRAS is not sufficient to confer sensitivity to cetuximab (22–24), and some patients with mutant KRAS are still sensitive to cetuximab (16, 25–28). These findings suggest that further investigation into the underlying mechanisms of cetuximab resistance and identification of a better predictor for cetuximab response are needed. Here, we investigate the role of extracellular methylation in EGFR signal transduction and unexpectedly discover that specific extracellular Arg methylations of EGFR render cancer cells resistant to cetuximab antibody therapy.
Results
PRMT1 methylates EGFR at R198 and R200.
Mass spectrometric (MS) analysis of immunopurified endogenous EGFR proteins from SKCO1 colorectal cancer cells demonstrated several methylated arginines on the extracellular domain of EGFR, including R29, R74, R198, R200, R285, and R497 (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI82826DS1). Among them, missense mutation of R198 and R285 were reported in colorectal adenocarcinoma (TCGA database; https://tcga-data.nci.nih.gov/tcga/), and both happen to be located on EGFR extracellular domain 2 (D2) (Figure 1A), suggesting functional role of these arginine residues in colorectal cancer biology. To understand whether methylation of EGFR extracellular domain arginines affect its function, we began by first interrogating which protein in PRMT family (PRMT1-8) is responsible for methylation of these arginines. Results from in vitro methylation screening assay by using D2 as substrate revealed that only PRMT1 generated strong methylation signal when coincubated with D2 (Figure 1B). Notably, among various functional domains of EGFR (Figure 1A), PRMT1 methylated only extracellular D2 (Figure 1B). Next, we individually mutated all 3 arginines, which were positive for methylation as determined by MS analysis, on D2 to lysine to determine which of these are methylated by PRMT1. Mutation of both R198 and R200, but not R285, to lysine reduced methylation signals compared with WT EGFR as demonstrated by an in vitro methylation assay (Figure 1C). Methylation signal was completely abolished for the R198/200K double mutant, suggesting that these 2 arginines in D2 are the major targets of PRMT1-mediated EGFR methylation in vitro (Figure 1D). PRMT1 frequently methylates arginine residues found within glycine-arginine–rich (GAR) domains (29, 30) and generates asymmetric dimethylated arginine. Sequence alignment between the GAR domains of known PRMT substrates and D2 of EGFR revealed a putative GAR domain between amino acids 196 and 204 with 2 arginine residues, R198 and R200 (Figure 1E), supporting our observation that R198/200 are the target sites of PRMT1 in vitro.
Figure 1. PRMT1 methylates EGFR at R198 and R200.

(A) Illustration of EGFR extracellular and intracellular domains that were individually subcloned with GST tag and were purified for in vitro methylation assay. (B) In vitro methylation assay showing methylation signal from each GST-tagged EGFR domain after incubation with purified GST-tagged PRMT1. Methylation signals were examined by fluorography. (C) In vitro methylation assay of WT EGFR and methylation-site single mutants. (D) In vitro methylation assay of WT EGFR and R198/200K double mutant. Data are representative of 3 independent experiments. (E) Sequence alignment of PRMT substrates, along with EGFR, showing a potential GAR motif on EGFR extracellular domain 2.
PRMT1 interacts with and methylates EGFR in ER/Golgi.
To analyze the methylation status of endogenous EGFR, we generated an anti–me-R198/200 methylation-specific antibody by using a synthesized asymmetric dimethylated EGFR peptide. Dot blot assay showed that this me-R198/200 antibody recognized asymmetric dimethylated R198/200 EGFR peptide but not unmodified R198/200 EGFR peptide, other methylation forms of R198/200 EGFR peptide, or asymmetric dimethylated histone H4 peptide (Figure 2A), validating the specificity of the antibody. The me-R198/200 antibody was later used to detect the methylation level of endogenous EGFR. We found that the methylation status of endogenous EGFR was proportional to PRMT1 expression, and the methylation signal detected by the me-R198/200 antibody was specifically depleted by methylated but not by unmodified peptides (Figure 2B), supporting that EGFR R198/200 methylation is regulated by PRMT1 in cells.
Figure 2. PRMT1 interacts with and methylates EGFR before it is transported to the cell membrane.

(A) Dot blot showing specificity of EGFR me-R198/200 Ab. H3R4, Histone H4 arginine 3 asymmetric dimethylated peptide; asym di-me, EGFR peptides asymmetric dimethylation on indicated sites; sym di-me, EGFR symmetric dimethylated R198/200 peptide; mono-me, EGFR mono methylated R198/200 peptide; scrambled, peptide with the same amino acid composition as the EGFR R198/200 peptide with the amino acids scrambled while maintaining the position of the 2 methyl-arginines. (B) Immunoblots comparing EGFR methylation level in SKCO1 cells exogenously expressing PRMT1 (left), PRMT1 shRNA (right), or control vector with EGFR methylation-specific antibody, me-R198/200 Ab. (C) Top: Immunoblots of indicated proteins of SKCO1 cells expressing control vector or PRMT1 shRNA in the absence or presence of tunicamycin (2 μM, 24 hr) or AMI-1 (100 μM). Bottom: In vitro methylation assay showing methylation signal of GST-GAR after incubation with purified GST-tagged PRMT1 in the absence or presence of AMI-1 (100 μM). Methylation signals were examined by fluorography. (D) Immunoblots of indicated proteins after ER isolation of SKCO1 cells expressing control vector or PRMT1 shRNA. Calnexin, ER marker; lamin b1, nuclear marker; HSP60, mitochondrial marker; GAPDH, cytosolic protein. (E) Reciprocal coimmunoprecipitation of SKCO1 cells with the indicated antibodies. (F) Duolink assay of endogenous EGFR and PRMT1 in SKCO1 cells. Red spots represent the interaction between PRMT1 and EGFR. Scale bars: 10 μm. Data are representative of 3 independent experiments.
PRMT1 is an intracellular protein and less likely to contact R198 and R200, which are located on the extracellular domain of EGFR, when EGFR is transported to the cell membrane. Protein modifications on the extracellular domain of transmembrane proteins, such as phosphorylation and glycosylation, can occur during biosynthesis before the transmembrane proteins are transported to cell membrane (5, 6, 8). Therefore, we speculated that PRMT1 methylates R198/200 through a similar mechanism, before EGFR is transported to the cell membrane. To investigate this possibility, we examined methylation status by treating cells with or without tunicamycin, an N-linked glycosylation inhibitor. During EGFR protein translation and maturation, glycans are added onto its extracellular domain before it is transported to the cell membrane. Addition of tunicamycin disrupted glycosylation, resulting in the accumulation and retention of immature, unglycosylated EGFR (130 kDa) in the ER/Golgi compartments (31). In the presence of tunicamycin, we detected EGFR methylation using the me-R198/200 antibody in both newly synthesized (130 kDa) and old (170 kDa) EGFR. EGFR methylation signals were reduced when we knocked down PRMT1 (Figure 2C, top). To rule out the possibility that PRMT1 methylates EGFR during cell lysis, PRMT inhibitor, AMI-1, was added into the cell lysis buffer (32). Detection of methylation signals both in the presence or absence of AMI-1 indicated that the methylation event occurred in cells before cell lysis. The efficacy of AMI-1 was validated by an in vitro methylation assay in which the addition of AMI-1 blocked methylation reaction of PRMT1 toward GST-GAR, a methyl-accepting substrate (Figure 2C, bottom). To further validate that newly synthesized EGFR is methylated, we isolated ER organelle and showed that the newly synthesized EGFR in the isolated ER-enriched fraction was indeed methylated as detected by the me-R198/200 antibody (Figure 2D). In contrast, knocking down of PRMT1 abolished methylated EGFR signals. Next, we validated the interaction between PRMT1 and EGFR by reciprocal coimmunoprecipitation (Figure 2E). Interestingly, a Duolink (proximity ligation) assay showed that the PRMT1-EGFR interacting complex colocalized with ER lumen proteins, calreticulin (Figure 2F), protein disulfide isomerase (PDI) (Supplemental Figure 2A), and Golgi lumen protein — stromal cell derived factor 4 (SDF4) (Supplemental Figure 2B) — suggesting that the methylation of EGFR by PRMT1 may occur in these subcellular compartments. To further validate whether a fraction of PRMT1 is indeed in the lumen of ER/Golgi, we performed immunofluorescence staining to detect colocalization of PRMT1 with PDI or SDF4. As shown in Supplemental Figure 2, C and D, PRMT1 localized to both the cytoplasm and nucleus. Among the cytoplasmic PRMT1, a fraction of PRMT1 colocalized with ER lumen protein, PDI (Supplemental Figure 2C), or Golgi lumen protein SDF4 (Supplemental Figure 2D). The colocalization between PRMT1 and ER or Golgi lumen markers are indicated by yellow signals in the merged images. ER-targeting signal peptide is commonly found at the N-terminus of proteins (33). Therefore, we deleted the N-terminal hydrophobic signal peptide (29 amino acids) of PRMT1 (34) and examined its subcellular distribution. N-terminal deletion resulted in an accumulation of PRMT1 in the nuclear region and reduced the colocalization between PRMT1 and PDI (Supplemental Figure 2C) or SDF4 (Supplemental Figure 2D), as indicated by the loss of yellow signals in the merged images. These results suggest that the N-terminal hydrophobic signal peptide is involved in ER/Golgi localization of PRMT1. Importantly, expression of WT PRMT1 increased EGFR methylation level, whereas expression of the N-terminal deletion PRMT1 mutant did not increase the methylation level of EGFR compared with vector control (Supplemental Figure 2E), suggesting that the N-terminal hydrophobic signal peptide may play important roles in regulating localization of PRMT1 and subsequent EGFR methylation level. Next, we asked whether the methyl group donor, SAM, exists in the ER/Golgi compartment. We examined the subcellular distribution of SAM by immunofluorescence with a commonly used anti-SAM antibody. SAM was detected in both the cytoplasm and nucleus. A fraction of SAM colocalized with ER (PDI; Supplemental Figure 2F) and Golgi (SDF; Supplemental Figure 2G) lumen proteins was observed in the ER/Golgi area. Taken together, the results suggest that PRMT1 likely interacts with and methylated EGFR at ER/Golgi before its translocation to the cell membrane.
PRMT1 upregulates EGFR signaling and cell proliferation in colorectal cancer cell lines.
Next, we asked how PRMT1 affects EGFR signaling. EGFR activation status was evaluated upon EGF stimulation by measuring specific tyrosine phosphorylation and 2 main downstream signaling, ERK and AKT, in both SKCO1 and GEO colorectal cancer cells expressing exogenous PRMT1 or vector control. Interestingly, EGFR activation was stronger in PRMT1-expressing cells than in vector control cells upon stimulation by EGF (Figure 3, A and B) and another EGFR ligand, TGFα, which is also highly expressed in colorectal cancer (ref. 35 and Supplemental Figure 3A). In contrast, exogenous expression of catalytically inactive mutant PRMT1 (VLD-AAA) (36) did not enhance EGFR activation or its downstream signaling, which indicates that upregulation of EGFR signaling by PRMT1 requires its enzymatic activity (Supplemental Figure 3B). In contrast, knockdown of PRMT1 by 2 different shRNAs severely blocked EGF-induced EGFR, ERK, and AKT activation (Figure 3, C and D). Although high PRMT1 expression has been shown to correlate with colorectal cancer progression (37), its role in colorectal cancer pathophysiology remains obscure. We therefore measured cell proliferation and anchorage-independent cell growth of stable transfectants that ectopically expressed PRMT1 with or without treatment of EGFR tyrosine kinase domain inhibitor, gefitinib. In line with upregulated EGFR signaling by PRMT1, gefitinib significantly inhibited cell proliferation (Figure 3, E and F) and anchorage-independent growth (Figure 3G) in both PRMT1-overexpressing and vector control (expressing endogenous PRMT1) cells, supporting the concept that PRMT1 upregulates EGFR signaling in response to ligand stimulation and increases cellular transformation.
Figure 3. PRMT1 upregulates EGFR signaling and cell proliferation in colorectal cancer cell lines.

(A and B) Immunoblots comparing pEGFR, pERK, and pAKT levels upon EGF stimulation for indicated time in SKCO1 (A) and GEO (B) cells expressing PRMT1 or control vector. (C and D) Immunoblots evaluating pEGFR, pERK and pAKT levels upon EGF stimulation for indicated time in SKCO1 cells expressing 2 different PRMT1 shRNAs or control vector. Blots shown are representative of 3 independent experiments. (E and F) Cell proliferation assay of SKCO1 (E) and GEO (F) cells expressing PRMT1 or vector control with or without gefitinib treatment. *P < 0.05 using Student’s t-test. (G) Anchorage-independent growth of 1,000 SKCO1 exogenously expressing PRMT1 and control vector with or without gefitinib treatment. **P < 0.005 using Student’s t-test. All quantitative data were generated from 5 replicates. Data are expressed as mean ± SD.
PRMT1 upregulates EGFR dimerization, activation, and EGFR-dependent cell proliferation through R198/200 methylation.
To understand whether PRMT1 upregulates EGFR signaling through R198/200 methylation, we constructed a full-length EGFR R198/200K mutant that cannot be methylated by PRMT1 for comparison with WT EGFR. Since both pEGFR and pERK were substantially changed by ectopic expression or knockdown of PRMT1 (Figure 3, A–D), we used them as markers to monitor EGFR signaling. Notably, EGFR and ERK phosphorylation were markedly reduced in GEO cells expressing the R198/200K mutant compared with those expressing the WT EGFR, suggesting the importance of EGFR R198 and R200 methylation for PRMT1-upregulated EGFR signaling (Figure 4A). To further confirm that stronger signaling activation of WT EGFR was a result of R198/200 methylation by PRMT1, we knocked down PRMT1 in both WT EGFR– and EGFR R198/200K–expressing HT29 colorectal cancer cells. Consistently, HT29 cells expressing exogenous WT EGFR had stronger EGFR and downstream ERK activation than those expressing exogenous EGFR R198/200K mutant (Figure 4B, left). Notably, knocking down of PRMT1 reduced signaling activation of exogenous WT EGFR but not exogenous EGFR R198/200K mutant (Figure 4B, middle and right). In line with EGFR activation, HT29 cells expressing exogenous WT EGFR also had higher cell proliferation rate (Figure 4C) compared with EGFR R198/200K-expressing cells. Knocking down of PRMT1 in WT EGFR–expressing cells substantially reduced the EGFR-dependent cell growth rate to a similar level observed in EGFR R198/200K–expressing cells, indicating that PRMT1 upregulates EGFR-dependent cell growth via R198/200 methylation.
Figure 4. PRMT1 upregulates EGFR dimerization and activation, and EGFR-dependent cell proliferation through R198/200 methylation.

(A) Immunoblot comparing EGFR and downstream ERK activation level of GEO cells expressing control vector, WT, and methylation-site mutant EGFR upon EGF stimulation. (B) Immunoblot comparing EGFR and downstream ERK activation level of HT29 cells expressing control vector, WT, and R198/200K mutant EGFR with or without PRMT1 knocking down upon EGF stimulation. Blots shown are representative of 5 independent experiments. (C) Cell proliferation assay of HT29 cells expressing control vector, WT, and R198/200K mutant EGFR with or without PRMT1 knocking down. (n = 6). Data are expressed as mean ± SD. (D) Left: Inactive tethered conformation of EGFR. The structure was prepared based on the crystal structure of the inactive human EGFR (PDB accession 1IVO) (http://www.rcsb.org/pdb/explore.do?structureId=1IVO). D1–D4 are color-coded; the weakly bound EGF is in cyan. R198 and R200 are highlighted. Right: Active dimerized form of EGFR (based on the crystal structure of human EGFR, PDB accession 1NQL). The second EGFR and EGF molecules are shown in gray and black, respectively. (E and F) Zoom onto R198 (E) and R200 (F) shown as stick figures. The molecular surface of EGFR (with the exception of the region 198-200) is colored in blue, positively charged atoms; red, negatively charged atoms; green, hydrophobic atoms; salmon, polar oxygens; marine, polar nitrogens; yellow, sulfur). EGFR domains are indicated on the surfaces, as is the location of D206. (G) Dimerization assay of SKCO1 cells exogenously expressing vector control and PRMT1. Anti-EGFR antibody was used to detect EGFR monomer and dimer. (H) Dimerization assay of SKCO1 cells expressing vector control and PRMT1 shRNA. (I) Dimerization assay of GEO cells expressing WT and R198/200K mutant EGFR with or without knocking down of PRMT1. Blots shown are representative of 4 independent experiments.
R198 and R200 are situated in the hinge region between D1 and D2. Available crystallographic structures show that EGFR dynamically transitions between an inactive monomeric tethered conformation and an active dimeric extended conformation (Figure 4D and refs. 38, 39). In the inactive form, the R198 side chain inserts into a narrow pocket provided by D1. Asymmetric dimethylation would increase the volume of R198 sufficiently to cause unfavorable steric clashes with D1 and, hence, disfavor the inactive conformation (Figure 4E, left). In the active form, the same binding pocket is substantially enlarged (Figure 4E, right) and provides sufficient space and hydrophobic surface patches (green) to accommodate asymmetric dimethylation. Compared with the inactive form, D1 and D2 are slightly rotated away from each other in the active form, reducing the contact surface between D1 and the rest of the molecule by ~150 Å. The relative orientation of domains D1 and D2 is conserved in all dimeric EGFR forms, including ligand-free forms from the drosophila EGFR homolog (40, 41), suggesting that our analysis holds for all dimeric conformations. Meanwhile, in the inactive form, R200 compensates for the charge of D206 (Figure 4F, left), and the backbone carbonyl of D206 binds to the backbone nitrogen of R200. In the active form, the side chain of D206 is rotated away and exposed to the solvent. The D206 backbone carbonyl forms a weak hydrogen bond with R200, bringing the R200 guanidinium moiety close to the hydrophobic surface of P219 (Figure 4F, right). Consequently, methylation of R200 appears to favor the active conformation because it provides a less charged and more hydrophobic environment for R200. Our structural analysis therefore suggests that R198/200 methylation predisposes EGFR to assume an active conformation and, hence, increases ligand-stimulated downstream signaling, supporting the observation of upregulated EGFR signaling and EGFR-dependent cell growth by R198/200 methylation.
The effect of EGFR R198/200 methylation by PRMT1 on EGFR signaling and subsequent cell growth prompted us to investigate how this extracellular modification affects intracellular downstream signaling of the receptor. Given that D2 of EGFR is critical for the receptor dimerization and subsequent downstream signaling activation (42), we further examined the effect of R198/R200 methylation on EGFR dimerization. Interestingly, upon EGF stimulation, endogenous EGFR of PRMT1-expressing SKCO1 cells showed higher receptor dimerization ability than cells expressing control vector (Figure 4G). In contrast, EGFR in PRMT1 shRNA–expressing cells had lower EGF-stimulated dimer formation than cells expressing control vector (Figure 4H). Consistently, loss of methylation of EGFR R198/200K mutant substantially reduced its dimerization ability compared with WT EGFR in GEO cells (Figure 4I). Likewise, knocking down of PRMT1 in WT EGFR–expressing cells greatly reduced the dimer formation of WT EGFR to a level similar to that observed for EGFR R198/200K–expressing cells, indicating that PRMT1 upregulates EGFR dimerization via R198/200 methylation (Figure 4I). Notably, while glycosylation was reported to affect the expression level of cell surface EGFR (43), our data indicated that methylation did not (Supplemental Figure 4). These results support a model that methylation at R198/200 of EGFR by PRMT1 enhances its EGF-induced dimerization ability, and they provide an explanation for PRMT1-upregulated EGFR signaling and cell proliferation.
Methylation of EGFR increases tumorigenesis in orthotopic colorectal cancer mouse model and correlates with poorer clinical outcomes of colorectal cancer patients.
Our current data support a model in which PRMT1 enhances EGFR dimerization and activation through methylation of EGFR at R198/200. Subsequently, upregulated EGFR signaling promotes cell proliferation and anchorage-independent growth of colorectal cancer cells. To further address the pathological relevance of this model, we examined whether PRMT1-mediated EGFR methylation contributes to tumorigenesis in an orthotopic colorectal cancer mouse model. One month after injection, HT29 cells expressing WT EGFR generated markedly larger tumors than those expressing R198/200K EGFR or vector control (Figure 5A). Knocking down PRMT1 substantially reduced tumor growth, supporting the positive role of EGFR R198/200 methylation in colorectal cancer tumorigenesis. To further address the clinical relevance of EGFR R198/200 methylation, we characterized me-R198/200 antibody for its ability to detect EGFR R198/200 methylation in IHC staining. GEO cells expressing WT EGFR, R198/200K EGFR, or vector control were fixed on slides by cytospin and stained by me-R198/200 antibody. As shown in Figure 5B, IHC staining signals were substantially stronger in WT EGFR–expressing cells than R198/200K EGFR– or vector-expressing cells. The staining signals in WT EGFR–expressing cells were blocked by synthesized asymmetric dimethylated R198/200 EGFR peptide but not by unmodified R198/200 EGFR peptides or asymmetric dimethylated histone H4 peptide, validating the specificity of the me-R198/200 antibody. Similarly, me-R198/200 antibody was able to detect EGFR methylation signals in patient tissue samples, and these methylation signals were blocked specifically by asymmetric dimethylated R198/200 EGFR peptide but not by other forms of peptides (Figure 5C). A retrospective study of clinical colorectal cancer specimens further indicated that EGFR R198/200 methylation level as detected by me-R198/200 antibody was elevated in tumor tissue compared with paired adjacent normal tissue. Also, higher EGFR R198/200 methylation level coincided significantly with poorer overall patient survival (Figure 5D) and higher recurrence rate (Figure 5E). Collectively, these results suggest that PRMT1-mediated EGFR R198/200 methylation contributes to tumorigenesis in vivo, and the methylation status of EGFR has the potential to serve as a predictive marker for patient prognosis.
Figure 5. Methylation of EGFR increases tumorigenesis in orthotopic colorectal cancer mouse model and correlates with poorer clinical outcomes of colorectal cancer patients.

(A) In vivo orthotopic colon tumor growth of HT29 cells expressing vector control, WT, or R198/200K mutant (Mut) EGFR with or without knockdown of PRMT1 (n = 5 per group). Data are expressed as mean ± SD. Top: Representative tumors from each group in the fourth week after inoculation. (B) Immunochemistry staining of GEO cells expressing WT, R198/200K EGFR, or vector control by me-R198/200 Ab competed without or with synthesized unmodified R198/200 EGFR peptide, asymmetric dimethylated histone H4R3 peptide, or asymmetric dimethylated R198/200 EGFR peptide. Scale bars: 50 μm. (C) Immunochemistry staining of colon cancer tissue by EGFR methylation antibody competed without or with indicated peptide. Scale bars: 25 μm. Images shown are representative of 3 independent experiments. (D) Kaplan-Meier plot of overall survival of 215 colorectal cancer cases with low or high methyl-EGFR level detected by me-R198/200 Ab. (E) Kaplan-Meier plot of recurrence rate of 120 colorectal cancer cases with low or high methyl-EGFR level detected by me-R198/200 Ab. P < 0.05 using Student’s t-test.
EGFR methylation enhances EGF binding to EGFR and correlates with higher recurrence rate of colorectal cancer patients after cetuximab treatment.
Clinically, cetuximab is used to treat metastatic colorectal cancer by disabling the interaction between EGF ligand and EGFR and attenuating EGFR signaling (44–46). Cetuximab binds exclusively to D3 of EGFR in its inactive conformation (Figure 4D). Upon binding, cetuximab occludes the EGF-binding site and prevents EGFR from adopting the active conformation required for ligand binding and dimerization (47). By predisposing EGFR to assume an active EGF-bound conformation, R198/200 methylation is expected to enhance the interaction between EGFR and EGF. Therefore, we asked whether R198/200 methylation of EGFR affects its affinity for EGF and the efficacy of cetuximab. Dissociation constant (Kd) between EGFR and EGF in the absence or presence of cetuximab was measured in the EGFR stable transfectants in HT29 and SW48 colorectal cancer cells by saturation binding assay. In the absence of cetuximab, WT EGFR in HT29 (Figure 6A) and SW48 (Supplemental Figure 5A) cells showed higher binding affinity (lower Kd) for EGF (Kd = 16.03 nM and Kd = 16.21 nM in HT29 and SW48, respectively) compared with the R198/200K mutant EGFR (Kd = 52.16 nM and Kd = 51.79 nM in HT29 and SW48, respectively). Knocking down PRMT1 in WT EGFR–expressing HT29 or SW8 cells greatly reduced the affinity of EGFR for EGF (Kd = 44.32 nM; Kd = 46.65 nM in HT29 and SW48, respectively) to a level comparable to that of the R198/200K mutant. The binding affinity between the R198/200K mutant EGFR and EGF was not affected by PRMT1 knockdown (Kd = 55.09 nM and Kd = 54.54 nM in HT29 and SW48, respectively). These results suggest that methylation of EGFR R198/200 by PRMT1 enhances its binding to EGF. In addition, in the presence of a relatively low concentration of cetuximab (1 μg/ml), at which the binding affinity between WT EGFR and EGF did not change significantly compared with the absence of cetuximab (Figure 6B; Kd from 17.86–17.25 nM, without and with cetuximab, respectively), the affinity of R198/200K mutant EGFR for EGF was markedly reduced (Kd from 47.28–94.33 nM, without and with cetuximab, respectively). Together, these results suggest that methylated EGFR at R198/200 responds better to EGF binding and is more resistant to cetuximab treatment.
Figure 6. EGFR methylation regulates EGF binding to EGFR and correlates with higher recurrence rate of colorectal cancer patients after cetuximab treatment.

(A) Top: Scatchard plot and binding curves (inset), which measured EGFR-EGF binding affinity of HT29 cells expressing WT or R198/200K mutant (Mut) EGFR with or without knocking down of PRMT1. Bottom: Bar graph of Kd values. Red, WT EGFR; blue, R198/200K mutant EGFR. (B) Top: Scatchard plot and binding curves (inset), which measured EGFR-EGF binding affinity of HT29 cells expressing WT or R198/200K mutant EGFR with or without cetuximab treatment. Bottom: Bar graph of Kd values. Red, WT EGFR; blue, R198/200K mutant EGFR. Data are expressed as mean ± SD. (C) Immunoblot assessing EGFR, ERK, and AKT activation levels of HT29 cells expressing control vector or PRMT1 upon EGF stimulation in the presence or absence of cetuximab. (D) Clonogenic assay of HT29 cells expressing control vector or PRMT1 shRNA under cetuximab treatment (n = 3). Data are expressed as mean ± SD. Expression levels of PRMT1 shown by immunoblot. Data shown are representative of 4 independent experiments. (E) Kaplan-Meier plot of recurrence rate of 41 colorectal cancer cases with WT KRAS treated with cetuximab with low or high total EGFR level. (F) Kaplan-Meier plot of recurrence rate of 41 colorectal cancer cases with WT KRAS treated with cetuximab with low or high methyl-EGFR level detected by me-R198/200 Ab. P < 0.05 using Student’s t-test. (G) Clonogenic assay of SKCO1 cells expressing control vector or PRMT1 shRNA, or reexpressing shRNA-resistant PRMT1 under cetuximab treatment (n = 5). Data are expressed as mean ± SD.
In line with the higher EGF binding affinity and higher EGFR methylation level, PRMT1-overexpressing HT29 cells demonstrated higher EGFR and ERK activation after EGF stimulation, and the upregulated pEGFR and pERK remained relatively strong, even in the presence of cetuximab in comparison to vector control cells (Figure 6C). To validate the role of EGFR methylation in cetuximab response, cells expressing exogenous PRMT1 shRNA or PRMT1 were treated with cetuximab, and their clonogenic ability was evaluated (Figure 6D and Supplemental Figure 5, B–E). The colony number was relatively lower in cells in which PRMT1 was knocked down than control cells in the presence of cetuximab (Figure 6D and Supplemental Figure 5, C and E). In contrast, cells expressing exogenous PRMT1 showed more resistance to cetuximab treatment (Supplemental Figure 5, B and D). To further investigate whether methyl-EGFR level is related to patient response to cetuximab, tumor tissues from cetuximab-treated metastatic colorectal cancer patients were collected and stained by me-R198/200 antibody. Consistent with previous clinical analyses (48–50), no association (P = 0.486) was found between total EGFR expression level and cetuximab response (Figure 6E); however, higher levels of methyl-EGFR in tumors from colorectal cancer patients correlated with higher recurrence rate (P = 0.034) after cetuximab treatment (Figure 6F). In addition, the expression level of methyl-EGFR correlated positively with PRMT1 expression (Table 1). Similarly, the correlation between the expression level of methyl-EGFR and PRMT1 was also observed in patients with head and neck cancer (Supplemental Table 1), another FDA-approved cancer type for cetuximab treatment. Importantly, higher methyl-EGFR level also correlated with poorer overall survival after cetuximab treatment in head and neck cancer patients (Supplemental Figure 5F). Together, the results support the notion that PRMT1-mediated EGFR R198/200 methylation contributes to cetuximab resistance in colorectal and head and neck cancer patients.
Table 1. Relationship between expression of PRMT1 and methyl-EGFR in WT KRAS colorectal cancer specimens from 41 cetuximab-treated patients.
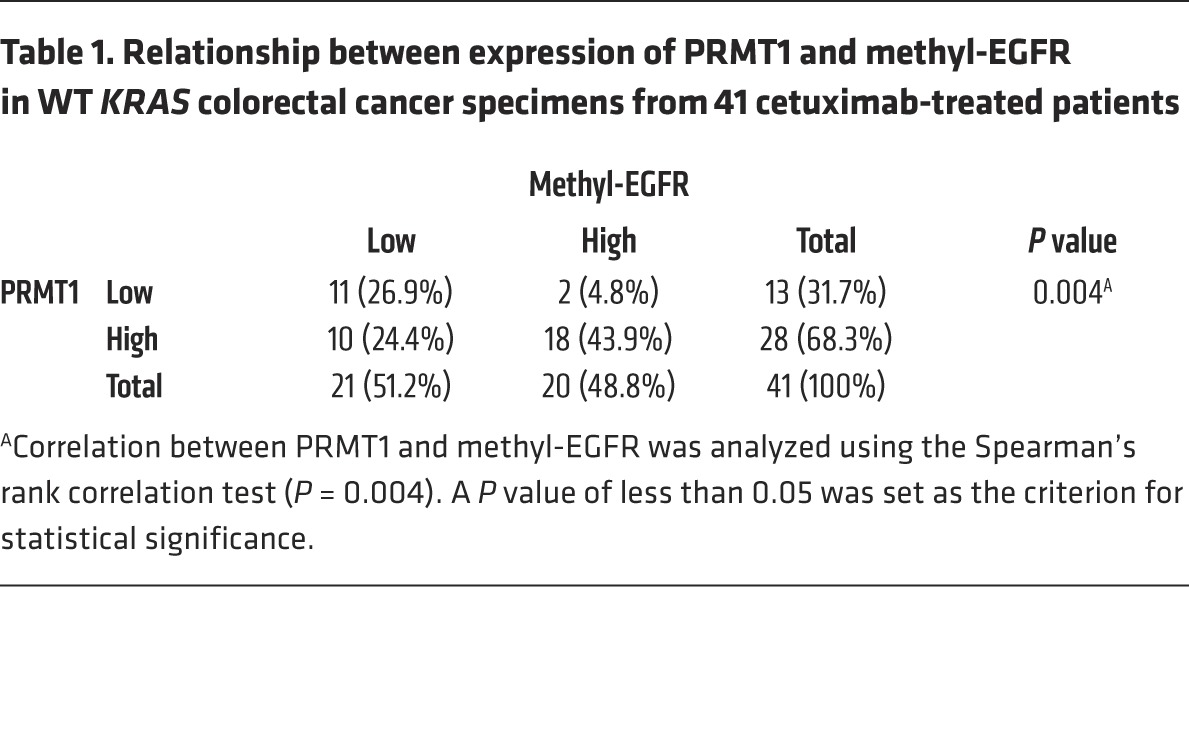
Clinically, the status of KRAS mutation has been used as a biomarker to predict patient response to cetuximab (21). We therefore asked whether methylation-mediated cetuximab resistance is affected by KRAS mutation. Interestingly, independently of KRAS status, knockdown of PRMT1 in all WT KRAS (HT29 and SW48) (Figure 6D and Supplemental Figure 5E) and mutant KRAS (SKCO1: KRAS G12V and GEO: KRAS G12A) (Figure 6G and Supplemental Figure 5C) cell lines sensitized cells to cetuximab treatment. Notably, knockdown of PRMT1 in KRAS G12V mutant and cetuximab-resistant SKCO1 cells rendered cells more sensitive to cetuximab than vector control cells. In contrast, reexpression of shRNA-resistant PRMT1 (rPRMT1) in PRMT1-knockdown cells restored the observed cetuximab resistance (Figure 6G), suggesting that KRAS mutation does not play a role in PRMT1-mediated cetuximab resistance.
Discussion
EGFR-targeted monoclonal antibodies have expanded the treatment options for colorectal cancer patients. Although these agents have great potential for individualized therapy, the reasons why some patients respond to treatment while others do not remain unclear. In this study, we demonstrate that PRMT1-mediated methylation of R198/200 on the extracellular domain of EGFR enhances receptor dimerization, EGFR signaling activation, and cell proliferation, and it reduces cellular response to cetuximab. Interestingly, although some studies have shown that mutant KRAS strongly represses EGF-stimulated activation of ERK phosphorylation in HCT116 and DLD1 colorectal cancer cell lines (51), our data demonstrate that ERK was still activated by EGF stimulation in GEO (G12A) and SKCO1 (G12V) KRAS mutant cell lines (Figure 3, A–D). The contradictory effect of EGF on ERK activation is somewhat expected, as different cancer cell lines can behave differently. Moreover, knockdown of PRMT1 sensitized cells to cetuximab treatment regardless of KRAS mutation status (Figure 6, D and G, and Supplemental Figure 5, C and E). High EGFR R198/200 methylation level correlated with higher recurrence rate in cetuximab-treated patients, implying that EGFR R198/200 methylation has the potential to serve as a predictive marker for cetuximab resistance in clinical colorectal cancer therapy.
It is worthwhile to mention that colorectal cancer patients carrying R497 polymorphism on EGFR extracellular domain 4 exhibit more unfavorable responses to cetuximab than those carrying K497 (52). Although we also observed endogenous EGFR R497 methylation from MS analysis (Supplemental Figure 1E), suggesting that this arginine methylation event may be another factor that contributes to cetuximab resistance, PRMT1 is not the methyltransferase for R497 methylation (D4), at least from our in vitro methylation assay (Figure 1B). Thus, identification of the PRMT that is responsible for R497 methylation would be critical to address this issue in the future. It is interesting to note that R198C mutation of EGFR was observed in colorectal and glioma patient samples. One possibility contributing to the positive effect of the R198C mutation on tumorigenesis may be that a substitution from Arg to Cys induces a similar enhancement in EGFR signaling as arginine methylation. The R198 contributes to stabilizing the inactive state by connecting D2 with D1 (through electro-static and van der Waals interactions, and exclusion of water molecules). In the inactive state, D1 and D2 have more contacts. There are then several ways to affect this contribution of R198. Methylation is one of them, since it creates steric hindrance and adds hydrophobic groups, but a loss of the Arg interactions through mutation into another residue, such as cysteine, may have the same effect. In addition, as demonstrated by a previous study, an Arg to Cys mutation on erythropoietin receptor led to constitutively activated, disulfide-linked homodimers (53). It is not yet clear whether the EGFR R198C mutation might promote tumorigenesis via a similar mechanism that increases formation of active homodimerization. Further investigations into the detailed mechanism regarding the effects of Arg to Cyst mutation on EGFR signaling would be needed in the future.
Methylation of EGFR may occur in the ER and Golgi during translation before it is translocated to cell surface membrane, a process similar to glycosylation of many cell surface receptor proteins, and therefore cannot be regulated by EGF stimulation, as shown in Figure 6C. This suggests that PRMT1 may predispose newly synthesized EGFR molecules to adopt the active EGF-binding conformation at the cell surface. The presence of PRMT1 and SAM in the ER/Golgi (Figure 2D and Supplemental Figure 2) supported the hypothesis that EGFR may be methylated by PRMT1 in these subcellular compartments. These results are consistent with those in plant cells in which SAM is incorporated into the Golgi vesicles and other subcellular compartment as detected by the incorporation of carboxyl [14C]- SAM into these subcellular organelles (54). In mammals, characterization of stacked Golgi fraction using multidimensional protein identification technology suggested that a SAM transporter and methyltransferase may exist in the Golgi proteome isolated from rat liver (55). Interestingly, the same report showed that several ER or Golgi resident proteins are Arg dimethylated on their luminal domain, suggesting the existence of SAM transporter as well as methylation machinery in ER/Golgi compartment that may play important roles in regulating cellular physiology. A systematic approach would be required to further address this mechanism in the future.
In addition, the stoichiometry of EGFR methylation was about 10.5 % in SKCO1 cells (Supplemental Figure 6). In line with previous reports showing that ERα methylation is a dynamic process, requiring PRMT1 for methylation and JMJD6 arginine demethylase to remove the methylated moiety (56, 57), global proteomic analysis also demonstrated that arginine methylation sites in human cells are regulated dynamically by an unknown Arg demethylase during transcriptional arrest (58). It is possible that the Arg demethylation process is a general event regulating cellular Arg methylation level.
We previously reported methylation on EGFR intracellular domain mediated by PRMT5 inhibits downstream ERK activation in breast cancer cells (3). Here, methylations on EGFR extracellular domain regulated by PRMT1 enhance receptor function in colorectal cancer cells. Interestingly, in colorectal cancer patients, elevated level of PRMT5 was observed and coincided with poor prognosis (59). A molecular switch that governs the tumor-suppressive or oncogenic activities of different PRMTs and their downstream target proteins in different cancer types remains to be further investigated. Several arginine methylated RTKs, such as EGFR and VEGFR-2, have been reported (3, 60). The current study demonstrates that arginine methylation on EGFR extracellular domain affects ligand-mediated signaling and may contribute to cetuximab resistance. Since many cell surface RTKs — including EGFR, HER2, and MET — are therapeutic targets, and since monoclonal antibodies against the extracellular domain of these RTKs are being used for therapy, this raises an interesting possibility that arginine methylation of this region on other RTK may also play a role in the regulation of their activities and in response to their corresponding monoclonal antibody therapeutics.
Overall, the role of PRMT1-mediated EGFR methylation in colorectal cancer tumorigenesis — and its correlation with poorer patient outcomes and cetuximab response by affecting the EGF-EGFR binding affinity and subsequent signaling activation, as demonstrated here — provide an insight into the response to EGFR-targeted therapy and also open an avenue toward the understanding of how arginine methylation regulates the function of RTKs.
Methods
Constructs, reagents, peptides, and antibodies.
EGFR (GI: 2811086) and PRMT1 (GI: 359338974) constructs were prepared as described previously (3). Four extracellular domains, juxtamembrane domain, kinase domain, and C-terminal tail of EGFR were further subcloned into pGEX vector for the preparation of truncated EGFR recombinant proteins. EGFR (R198/200K) mutagenesis was generated using the QuickChange Site-Directed Mutagenesis Kit according to the manufacturer’s protocol (Stratagene). EGF (Sigma-Aldrich) was prepared according to the manufacturers’ instructions. Unmodified (Ac-QCSGRCRGKSPSD-C), asymmetric di-methylated (Ac-QCSG[asymmetric dimethyl-R]C[asymmetric dimethyl-R]GKSPSD-C), symmetric di-methylated (Ac-QCSG[symmetric dimethyl-R]C[symmetric dimethyl-R]GKSPSD-C), and mono methylated (Ac-QCSG[mono methyl-R]C[mono methyl-R]GKSPSD-C) EGFR peptides were chemically synthesized by LifeTein for antibody production in mice and dot blot and peptide competition assays. Anti-EGFR antibody (catalog 06-847, Millipore) was used to detect full-length EGFR. Antibodies against phospho–Tyr 1086 (catalog ab5650, Abcam) and –Tyr 1148 (catalog ab5651, Abcam) were used for detection of EGFR activation. Antibodies to ERK (catalog 06-182, Millipore) and phospho-ERK (catalog 9106, Cell Signaling Technology) were used to detect the EGFR downstream signaling activation. Anti-PRMT1 (catalog 2449, Cell Signaling Technology) was used to detect PRMT1 level after overexpression or knockdown. Anti-SAM antibody was used to detect location of SAM (catalog SAM88-A50, Eagle Biosciences). Anti-tubulin antibody was purchased from Sigma-Aldrich (catalog T5168).
Cell culture.
SKCO1 (KRAS G12V) and SW48 (KRAS WT) were purchased from ATCC. GEO (KRAS G12A) and HT29 (KRAS WT) cells were gifts from Zhen Fan (MD Anderson Cancer Center). GEO, HT29, and SW48 cells were cultured in DMEM/F-12 with 10% FCS. SKCO1 cells were cultured in MEM with 10% FCS. All cell lines were characterized as mycoplasma negative and validated by STR DNA fingerprinting using the AmpF_STR Identifiler kit according to manufacturer’s instructions (catalog 4322288, Applied Biosystems). The STR profiles were compared with known ATCC fingerprints (www.ATCC.org) and with the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/) (Nucleic Acids Research 37:D925-D932, PMCID: PMC2686526). The STR profiles matched known DNA fingerprints or were unique. Before EGF stimulation, 80% confluent cells were serum-starved for 20 hours and then stimulated with 50 ng/ml EGF for indicated time or 50 ng/ml TGFα for 20 minutes.
shRNA construct and transfection.
Lentiviral-based pLKO.1 PRMT1 shRNA vector was obtained from Academia Sinica. The pLKO.1 scrambled shRNA vector was purchased from Addgene. The PRMT1-targeting shRNA sequences used in the lentiviral construct were: 5′-CCGGCCGGCAGTACAAAGACTACAACTCGAGTTGTA GTCTTTGTACTGCCGGTTTTTG-3′ (shRNA #1) and 5′-CCGGGCAAGTGAAGCGGAAT GACTACTCGAGTAGTC ATTC CGCTTCACTTGCTTTTTG-3′ (shRNA #2). For lentiviral production, PLKO.1 PRMT1 shRNA vector, packaging (pCMV-dr8.Z dvpr), and envelope (pCMV-VSV-G) plasmids were cotransfected into 293T cells using Lipofectamine Reagent (Invitrogen). After 48-hour transfection, colon cancer cells were infected with viral particles. Stable knockdown clones were selected by culturing cells in medium with 4 μg/ml puromycin for 1 month.
Mass spectrometry.
EGFR was isolated by immunoprecipitation with anti-EGFR antibody and then was analyzed by SDS-PAGE. The protein band corresponding to EGFR was excised and subjected to in-gel digestion with trypsin. After isolation by immobilized metal affinity chromatography, the enriched methyl peptides were analyzed by microliquid chromatography/tandem mass spectrometry.
In vitro methylation assay.
To prepare recombinant PRMT1 and EGFR fragments for in vitro methylation assay, the cDNA of PRMT1 or EGFR fragments were subcloned into pGEX vector (GE Healthcare), and GST-tagged recombinant proteins were expressed in E. coli BL21 (DE3) strain by 1 mM IPTG (isopropyl-β-D-thiogalactopyranoside) induction. GST-tagged recombinant proteins were then purified using glutathione sepharose 4B (GE Healthcare) and dialyzed against phosphate-buffered saline. To perform in vitro methylation assay, GST-tagged recombinant PRMT1 and EGFR fragments were incubated together in the presence of 2.2 Ci S-adenosyl-L-[methyl-3H] methionine (85 Ci/mmol from a 0.55 mCi/ml stock solution; MP Biomedicals) for 1 hour at 30°C in a final volume of 50 μl of phosphate-buffered saline. After incubation, samples were separated by SDS-PAGE and methylation levels were examined by fluorography.
In vivo protein interaction by Duolink assay.
Cells were seeded in 8-well chamber slides. When harvesting cells, cells were washed with cold PBS twice and fixed with 4% paraformadehyde at 4°C for 2 hours. After 2 more PBS washes, cells were permeabilized by cold 0.2% Triton X-100 (Sigma-Aldrich)for 30 minutes at room temperature and subjected to Doulink assay (Olink Bioscience) according to the manufacturers’ instructions.
Cell proliferation assay.
Cells were seeded in 6-well plates (triplicate), and fresh medium (with or without gefitinib or cetuximab) was added every day. Cells were then trypsinized, and cell numbers were counted on a daily basis.
Anchorage-independent growth assay for colony formation.
The base layer of cell growth matrix containing DMEM/F12 medium, 10% FBS, and 0.5% agar was paved in 6-well plates (1.5 ml per well). After solidification of the base layer, the top layer (1.5 ml per well) containing DMEM/F12 medium, 10% FBS, 0.35% agarose, and cells was plated. Culture medium (1 ml) was added to each well and changed every 3 days. After 4-week culture, colonies were stained by 0.005% crystal violet. Colonies with a diameter larger than 0.5 mm were counted.
Dimerization assay.
Cells were starved in serum-free medium for 24 hours. After starvation, cold PBS containing 50 ng/ml EGF was added onto plates for 30 minutes at 4°C. Then, cells were washed with cold PBS (137 mM NaCl, 0.67 mM KCl, 8 mM Na2HPO4, 1.4 mM KH2PO4) 3 times and incubated for 2 hours at 4°C with 5 mM cross linker BS3 (bis[sulfosuccinimidyl] suberate; Thermo Fisher Scientific) in PBS. After washing 3 times with cold PBS, cross-linking reactions were stopped by incubating cells in 50 mM Tris buffer (pH 7.5) for 15 minutes at ambient temperature. Cells were subsequently lysed and cell lysates analyzed by Western blotting.
Orthotopic colon cancer mouse model.
Nude female mice at 4–5 weeks of age were maintained at the MD Anderson Animal Facility for 1 week prior to injection of cancer cells. The cecum was exteriorized through a small midline laparotomy, and 107 HT29 cells expressing WT EGFR, EGFR methylation site mutant, or vector control with or without knockdown of PRMT1 were injected into the cecal wall. After injection, the abdominal wall was closed by wound clips. One month after surgery, tumors were harvested and tumor weights were measured.
IHC staining.
IHC of methylated EGFR was performed using homemade me-R198/200 antibody. Colorectal cancer tissue microarrays were purchased from National Cancer Institute Cancer Diagnosis Program. Colorectal cancer samples from patients treated with cetuximab were collected from MD Anderson Cancer Center (n = 41). Head and neck cancer samples from patients treated with cetuximab were collected from Taipei Veterans General Hospital (n = 38) and University of Pittsburgh Medical Center (n = 21). Samples were deparaffinized and rehydrated. Antigen retrieval was performed by using 0.01 M sodium-citrate buffer (pH 6.0) in a microwave oven. The sections were treated with 1% hydrogen peroxide in methanol for 30 minutes to block endogenous peroxidase activity. After 1 hour of preincubation in 10% normal serum to prevent nonspecific staining, the samples were incubated with primary antibodies at 4°C overnight. The sections were then treated with biotinylated secondary antibody, followed by incubations with avidinbiotin peroxidase complex solution for 1 hour at room temperature. Color was developed with the 3-amino-9-ethylcarbazole solution. Counterstaining was carried out using Mayer’s hematoxylin. All immunostained slides were scanned on the Automated Cellular Image System III (ACIS III) for quantification by digital image analysis. A total score of protein expression was calculated automatically from the percentage of immunopositive cells and immunostaining intensity.
Saturation binding assay.
ELISA 96-well plates were captured with 3 μg/ml anti-EGFR antibody (Abcam) in 0.2 M sodium phosphate buffer (pH 6.5) at 100 μl/well overnight at room temperature. The plates were then rinsed 3 times with PBS with 0.05% Tween-20 (PBST) and blocked with 200 μl/well of 1% BSA solution at 37°C for 2 hours. After rinsing 3 times with PBST, 100 μl/well of HT29-RIPA lysates or RIPA buffer only as a negative control were added and incubated at 37°C for 1.5 hours. The plates were then washed with 400 μl/well of PBST 3 times, followed by addition of recombinant human biotin–EGF at a series of diluted concentrations in RIPA buffer. After incubation at 37°C for 1.5 hours, wells were washed with 400 μl/well of PBST 3 times, added by 100 μl/well of streptavidin-conjugated HRP (1:2,000 in blocking buffer), and incubated for 30 minutes at room temperature. The wells were washed again with PBST 3 times, and 100 μl/well of TMB as a peroxidase substrate were added and incubated for 30 minutes at room temperature. The reaction was terminated by addition of 50 μl/well of stop solution. The optical density was determined at 450 nm, corrected by subtraction of readings at 570 nm, using a BioTek Synergy Neo multi-mode reader. The Kd was estimated by the above binding data and then transformed to create a Scatchard plot with GraphPad Prism program (version 6; Prism Software Inc.).
Clonogenic assay.
Cells (5,000 per well) were seeded in 24-well plates. Culture medium was changed every 3 days. After 10 days of culturing, cells were washed by cold PBS twice and fixed by 4% paraformadehyde for 1 hour. Cells then were stained by 0.005% crystal violet at 4°C overnight. After ddH2O wash, colonies with a diameter larger than 0.5 mm were counted.
Statistics.
Statistical analyses were performed using SPSS software. The association between the expression level of methyl-EGFR and PRMT1 was analyzed by Spearman’s rank correlation test. Survival curves were plotted using Kaplan-Meier method, and log-rank tests were performed to evaluate prognostic differences between groups for categorical variables. Differences were analyzed by a 2-tailed Student’s t test, with a P value < 0.05 being considered as statistically significant.
Study approval.
All animal experiments were carried out in accordance with approved protocol from Institutional Animal Care and Use Committee (IACUC) at MD Anderson Cancer Center (068706139) and the study adhered to NIH guidelines for the use of experimental animals. Human colorectal cancer and head and neck cancer tissues with cetuximab treatment were obtained under protocols approved by the University of Texas MD Anderson Cancer Center IRB (PA11-0959).
Author contributions
HWL and MCH designed and conceived the study. HWL and MCH wrote the manuscript. HWL, JMH, WX, YNW, CKC, PHT, HY, YFF, HJL, and HHL performed the experiments. HLW generated me-R198/200 Ab. WCC and CHC performed MS analysis. STA and JEL performed structural analysis. MPM, SK, SKT, MHY, MS, and JRG provided clinical tissue samples and related medical records.
Supplementary Material
Acknowledgments
We thank Jennifer L. Hsu for critical reading of the manuscript; Su Zhang, Jian-Guang Shi, Zhen-Bo Han, and Jin-Fong Lee for technical assistance; Zhen Fan for providing GEO and HT29 colorectal cancer cell line; Xiangbo Wan for providing patient samples; The National Cancer Institute for providing colorectal tissue microarrays; The Cancer Center Support Grant–funded (CA016672) Characterized Cell Line Core for performing STR DNA fingerprinting; and ShRNA/ORFeome Core Facility for providing shRNA constructs. Funding was provided by NIH (CA109311, CA099031, and CCSG CA16672 to M.C. Hung; P50CA097190 to J.R. Grandis); Cancer Prevention and Research Institute of Texas (RP150245); The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund (to M.C. Hung); Ministry of Science and Technology, International Research-intensive Centers of Excellence in Taiwan (I-RiCE; MOST 104-2911-I-002-302); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW104-TDU-B-212-124-002); Center for Biological Pathways; and the American Cancer Society (to J.R. Grandis). Research by S.T. Arold reported in this publication was supported by KAUST.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(12):4529–4543. doi:10.1172/JCI82826.
See the related Commentary beginning on page 4320.
References
- 1.Patel R, Leung HY. Targeting the EGFR-family for therapy: biological challenges and clinical perspective. Curr Pharm Des. 2012;18(19):2672–2679. doi: 10.2174/138161212800626148. [DOI] [PubMed] [Google Scholar]
- 2.Sasaki T, Hiroki K, Yamashita Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed Res Int. 2013;2013: doi: 10.1155/2013/546318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu JM, et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. 2011;13(2):174–181. doi: 10.1038/ncb2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen LK, Kolch W, Kholodenko BN. When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun Signal. 2013;11: doi: 10.1186/1478-811X-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bishayee S. Role of conformational alteration in the epidermal growth factor receptor (EGFR) function. Biochem Pharmacol. 2000;60(8):1217–1223. doi: 10.1016/S0006-2952(00)00425-1. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa HO, Takeuchi H, Haltiwanger RS, Irvine KD. Four-jointed is a Golgi kinase that phosphorylates a subset of cadherin domains. Science. 2008;321(5887):401–404. doi: 10.1126/science.1158159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yalak G, Vogel V. Extracellular phosphorylation and phosphorylated proteins: not just curiosities but physiologically important. Sci Signal. 2012;5(255): doi: 10.1126/scisignal.2003273. [DOI] [PubMed] [Google Scholar]
- 8.Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010;36(6 pt 1):423–435. doi: 10.1016/j.diabet.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Yoshimatsu M, et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer. 2011;128(3):562–573. doi: 10.1002/ijc.25366. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13(1):37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 11.Cha B, Jho EH. Protein arginine methyltransferases (PRMTs) as therapeutic targets. Expert Opin Ther Targets. 2012;16(7):651–664. doi: 10.1517/14728222.2012.688030. [DOI] [PubMed] [Google Scholar]
- 12.Wei H, Mundade R, Lange KC, Lu T. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle. 2014;13(1):32–41. doi: 10.4161/cc.27353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng L, et al. Anti-EGFR MoAb treatment in colorectal cancer: limitations, controversies, and contradictories. Cancer Chemother Pharmacol. 2014;74(1):1–13. doi: 10.1007/s00280-014-2489-6. [DOI] [PubMed] [Google Scholar]
- 14.Funakoshi T, Latif A, Galsky MD. Safety and efficacy of addition of VEGFR and EGFR-family oral small-molecule tyrosine kinase inhibitors to cytotoxic chemotherapy in solid cancers: a systematic review and meta-analysis of randomized controlled trials. Cancer Treat Rev. 2014;40(5):636–647. doi: 10.1016/j.ctrv.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Krawczyk PA, Kowalski DM. Genetic and immune factors underlying the efficacy of cetuximab and panitumumab in the treatment of patients with metastatic colorectal cancer. Contemp Oncol (Pozn) 2014;18(1):7–16. doi: 10.5114/wo.2013.38566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Custodio A, Feliu J. Prognostic and predictive biomarkers for epidermal growth factor receptor-targeted therapy in colorectal cancer: Beyond KRAS mutations. Crit Rev Oncol Hematol. 2013;85(1):45–81. doi: 10.1016/j.critrevonc.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Lievre A, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26(3):374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 18.De Roock W, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19(3):508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 19.Van Cutsem E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 20.Bokemeyer C, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27(5):663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 21.Morton RF, Hammond EH. ASCO provisional clinical opinion: KRAS, Cetuximab, and Panitumumab-clinical implications in colorectal cancer. J Oncol Pract. 2009;5(2):71–72. doi: 10.1200/JOP.0924603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laurent-Puig P, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–5930. doi: 10.1200/JCO.2008.21.6796. [DOI] [PubMed] [Google Scholar]
- 23.Fabian P, Berkovcova J. [Molecular predictive markers of EGFR-targeted therapy in metastatic colorectal cancer] Cesk Patol. 2011;47(4):154–158. [PubMed] [Google Scholar]
- 24.Silvestris N, et al. KRAS mutations and sensitivity to anti-EGFR monoclonal antibodies in metastatic colorectal carcinoma: an open issue. Expert Opin Biol Ther. 2009;9(5):565–577. doi: 10.1517/14712590902870394. [DOI] [PubMed] [Google Scholar]
- 25.Messner I, et al. KRAS p.G13D mutations are associated with sensitivity to anti-EGFR antibody treatment in colorectal cancer cell lines. J Cancer Res Clin Oncol. 2013;139(2):201–209. doi: 10.1007/s00432-012-1319-7. [DOI] [PubMed] [Google Scholar]
- 26.Mao C, Huang YF, Yang ZY, Zheng DY, Chen JZ, Tang JL. KRAS p.G13D mutation and codon 12 mutations are not created equal in predicting clinical outcomes of cetuximab in metastatic colorectal cancer: a systematic review and meta-analysis. Cancer. 2013;119(4):714–721. doi: 10.1002/cncr.27804. [DOI] [PubMed] [Google Scholar]
- 27.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30(29):3570–3577. doi: 10.1200/JCO.2012.42.2592. [DOI] [PubMed] [Google Scholar]
- 28.De Roock W, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304(16):1812–1820. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 29.Boisvert FM, Rhie A, Richard S, Doherty AJ. The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle. 2005;4(12):1834–1841. doi: 10.4161/cc.4.12.2250. [DOI] [PubMed] [Google Scholar]
- 30.Yu Z, et al. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 2012;22(2):305–320. doi: 10.1038/cr.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008;68(10):3803–3809. doi: 10.1158/0008-5472.CAN-07-6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng D, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases. J Biol Chem. 2004;279(23):23892–23899. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- 33.Hassdenteufel S, Klein MC, Melnyk A, Zimmermann R. Protein transport into the human ER and related diseases, Sec61-channelopathies. Biochem Cell Biol. 2014;92(6):499–509. doi: 10.1139/bcb-2014-0043. [DOI] [PubMed] [Google Scholar]
- 34.Scorilas A, Black MH, Talieri M, Diamandis EP. Genomic organization, physical mapping, and expression analysis of the human protein arginine methyltransferase 1 gene. Biochem Biophys Res Commun. 2000;278(2):349–359. doi: 10.1006/bbrc.2000.3807. [DOI] [PubMed] [Google Scholar]
- 35.Troiani T, et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19(24):6751–6765. doi: 10.1158/1078-0432.CCR-13-0423. [DOI] [PubMed] [Google Scholar]
- 36.Herrmann F, Fackelmayer FO. Nucleo-cytoplasmic shuttling of protein arginine methyltransferase 1 (PRMT1) requires enzymatic activity. Genes Cells. 2009;14(3):309–317. doi: 10.1111/j.1365-2443.2008.01266.x. [DOI] [PubMed] [Google Scholar]
- 37.Mathioudaki K, Papadokostopoulou A, Scorilas A, Xynopoulos D, Agnanti N, Talieri M. The PRMT1 gene expression pattern in colon cancer. Br J Cancer. 2008;99(12):2094–2099. doi: 10.1038/sj.bjc.6604807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogiso H, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110(6):775–787. doi: 10.1016/S0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 39.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11(2):507–517. doi: 10.1016/S1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 40.Alvarado D, Klein DE, Lemmon MA. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature. 2009;461(7261):287–291. doi: 10.1038/nature08297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarado D, Klein DE, Lemmon MA. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell. 2010;142(4):568–579. doi: 10.1016/j.cell.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464(7289):783–787. doi: 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- 43.Partridge EA, et al. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306(5693):120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 44.Blick SK, Scott LJ. Cetuximab: a review of its use in squamous cell carcinoma of the head and neck and metastatic colorectal cancer. Drugs. 2007;67(17):2585–2607. doi: 10.2165/00003495-200767170-00008. [DOI] [PubMed] [Google Scholar]
- 45.Martinelli E, De Palma R, Orditura M, De Vita F, Ciardiello F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin Exp Immunol. 2009;158(1):1–9. doi: 10.1111/j.1365-2249.2009.03992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bou-Assaly W, Mukherji S. Cetuximab (erbitux) AJNR Am J Neuroradiol. 2010;31(4):626–627. doi: 10.3174/ajnr.A2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Cunningham D, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 49.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22(7):1201–1208. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 50.Vallbohmer D, et al. Molecular determinants of cetuximab efficacy. J Clin Oncol. 2005;23(15):3536–3544. doi: 10.1200/JCO.2005.09.100. [DOI] [PubMed] [Google Scholar]
- 51.van Houdt WJ, et al. Oncogenic KRAS desensitizes colorectal tumor cells to epidermal growth factor receptor inhibition and activation. Neoplasia. 2010;12(6):443–452. doi: 10.1593/neo.92088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsieh YY, Tzeng CH, Chen MH, Chen PM, Wang WS. Epidermal growth factor receptor R521K polymorphism shows favorable outcomes in KRAS wild-type colorectal cancer patients treated with cetuximab-based chemotherapy. Cancer Sci. 2012;103(4):791–796. doi: 10.1111/j.1349-7006.2012.02225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watowich SS, Hilton DJ, Lodish HF. Activation and inhibition of erythropoietin receptor function: role of receptor dimerization. Mol Cell Biol. 1994;14(6):3535–3549. doi: 10.1128/MCB.14.6.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ibar C, Orellana A. The import of S-adenosylmethionine into the Golgi apparatus is required for the methylation of homogalacturonan. Plant Physiol. 2007;145(2):504–512. doi: 10.1104/pp.107.104679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu CC, et al. Organellar proteomics reveals Golgi arginine dimethylation. Mol Biol Cell. 2004;15(6):2907–2919. doi: 10.1091/mbc.E04-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poulard C, Rambaud J, Hussein N, Corbo L, Le Romancer M. JMJD6 regulates ERalpha methylation on arginine. PLoS One. 2014;9(2): doi: 10.1371/journal.pone.0087982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le Romancer M, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31(2):212–221. doi: 10.1016/j.molcel.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 58.Sylvestersen KB, Horn H, Jungmichel S, Jensen LJ, Nielsen ML. Proteomic analysis of arginine methylation sites in human cells reveals dynamic regulation during transcriptional arrest. Mol Cell Proteomics. 2014;13(8):2072–2088. doi: 10.1074/mcp.O113.032748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho EC, et al. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012;31(7):1785–1797. doi: 10.1038/emboj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rahimi N, Costello CE. Emerging roles of post-translational modifications in signal transduction angiogenesis. Proteomics. 2014;15(2–3):300–309. doi: 10.1002/pmic.201400183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.