

Abstract
Conventional wisdom holds that methylation of RTKs should be restricted to intracellular sites. Alterations — such as deletion, mutation, and proteolytic cleavage events — to the extracellular ligand binding and dimer interface domains of the EGFR can induce EGFR dimer formation, leading to aberrant receptor activation and oncogenic activity. Recently, the extracellular domain of EGFR was also shown to be methylated, suggesting that posttranslational protein methylation events directed to the extracellular dimer interface provide another mechanism to regulate the EGFR activation state by modulating receptor dimerization. Critically, these methylation events abrogate response to conformation-specific therapeutic antibodies such as cetuximab. In this issue of the JCI, Liao et al. investigate the role of protein arginine methyltransferase I (PRMT1) in regulating EGFR function in colorectal cancer. The authors provide evidence that methylation of R198 and R200 within the dimer interface enhances growth factor ligand binding and cetuximab resistance through induction and stabilization of the active EGFR dimer conformation. Delineation of these and other subtleties involved in oncogenic RTK activation and their response to targeted therapies should facilitate the development of improved antibody-based treatments.
EGFR: alterations to the extracellular domain drive oncogenic signaling
The EGFR is a critical oncoprotein and a very well characterized RTK signaling system. Mutations that affect the intracellular catalytic domain result in oncogenic activity, and these have been exploited to successfully deliver precision therapeutics to patients who harbor these mutations (1, 2). An emerging body of evidence now demonstrates that a diverse collection of molecular alterations affecting the extracellular domain — including genomic, transcript, proteolytic (3), and posttranslational processing (Table 1 and references therein) — can also drive oncogenic activity. As these alterations have not yet been successfully targeted by precision therapeutics, a better understanding for how these diverse modifications drive EGFR activation will be critical to effectively treat patients whose tumors express these variants.
Table 1. Oncogenic alterations of the EGFR ectodomain.
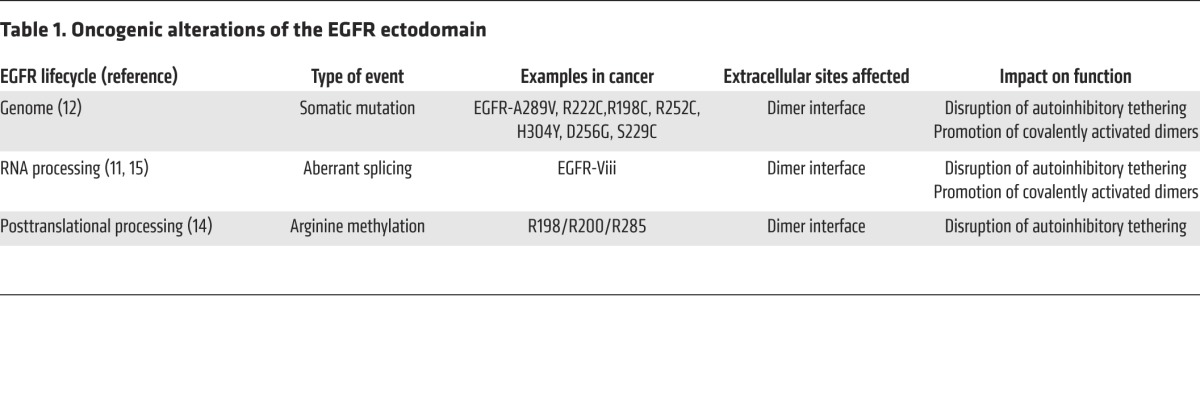
Structural and mechanistic studies have detailed the mechanism of EGF-ligand mediated activation through binding to the extracellular domain of EGFR. In the absence of bound ligand, EGFR is held in an autoinhibited conformation that is mediated by intramolecular interactions between domains 2 (CR1) and 4 (CR2) (4). A triangular salt bridge formed by Y270, D587, and K609 is a key molecular determinant for autoinhibition and is an element shared by other members of the ErbB family of RTK (5). This so-called tethered conformation occludes critical contacts that are required for dimerization, including a dimerization arm that contains the major energetic binding interactions in domain 2 (4). EGF binds to sites on both domains 1 (L1) and 3 (L2) that are distal to the dimerization domain, thereby stabilizing the receptor in the so-called extended conformation and inducing dimerization and receptor activation (6–8). Importantly, as activation by EGF requires binding to both domains 1 and 3, high-affinity binding occurs only in the extended conformation; otherwise, these sites are too far apart in the tethered conformation to accommodate binding at both sites. In the absence of ligand, it is estimated that only 5% of receptors are in the extended conformation. Therefore, ligand binding shifts the equilibrium to increase the size of this population (8).
The anti-EGFR antibody cetuximab binds exclusively to domain 3, providing a steric block of ligand binding (9). Additionally, cetuximab binding prevents EGFR from adopting the extended conformation and domain 2 from adopting the conformation required for dimerization. Therefore, cetuximab inhibits EGFR by both blocking ligand binding directly and preventing the receptor from adopting the conformation required for high-affinity ligand binding.
Previous studies have demonstrated that mutations and splicing events occurring in extracellular domains 2 and 4 disrupt autoinhibitory contact regions and promote uncontrolled ligand-independent receptor activation in cancer (Figure 1). For example, the EGFR splice variant III (EGFRvIII), which is commonly expressed in gliobastoma (GBM), disrupts domain 2, resulting in loss of key contacts, including Y270, that mediate transformation to the tethered conformation (10, 11). Moreover, genomic mutations have been characterized that affect the same receptor regions, including mutation of Y270 itself in the salt bridge triad and mutations that affect domains 2 and 4 (10), and confer oncogenicity (12). Proteolytic processing of the extracellular domain of EGFR is also observed in cancer (3) and may carry similar oncogenic consequences, as has been clearly shown for HER2 (13).
Figure 1. Ribbon diagram of the extracellular domain of EGFR.
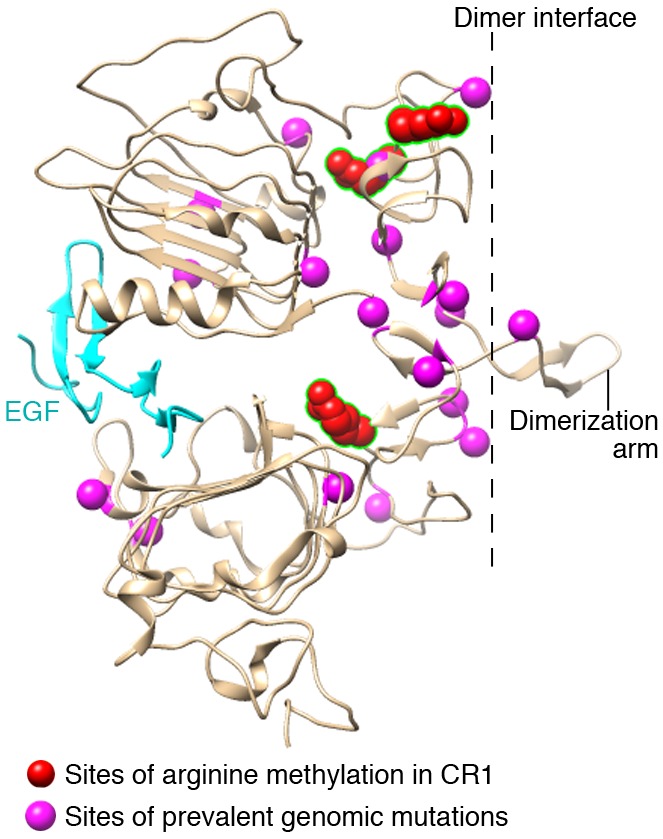
The diagram is based on the crystal structure of the inactive human EGFR (PBD accession 1IVO; http://www.ncbi.nlm.nih.gov/) and resolved using the extensible molecular modeling system Chimera (https://www.cgl.ucsf.edu/chimera/). EGF is shown in cyan. The position of mutations observed in cancer is shown in magenta and compiled using data deposited in cBioPortal (http://www.cbioportal.org/). Sites of arginine methylation within domain 2 (CR1; R198, R200, and R285) are shown in red. Numbering scheme derived from sequence without leader peptide. The dimerization interface and dimerization arm are noted.
Posttranslational modification of EGFR in colorectal cancer
In this issue, Liao et al. demonstrate that posttranslational modifications also affect these same extracellular receptor hotspot sites and can cause ligand-independent activation of EGFR by relieving autoinhibition (14). Liao and colleagues provide evidence of arginine methylation at six extracellular sites: R29, R74, R198, R200, R285, and R497. Two of these affect arginine residues that are commonly mutated in colorectal cancer: R198 and R285. The authors demonstrate that protein arginine methyl transferase 1 (PRMT1), which is expressed in the Golgi body, is responsible for selective methylation at both R198 and R200. Importantly, Liao et al. demonstrated that PRMT1 expression was elevated in colorectal cancer and associated with poor prognosis, providing functional and clinical relevance of these events.
Using an array of biochemical assays, Liao and colleagues demonstrated that arginine methylation at R198 and R200 increases EGF binding affinity, thereby stimulating EGF-induced receptor activation and EGFR-dependent cell proliferation. R198 and R200 are situated at the hinge region between domains 1 and 2, and methylation at these sites is predicted to disrupt autoinhibitory tethering and foster the extended conformation (14). Therefore, methylation is predicted to shift EGFR equilibrium in favor of the EGF-competent binding conformation.
Liao and colleagues further demonstrated that the arginine methylation at these sites confers resistance to cetuximab. Cetuximab binds to the tethered — but not the extended — conformation; thus, methylation shifts the equilibrium away from the EGFR conformation that is favored for cetuximab binding. Moreover, the affect of EGFR methylation on cetuximab binding was further supported by clinical data that correlated elevated levels of methylated EGFR in colorectal cancer patients with a higher recurrence rate following cetuximab treatment (14).
Together, the study by Liao and colleagues support the concept that a multitude of molecular alterations, stemming from genomics, transcriptomics, and posttranslational processing, converge to commonly activate EGFR by relieving autoinhibition and to foster receptor dimerization. This understanding of EGFR regulation carries important clinical implications, since expression levels of these EGFR variants may direct patient treatment decisions for existing EGFR antibodies and also may instruct the design of next-generation antibody therapeutics to more selectively and efficaciously inhibit them.
Footnotes
Conflict of interest: David M. Epstein and Elizabeth Buck have first-tier conflicts as co-founders of ASET Therapeutics, LLC, a private biotech company focused on discovering and developing small molecule inhibitors as potential precision therapeutic agents to target variants of EGFR and other RTKs. Elizabeth Buck is an employee of ASET Therapeutics, and both David M. Epstein and Elizabeth Buck are named inventors on pending patent applications assigned to ASET Therapeutics.
Reference information:J Clin Invest. 2015;125(12):4320–4322. doi:10.1172/JCI85001.
See the related article beginning on page 4529.
References
- 1.Russo A, et al. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget. 2015;6(29):26814–26825. doi: 10.18632/oncotarget.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mathisen MS, Kantarjian HM, Cortes J, Jabbour EJ. Practical issues surrounding the explosion of tyrosine kinase inhibitors for the management of chronic myeloid leukemia. Blood Rev. 2014;28(5):179–187. doi: 10.1016/j.blre.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Chen M, Chen LM, Lin CY, Chai KX. The epidermal growth factor receptor (EGFR) is proteolytically modified by the Matriptase-Prostasin serine protease cascade in cultured epithelial cells. Biochim Biophys Acta. 2008;1783(5):896–903. doi: 10.1016/j.bbamcr.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11(2):507–517. doi: 10.1016/S1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 5.Bouyain S, Longo PA, Li S, Ferguson KM, Leahy DJ. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc Natl Acad Sci U S A. 2005;102(42):15024–15029. doi: 10.1073/pnas.0507591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogiso H, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110(6):775–787. doi: 10.1016/S0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 7.Garrett TP, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110(6):763–773. doi: 10.1016/S0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 8.Burgess AW, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12(3):541–552. doi: 10.1016/S1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Brennan CW, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87(21):8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JC, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006;3(12): doi: 10.1371/journal.pmed.0030485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pedersen K, et al. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol Cell Biol. 2009;29(12):3319–3331. doi: 10.1128/MCB.01803-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao H-W, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125(12):4529–4543. doi: 10.1172/JCI82826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ymer SI, et al. Glioma specific extracellular missense mutations in the first cysteine rich region of epidermal growth factor receptor (EGFR) initiate ligand independent activation. Cancers (Basel) 2011;3(2):2032–2049. doi: 10.3390/cancers3022032. [DOI] [PMC free article] [PubMed] [Google Scholar]