

Abstract
The small intestine is a dynamic and complex organ that is characterized by constant epithelium turnover and crosstalk among various cell types and the microbiota. Lymphatic capillaries of the small intestine, called lacteals, play key roles in dietary fat absorption and the gut immune response; however, little is known about the molecular regulation of lacteal function. Here, we performed a high-resolution analysis of the small intestinal stroma and determined that lacteals reside in a permanent regenerative, proliferative state that is distinct from embryonic lymphangiogenesis or quiescent lymphatic vessels observed in other tissues. We further demonstrated that this continuous regeneration process is mediated by Notch signaling and that the expression of the Notch ligand delta-like 4 (DLL4) in lacteals requires activation of VEGFR3 and VEGFR2. Moreover, genetic inactivation of Dll4 in lymphatic endothelial cells led to lacteal regression and impaired dietary fat uptake. We propose that such a slow lymphatic regeneration mode is necessary to match a unique need of intestinal lymphatic vessels for both continuous maintenance, due to the constant exposure to dietary fat and mechanical strain, and efficient uptake of fat and immune cells. Our work reveals how lymphatic vessel responses are shaped by tissue specialization and uncover a role for continuous DLL4 signaling in the function of adult lymphatic vasculature.
Introduction
The small intestinal epithelium provides a barrier between the microorganism-rich intestinal lumen and the underlying stroma. Epithelial cells are constantly renewed from the stem cell niche at the crypt bottoms and migrate until reaching villus tips after differentiation (1). Sharp morphogen gradients control this polarized structure, including high Wnt and Notch signaling in the crypts and high TGF-β/BMP and Hh signaling in villi (1). Furthermore, the small intestine is unique in its role in immunity, as the gut harbors billions of microbes, and retinoic acid–driven fine-tuning of specialized immune cells ensures proper intestinal homeostasis (2). The intestine is a site of strong mechanical forces as well, as continuous piston-like villus contractions and peristaltic movement of the gut wall promote both absorption and movement of food particles (3).
The intestinal vasculature plays a key role in the regulation of nutrient absorption and gut immune function. Amino acids, carbohydrates, and short- and medium-chain fatty acids are taken up by blood capillaries in the villi for systemic distribution. Furthermore, retinoic acid–imprinted tolerogenic regulatory T cells home to the gut via intestinal microvasculature, and they play a key role in intestinal tolerance toward dietary antigens and the microbiota (4). Specialized lymphatic vessels in the villi, called lacteals, absorb long-chain fatty acids and fat-soluble vitamins. In addition to fat transport, lymphatic vessels of the gut serve as a main route for transport of microbial antigens and antigen-presenting cells to the mesenteric lymph node, including dietary vitamin A/retinoic acid–programmed CD103+ DCs. Such DCs migrate to the draining mesenteric lymph nodes, where they present retinoic acid along with processed antigen to T cells to endow them with gut-homing properties (2, 4).
Lacteals form during late embryogenesis and in the early postnatal period and expand into the villus by P7 (5, 6). Signaling via VEGFR3 and its coreceptor neuropilin 2 (NRP2) plays a key role in inducing postnatal sprouting and lacteal growth (6, 7). Disruption of intestinal lymphatic vasculature in adult mice leads to acute failure of immune surveillance systems in the small intestine and rapid lethality, underscoring the importance of intestinal lymphatic vessels in immune homeostasis (8). Impairment of lymphatic vascular function has also recently been implicated in the pathology of inflammatory bowel disease, where blocking lymphangiogenesis led to further disease exacerbation (9, 10). Mice with targeted inactivation of the transcription factor T-box transcription factor 1 (TBX1) have severe hypoplasia of mesenteric lymphatic vessels (11), and patterning of intestinal lymphatic vessels is perturbed in mice mutant for Pik3r1, encoding the p85α, p55α, and p50α subunits of PI3K (12). Human dermal and small intestinal lymphatic endothelial cells (LECs) have related but distinct gene expression profiles (13); however, to date, few studies reported mechanisms regulating adult small intestinal lymphatic vessels. Yet pathological conditions arise in adults in the context of highly specialized organ microenvironments; therefore, there is a marked significant need for a better understanding of the principles of adult lymphatic vasculature organization and maintenance in a tissue-specific context. Here, we provide a detailed 3D analysis of the adult small intestinal stroma and show that, unlike other adult lymphatic vascular beds, lacteals undergo continuous remodeling. We further report that such remodeling and maintenance of lacteals relies on delta-like 4 (DLL4), which promotes both the survival and regeneration of these vessels.
Results
Small intestinal stroma organization.
Although intestinal epithelial and immune cells are being intensely studied (1, 14–19), less focus has been given to other cell types in the small intestine. Using a whole-mount staining approach, we analyzed the 3D organization of the stroma of small intestinal villi. In agreement with their key role in nutrient absorption, villus blood capillaries, identified by staining for VEGFR2, form a well-organized cage-like structure in apposition to intestinal epithelial cells (Figure 1A). Intestinal lymphatic capillaries called lacteals — stained with LYVE1 (Figure 1B) but also positive for VEGFR2 (Figure 1A and Supplemental Figure 1B; supplemental material available online with this article; doi:10.1172/JCI82045DS1) and VEGFR3 (Supplemental Figure 1A) — were located in the villus center and are spatially separated from blood vessels (Figure 1B). As expected, NG2 staining identified pericytes closely associated with villus blood capillaries (Figure 1C) and a dense network of cells between villus epithelial cells and blood capillaries, which are most likely subepithelial myofibroblasts (20). Although previous studies in tissue sections suggested the presence of vascular smooth muscle cells (SMCs) on lacteals and blood capillaries (21, 22), we failed to confirm this observation in our 3D analysis (Figure 1, D and E). Instead, in agreement with high villus contractility (23), we observed tree-like smooth muscle fibers (Figure 1, D–F), identified by α–smooth muscle actin (αSMA) and desmin staining (Supplemental Figure 1C). αSMA+/desmin+ cells have been previously observed in intestinal villi (24), and αSMA–desmin+ cells most likely are subepithelial myofibroblasts (21). The SMC network extends throughout the entire villus length and is focally in close proximity to lacteals (Figure 1, E and F), whereas the terminal ends of SMC fibers are closely associated with villus blood capillaries (Figure 1G). Further analysis revealed that immune cells in the villus were closely associated with the SMC fibers, including CD3+FOXP3+ regulatory T cells (Supplemental Figure 1D). F4/80+ macrophages were observed throughout the intestinal stroma; however, CD11chi DCs, sometimes sampling the intestinal lumen, were found at discrete locations along SMC fibers (Figure 2A), suggesting a scaffolding role for this network. Cells in the intestinal stroma are attached to and interact with the extracellular matrix (ECM). Interestingly, the highest levels of the ECM protein fibronectin were detected in intestinal villi, especially on SMC fibers (Figure 2B). Integrins transduce signals from the ECM to promote cellular signaling and migration (25). Integrin α9 is highly expressed in LECs, and mechanosignaling, through β1 integrins, promotes VEGFR3 phosphorylation and LEC survival, proliferation, and migration (26). However, we found that in intestinal stroma, the highest levels of both fibronectin-binding integrins are found in villus SMCs, with weaker expression on lacteal LECs (Supplemental Figure 1E). As reported previously for other vascular beds, blood endothelial cells (BECs) and LECs expressed comparable levels of Itgb1, while Itga9 was preferentially expressed on LECs (Supplemental Figure 1F and ref. 27). Tenascin C, a matricellular protein induced upon tissue stretching, injury, and inflammation (28), was abundantly expressed in the villus and was almost undetectable in the submucosa (Figure 2C). At the same time, another ECM component, periostin, important for Wnt signaling (29), was present mostly in pericryptal fibroblasts, as previously reported (Figure 2C and ref. 30). Taken together, our data revealed a highly specialized and unique organization of the intestinal stromal compartment in adult animals.
Figure 1. Unique organization of small intestinal stroma.

Confocal microscope images of adult mouse small intestinal villi after whole-mount immunostaining. (A) A dense blood capillary network (VEGFR2, green) lies in close proximity to intestinal epithelial cells (E-cadherin, red). (B) Adult lacteals (LYVE1, red) and villus blood vessels (PECAM1, green) in adult small intestine. (C) Pericytes (NG2, green, arrowheads) are closely associated with villus blood capillaries (VEGFR2, red). (D) αSMA (red) staining reveals a network of SMCs in the villi, located inside the blood capillary network (green). MM, muscularis mucosa; CM, circular muscle; LM, longitudinal muscle. (E) A subset of villus SMCs (αSMA, cyan) are in close proximity to lacteals (LYVE1, red). (F) Higher-magnification view of near lacteal villus SMCs (αSMA, cyan). (G) Terminal ends of villus SMCs (αSMA, red) interact with villus blood vessels (PECAM1, green). Nuclei are stained with DAPI (blue). Ep, epithelial cells. Scale bars: 50 μm, A–D; 20 μm, E; 10 μm, F; 5 μm, G.
Figure 2. Immune cells and specialized ECM organization in adult small intestinal villi.
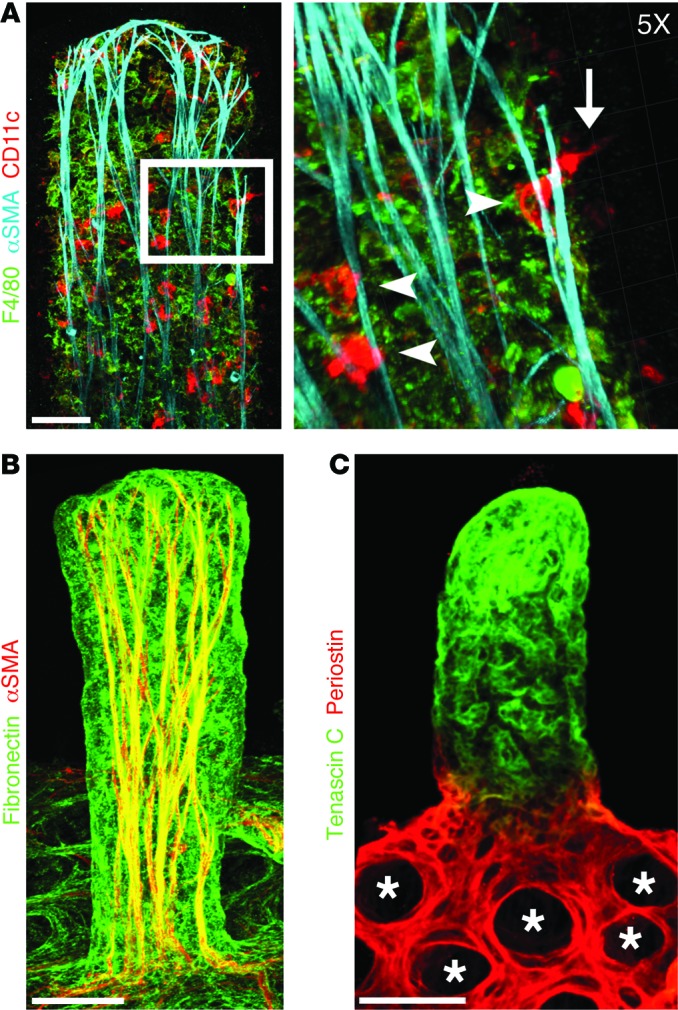
Confocal microscope images of adult mouse small intestinal villi after whole-mount immunostaining. (A) DCs (CD11c, red) are closely associated with SMC fibers (αSMA, cyan). Inset: ×5 3D Surpass projection of DCs (CD11c, red) interacting with SMC fibers (arrowheads). DCs associated with SMC fibers can be observed sampling the intestinal lumen (arrow). Macrophages are shown in green (F4/80). (B) Fibronectin (green) is highly expressed on villus SMCs (αSMA, red). (C) Expression of periostin (red) and tenascin C (green) is restricted to the crypts (*) and villi, respectively. Scale bars: 50 μm, A–C.
Adult lacteals are continuously regenerating.
(Lymph)angiogenesis is initiated in response to a reduced oxygen/nutrient supply, increased inflammation, or interstitial fluid pressure (26, 31). It is characterized by coordinated migration and increased proliferation of LECs or BECs in response to VEGFA or VEGFC until metabolic or fluid drainage requirements of the tissue or organ are met. It is necessary for proper embryonic development and is a feature of many pathological conditions (31, 32). In contrast, most adult vascular networks are quiescent and growth factor independent (33, 34). Examination revealed that filopodia were present in 75% of lacteals but were absent in submucosal lymphatics (Figure 3A and data not shown). As expected, lymphatic capillaries in other organs, such as skin, were blind ended and devoid of filopodia, which is typical for quiescent lymphatic vessels (Figure 3A). During embryonic development, lymphatic sprouting and filopodia formation are associated with LEC proliferation (31, 32). Therefore, we analyzed small intestinal LEC proliferation using the proliferation marker Ki67 and lymphatic endothelial marker PROX1 (35). As expected, strong Ki67 staining could be observed in intestinal crypt epithelial cells; however, virtually no Ki67+ LECs were detected in the intestinal submucosa, and only 2.5% of villus LECs were Ki67+ (Figure 3B). In addition, as expected, only 0.2% of LECs observed in adult ear skin were Ki67+ (Figure 3C). In contrast, analysis of E16.5–E17.5 embryonic skin, where there is active ongoing lymphangiogenesis, demonstrated that approximately 30% of LECs are Ki67+ (Figure 3D).
Figure 3. Lacteal regeneration in the adult small intestine.

(A) Adult lacteals, but not dermal lymphatic capillaries (LYVE1, red), display filopodia. Filopodia/lymphatic capillary (mean ± SD) in adult intestine and ear skin; n = 3–4. S.I., small intestine. (B) A small proportion of lacteal LECs are Ki67+ (PROX1+Ki67+DAPI+, white nuclei, arrowhead), while submucosal LECs (PROX1+DAPI+, pink nuclei, arrow) are quiescent. Percentage (mean ± SD) of Ki67+ LECs in the submucosal and lacteal lymphatic vessels of the small intestine; n = 8. (C) Adult ear skin LECs are quiescent (PROX1+DAPI+, pink nuclei). Percentage (mean ± SD) of Ki67+ LECs in adult ear skin; n = 4. (D) Embryonic skin LECs are actively proliferating (PROX1+Ki67+, yellow nuclei, arrowheads). Quiescent LECs are Ki67– (PROX1+DAPI+, pink nuclei, arrows). Percentage (mean ± SD) of Ki67+ LECs in E16.5–E17.5 embryonic skin; n = 3. (E) Relative lacteal length is constant along the entire small intestine. Whole-mount immunostaining of lacteals (LYVE1, red) and intestinal blood capillaries (PECAM1, green) from different intestinal segments of adult C57BL/6 mice. (F) The number of lacteals per villus (mean ± SD; n = 4) decreases from the duodenum to the ileum. (G) Quantification of relative lacteal length in the indicated parts of small intestine. Lacteal length/blood capillary cage length (mean ± SD); n = 4. (H) Quantification of lacteal filopodia in different parts of intestine. Filopodia/lacteal (mean ± SD); n = 4. Scale bars: 20 μm, A–D; 100 μm, E. *P < 0.05, 2-tailed unpaired Student’s t test with Welch’s correction. duo, duodenum; jej, jejunum.
Using the whole-mount staining approach, we also characterized lacteals in different intestinal compartments. In the mouse small intestine, villus length decreased from the proximal duodenum to the distal ileum (Figure 3E). Villi in the duodenum and jejunum frequently had on average two lacteals per villus, while most villi in the distal part of the intestine contained one lacteal (Figure 3, E and F). Such organization likely reflects the fact that most nutrient absorption occurs in the duodenum and jejunum. We next measured lacteal length as a percentage of the blood capillary cage length and the number of filopodia/lacteal. Interestingly, relative lacteal length was constant in the different intestinal segments (60%–70% of villus length), and the number of lacteal filopodia per villus remained similar along the length of the intestine (Figure 3, E, G, and H). Taken together, these observations suggested that adult lacteals, similar to intestinal epithelial cells, are in a constant state of regeneration, albeit much less proliferative than embryonic lymphatic vessels.
DLL4 maintains lacteal length and filopodia.
Notch signaling regulates many aspects of BEC biology, including arteriovenous differentiation and angiogenesis (36, 37). The role of Notch during sprouting lymphangiogenesis remains controversial: Notch signaling blockade was shown to both inhibit and promote lymphatic growth, suggesting a highly context-dependent role of this pathway (38–41). In BECs, the Notch ligand DLL4 is highly expressed in tip cells, where it regulates sprouting through the induction of Notch signaling in stalk cells (42–46); however, the expression pattern of DLL4 in the small intestine vasculature has not to our knowledge been studied. As expected, the highest levels of DLL4 were observed in arterioles (Figure 4A) and intermediate “salt-and-pepper” levels were found in blood capillaries of intestinal villi (Figure 4B), while venules, identified by staining for the venous marker endomucin (47), expressed low levels of DLL4 (Figure 4A). Of interest, high levels of DLL4 were observed in tip cells of sprouting lacteals (Figure 4C), while lower but detectable levels of DLL4 were also present in stalk cells and throughout the entire lacteal length (Figure 4C and Supplemental Figure 2) and submucosal lymphatics (data not shown). Although DLL4 is a membrane-bound protein, mostly punctate perinuclear staining was observed, similar to previous observations (42, 48).
Figure 4. DLL4 expression and Notch signaling are high in adult lacteals.

(A) DLL4 (white) displays perinuclear (arrows) and surface localization on villus arterioles. Endomucin (green) marks villus venules. Endothelial cells were identified by staining for zonula occludens 1 (ZO1, red). (B) DLL4 (white) is highly expressed on tip cells of villus blood capillaries (PECAM1, green). (C) DLL4 (white) is highly expressed on tip cells of villus lacteals (LYVE1, red); DAPI, blue. Dotted staining outside of lacteals is likely due to shedding from intestinal epithelial cells, where DLL4 was previously reported to be highly expressed (82). Relative DLL4 intensity (mean ± SD) in lacteal tip cells compared with neighboring stalk cells; n = 3. (D) Villus arterioles (VEGFR2, blue, arrowheads) have higher levels of Notch reporter (Venus, green) compared with venules (MAdCAM-1, red, arrows) in CBF1:H2B-Venus mice. Percentage (mean) Venus intensity of MAdCAM-1–negative arterioles versus MAdCAM-1–positive venules; n = 2. (E) Whole-mount immunostaining of lacteals (LYVE1, blue) and Venus (green) from adult CBF1:H2B-Venus mice. All LEC nuclei (PROX1, red) were also Venus+. Scale bars: 50 μm, A and D; 10 μm, B and C; 20 μm, E. **P < 0.01, 2-tailed unpaired Student’s t test.
To further monitor Notch activity in intestinal lacteals, we analyzed CBF1:H2B-Venus mice, where CBF1 binding sites drive expression of histone H2B–Venus (49). In agreement with DLL4 expression patterns and consistent with the previously described Notch activity in these vessels, arterioles were highly Venus+, while venule BECs, stained for mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1), expressed lower levels or no transgene (Figure 4D and refs. 50, 51). Importantly, lacteals were intensely positive for Venus, suggesting a high level of Notch signaling (Figure 4E), although potential differences in Notch signaling between tip and stalk cells are likely blunted because of the long reporter protein half-life (52). These results thus demonstrated that lacteals in the adult small intestine have continuous Notch signaling, which may be at least in part mediated by DLL4.
To investigate the role of DLL4 in lacteals, we generated Dll4fl/fl Prox1-CreERT2 mice (hereafter referred to as Dll4ΔLEC mice), in which Dll4 deletion is induced in lymphatic vessels upon injection of tamoxifen. Adult Dll4ΔLEC and control mice were injected with tamoxifen, and the intestine was examined 3 and 10 weeks later. Efficient Dll4 deletion was confirmed by both DLL4 immunostaining (Figure 5A) and quantitative reverse transcription PCR (RT-qPCR) for Dll4 on sorted LECs from control and Dll4ΔLEC intestine (Figure 5B). Quantification demonstrated a significant decrease in the number of filopodia in lacteals of Dll4ΔLEC compared with control mice (Figure 5C). In addition, 3 weeks after inactivation, Dll4ΔLEC mice had a tendency toward shorter lacteals, and after 10 weeks of deletion Dll4ΔLEC lacteals were significantly shorter than those of control mice (Figure 5D). In contrast, no difference in the skin lymphatic vascular organization or density was observed (Supplemental Figure 3). Control and Dll4ΔLEC mice had a similar number of Ki67+ intestinal LECs (Supplemental Figure 4A), suggesting that a decrease in lacteal LEC proliferation did not account for reduced lacteal length in Dll4ΔLEC mice. In addition, lacteals of control and Dll4ΔLEC mice had a similar cell density, but fewer total LECs (Supplemental Figure 4, B and C), suggesting that Dll4 inactivation impairs LEC survival and does not simply reduce cell size.
Figure 5. DLL4 is required for lacteal length maintenance.

(A) Loss of DLL4 protein in Dll4ΔLEC lacteals. Whole-mount immunofluorescence staining for DLL4 (red) and VEGFR3 (green) in WT and Dll4ΔLEC lacteals. (B) Efficient loss of Dll4 mRNA in Dll4ΔLEC LECs. Dll4 expression (mean ± SD) in sorted intestinal LECs from WT and Dll4ΔLEC mice analyzed by RT-qPCR; n = 3. (C) Dll4 inactivation reduces lacteal filopodia (LYVE1, red). LYVE1 is intentionally overexposed to highlight lacteal filopodia (white dots). Filopodia/lacteal (mean ± SD) of control and Dll4ΔLEC mice 10 weeks after tamoxifen injection; n = 4–5. (D) Lacteals are shorter in Dll4ΔLEC mice. Representative images of villus blood capillaries (PECAM1, green) and lacteals (LYVE1, red) from control and Dll4ΔLEC mice after 10 weeks of Dll4 deletion. Lacteal length (mean ± SD) binned for given lengths 3 weeks (top) and 10 weeks (bottom) after tamoxifen injection; n = 3. Scale bars: 10 μm, A; 20 μm, C; 100 μm, D. *P < 0.05, ***P < 0.001, 2-tailed unpaired Student’s t test.
To compare the roles of DLL4 in blood and lymphatic intestinal capillaries, we analyzed the intestine of mice with inducible inactivation of Dll4 in blood vessels using Dll4fl/fl Pdfgb-CreERT2 mice. As observed previously in other vascular beds (44, 46, 53, 54), loss of Dll4 resulted in a dramatic increase in filopodia, vessel branching, and the formation of sheet-like endothelial structures (Supplemental Figure 4D). With these results, we conclude that continuous DLL4 signaling is required for maintenance of lacteals, and the outcomes of DLL4 inactivation differ substantially in blood versus lymphatic capillaries.
DLL4 promotes lacteal LEC tip cell phenotype, survival, and efficient migration.
In BECs DLL4 expression confers competence to preferentially migrate to the tip cell position, as determined by competition assays in mosaic Dll4+/+/Dll4–/– embryoid bodies (42). Furthermore, LEC DLL4 overexpression promoted migration to the tip cell position in LEC spheroids in vitro (40). To study the in vivo function of DLL4 in lymphatic endothelial tip cell positioning, proliferation, and survival, we generated Dll4fl/fl Prox1-CreERT2 Rosa26-EYFP mice, in which Cre-mediated recombination induces both deletion of Dll4 and YFP expression in LECs, while in control Prox1-CreERT2 Rosa26-EYFP mice, only YFP is expressed. We injected adults of both strains with a reduced concentration of tamoxifen to induce mosaic Cre activation and then examined lacteals by whole-mount immunostaining after 3 weeks. Control lacteals had, on average, 40% YFP+ LECs; however, only 20% of LECs expressed YFP in Dll4ΔLEC animals (Figure 6, A and B). Further analysis of YFP+ LEC position in lacteals revealed that YFP+ cells in control animals were nearly twice as likely to be present in the tip cell position than YFP+ cells in Dll4ΔLEC mice, even after taking into account the reduced number of Dll4-deficient LECs (Figure 6, A and C). Quantification of the position of YFP+ LECs along the length of the lacteal also showed that YFP+ LECs in Dll4ΔLEC mice were found significantly lower on the lacteal than in control animals (Figure 6, A and D). Taken together, these results demonstrate a complex role for DLL4 in lacteals, where expression of DLL4 promotes both LEC survival and migration to the tip cell position.
Figure 6. DLL4 is required for LEC lacteal tip cell position and survival.

(A) DLL4 expression is important for tip cell competence. Control (Prox1-CreERT2 Rosa26-EYFP) and Dll4ΔLEC (Dll4fl/fl Prox1-CreERT2 Rosa26-EYFP) mice were injected with a reduced amount of tamoxifen to induce mosaic Cre activation. Representative images from WT and Dll4ΔLEC mosaic lacteals (YFP, green; PROX1, red; LYVE1, blue.) (B) Decreased number of Dll4-deficient LECs. Percentage (mean ± SD) of YFP+ LECs in mosaic Cre recombination control and Dll4ΔLEC Rosa26-YFP mice; n = 4. (C) Quantification of recombined YFP+ control or Dll4ΔLEC cells in tip position. Percentage (mean ± SD) of YFP+ lacteal tip cells in mosaic Cre recombination control and Dll4ΔLEC Rosa26-YFP mice; n = 4. (D) Quantification of recombined YFP+ control or Dll4ΔLEC cells position on vertical axis of the lacteal. Position of YFP+ lacteal cells in mosaic Cre recombination control and Dll4ΔLEC Rosa26-YFP mice as a percentage (mean ± SD) of lacteal length (lacteal base = 0, lacteal tip cell = 100); n = 4. (E) DLL4 depletion in vitro promotes apoptosis. Quantification of caspase-3+ LECs (caspase-3, red; β-catenin, green; DAPI, blue) 48 hours after transfection with two different siRNAs targeting DLL4. Mean percentage of caspase-3+ LECs. The graph represents pooled data of 2 independent experiments. Scale bars: 25 μm, A; 20 μm, E. *P < 0.05, 2-tailed unpaired Student’s t test.
We investigated whether lacteal DLL4 promotes LEC survival by costaining for PROX1 and caspase-3 on intestine sections from WT and Dll4ΔLEC mice. Epithelial cells at villus tips undergo apoptosis and slough off into the gut lumen. We found that although caspase-3+ differentiated epithelial cells could be readily detected in many villi, the number of caspase-3+ LECs in both the WT and Dll4ΔLEC mice was extremely low, making in vivo evaluation difficult (0.13% and 0.16%, respectively; Supplemental Figure 4E and data not shown). To further investigate the direct role of DLL4 in cell survival, we analyzed LEC apoptosis after DLL4 knockdown in vitro (Supplemental Figure 4F), which also decreased expression of the Notch target genes HES1, HEY1, and NRARP (Supplemental Figure 4G). While less than 0.5% of LECs transfected with control siRNA were caspase-3+, the number of caspase-3+ LECs was significantly increased after DLL4 inactivation (Figure 6E). Taken together with the in vivo data, demonstrating attrition of Dll4-deficient cells (Figure 6, A and B, and Supplemental Figure 4C), these results suggest that decreased cell survival of Dll4-deficient LECs is one of the mechanisms for decreased lacteal length.
We next analyzed whether the decreased number of Dll4ΔLEC lacteal tip cells could be due to a defect in LEC migration. Time-lapse microscopy was performed for 20 hours after “wounding” a monolayer of LECs transfected with control or DLL4-specific siRNAs and additionally treated with either BSA or VEGFC (Figure 7A and Supplemental Videos 1–4). Treatment with VEGFC increased the average total distance traveled by each cell; however, the average total distance remained unchanged between LECs transfected with control or DLL4-specific siRNA (Figure 7, A and B). In contrast, the actual distance traveled by siDLL4-transfected cells was decreased (Figure 7, A and C) and resulted in significant decreases in directionality (Figure 7D) in both BSA- and VEGFC-treated cells. These results suggest that, along with a decrease in LEC survival, inefficient LEC migration likely accounts for the decreased number of Dll4-deficient lacteal tip cells observed in vivo.
Figure 7. DLL4 is necessary for efficient LEC migration.

(A) Individual LEC migration was tracked for 20 hours after a wound was made in a LEC monolayer transfected with either control or DLL4-specific siRNA and treated with BSA or VEGFC. Tracks in red or black represent LECs that traveled less than or more than the average actual distance (avg. dist.), respectively. Data shown are representative of 3 independent experiments. (B) Total LEC track distance (mean ± SD) in arbitrary units (a.u.); n = 3. (C) Actual LEC distance (mean ± SD) traveled in arbitrary units; n = 3. (D) Directionality (actual/total distance; mean ± SD); n = 3. *P < 0.05, **P < 0.01, 2-tailed unpaired Student’s t test.
VEGFR3 signaling promotes DLL4 expression in lacteals.
VEGFR2 and VEGFR3 are essential for embryonic angiogenesis and lymphangiogenesis, respectively (31, 55). VEGFA/VEGFR2 signaling induces DLL4 in BECs (46, 56), and VEGFA and VEGFC induce DLL4 expression in cultured LECs (Supplemental Figure 5A and ref. 40). Both receptors are highly expressed in lacteals (Supplemental Figure 1, A and B, and Supplemental Figure 5B). To determine whether ligand-dependent activation of VEGFR2 or VEGFR3 contributes to DLL4 expression in lacteals, we treated adult mice with the VEGFR2- or VEGFR3-blocking antibodies DC101 (57) and mF4-31C1 (58), respectively.
Treatment with either blocking antibody for 7 days significantly decreased lacteal DLL4 protein in vivo (Figure 8A), while treatment with VEGFR3 blocking antibodies also increased membrane staining for lacteal VEGFR3 (Supplemental Figure 5B). To further study the role of VEGFR2 and VEGFR3 in DLL4 regulation, we knocked down both receptors, individually and simultaneously, in primary LECs (Supplemental Figure 5C) and measured DLL4 levels following VEGFC treatment (Figure 8B). As expected, DLL4 was upregulated following VEGFC treatment in LECs transfected with control siRNA. However, inactivation of either VEGFR2 or VEGFR3 prevented DLL4 induction in response to VEGFC (Figure 8B). In addition, treating LECs with the VEGFR3-specific ligand VEGFC C156S also induced DLL4 expression, albeit to a lesser extent than with WT VEGFC (Figure 8C). Thus, we conclude that ligand-dependent VEGFR3 and VEGFR2 signaling is the main inducer of DLL4 expression in lacteals. Consistent with these observations, VEGFR2- and VEGFR3-blocking antibodies reduced the number of lacteal filopodia (Figure 8D). Further analyses of lacteal length demonstrated that blocking VEGFR3 signaling resulted in a slight but significant reduction in lacteal length, while a tendency to have shorter lacteals was observed in DC101-treated animals (Figure 8E). Thus, we conclude that active VEGFR3 and, to a lesser extent, VEGFR2 signaling are necessary for lacteal length and filopodia maintenance in adult small intestinal villi.
Figure 8. DLL4 expression is dependent on active VEGFR3 and VEGFR2 signaling.

(A) Whole-mount immunostaining of DLL4 (white, arrow) on lacteals (LYVE1, red) of mice treated with control IgG, αVEGFR2, or αVEGFR3 blocking antibodies. Perinuclear DLL4 intensity/lacteal volume (mean ± SD); n = 3. (B) Serum-starved LECs transfected with control, VEGFR2, VEGFR3, and VEGFR2/VEGFR3 (R2+R3) siRNA were treated for 24 hours with either BSA or VEGFC and analyzed by Western blotting. Data are representative of 2 independent experiments. (C) Serum-starved LECs were treated with BSA, VEGFC, and VEGFC C156S for 24 hours and analyzed by Western blotting. Data are representative of 2 independent experiments. (D) Quantification of lacteal filopodia after αVEGFR2 or αVEGFR3 treatment. LEC filopodia/villus (mean ± SD); n = 3. (E) Representative whole-mount images of intestinal villus blood capillaries (PECAM1, green) and lacteals (LYVE1, red) after αVEGFR2 or αVEGFR3 treatment. Lacteal length (mean ± SD) binned for given lengths after 2 weeks of antibody administration; n = 3. Scale bars: 10 μm, A; 50 μm, E. *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed unpaired Student’s t test (E) or 1-way ANOVA with Bonferroni correction (A and D).
Lymphatic DLL4 is necessary for lacteal function.
One of the primary roles of lacteals is the transport of dietary long-chain fatty acids, secreted from enterocytes in the form of lipoprotein particles called chylomicrons (59). To study chylomicron transport, we gavaged Dll4ΔLEC and control animals with olive oil and measured accumulation of plasma triglycerides (TGs) and free fatty acids (FFAs) at different time points. Dll4ΔLEC mice had a significantly blunted increase in plasma TG and FFA levels compared with control animals (Figure 9A).
Figure 9. Lymphatic DLL4 is necessary for fat uptake and discontinuous lacteal adherens junctions.

(A) Quantification of plasma TGs and FFAs in olive oil–gavaged WT and Dll4ΔLEC mice. Plasma TG and FFA (mean ± SD) at indicated time points in WT and Dll4ΔLEC mice; n = 3. (B) Representative whole-mount images of submucosal lymphatic capillaries (LYVE1, green; VE-cadherin, red) from control and Dll4ΔLEC mice. Arrows indicate button-like junctions that are present in both WT and Dll4ΔLEC submucosal lymphatic capillaries. (C) Top: Representative whole-mount images of lacteals (LYVE1, green; VE-cadherin, red) from control and Dll4ΔLEC mice. White arrowheads, continuous junctions; white arrows, discontinuous junctions. Bottom: Representative high-magnification images of single lacteal LECs (VE-cadherin, black) from control and Dll4ΔLEC mice. (D) Quantification of lacteals with continuous, discontinuous, or mixed staining for VE-cadherin from control and Dll4ΔLEC mice. Percentage (mean ± SD) of lacteals in each group from control and Dll4ΔLEC mice; n = 5. Scale bars: 10 μm, B and C (top); 5 μm, C (bottom). *P < 0.05, **P < 0.01, ***P < 0.001, 2-tailed unpaired Student’s t test.
To gain a better mechanistic understanding of lacteal regression and impaired function, we analyzed the expression of previously identified targets and downstream effectors of DLL4/Notch signaling in LECs. However, despite efficient inactivation of Dll4 (Figure 5, A and B), we did not observe significant changes in the levels of ephrin B2 (Efnb2), Vegfr3, or Lyve1 (Figure 5D, Supplemental Figure 6, A and B; ref. 38), suggesting different underlying mechanisms. Mature lymphatic capillaries display a unique junctional organization, in which borders of oak leaf–shaped endothelial cells are discontinuously sealed by button-like junctions, containing both adherens and tight junction proteins, such as VE-cadherin and claudin-5, respectively (60). Such junctional organization is functionally important, as flap-like openings between adjacent “buttons” allow unimpeded access of interstitial fluid and immune cells into the lymphatic capillary lumen (60, 61). Of interest, actively growing lymphatic vessels display continuous cell-cell junctions, which likely reduce their uptake and transport capacity (60, 62). At the same time, lymphatic collecting vessels have continuous cell-cell junctions, important for avoiding lymph leakage during transport from capillaries to lymph nodes (60). To better understand the molecular mechanisms of impaired chylomicron transport in Dll4ΔLEC mice, we analyzed the organization of cell-cell junctions in intestinal lymphatic vessels by staining for VE-cadherin and LYVE1. Submucosal lymphatic vessels in both control and Dll4ΔLEC mice displayed the expected oak leaf–shaped pattern and button-like staining of LYVE1, with discontinuous localization of VE-cadherin (Figure 9B and ref. 60). Interestingly, lacteals in WT mice showed a mix of both continuous and discontinuous junctions (Figure 9C), demonstrating that they have features of both sprouting and quiescent lymphatic capillaries. Importantly, loss of DLL4 resulted in formation of continuous VE-cadherin junctions in all cells, which, together with reduced lacteal surface, likely explains impaired chylomicron uptake in these animals (Figure 9, C and D).
Discussion
Intestinal lymphatic vasculature plays a key role in the absorption and transport of dietary fat and in the regulation of immune surveillance in the gut. It is also likely that intestinal lymphatic vessel dysfunction contributes to several common diseases of industrialized societies, such as metabolic syndrome, inflammatory bowel diseases, and colon cancer. Furthermore, intestinal lymphatic vasculature is an attractive emerging target for mucosal vaccination and drug delivery, by virtue of direct access to the lymph node and bypass of liver metabolism. Despite the important physiological and pathophysiological roles of intestinal lymphatics, surprisingly little is known about the mechanisms that regulate their maintenance and function in the adult state. Here we report that lymphatic vessels in adult small intestinal villi are constantly regenerating and that continuous Notch signaling is necessary to maintain the optimal length, junctional organization, and function of lacteals (Figure 10).
Figure 10. DLL4 promotes continuous lacteal regeneration in the adult small intestine.

(A) During normal homeostasis the intestinal villi are subject to many stressors, including dietary fat, the gut microbiota, and villus movements, which lacteals encounter as chylomicrons, TLR ligands, and mechanical stress, respectively. In normal mice, VEGFC/D signaling, through VEGFR2 and VEGFR3, promotes lacteal DLL4 expression and Notch signaling, thus maintaining LEC survival, sprouting, and migration to replace damaged cells. Furthermore, LECs in lacteals have both continuous and discontinuous adherens junctions, balancing the need for continuous regeneration with the need for efficient chylomicron uptake. (B) In the absence of DLL4, lacteals are unable to undergo remodeling and have impaired survival and migration and thus are not able to maintain the normal length in villi. Lymphatic Dll4 deletion also promotes a shift from mixed adherens junctions to mostly continuous junctions, contributing to inefficient chylomicron uptake and transport. MLN, mesenteric lymph node.
Detailed characterization of adult lacteals revealed that they undergo continuous and slow remodeling (Figure 3). Unlike most other capillary beds, the vessels of small intestinal villi are continuously exposed to high concentrations of lipoprotein particles and other biologically active dietary and microbial products, such as retinoic acid (2) or Toll-like receptor ligands (63); gradients of osmolarity (64); and high mechanical strain due to peristalsis and villus contractions (23). Furthermore, the adult intestine is in a constant state of renewal, as the mouse epithelium is wholly replaced every 5 days (65). Thus, we believe that lacteal remodeling reflects a need for continuous lacteal regeneration due to the unique small intestine microenvironment, which itself is constantly renewing.
Mechanistically, we show that DLL4/Notch signaling, acting downstream of VEGFR3 and VEGFR2, mediates part of such a maintenance and regeneration program in lacteals. Although mechanical signals were shown to induce LEC proliferation during embryonic lymphangiogenesis through β1 integrins (26), in the adult intestinal villi Itgb1 is expressed at much lower levels in LECs than villus SMCs (Supplemental Figure 1E), perhaps accounting for lower levels of LEC proliferation in adult lacteals compared with embryonic lymphatic vessels. The role of Notch signaling in mammalian lymphatic vasculature has been investigated previously in several contexts. Developmental studies suggested that this pathway limits LEC differentiation and regulates skin lymphatic vessel sprouting (39, 41). Treatments using DLL4- and NOTCH1-blocking antibodies impaired both postnatal lymphangiogenesis and recruitment of SMCs to collecting lymphatic vessels (38), while studies of adult ear lymphangiogenesis using DLL4-Fc fusion protein suggested an opposite effect, due to increased responsiveness of LECs to VEGFA (40). Thus, the role of Notch signaling in lymphatic vasculature seems to be highly context dependent, as in the blood vasculature. Indeed, it was recently demonstrated that Notch activation, which in the pup retina leads to vessel quiescence and normalization (44, 66), instead increased bone marrow blood vessel density in young mice (67). Therefore, further investigation is needed to elucidate specific mechanisms determining endothelial response to Notch signaling.
Here, using genetic ablation of Dll4 in LECs, we demonstrate that DLL4 in adult lacteals promotes efficient migration and acquisition of the tip cell position, in agreement with in vitro mosaic LEC sprouting competition assays by Zheng et al. (40), and LEC survival, which is broadly in agreement with the role proposed by Niessen et al. (38). However, we did not observe significant changes in the expression or localization of Efnb2, Vegfr3, or Lyve1 in lacteals (Figure 5D and Supplemental Figure 6, A and B), nor defects in collecting lymphatic vessels of Dll4ΔLEC mice (J. Bernier-Latmani, unpublished observation). Rather, we show that the expression of DLL4 itself is induced through the activation of VEGFR3/VEGFR2 signaling, which places VEGFR3/VEGFR2 upstream of the DLL4/Notch signaling cascade (Figure 10). The exact molecular details of DLL4/Notch signaling in LECs remains to be investigated, and they may indeed be highly organ- or context-specific. Here, we show that one of the main consequences of genetic Dll4 inactivation is stabilization of lymphatic endothelial cell-cell junctions. Previous studies of BECs demonstrated that dynamic remodeling of VE-cadherin is a prerequisite for efficient tip cell positioning (68). Furthermore, stabilization of endothelial cell-cell contacts in mice bearing VE-cadherin fused with α-catenin strongly impairs formation of lymphatic vessels during embryogenesis (69). Thus, abnormal stabilization of adherens junctions may both underlie the inability of Dll4-deficient LECs to efficiently migrate to occupy the tip cell position and, at the same time, lead to the impairment of chylomicron uptake and reduction of lacteal transport capacity. Therefore, these observations suggest that an equilibrium must be maintained in adult lacteals between adherens junction plasticity and stabilization to allow efficient uptake of chylomicrons while also maintaining lacteal length through regeneration.
Our study also highlights intriguing similarities and differences in the usage of the Notch pathway by blood and lymphatic vessels. In both vessel types, DLL4 confers competence to preferentially migrate to the tip cell position (Figure 6, A and C, and refs. 40, 42); however, vessel hypersprouting and increased proliferation in the absence of DLL4 occurs only in BECs but not lacteal LECs. The latter, in contrast, show impaired survival and migration. Theoretically, such dissimilar behavior may be due to tissue-specific differences in production and gradients of angiogenic versus lymphangiogenic growth factors. Indeed, in our experiments we observed a preference for Dll4-deficient cells to remain in the lower part of the lacteal (Figure 6D), which is likely a result of the cell migration defect (Figure 7). Such nonmigrating cells might also reside in areas with low lymphangiogenic growth factor concentrations, and thus, with time, they will be depleted due to impaired cell survival. Alternatively, it is possible that the intracellular signaling downstream of Notch receptors differs in blood and LECs, possibly in a context-dependent manner. Since the in vitro responses of BECs and LECs to DLL4 inhibition are rather comparable (40, 42), it will be important in the future to establish the microenvironmental determinants of such distinct responses in vivo, for example by comparing the role of DLL4 in lymphangiogenic responses in different adult and embryonic tissues. Of interest, previous studies (70–72) demonstrated differential requirements for lymphatic vascular growth during the early postnatal period versus adulthood, and we found that inactivation of Dll4 at P1 did not inhibit lacteal expansion by P7 (data not shown). Taken together, these results further support the idea that distinct mechanisms are involved in lymphangiogenic vessel expansion versus regeneration in adult tissues.
Our results also raise important clinically relevant questions. Previous studies demonstrated that anti-angiogenic treatments (soluble VEGFR1; anti-VEGF and anti-VEGFR2 blocking antibodies) affect normal blood vessels in the organs with fenestrated endothelial cells, including the intestine, in addition to pathological vasculature (33, 34). As therapeutic agents targeting DLL4 signaling in tumor vasculature are in clinical trials (73), our study highlights the need to consider potential side effects of such treatment on intestinal vessel function. Last, we have shown one molecular mechanism of lacteal maintenance and how this directly affects lipid uptake into the bloodstream through lymphatic vessels. Further studies of such mechanisms may be of use for delivery of oral drugs, and the ability to shunt the route of orally administered drugs to either the lymphatic or blood system would be a major advance in drug delivery technology (74).
Methods
Animal models.
Dll4fl/fl, Pdgfb-CreERT2, Prox1-CreERT2, Rosa26-EYFP, and CBF1:H2B-Venus mice were previously described (27, 49, 75–77). Cre-mediated deletion was induced in 8- to 12-week-old mice either by i.p. (50 mg/kg mouse) or subcutaneous injection (100 mg/kg mouse) of tamoxifen in sunflower oil (Sigma-Aldrich). Mosaic deletion in Dll4fl/fl Prox1-CreERT2 Rosa26-EYFP and Prox1-CreERT2 Rosa26-EYFP mice was induced by i.p. injection of tamoxifen (7.5 mg/kg mouse). DC101 and mF4-31C1 blocking antibodies, which block the interaction of ligands with VEGFR2 and VEGFR3, respectively, were described previously (57, 58). Anti-VEGFR2, anti-VEGFR3, and control rat anti-HRP IgG (BioXCell) were delivered by i.p. injections (40 mg/kg mouse) every 3 days. For fat gavage and blood sampling, mice were injected with tamoxifen as described above 3 weeks prior to treatment. Mice were gavaged with 200 μl olive oil (Sigma-Aldrich) after being fasted for 6 hours. Blood was sampled at 0, 1, 2, and 3 hours after gavage by tail vein collection in EDTA-coated tubes. TG and FFA analysis was performed on a Cobas C111 (Roche Diagnostics).
Mouse tissue collection, staining procedures, and image acquisition.
Small intestine whole-mount staining was performed as in Stappenbeck et al. (78) with modifications. Prior to sacrifice anesthetized mice were perfused with 4% paraformaldehyde (PFA) by cardiac puncture. The small intestine was then quickly dissected in ice-cold PBS, cleaned, cut longitudinally, and pinned on silicon plates. Intestines were then fixed overnight at 4°C in 15% picric acid, 0.5% PFA, and 0.1 M sodium phosphate. Samples were washed 3 times in ice-cold PBS for 5 minutes and subsequently washed 3 hours with 10% sucrose in PBS and overnight in 20% sucrose, 10% glycerol in PBS. Following the last wash step, samples were stored in 0.1% sodium azide in PBS. Epithelial cells were removed for whole mount immunostaining with anti-DLL4, -NG2, -endomucin, –tenascin C, and -periostin antibodies, using 10 minutes of incubation in 1 mM EDTA on ice after PFA perfusion fixation and dissection. For ear whole-mount and intestinal sections, tissues were dissected and fixed in cold 4% PFA. Whole-mount and paraffin section immunostaining was performed as previously described (79). Primary antibodies are listed in Supplemental Table 1. Alexa Fluor 488, 555, and 647 fluorochrome–conjugated secondary antibodies (Invitrogen) were used for signal detection. Nuclei were detected with DAPI. Confocal images were obtained using either a Zeiss LSM 510 META or Leica SP5 TANDEM microscope, and standard fluorescence images were obtained using a Zeiss Axio Imager Z1 and analyzed using Imaris (Bitplane), ImageJ (http://imagej.nih.gov/ij/), and Photoshop (Adobe) software.
Intestinal LEC sorting.
LECs were isolated from adult intestine using cell sorting. Briefly, mice were sacrificed, and the intestine was dissected and flushed with ice-cold PBS. Peyer’s patches were removed and the intestine was cut into 1 cm pieces, which were put in a 10 mM EDTA solution with agitation at 37°C for 30 minutes to remove epithelial cells. The remaining tissue was then digested with Collagenase IV (3 mg/ml) in complete DMEM (Gibco; ThermoFisher Scientific) containing CaCl2 (2 mM) and 50 μg/ml DNase I (with constant stirring at 37°C) for 20 minutes and washed with medium. The cell suspension was incubated with labeled antibodies listed in Supplemental Table 1. FACS sorting was performed on a BD FACSAria IIu (BD Biosciences).
RNA isolation and RT-qPCR.
Total RNA was isolated using the QIAGEN RNeasy Plus Micro Kit. Reverse transcription was performed using Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics). Alternatively, mRNA was amplified using Ovation Pico WTA System V2 (NuGEN). RT-qPCR analyses were performed using StepOnePlus (Applied Biosystems) using SYBR Fast PCR Master Mix (Kapa Biosystems). Analysis of gene expression was carried out using the comparative Ct (ΔΔCt) method as described by the manufacturer. Sequences of PCR primers are provided in Supplemental Table 2.
Cell culture and treatment.
Primary LECs were isolated as described previously (13) and cultured in complete EBM-2 (Lonza). For VEGFC treatment, LECs were plated (on coverslips for cell staining) and serum starved for 16 hours prior to treatment with BSA, VEGFC (100 ng/ml), or VEGFC C156S (300 ng/ml, R&D Systems) for either 24 hours (Western blotting) or 48 hours (cell staining). For apoptosis analysis, cells were plated on coverslips and transfected with control or DLL4-specific siRNA and grown for 48 hours. Coverslips were fixed in 4% PFA prior to immunostaining. For migration analysis, cells were plated on glass-bottomed plates and transfected with control or DLL4-specific siRNA and grown for 48 hours, after which the monolayer was scratched using a pipette tip and medium containing BSA or VEGFC was added. Cells along the scratched area were recorded by time-lapse microscopy every 15 minutes for 20 hours using an Axio Observer Z1 microscope with stage-top incubator. Antibodies used for cell staining or Western blotting are listed in Supplemental Table 1, and siRNAs are listed in Supplemental Table 3.
Quantification.
Lacteal length was measured using images of villus PECAM1 and LYVE1 immunostaining and the lymphatic vessels density plugin for ImageJ (80). Lacteal length was measured and normalized by the blood capillary cage length. Quantification of lacteal filopodia and VE-cadherin–stained junctions was performed by examination with a Zeiss Axio Imager Z1 with ×63 objective by an observer blinded to the experimental protocol. For both quantifications at least 50 lacteals were analyzed per sample. Dermal lymphatic capillary length and branchpoint quantification was performed using AngioTool (https://ccrod.cancer.gov/confluence/display/ROB2/Home; ref. 81) after input of capillary network overlays drawn in Photoshop. Lacteal DLL4 expression was quantified using 3D reconstruction by masking the DLL4 signal to lacteal LYVE1 staining and quantifying perinuclear DLL4 using the Spots module in Imaris. A similar protocol was also used to quantify YFP expression in CBF1:H2B-Venus mice. Quantification of LEC migration was performed manually using the Track Points application in the MetaMorph software package. Statistical analysis and graphing of migration data were performed using the Chemotaxis and Migration Tool plugin for ImageJ (Ibidi).
Statistics.
Two-tailed unpaired Student’s t test or 1-way ANOVA was performed to determine statistical significance by calculating the probability of difference between two means, with a Bonferroni correction performed to account for multiple testing or Welch’s correction to account for differences in variance where noted. P values less than 0.05 were considered statistically significant. Data are shown as mean ± SD.
Study approval.
Animal experiments were approved by the Animal Ethics Committee of Vaud, Switzerland, and the CNIC and Madrid Council Ethics Committee.
Author contributions
JBL and TVP designed the study; JBL, CC, MB, MJ, SD, SR, CSD, and SS conducted experiments and acquired and analyzed data; OD, RB, FR, and SAL provided reagents; JBL and TVP wrote the manuscript. All authors discussed the results.
Supplementary Material
Acknowledgments
We thank Holger Gerhardt for useful discussions; Taija Mäkinen and Marcus Fruttiger for providing Prox1-CreERT2 and Pdgfb-CreERT2 mice, respectively; Eli Lilly for the gift of DC101 and mF4-31C1 antibodies; Kari Alitalo and Michael Jeltsch for VEGFC protein; Jean-Marie Calmes for help with LEC isolation; Céline Beauverd for mouse genotyping, colony maintenance, and help with immunohistochemistry; Amélie Sabine for help with artwork; François Renevey, Maria Broggi, and Camilla Jandus for experimental assistance; and Fiona Pigny, Marc LeCoultre, Nina Dumauthioz, and Cristina Mauri for participation at various stages of this work. The Animal, Cellular Imaging, Genomic Technologies, Mouse Metabolic, and Mouse Pathology Facilities of UNIL and the Gene Expression Core Facility of EPFL are gratefully acknowledged. This work was supported by the Swiss National Science Foundation (PPP0033-114898, CRSII3-141811, and 31003A-156266); Fondation MEDIC; the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/ under REA grant agreement 317250; and a Theodor and Gabriela Kummer MD-PhD fellowship (to C. Cisarovsky).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(12):4572–4586. doi:10.1172/JCI82045.
References
- 1.Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154(2):274–284. doi: 10.1016/j.cell.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35(1):13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gayer CP, Basson MD. The effects of mechanical forces on intestinal physiology and pathology. Cell Signal. 2009;21(8):1237–1244. doi: 10.1016/j.cellsig.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol. 2012;5(3):232–239. doi: 10.1038/mi.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim KE, Sung HK, Koh GY. Lymphatic development in mouse small intestine. Dev Dyn. 2007;236(7):2020–2025. doi: 10.1002/dvdy.21200. [DOI] [PubMed] [Google Scholar]
- 6.Xu YL, et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J Cell Biol. 2010;188(1):115–130. doi: 10.1083/jcb.200903137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tammela T, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454(7204):656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 8.Jang JY, et al. Conditional ablation of LYVE-1+cells unveils defensive roles of lymphatic vessels in intestine and lymph nodes. Blood. 2013;122(13):2151–2161. doi: 10.1182/blood-2013-01-478941. [DOI] [PubMed] [Google Scholar]
- 9.Jurisic G, Sundberg JP, Detmar M. Blockade of VEGF Receptor-3 aggravates inflammatory bowel disease and lymphatic vessel enlargement. Inflamm Bowel Dis. 2013;19(9):1983–1989. doi: 10.1097/MIB.0b013e31829292f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Alessio S, et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest. 2014;124(9):3863–3878. doi: 10.1172/JCI72189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L, et al. Tbx1 regulates Vegfr3 and is required for lymphatic vessel development. J Cell Biol. 2010;189(3):417–424. doi: 10.1083/jcb.200912037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mouta-Bellum C, et al. Organ-specific lymphangiectasia, arrested lymphatic sprouting, and maturation defects resulting from gene-targeting of the PI3K regulatory isoforms p85α, p55α, and p50α. Dev Dyn. 2009;238(10):2670–2679. doi: 10.1002/dvdy.22078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norrmen C, et al. Liprin β1 is highly expressed in lymphatic vasculature and is important for lymphatic vessel integrity. Blood. 2010;115(4):906–909. doi: 10.1182/blood-2009-03-212274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 15.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8(6):411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 16.Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31(3):401–411. doi: 10.1016/j.immuni.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9(5):356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 18.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8(6):435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140(6):859–870. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terada N, Ohno N, Murata S, Katoh R, Stallcup W, Ohno S. Immunohistochemical study of NG2 chondroitin sulfate proteoglycan expression in the small and large intestines. Histochem Cell Biol. 2006;126(4):483–490. doi: 10.1007/s00418-006-0184-3. [DOI] [PubMed] [Google Scholar]
- 21.Mifflin RC, Pinchuk, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G684–G696. doi: 10.1152/ajpgi.00474.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell DW, Pinchuk, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol. 2011;73:213–237. doi: 10.1146/annurev.physiol.70.113006.100646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Womack WA, Barrowman JA, Graham WH, Benoit JN, Kvietys PR, Granger DN. Quantitative assessment of villous motility. Am J Physiol. 1987;252(2 pt 1):G250–G256. doi: 10.1152/ajpgi.1987.252.2.G250. [DOI] [PubMed] [Google Scholar]
- 24.Madison B, Braunstein K, Kuizon E, Portman K, Qiao X, Gumucio D. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development. 2005;132(2):279–289. doi: 10.1242/dev.01576. [DOI] [PubMed] [Google Scholar]
- 25.Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15(12):802–812. doi: 10.1038/nrm3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Planas-Paz L, Strilic B, Goedecke A, Breier G, Fassler R, Lammert E. Mechanoinduction of lymph vessel expansion. EMBO J. 2012;31(4):788–804. doi: 10.1038/emboj.2011.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bazigou E, et al. Integrin-alpha 9 Is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev Cell. 2009;17(2):175–186. doi: 10.1016/j.devcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiquet-Ehrismann R, Chiquet M. Tenascins: regulation and putative functions during pathological stress. J Pathol. 2003;200(4):488–499. doi: 10.1002/path.1415. [DOI] [PubMed] [Google Scholar]
- 29.Malanchi I, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481(7379):85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 30.Kikuchi Y, et al. Periostin is expressed in pericryptal fibroblasts and cancer-associated fibroblasts in the colon. J Histochem Cytochem. 2008;56(8):753–764. doi: 10.1369/jhc.2008.951061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8(6):464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 32.Schulte-Merker S, Petrova TV. Lymphatic vascular morphogenesis in development, physiology, and disease. J Cell Biol. 2011;193(4):607–618. doi: 10.1083/jcb.201012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamba T, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290(2):H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y, et al. Anti-VEGF- and anti-VEGF receptor-induced vascular alteration in mouse healthy tissues. Proc Natl Acad Sci U S A. 2013;110(29):12018–12023. doi: 10.1073/pnas.1301331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98(6):769–778. doi: 10.1016/S0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- 36.Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21(20):2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 37.Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by Notch. Dev Cell. 2009;16(2):196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 38.Niessen K, et al. The Notch1-Dll4 signaling pathway regulates mouse postnatal lymphatic development. Blood. 2011;118(7):1989–1997. doi: 10.1182/blood-2010-11-319129. [DOI] [PubMed] [Google Scholar]
- 39.Fatima A, et al. Murine Notch1 is required for lymphatic vascular morphogenesis during development. Dev Dyn. 2014;243(7):957–964. doi: 10.1002/dvdy.24129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng W, et al. Notch restricts lymphatic vessel sprouting induced by vascular endothelial growth factor. Blood. 2011;118(4):1154–1162. doi: 10.1182/blood-2010-11-317800. [DOI] [PubMed] [Google Scholar]
- 41.Murtomaki A, et al. Notch1 functions as a negative regulator of lymphatic endothelial cell differentiation in the venous endothelium. Development. 2013;140(11):2365–2376. doi: 10.1242/dev.083865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakobsson L, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12(10):943–953. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 43.del Toro R, et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood. 2010;116(19):4025–4033. doi: 10.1182/blood-2010-02-270819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hellstrom M, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445(7129):776–780. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 45.Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445(7129):781–784. doi: 10.1038/nature05577. [DOI] [PubMed] [Google Scholar]
- 46.Suchting S, et al. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A. 2007;104(9):3225–3230. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.dela Paz NG, D’Amore PA. Arterial versus venous endothelial cells. Cell Tissue Res. 2009;335(1):5–16. doi: 10.1007/s00441-008-0706-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stenzel D, et al. Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep. 2011;12(11):1135–1143. doi: 10.1038/embor.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nowotschin S, Xenopoulos P, Schrode N, Hadjantonakis AK. A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Dev Biol. 2013;13: doi: 10.1186/1471-213X-13-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gale NW, et al. Haploinsufficiency of Δ-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101(45):15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shutter JR, et al. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000;14(11):1313–1318. [PMC free article] [PubMed] [Google Scholar]
- 52.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20(1):87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 53.Benedito R, et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484(7392):110–114. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 54.Benedito R, et al. Loss of Notch signalling induced by Dll4 causes arterial calibre reduction by increasing endothelial cell response to angiogenic stimuli. BMC Dev Biol. 2008;8: doi: 10.1186/1471-213X-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeltsch M, Leppanen V-M, Saharinen P, Alitalo K. Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb Perspect Biol. 2013;5(9): doi: 10.1101/cshperspect.a009183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lobov IB, et al. Δ-Like ligand 4 (DII4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A. 2007;104(9):3219–3224. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prewett M, et al. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999;59(20):5209–5218. [PubMed] [Google Scholar]
- 58.Pytowski B, et al. Complete and specific inhibition of adult lymphatic regeneration by a novel VEGFR-3 neutralizing antibody. J Natl Cancer Inst. 2005;97(1):14–21. doi: 10.1093/jnci/dji003. [DOI] [PubMed] [Google Scholar]
- 59.Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest. 2014;124(3):929–935. doi: 10.1172/JCI71610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baluk P, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204(10):2349–2362. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pflicke H, Sixt M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J Exp Med. 2009;206(13):2925–2935. doi: 10.1084/jem.20091739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol. 2012;180(6):2561–2575. doi: 10.1016/j.ajpath.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10(2):131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 64.Hallback DA, Jodal M, Mannischeff M, Lundgren O. Tissue osmolality in intestinal villi of four mammals in vivo and in vitro. Acta Physiol Scand. 1991;143(3):271–277. doi: 10.1111/j.1748-1716.1991.tb09232.x. [DOI] [PubMed] [Google Scholar]
- 65.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15(1):19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- 66.Izumi N, Helker C, Ehling M, Behrens A, Herzog W, Adams RH. Fbxw7 controls angiogenesis by regulating endothelial Notch activity. PLoS One. 2012;7(7): doi: 10.1371/journal.pone.0041116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature. 2014;507(7492):376–380. doi: 10.1038/nature13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bentley K, et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16(4):309–321. doi: 10.1038/ncb2926. [DOI] [PubMed] [Google Scholar]
- 69.Dartsch N, Hägerling R, Kiefer F, Vestweber D. Fusing VE-cadherin to α-catenin impairs fetal liver hematopoiesis and lymph but not blood vessel formation. Mol Cell Biol. 2014;34(9):1634–1648. doi: 10.1128/MCB.01526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karpanen T, et al. Lymphangiogenic growth factor responsiveness is modulated by postnatal lymphatic vessel maturation. Am J Pathol. 2006;169(2):708–718. doi: 10.2353/ajpath.2006.051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yao LC, et al. Pulmonary lymphangiectasia resulting from vascular endothelial growth factor-c overexpression during a critical period. Circ Res. 2014;114(5):806–822. doi: 10.1161/CIRCRESAHA.114.303119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Makinen T, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001;7(2):199–205. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- 73.Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the notch signaling pathway in cancer. Clin Cancer Res. 2015;21(5):955–961. doi: 10.1158/1078-0432.CCR-14-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. doi: 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- 75.Koch U, et al. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J Exp Med. 2008;205(11):2515–2523. doi: 10.1084/jem.20080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46(2):74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- 77.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1: doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A. 2002;99(24):15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sabine A, et al. Mechanotransduction, PROX1, and FOXC2 cooperate to control Connexin37 and Calcineurin during lymphatic-valve formation. Dev Cell. 2012;22(2):430–445. doi: 10.1016/j.devcel.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 80.Shayan R, et al. A system for quantifying the patterning of the lymphatic vasculature. Growth Factors. 2007;25(6):417–425. doi: 10.1080/08977190801932550. [DOI] [PubMed] [Google Scholar]
- 81.Zudaire E, Gambardella L, Kurcz C, Vermeren S. A computational tool for quantitative analysis of vascular networks. PLoS One. 2011;6(11): doi: 10.1371/journal.pone.0027385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pellegrinet L, et al. Dll1- and Dll4-mediated Notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology. 2011;140(4):1230–1240.e1237. doi: 10.1053/j.gastro.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.