

Abstract
Mitochondria are critical for respiration in all tissues; however, in liver, these organelles also accommodate high-capacity anaplerotic/cataplerotic pathways that are essential to gluconeogenesis and other biosynthetic activities. During nonalcoholic fatty liver disease (NAFLD), mitochondria also produce ROS that damage hepatocytes, trigger inflammation, and contribute to insulin resistance. Here, we provide several lines of evidence indicating that induction of biosynthesis through hepatic anaplerotic/cataplerotic pathways is energetically backed by elevated oxidative metabolism and hence contributes to oxidative stress and inflammation during NAFLD. First, in murine livers, elevation of fatty acid delivery not only induced oxidative metabolism, but also amplified anaplerosis/cataplerosis and caused a proportional rise in oxidative stress and inflammation. Second, loss of anaplerosis/cataplerosis via genetic knockdown of phosphoenolpyruvate carboxykinase 1 (Pck1) prevented fatty acid–induced rise in oxidative flux, oxidative stress, and inflammation. Flux appeared to be regulated by redox state, energy charge, and metabolite concentration, which may also amplify antioxidant pathways. Third, preventing elevated oxidative metabolism with metformin also normalized hepatic anaplerosis/cataplerosis and reduced markers of inflammation. Finally, independent histological grades in human NAFLD biopsies were proportional to oxidative flux. Thus, hepatic oxidative stress and inflammation are associated with elevated oxidative metabolism during an obesogenic diet, and this link may be provoked by increased work through anabolic pathways.
Introduction
Hepatic mitochondria support a broad range of metabolic activities, many of which are unique to liver and contribute to the pathophysiology of insulin resistance, diabetes, and fatty liver disease. The tricarboxylic acid (TCA) cycle is a mitochondrial pathway that oxidizes acetyl-CoA to CO2 and produces NADH and flavin adenine dinucleotide hydroquinone (FADH2) to support ATP synthesis via the respiratory chain. A closely linked, but distinct mitochondrial function is its anaplerotic and cataplerotic pathways, which allow 4-carbon intermediates to move into (anaplerosis) and out of (cataplerosis) the TCA cycle without undergoing oxidation. Though all tissues support minor activities of anaplerosis and cataplerosis, these pathways are a major function of liver TCA cycle, where anaplerosis and cataplerosis are required for shuttling reducing equivalents (RE) and biosynthetic substrates related to ureagenesis, amino acid synthesis, lipogenesis, and most prominently, gluconeogenesis (GNG) (1). In this study, we examined whether anaplerosis and cataplerosis contribute to oxidative stress and inflammation during diet-induced fatty liver and insulin resistance.
Hepatic GNG is a notable anaplerotic pathway because of its role in the pathology of insulin resistance and diabetes (2) and its high flux relative to other hepatic pathways (3). Except for glycerol, all other gluconeogenic substrates require anaplerosis (1). The carboxylation of pyruvate to the TCA cycle intermediate oxaloacetate (OAA) by mitochondrial pyruvate carboxylase (PC) is the archetypal anaplerotic pathway, though others exist (4). Cataplerosis occurs by subsequent transport of OAA (as malate or aspartate) from the mitochondria and decarboxylation to phosphoenolpyruvate (PEP) by PEP carboxykinase (PEPCK-C) (5, 6). This seminal step in GNG is regulated mainly by transcription of the PEPCK-C gene (phosphoenolpyruvate carboxykinase 1 [Pck1]) through hormonal and nutritional mechanisms (5, 6). Inasmuch as mitochondrial TCA cycle intermediates do not accumulate, the pathways of anaplerosis and cataplerosis operate at precisely the same flux.
In liver, flux through anaplerosis and cataplerosis is greater than the oxidation of acetyl-CoA in the TCA cycle (7). In principle, the oxidative and anaplerotic/cataplerotic pathways of the TCA cycle can operate independently, but in practice, they are mediated by mutual metabolic mechanisms. First, anaplerosis/cataplerosis and substrate oxidation both produce and require OAA (1, 8). Second, acetyl-CoA is oxidized by the TCA cycle and also potently induces anaplerosis by allosteric activation of PC (9). Accordingly, β-oxidation and the generation of acetyl-CoA increase anaplerotic capacity (10). Finally, GNG requires a continuous supply of high-energy cofactors (NADH and ATP) derived from TCA cycle activity and respiration. Feedback from redox state and energy charge induces oxidative metabolism when GNG is stimulated (11). Thus, hepatic anaplerotic/cataplerotic pathways are energetically backed by elevated oxidative metabolism in liver.
Hepatic insulin resistance and fatty liver disease may be linked to oxidative metabolism through the generation of oxidative stress (12). During nonalcoholic fatty liver disease (NAFLD), mitochondria have impaired respiratory coupling (13–15), but elevated TCA cycle function (7, 13), which may increase the reductive load on respiration and the potential for ROS formation. Thus, we examined the link between anaplerosis/cataplerosis and oxidative metabolism to determine whether the anaplerotic workload contributes to oxidative stress and inflammation during NAFLD. We report that increased lipid delivery, either exogenously or by high-fat diet (HFD), amplified anaplerosis/cataplerosis and caused a rise in oxidative stress and inflammation in proportion to oxidative flux. Preventing the induction of anaplerosis/cataplerosis by genetic knockdown of PEPCK-C also altered redox and metabolite profiles, which prevented the induction of oxidative flux and stimulated antioxidant capacity. Likewise, metformin treatment prevented the rise in oxidative flux and blocked the induction of anaplerotic flux. Finally, inflammatory scores of human liver biopsies correlated with hepatic oxidative flux. Thus, obligate induction of oxidative metabolism by increased anabolism may contribute to oxidative stress and inflammation during a HFD and NAFLD.
Results
Validation of tracer methodology for evaluating mitochondrial fluxes.
We first examined the validity of a tracer approach ([U-13C]propionate and 2H2O) used to measure hepatic anaplerosis and TCA cycle flux (7, 13, 16–18). Neither hepatic glucose production (Figure 1A) nor oxygen consumption (Figure 1B) were perturbed in perfused livers exposed to propionate at a concentration used in ex vivo experiments (0.1 mM). Endogenous glucose production was also unaltered when propionate was infused at rates equal to tracer protocols (Figure 1C) and resulted in approximately 3% enrichment in the C2 of glucose (Figure 1D). To determine whether the propionate alters flux through anaplerosis, GNG, or pyruvate cycling, we perfused livers with [U-13C]lactate/pyruvate and nonesterified fatty acids (NEFA) in the presence and absence of propionate. Carbon-13 NMR analysis of glucose confirmed that propionate did not alter the isotopomer distributions in glucose (Figure 1E). Flux analyses using simple analytical expressions assumed 100% equilibration between OAA and fumarate isotopomers (i.e., randomization or backward scrambling), but this equilibration may be closer to 80% to 85% (19, 20). Metabolic modeling revealed that incomplete randomization causes the relative flux of pyruvate cycling to be underestimated and GNG to be overestimated. Tracers that entered the TCA cycle downstream of fumarate (e.g., [U-13C]lactate/pyruvate/alanine) were more vulnerable to this artifact than tracers that entered upstream of fumarate (e.g., [U-13C]propionate) (Figure 1F). As predicted, [U-13C]lactate/[U-13C]pyruvate overestimated GNG and underestimated pyruvate cycling compared with [U-13C]propionate using simple analytical expressions (Figure 1G). However, when a regression model including randomization was used, data from [U-13C]lactate/[U-13C]pyruvate–perfused livers gave values identical to those from [U-13C]propionate and simple equations (Figure 1G). Hence, in contrast to a recent report (21), but in agreement with another (22), these findings demonstrate that tracer doses of propionate do not perturb glucose production or anaplerosis and explain why [U-13C]pyruvate/lactate/alanine tracers provide lower estimates of pyruvate cycling when incomplete randomization is not modeled (23).
Figure 1. Propionate tracers do not perturb basal hepatic flux.

Propionate (0.8 μmol•min–1) did not alter (A) glucose production or (B) O2 consumption in livers perfused with gluconeogenic substrates and NEFA (n = 4). Propionate infusion (0.5 μmol•min–1) into conscious and unrestrained mice did not alter (C) endogenous glucose production (n = 3) and (D) resulted in tracer level (<4%) glucose enrichment (n = 4–8). (E) Isotopomers of glucose formed by [U-13C]lactate/pyruvate during liver perfusion and reported in the 13C NMR spectrum of glucose C2 were not altered by the addition of propionate (n = 3). Glucose isotopomers in carbons 1–3 (black circles are 13C) that contribute to the NMR signal are indicated above the corresponding signal. (F) Modeling the effect of incomplete OAA/fumarate equilibration in the TCA cycle predicted that [U-13C]lactate/pyruvate (white circles) would underestimate pyruvate cycling (PK+ME) and overestimate GNG more severely than [U-13C]propionate (gray circles). The highlighted area around 80% to 85% is the experimentally expected degree of randomization (3, 19). (G) Relative fluxes reported by [U-13C]lactate/pyruvate underestimated pyruvate cycling and overestimated GNG relative to [U-13C]propionate when simple equations (64) were used (left panel), but gave identical values when randomization was fit using a regression model (right panel) (n = 3–4). Data are shown as mean ± SEM. Statistical differences were detected by 2-tailed t test. *P < 0.05; **P < 0.001.
Oxidative metabolism is linked to anaplerosis/cataplerosis.
Next we examined whether oxidative flux measured by 2H and 13C NMR isotopomer analysis was consistent with measured oxygen consumption. To examine the precision of the method across a range of gluconeogenic and oxidative conditions, livers from fed or fasted mice were perfused with long- or medium-chain NEFA. Fluxes through pathways that generate or consume NADH/FADH2 (TCA cycle, ketogenesis, etc.) were used to calculate putative oxygen consumption (Equations 1 and 2), and this estimate was compared with measured oxygen consumption. Tracer estimation of oxygen consumption correlated strongly with analytically measured oxygen consumption (Figure 2A). Consistent with the energetic requirements of GNG, the rate of anaplerosis (Figure 2B) also correlated with measured O2 consumption. Thus, flux determined by tracer analysis reflects hepatic energy metabolism as indicated by hepatic oxygen uptake, and rates of anaplerosis/GNG correlate with hepatic energy demand.
Figure 2. Oxidative metabolism is linked to anaplerosis and GNG in liver.

(A) Oxygen consumption in fed, fasted, or octanoate-perfused mouse liver correlated with oxygen consumption calculated from isotopomer analysis using Equations 1 and 2 (see Methods). (B) Fasting oxygen consumption increased in proportion to anaplerosis (n = 4 × 3 repeated measures). A separate group of livers from fed mice (n = 4) was perfused with either low or high NEFA (0.2 mM or 0.8 mM). High NEFA increased oxidative metabolism measured by (C) ketone output and TCA cycle flux. High NEFA also increased (D) anaplerosis and GNG. (E) Addition of insulin suppressed glycogenolysis, but not anaplerosis or GNG regardless of NEFA concentration (n = 3–4). Data are shown as mean ± SEM. Statistical differences were detected by a 2-tailed t test. *P < 0.05; #P < 0.05 versus insulin perfusion.
Increasing NEFA causes an autonomous rise in hepatic oxidation and anaplerosis/cataplerosis.
Lipolysis is increased during insulin resistance, so we tested whether elevated NEFA alters anaplerosis/cataplerosis and oxidative flux in the TCA cycle. Livers from fed mice perfused with high NEFA (0.8 mM) had increased hepatic ketogenesis and oxidative flux in the TCA cycle (Figure 2C) compared with livers perfused with low NEFA (0.2 mM). In addition, high NEFA caused an increase in GNG mediated by elevated anaplerosis/cataplerosis (Figure 2D) despite no change in gluconeogenic substrate concentration (1.5 mM lactate and 0.15 mM pyruvate). Insulin was then administered to determine whether insulin action can suppress the induction of oxidative metabolism and GNG by NEFA. Insulin potently suppressed glycogenolysis, but did not prevent the rise in GNG, anaplerosis, or oxygen utilization caused by high NEFA (Figure 2E). Thus, increased NEFA per se is sufficient to increase ex vivo hepatic oxidative metabolism and anaplerosis/GNG.
To determine whether NEFA induces anaplerosis/cataplerosis and oxidative metabolism in vivo, hepatic fluxes were measured in rats after 6 hours of intralipid infusion. Intralipid increased circulating NEFA, triglycerides, ketones, and insulin (Table 1). These conditions stimulated an increase in ketone turnover and oxidative flux in the TCA cycle (Figure 3A). In agreement with previous findings (24), elevated NEFA also increased GNG (Figure 3B), which was facilitated by a marked increase in anaplerosis/cataplerosis (Figure 3B). Overall, isotopomer analysis indicated elevated oxygen consumption that rose in proportion to plasma NEFA (Figure 3C). Thus, in agreement with ex vivo liver perfusions, elevated NEFA is sufficient to induce hepatic energy metabolism and cause a rise in anaplerosis and GNG in vivo.
Table 1. Metabolic characteristics of 4-hour–fasted rats infused with intralipid.
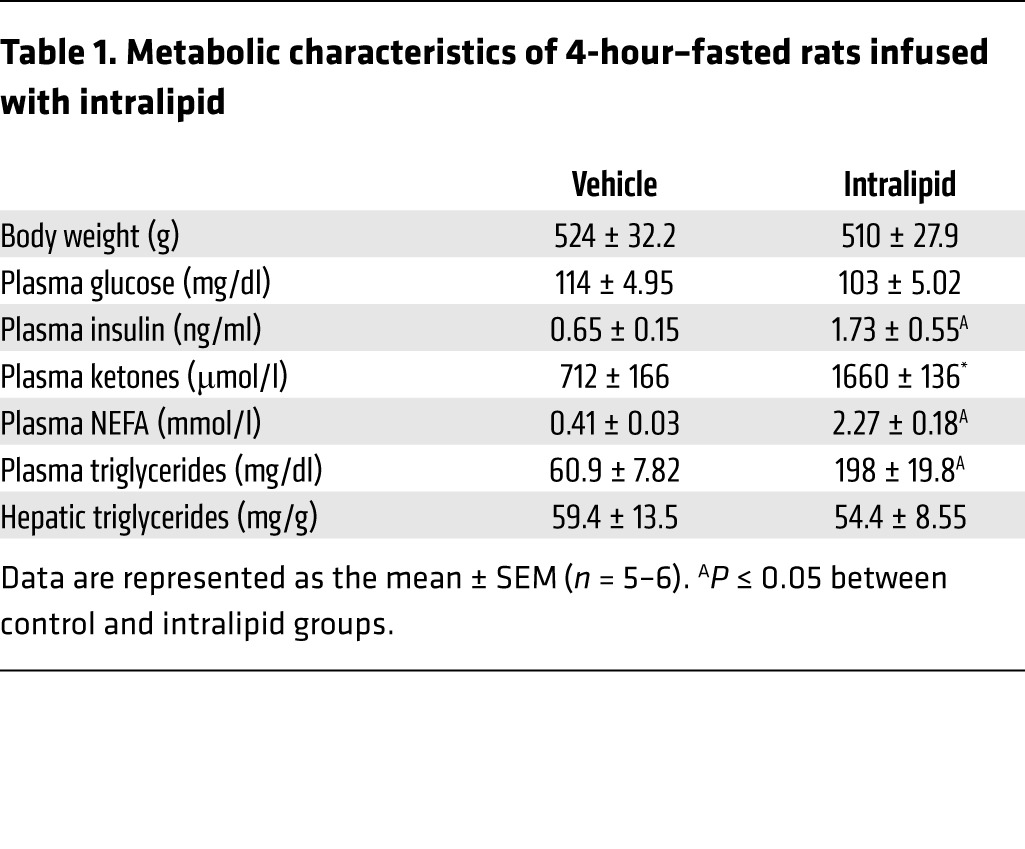
Figure 3. The induction of oxidative metabolism by NEFA requires increased anaplerosis/cataplerosis to cause oxidative stress and inflammation.

Increasing circulating NEFA by intralipid infusion (n = 4–6) caused a rise in fat oxidation indicated by a rise in (A) ketogenesis and TCA cycle flux and (B) GNG and anaplerosis measured by tracer methods. (C) Calculated oxygen consumption was increased by intralipid infusion in proportion to the rise in circulating NEFA. Oxidative stress, as indicated by (D) Sod2 mRNA and (E) lipid peroxidation, increased in proportion to the rise in oxygen consumption. The inflammatory response, as indicated by (F) Tnfa and (G) Il6 mRNA, was elevated by NEFA in proportion to the rise in oxygen consumption. (H) Administration of an inhibitor of PEPCK, 100 μM mercaptopicolinate, blocked the induction of ROS in H4IIE rat hepatoma cells treated with high NEFA (n = 3). Data are shown as mean ± SEM. Statistical differences were detected by 2-tailed t tests, except for H, which used a 2-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001.
Oxidative stress and inflammation are associated with increased oxidative flux.
Since oxidative metabolism is associated with increased ROS (25), we tested whether the marked rise in oxidative metabolism also induces oxidative stress. Mitochondrial superoxide dismutase 2 (Sod2) mRNA (Figure 3D) and oxidative damage indicated by malondialdehyde levels (Figure 3E) were increased in proportion to the induction of calculated hepatic oxygen consumption that occurred during intralipid infusion. Consistent with oxidative damage, Tnfa (Figure 3F) and Il6 (Figure 3G) were also elevated in proportion to the predicted oxygen consumption. Since GNG mediated by TCA cycle cataplerosis is a significant consumer of hepatic energetics (16), we examined whether suppressing cataplerosis reduces oxidative stress in vitro. H4IIE cells incubated in the presence of high NEFA developed a 2-fold increase in ROS detected by 2′,7′-dichlorofluorescein, which is similar to results reported in primary hepatocytes (26). However, this effect was attenuated when anaplerotic/cataplerotic flux was blocked by the PEPCK inhibitor mercaptopicolinic acid (Figure 3H). These data suggest that induction of hepatic anaplerosis/cataplerosis may play a role in increased oxidative stress associated with acute exposure of liver to substrate.
Suppression of cataplerotic flux during HFD protects against elevated GNG and hepatic insulin resistance.
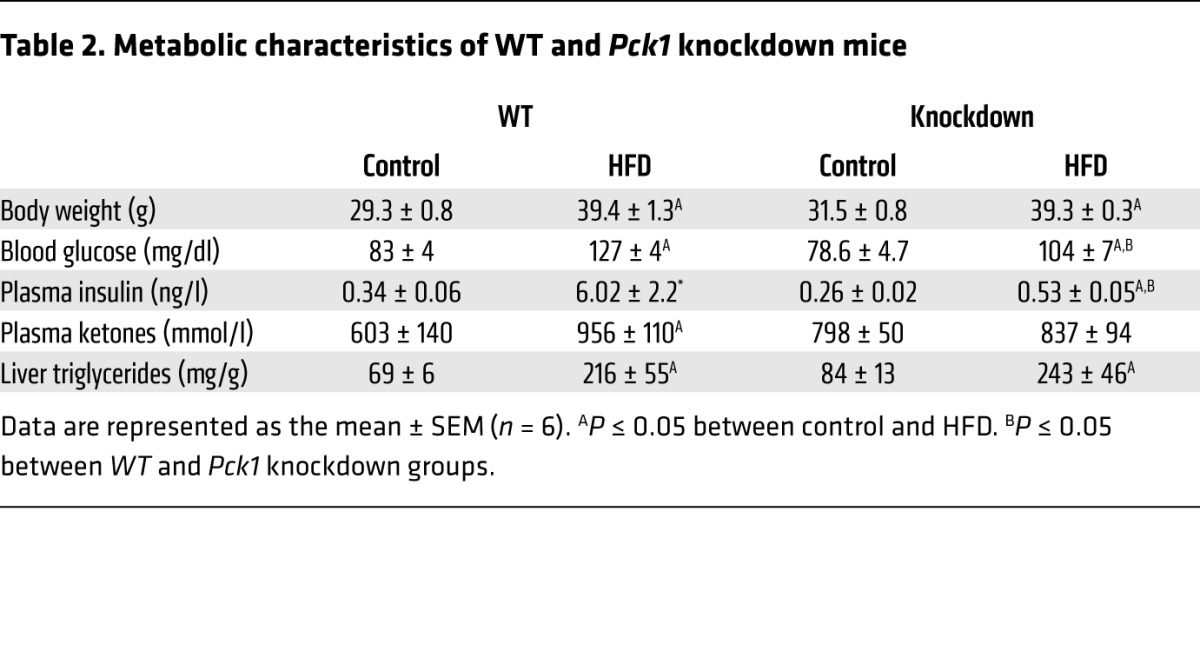
To determine whether TCA cycle pathways contribute to liver pathology during obesity in vivo, we suppressed anaplerosis/cataplerosis using a genetic knockdown of Pck1 (PEPCK-C) and 16 weeks of a HFD. Knockdown mice (Pck1lox+neo/lox+neo) had less than 50% hepatic Pck1 mRNA compared with WT mice (Pck1f/f) (Figure 4A). Knockdown mice and WT littermates were phenotypically similar on a control diet (Table 2), as we previously reported (27, 28). However, despite body weights and liver fat similar to those of WT mice, glucose and insulin concentrations were lower in knockdown mice on a HFD, suggesting improved regulation of hepatic fluxes (Table 2). As previously reported (28), knockdown of Pck1 did not reduce anaplerotic/cataplerotic flux on a control diet (Figure 4B). However, the knockdown was sufficient to prevent the rise in anaplerotic/cataplerotic flux caused by a HFD (Figure 4B). Likewise, Pck1 knockdown did not cause lower fasting basal endogenous glucose production (Figure 4C) or GNG (Figure 4D) on a control diet, but it was sufficient to prevent the rise in these fluxes during a HFD.
Figure 4. Preventing the induction of anaplerosis/cataplerosis during a HFD protected against elevated GNG and hepatic insulin resistance.

(A) Pck1 mRNA in overnight-fasted WT, Pck1 knockdown (Pck1lox+neo/lox+neo), and liver-specific knockout mice (Pck1lox/lox Alb-Cre) (n = 8–12). (B) Anaplerosis, (C) endogenous glucose production, and (D) GNG remained normal in knockdown mice on a HFD (n = 4–9). (E) Gluconeogenic genes were decreased in knockdown mice on a HFD (n = 4–5). (F) The rate of glucose disposal during a hyperinsulinemic-euglycemic clamp (n = 4–8) was reduced by a HFD in WT and knockdown mice. (G) Knockdown mice on a HFD maintained normal suppression of hepatic glucose production during the clamp. (H) Western blot analysis of p-Akt/Akt ratios in liver before (basal) and 3 minutes after portal insulin injection (+insulin) and (I) fold induction of p-Akt/Akt by insulin (n = 4). Ctrl, control. Data are shown as mean ± SEM. Statistical differences were detected by 2-way ANOVA (A–D, F, and G), 2-tailed t test (E), and 1-tailed paired t test (H and I). *P < 0.05; **P < 0.01.
Table 2. Metabolic characteristics of WT and Pck1 knockdown mice.
Reduced gluconeogenic flux occurred in conjunction with lower insulin levels and gluconeogenic gene expression (Figure 4E), suggesting that knockdown mice preserved hepatic insulin sensitivity during a HFD. Hence, we examined hepatic insulin sensitivity by hyperinsulinemic-euglycemic clamp and Akt phosphorylation. WT mice on a HFD had lower glucose infusion rates (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI82204DS1) and impaired glucose disposal (Figure 4F) and did not suppress glucose production (Figure 4G) compared with mice on a control diet. Knockdown mice had slightly reduced glucose disposal compared with WT mice, which was further impaired by a HFD (Figure 4F). However, in contrast to WT mice, knockdown mice on a HFD retained the ability to suppress glucose production during insulin clamp (Figure 4G). Hepatic AKT phosphorylation (Supplemental Figure 2) was also robustly induced by acute insulin injection in HFD knockdown mice, but not in WT mice (Figure 4, H and I). These data indicate that preventing the rise in anaplerosis/cataplerosis and oxidative flux during a HFD allowed knockdown livers to retain insulin action despite the same degree of peripheral insulin resistance (perhaps worse) and hepatic steatosis as that seen in WT mice.
Suppression of cataplerotic activity protects against elevated oxidative metabolism via metabolic mechanisms.
Previous calculations (16) and ex vivo experiments (Figure 2 and Table 3) demonstrated that GNG is a substantial consumer of hepatic energy metabolism, so we tested whether preventing the induction of the anaplerotic/cataplerotic pathway would obviate the rise in oxidative flux. Substrate oxidation in the TCA cycle was elevated by a HFD in WT mice as we previously reported (13), but not in knockdown mice (Figure 5A). Ketogenesis was elevated in WT mice on a HFD and tended to be higher in knockdown mice on a control diet (Figure 5B). Based on flux measurements, calculated hepatic oxygen consumption rose in WT mice on a HFD but remained normal in knockdown mice (Figure 5C). Despite decreased oxidative metabolism, ATP, ADP, and AMP concentrations were normal (Figure 5D), indicating that the decrease in oxidative flux was appropriate for maintaining normal energy charge in knockdown mice. Thus, a rise in anaplerosis/cataplerosis during a HFD appears to instigate increased oxidative metabolism.
Table 3. Energetic demand of GNG in perfused liver.
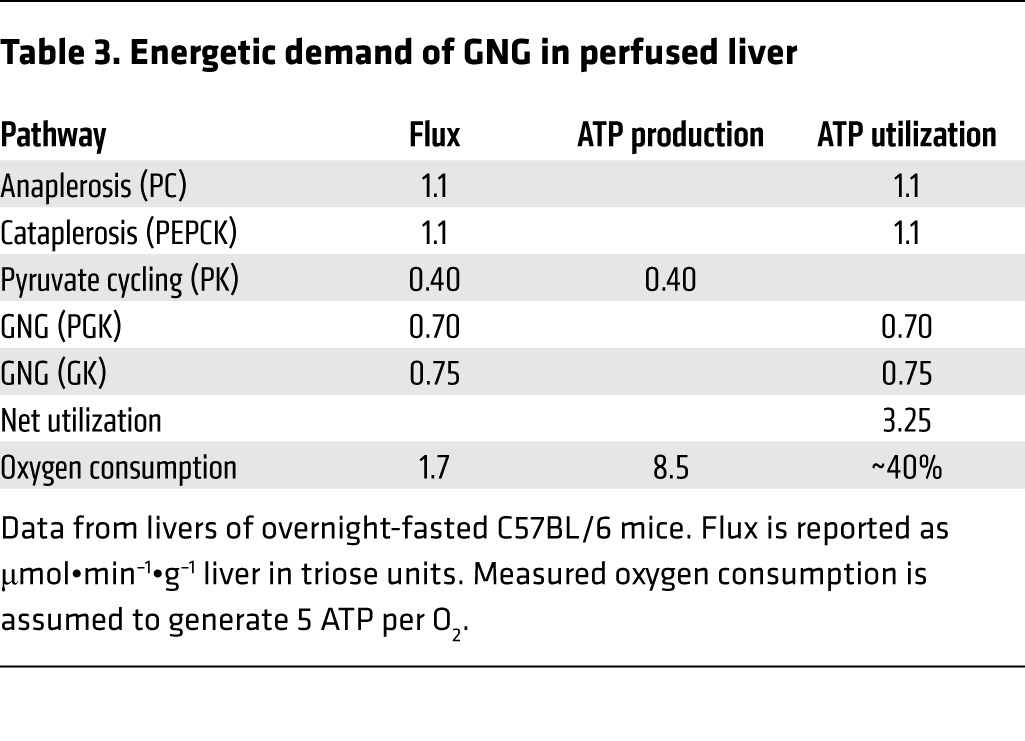
Figure 5. Preventing the induction of anaplerosis/cataplerosis during a HFD prevented the rise in oxidative flux through metabolic mechanisms.

(A) TCA cycle flux measured by isotopomer analysis of plasma glucose remained normal in knockdown mice during a HFD. (B) Ketogenesis measured by apparent ketone turnover. (C) Calculated oxygen consumption increased in WT mice, but not knockdown mice, on a HFD (n = 4–9 for A–C). (D) ATP, ADP, and AMP measured by LC-MS. (E) Expression of genes related to oxidative metabolism was normal or elevated in knockdown mice. (F) Mitochondrial NAD+/NADH measured by plasma acetoacetate/β-hydroxybutyrate ratio was reduced in liver of knockdown mice. (G) Cytosolic NAD+/NADH measured by pyruvate/lactate ratio was reduced in liver of knockdown mice. (H) Hepatic citrate, succinate, and OAA were elevated in knockdown liver (n = 6–9 for D–H). Data are shown as mean ± SEM. Statistical differences were detected by 2-way ANOVA (A–C and E) and 2-tailed t test (D and F–H). *P < 0.05; **P < 0.01; ***P < 0.001.
It is unlikely that reduced oxidative flux in the TCA cycle of knockdown mice on a HFD was mediated by gene expression, inasmuch as oxidative genes were upregulated, but flux was reduced (Figure 5E). Putative metabolic regulation of TCA cycle oxidation is governed by redox state, product inhibition, and energy charge (reviewed in ref. 11); thus, we investigated these factors in WT and knockdown mice. Mitochondrial NAD+/NADH estimated from plasma acetoacetate/β-hydroxybutyrate (29) was reduced in knockdown liver (Figure 5F), which is known to inhibit the forward reactions of the TCA cycle (11). This finding was also supported by a reduced cytosolic redox state determined from the liver lactate/pyruvate ratio (Figure 5G and Supplemental Table 1). Several TCA cycle metabolites are known to inhibit upstream enzymes. Citrate, succinyl-CoA, and OAA inhibit citrate synthase, α-ketoglutarate dehydrogenase, and succinate dehydrogenase (complex II), respectively (11). Citrate and succinate concentrations were modestly elevated, but OAA was 5-fold higher in knockdown liver (Figure 5H and Supplemental Table 1). These data are consistent with feedback inhibition of the TCA cycle in response to restricted cataplerosis via effects on redox state and product inhibition.
Suppression of cataplerotic and oxidative flux in the hepatic TCA cycle protects against oxidative stress and inflammation.
Since mitochondrial oxidative metabolism induces free radical production, we determined whether preventing the rise in anaplerosis/cataplerosis reduces hepatic oxidative stress and inflammation during a HFD. A quantitative real-time PCR (qPCR) array of 84 genes related to oxidative stress indicated that most classes of superoxide dismutases, peroxidases, and peroxiredoxins (antioxidant pathways) were elevated by HFD in WT but not knockdown liver (Figure 6A). A fluorescent probe (dihydroethidium [DHE] bromide) was used to confirm the induction of ROS in liver. WT mice, but not knockdown mice, had a substantial increase in fluorescence intensity in liver after a HFD (Figure 6B). Consistent with less ROS, knockdown mice were also protected from HFD-induced lipid peroxidation (Figure 6C).
Figure 6. Preventing the induction of anaplerosis/cataplerosis protected against hepatic oxidative stress and inflammation during a HFD.

Oxidative stress, indicated by (A) a qPCR gene array, (B) histological DHE ROS staining, and (C) lipid peroxidation (n = 6), was increased by a HFD in WT, but not knockdown mice. TBARS, thiobarbituric acid reactive substances. (D) The Q/QH2 ratio, estimated from the fumarate/succinate ratio, was oxidized in knockdown mice (n = 6–8). (E) The calculated free energy of complexes I and II was more negative in knockdown mice (n = 6–8). (F) The NADP+/NADPH ratio, estimated from the pyruvate/malate ratio, was reduced in knockdown mice (n = 6–8). (G) TCA cycle intermediates with antioxidant properties were increased in knockdown mice (n = 3–4) as were (H) antioxidant genes (n = 6). Knockdown mice were protected from the inflammatory response of a HFD as indicated by (I) fewer inflammatory infiltrates in H&E-stained tissue (n = 6–8) and lower expression of (J) Tnfa and (K) Il6 mRNA (n = 6). (L) NF-κB S536 phosphorylation was reduced in knockdown liver (n = 5–6). Original magnification, ×10 (I); ×20 (B). Data are shown as mean ± SEM. Statistical differences were detected by 2-way ANOVA (J and K), 2-tailed t test (C–H), or 1-tailed t test (I and L). *P < 0.05; **P < 0.01; ***P < 0.001.
To explore the mechanism of lower oxidative stress in knockdown mice, we examined mitochondrial inner-membrane redox and ROS-scavenging potential. Coenzyme Q10 (Q) was reduced to QH2 in the inner-membrane by NADH in complex I (NADH + Q → QH2 + 4H+) or by FADH2 in complex II/SDH (succinate + Q → fumarate + QH2). Hence, the succinate/fumarate ratio indicates inner-membrane Q/QH2 (30). In contrast to the reduced redox state of the mitochondrial matrix and cytosol (Figure 5, F and G), the Q/QH2 redox state was more oxidized in knockdown liver (Figure 6D). In conjunction with reduced mitochondrial NAD+/NADH, the oxidized Q/QH2 dictated a more negative ΔG for complexes I and II (31) in the knockdown mice (Figure 5E). This finding indicates that the reverse transport mechanism of ROS formation may be less favorable in knockdown mice than in WT mice.
ROS scavenging capacity was evaluated by examining free NADP+/NADPH using the malic enzyme–associated malate/pyruvate redox couple (32). Knockdown mice on a HFD had 3-fold reduced NADP+/NADPH compared with WT mice (Figure 6F). These conditions are favorable for the reduced form of glutathione (GSH) and the clearance of peroxides by GSH reductase antioxidant systems. In addition to influencing various redox systems, altered TCA cycle metabolites can activate antioxidant programs. Fumarate and α-ketoglutarate interact with essential hepatic antioxidant transcription factors, such as Nrf1, Nrf2, and If1. These metabolites (Figure 6G), transcription factors, and several of their target antioxidant enzymes (Figure 6H) were elevated, even in the absence of a HFD (Figure 6H). Thus, in addition to a reduced oxidative load, the knockdown liver has improved antioxidant capacity.
In accordance with less oxidative damage, knockdown mice had reduced inflammatory cell infiltration observed by H&E staining (Figure 6I). Hepatic inflammatory markers Tnfa and Il6 were also substantially elevated by a HFD in WT mice, but not in knockdown mice (Figure 6, J and K), and inflammatory signaling was confirmed by reduced NF-κB S536 phosphorylation (Figure 6L and Supplemental Figure 3). Since Pck1 is expressed in many extrahepatic tissues, we confirmed hepatocyte autonomy of the antiinflammatory effect in liver-specific PEPCK-C knockout mice (Supplemental Figure 4). These data indicate that preventing the rise in anaplerosis/cataplerosis during a HFD protects against oxidative stress and inflammation in liver, either by obviating increased oxidative metabolism or by activating antioxidant programs.
Metformin recapitulates the metabolic effects of PEPCK knockdown during a HFD.
The antidiabetic actions of biguanides have been linked to several mechanisms, including the suppression of hepatic energetics (33). Thus, we examined whether metformin treatment suppresses oxidative flux and inflammation in HFD mice similarly to direct suppression of anaplerosis/cataplerosis in Pck1 knockdown mice. As expected, 2 weeks of metformin reduced fasting GNG (Figure 7A) and anaplerosis/cataplerosis (Figure 7B) in mice on a HFD. Consistent with suppressed hepatic energetics, TCA cycle flux (Figure 7C) and calculated oxygen consumption (Figure 7D) were markedly suppressed in metformin-treated mice. Hepatic Il6 (Figure 7E) and Tnfa (Figure 7F) expressions were reduced in metformin-treated mice. Thus, either inhibition of oxidative metabolism by metformin or by genetic suppression of anaplerosis/cataplerosis is sufficient to improve inflammatory markers in the liver of HFD mice.
Figure 7. Metformin suppressed oxidative metabolism and anaplerosis and lowered inflammation in livers of mice on a HFD.

Mice were treated with metformin during the last 4 weeks of a 16-week HFD. Metformin treatment reduced (A) GNG by suppressing (B) anaplerosis. Oxidative metabolism was suppressed as indicated by reduced (C) TCA cycle flux and (D) calculated oxygen consumption. Markers of inflammation (E) Il6 and (F) Tnfa were reduced in proportion to oxygen consumption. Data are shown as mean ± SEM (n = 4–5). Statistical differences were detected by 2-tailed t test. *P < 0.05; **P < 0.01; ***P < 0.001.
NAFLD activity scores correlate with calculated hepatic oxygen consumption in humans.
To determine whether activation of oxidative metabolism is an important factor in the development of NAFLD in humans, we compared oxidative flux to NAFLD activity score (NAS, ref. 34) and inflammatory scores (Ishak, ref. 35) in liver biopsies. Estimated oxygen consumption calculated from fluxes recently reported in NAFLD subjects (7) correlated strongly with biopsy NAS and inflammatory scores in those same subjects (Figure 8A). Thus, higher oxidative metabolism was associated with poorer histological grading in humans under evaluation for NAFLD. On the whole, the data indicate that increased flux through anaplerotic/cataplerotic pathways not only contributes to dysregulation of downstream nutrients (e.g., glycemia and lipidemia), but also triggers oxidative metabolism and altered antioxidant capacity that contribute to the development of oxidative stress, inflammation, and insulin resistance (Figure 8B and Supplemental Table 2).
Figure 8. Histological data support a role for oxidative metabolism in NAFLD in humans.

(A) Liver biopsies obtained from 8 individuals with suspected NAFLD were given NAS and Ishak scores. These scores correlated with oxygen consumption calculated from previously reported fluxes (7). (B) The data support a role for oxidative metabolism in facilitating increased anaplerotic work, collateral oxidative stress, and inflammation during NAFLD and hepatic insulin resistance. Correlations were detected by 1-tailed Pearson tests.
Discussion
Liver mitochondria oxidize substrate, produce ATP, and contain highly active anaplerotic and cataplerotic pathways used to support biosynthesis. Hence, we examined whether elevated anaplerotic/cataplerotic workload provokes oxidative metabolism and therefore oxidative stress and inflammation during obesity and insulin resistance. Multiple tracers were used to measure GNG, anaplerosis/cataplerosis, ketogenesis, and TCA cycle oxidation; and this approach was validated by its agreement with hepatic oxygen consumption. NEFA autonomously increased oxidative metabolism, amplified anaplerosis, and caused a rise in oxidative stress and inflammation. Preventing the induction of anaplerosis/cataplerosis and/or oxidative flux averted oxidative stress and inflammation and protected hepatic insulin sensitivity during a HFD. Finally, liver histology in humans who were previously evaluated with tracers of hepatic metabolism demonstrated a positive correlation between oxidative flux and NAS/necroinflammation. Thus, anaplerosis and cataplerosis impinge on oxidative metabolism and are therefore linked to oxidative stress and inflammation during NAFLD.
Mitochondrial function during NAFLD.
Obesity and NAFLD are accompanied by broad effects on mitochondrial metabolism that include increased or decreased activity, depending on the context. In vitro defects in respiration (13–15, 36, 37) and morphology (36) are apparent in severe models of obesity and insulin resistance. Under moderate conditions, the effect of obesity and insulin resistance on hepatic respiration is less clear. In vivo ATP turnover by 31P magnetic resonance (MR) saturation transfer was reduced by 40% in humans with diabetes (38). In contrast, in vivo splanchnic oxygen consumption was increased by 45% in obese subjects, suggesting increased respiration (39), and more recently, mitochondrial respiration was found to be increased in obese humans (40). A uniform conclusion about β-oxidation is similarly difficult to ascertain across studies. Global suppression of β-oxidation was sufficient to cause hepatic lipid accumulation and insulin resistance, but without elevated GNG or activation of inflammatory pathways common to obesity (41). In vitro hepatic palmitate oxidation is also impaired in hyperphagic genetic models of obesity (15), which we found manifested as reduced in vivo ketogenesis in ZDF rats and very long-term (32 weeks) HFD mice (13, 42). Cotter and coworkers more carefully investigated ketogenesis and found that 3-hydroxymethylglutaryl-CoA synthase (HMGCS2) loss of function suppressed ketogenesis, increased steatosis, and caused liver inflammation (43). However, tracer analysis indicated that impaired ketogenesis may have caused increased activity of the TCA cycle (43), consistent with our previous findings (13, 42). In principal, the high reductive yield of the TCA cycle (46 NADH/palmitate) compared with β-hydroxybutyrate formation (10 NADH/palmitate) allows the energetics of a 30% reduction in ketogenesis to be recaptured by a mere 7% increase in the TCA cycle. Thus, it is remarkable that moderate defects in β-oxidation may not limit energy capture by liver. Nonetheless, defects in β-oxidation are not apparent in moderate examples of obesity. We found that 16 weeks of a HFD in mice caused obesity and increased ketogenesis (Figure 4 and ref. 13). Others reported that obese humans have elevated 11C-palmitate oxidation (44), consistent with our finding that humans with NAFLD have increased TCA cycle flux (7). On balance, mitochondrial respiration and oxidation appear to evolve from a hyperactive but perhaps inefficient state during moderate obesity and insulin resistance to a more generalized mitopathy during severe obesity/insulin resistance and/or liver disease.
Energetic requirements downstream of anaplerosis/cataplerosis induce oxidative metabolism during obesity.
We focused our study on anaplerosis/cataplerosis, a mitochondrial pathway that is required for GNG. Inasmuch as this mitochondrial pathway is elevated during obesity and insulin resistance, it may provide a partial explanation for the spectrum of mitochondrial function observed during these conditions. Mitochondrial anaplerosis/cataplerosis and oxidative function are coupled by ATP/GTP consumption at PC (anaplerosis), PEPCK (cataplerosis), and phosphoglycerate kinase (GNG). In perfused livers, where oxygen consumption was measured directly, these pathways consumed roughly 40% of hepatic energy production (Table 3), which is very similar to the estimate made by Landau in human liver (16). Hence, individuals with a 60% increase in GNG (2) should have a roughly 30% increase in hepatic oxidative metabolism. Indeed, splanchnic oxygen consumption was approximately 45% higher in obese humans compared with lean subjects (39). When we prevented the rise in anaplerosis/cataplerosis during a HFD, the calculated oxygen consumption remained constant, consistent with our (28, 45) and others’ (8, 27) reports that the PEPCK pathway links downstream energy utilization and oxidative metabolism.
The mechanisms for this metabolic regulation are partially understood. First, a product of β-oxidation, acetyl-CoA, is also an essential allosteric activator of PC (9). Indeed, raising NEFA increased anaplerosis/cataplerosis linearly with oxidative flux in mouse liver and in rats infused with intralipid. Second, PC activity may also be modified by energy charge. Owen and Halestrap (46) found that mild inhibition of respiration, similar to the effects of metformin, was sufficient to suppress anaplerosis by reducing ATP energy charge. Indeed, we found that metformin suppressed oxidative metabolism and reduced PC flux (anaplerosis). Third, inducing GNG reduces energy charge, activates AMPK, and increases oxidative metabolism. When PEPCK was absent, the AMPK response was negated (47). Consistent with this mechanism, knockdown mice had elevated energy charge. Fourth, decreasing flux through ATP-demanding pathways also decreases oxidative phosphorylation and reduces mitochondrial redox state (i.e., lower NAD+/NADH), which in turn suppresses TCA cycle function (11). Indeed, acetoacetate/β-hydroxybutyrate ratios indicated reduced redox state in the mitochondrial matrix of knockdown mice. Finally, a reduction in GNG/cataplerosis results in a buildup of TCA cycle intermediates, which in turn inhibit the forward reactions of the TCA cycle (11). Knockdown liver had elevated TCA cycle intermediates, particularly OAA and citrate, which inhibit the forward reactions of SDH/complex II and citrate synthase, respectively (11). Interestingly, knockdown mice on a control diet tended to have a modestly elevated ketone turnover despite no effects on other fluxes. This may occur through a reduced mitochondrial redox state, lower insulin, and elevated Ppara and Hmgcs, which allow the knockdown mice to sense and compensate for a vulnerability to hypoglycemia. In short, the activation of anaplerosis/cataplerosis is required for increased hepatic oxidation, and oxidative capacity is required to support anaplerosis/cataplerosis.
Anaplerosis/cataplerosis can instigate oxidative stress and inflammation.
The energetic requirements downstream of anaplerosis/cataplerosis in pathways, such as GNG, dictate concomitant substrate oxidation and electron transport. In the setting of limited mitochondrial capacity (14, 15), this reductive pressure may exacerbate ROS formation (25) in part by direct leak from the TCA cycle at succinate dehydrogenase/complex II (48) and αKG dehydrogenase (49). Indeed, we found that oxidative flux correlated with oxidative stress during acute NEFA delivery and chronically during a HFD. This finding is consistent with studies performed in cell lines that reported β-oxidation–dependent ROS formation is sufficient to induce insulin resistance (50) by activating JNK-mediated phosphorylation of IR substrate (IRS) (51). ROS formation may be exacerbated in vivo where increased oxygen extraction across the lobular sinusoid can increase localized hypoxia and ROS formation during a HFD (14). Most importantly, we found that calculated oxygen consumption in humans, previously shown to have elevated anaplerosis (7), correlated with poorer NAS and necroinflammatory scores. These data are similar to results by Egnatchik et al. (26, 52), who found that exposure of liver cells to elevated NEFA caused increased TCA cycle flux and oxidative stress.
Suppressing the rise in anaplerosis/cataplerosis by knocking down PEPCK or indirectly by inhibiting oxidative metabolism with metformin (or phenformin in Egnatchik’s study, ref. 52) was sufficient to prevent oxidative stress and inflammation. ROS formation is dictated primarily by mitochondrial redox systems, including matrix/membrane NAD+/NADH and the inner membrane ubiquinone cycle (Q/QH2) (reviewed in ref. 53). In contrast to the reduced NAD+/NADH redox state of the mitochondria, knockdown mice had a more oxidized Q/QH2. These are unusual conditions, inasmuch as electron flow should proceed spontaneously to Q via complex I (53). However, based on similar effects, Veech and colleagues concluded that ketones improve cardiac function by lowering the free energy of complexes I and II (31). In knockdown liver, lower free energy of electron transport through complexes I and II also indicates less “reducing pressure” required for reverse electron transport (53). This possibility was demonstrated by Brand and colleagues, who found that suppressing complex II resulted in an oxidized state of the Q pool and slowed rates of reverse electron transport/superoxide production, independent of mitochondrial NAD+/NADH (54). Thus, the reduced NAD+/NADH matrix, elevated energy charge, and TCA cycle intermediate profile in knockdown liver may suppress TCA cycle flux (including SDH/complex II) and decrease the reduction state of the Q pool.
Anaplerosis/cataplerosis can mediate molecular defenses against hepatocellular stress.
Anaplerosis/cataplerosis may also initiate secondary events that confer improved antioxidant capacity. Modulation of Pck1 expression caused an upregulation of Ppara, as previously reported (27), which may provide hepatocellular protection through increased Ucp2 or by activation of antiinflammatory pathways (55). Similarly, despite no change in anaplerotic/cataplerotic flux, knockdown mice on a control diet developed increased TCA cycle intermediates such as fumarate and α-ketoglutarate, likely due to increased metabolic pressure through these pathways. These intermediates induce Nrf1/2 transcription factors that bind to the antioxidant response element and protect against oxidative stress during elevated β-oxidation and steatosis (56, 57). Indeed, antioxidant genes (Gpx2, Sod1, and Sod2) were elevated in knockdown mice. Hepatic α-ketoglutarate may also activate IF1 via HIF1a and prevent ROS production by inhibiting respiration (58), consistent with reduced oxidative metabolism in knockdown mice on a HFD. Altered metabolite profiles were also linked to a change in hepatocellular redox state that translated to a reduced pyruvate/malate ratio and hence reduced NADP+/NADPH, indicated by the malic enzyme equilibrium (32). This redox condition is favorable for neutralizing peroxides. The induction of antioxidant mechanisms in knockdown mice may prime the liver for defense against oxidative stress in these mice during a HFD.
Remarkably, loss of cataplerotic capacity also protected hepatic insulin sensitivity despite comparable obesity, peripheral insulin resistance, and hepatic steatosis. This result is similar to the recovery of insulin signaling in db/db mice when PEPCK-C was knocked down (59) and is consistent with the loss of hepatic insulin sensitivity with the overexpression of PEPCK-C (60). Protection from elevated GNG could be explained by lower capacity in the knockdown mice, but protected insulin action likely stems from secondary factors such as reduced oxidative stress, inflammation, and/or altered redox signaling (53, 61).
Validation of methodology.
In contrast to one report (21), tracer amounts of [U-13C]propionate resulted in only 3% glucose enrichment and did not stimulate glucose production or anaplerosis. This agrees with our previous experience, where we optimized propionate administration in mice to avoid elevated glucose or insulin levels (62). Recently, investigators from the Vanderbilt Mouse Metabolic Phenotyping Center, Vanderbilt University, used an identical tracer approach, but an independent mass spectrometry platform, and found fluxes that were similar to those we previously reported; they also found that tracer amounts of [U-13C]propionate had no effect on hepatic metabolic flux in mice when fit to a complete regression model (22). Notably, stoichiometric analysis of oxygen consumption correlated strongly with flux measurements using 13C and 2H isotopomer analysis. A similar retrospective analysis of fluxes determined in humans (7) provided a theoretical oxygen consumption (1.7 mmol•min–1 ) that was very close to previously reported splanchnic oxygen uptake (2.3 mmol•min–1) (39). Metabolic modeling demonstrated that the obligate transmission of [13C]propionate through the symmetrical intermediate fumarate made it less vulnerable to incomplete equilibration of OAA and fumarate in the TCA cycle compared with [13C]lactate/alanine/pyruvate. Indeed, application of [13C]pyruvate provides lower estimates of pyruvate cycling using simple equations (23) than when backward scrambling is modeled (3). We confirmed the effect using 2 identical liver perfusion conditions, with the only difference being in whether lactate/pyruvate or propionate was labeled. Simple equations gave 60% lower estimates of pyruvate cycling from [U-13C]lactate/pyruvate than when a full regression model containing randomization was used. In contrast, [U-13C]propionate gave similar values using the simple equations and regression models with randomization. Ideally, regression models would always be used to fit isotopomer data to complex pathways, but in some cases, simple equations derived from complex models can be sufficient (and far more portable) if assumptions are valid or have limited effects when violated.
Summary.
Increased hepatic anaplerotic/cataplerotic flux not only contributes to impaired regulation of circulating nutrients (e.g., glycemia and lipidemia), but may also initiate oxidative metabolism during obesity and insulin resistance (Figure 8B). In the setting of NAFLD, constitutive oxidative metabolism may cause collateral oxidative stress and inflammatory events that reinforce insulin resistance and hepatocellular damage.
Methods
Chemicals
[3, 4-13C2] glucose (98%) was purchased from Omicron Biochemicals. [3, 4-13C2] ethyl acetoacetate (98%), [1,2-13C2] sodium β-hydroxybutyrate (98%) and [U- 13C4] sodium β-hydroxybutyrate (98%) were purchased from Isotec. [U-13C3]propionate and deuterium oxide (99%) were purchased from Cambridge Isotopes. Other common chemicals were obtained from Sigma-Aldrich.
Animals and diets
Male C57BL/6 mice (~25–30 g) and Wistar rats (~175–200 g) were purchased from Charles River Laboratories and maintained on a control diet with 10% calories from fat (Harlan Teklad TD06416). Whole-body PEPCK knockdown (Pck1lox+neo/lox+neo, knockdown), liver-specific knockout (Pck1lox/lox Alb-Cre), and WT (Pck1lox/lox) mice were generated as previously described (27). Animals were maintained on a standard lab chow, a semisynthetic control diet (10% calories from fat; Harlan Teklad TD06416), or a HFD (60% calories from fat; Harlan Teklad TD06414) for 16 weeks beginning at 4 to 6 weeks of age. After 12 weeks on a HFD, a subset of mice were provided a HFD mixed with 0.5% (w/w) metformin (Sigma-Aldrich) for 4 weeks. Experiments were carried out in either overnight-fasted (~16 hours) or ab libitum–fed mice.
Liver perfusions
Mouse livers were perfused as previously described (28, 45). Briefly, livers were exposed by laparotomy, and the portal vein was cannulated. Perfusion was initiated, and the hepatic vein was dissected simultaneously. The liver was isolated and perfused at 8 ml/min with a nonrecirculating buffer in a double-jacketed vessel maintained at 37°C for 60 minutes. Perfusate consisted of a well-oxygenated (~500 mm Hg) Krebs-Henseleit bicarbonate buffer containing 1.5 mM lactate, 0.15 mM pyruvate, 0.25 mM glycerol, 0.1 mM [U-13C3] propionate, 3% v/v D20, and a 0.2 mM or 0.8 mM mix of NEFA (40% palmitic, 35% oleic acid, 15% linoleic, and 10% palmitoleic) bound to fatty acid–free BSA. Several variations of this design were used to test the responsiveness of the tracer approach. Where designated, NEFA was replaced with 0.2 mM octanoate to increase fat oxidation. Where designated, [U-13C3] lactate and [U-13C3] pyruvate were used and propionate was not 13C enriched. Where designated, insulin was infused directly into the portal line using a syringe pump to deliver the equivalent of 10 ng/ml. Aliquots of effluent perfusate were collected every 15 minutes for glucose, ketone, and oxygen measurements. Oxygen concentration was measured using a blood gas analyzer, and oxygen consumption (MVO2) was determined as the difference between oxygen concentration in the afferent and efferent perfusate.
Tracer infusions
Rats.
Jugular vein catheters were surgically implanted, and rats were allowed to recover for 5 days. On day 6, rats were fasted for 4 hours and were infused with 20% fat emulsion (Intralipid, Fresenius Kabi) and heparin (1.3 ml/h for 5 hours) to achieve a 4- to 5-fold increase in the plasma free fatty acid levels. A 2.5% glycerol infusion served as control for intralipid infusions to account for the 2.5% glycerol in the intralipid mixture. After an initial 2-hour infusion of glycerol or intralipid, stable isotope tracers were coinfused as previously described (42). Briefly, rats received an intraperitoneal injection (20 μl/g rat) containing [U-13C3] propionate (5 mg/ml) dissolved in D2O. A bolus of infusion for 10 minutes (2.25 ml/h) was administered, followed by continuous infusion for 80 minutes (0.5 ml/h) containing ~20 mM each of [3,4-13C2] glucose, [3,4-13C2] acetoacetate, and [1,2-13C2] β-hydroxybutyrate. At the end of the infusion, rats were anesthetized, whole blood was collected from vena cava, and tissues were collected and stored at –80°C until further analysis.
Mice.
An indwelling jugular vein catheter was implanted, and mice were allowed to recover to their presurgical weights. Following an overnight fast (~16 hours), mice were infused with a mixture of stable isotope tracers in a 2-phase manner of 90 minutes each, as previously described (13). Briefly, mice were infused with [3,4-13C2] acetoacetate and [U-13C4] sodium β-hydroxybutyrate as a bolus (8.8 and 6.7 μmol/h) for 10 minutes and as a continuous infusion (3.5 and 2.7 μmol/h) for another 80 minutes. Approximately 50 μl of blood was collected for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis of ketone turnover (63).
Mice then received an intraperitoneal injection of isotonic D2O (99%; 28 μl/ g body weight) followed by an infusion of [U-13C3]propionate (50 mg/ml) and [3,4-13C2] glucose (3.72 mg/ml) at a 0.3 ml/h bolus for 10 minutes and a 0.06 ml/h continuous infusion for another 80 minutes. Mice were anesthetized, whole blood was rapidly collected from the descending aorta, and tissues were collected and stored at –80°C until further analysis.
Isotopomer analysis
Glucose and TCA cycle metabolism.
Briefly, blood glucose from rats and mice and glucose isolated from perfusate was converted to 1,2-isopropylidene glucofuranose (monoacetone glucose [MAG]). MAG was analyzed by 2H and 13C isotopomer analysis on a 14T spectrometer equipped with a 3-mm broad-band probe, and peak areas were analyzed (ACD/Labs 11.0). The 2H signals in the H2, H5, and H6s positions of MAG were used to determine fractional rates of glycogenolysis and GNG as previously detailed (13). The 13C NMR multiplets in the C3 and C4 resonance were used to determined [3,4-13C2]glucose enrichment. Glucose production, measured by glucose assay in liver perfusions (28, 45), or [3,4-13C2]glucose turnover was used to determine rates of glycogenolysis and GNG (13).
Isotopomers of C1, C2, and C3 of MAG were evaluated as the doublet 1,2 (D12 signifies 13C in C1 and C2), doublet 2,3 (D23 signifies 13C in C2 and C3), and quartet (Q signifies 13C in C1, C2 and C3) (Figure 1E). These isotopomers were used to calculate rates of anaplerosis, GNG, and pyruvate cycling relative to TCA cycle flux as previously described (64), using a principle similar to that described by Landau (16, 19). Absolute rates were obtained by normalizing the relative rate of GNG to the absolute rate of GNG determined by 2H analysis (13).
Ketone turnover.
In rats, steady-state [3,4-13C2]acetoacetate/β-hydroxybutyrate and [1,2-13C2] β-hydroxybutyrate/acetoacetate enrichments were examined by isotopomer analysis of their 13C NMR resonances and multiplets as previously described (42). In mice, [3,4-13C2]acetoacetate/β-hydroxybutyrate and [U-13C4]β-hydroxybutyrate/acetoacetate enrichments were examined by LC-MS/MS mass isotopomer analysis, as previously described (63).
Estimated hepatic oxygen consumption
Oxygen consumption by liver was estimated from the net production of RE (NADH or FADH2) produced via pathway fluxes as previously described (65) with some modifications. The β-oxidation of palmitate generated 8 acetyl-CoA and 14 RE (or 1.75 RE per acetyl-CoA), and oxidation of acetyl-CoA in the TCA cycle produced an additional 4 RE. Therefore, TCA cycle flux generated 5.75 RE. Ketogenesis consumed 2 acetyl-CoA per ketone; thus, acetoacetate production generated 3.5 RE, while its reduction to β-hydroxybutyrate generated 2.5 RE. The glycerol 3-phosphate dehydrogenase reaction generated 1 RE during GNG from glycerol. The glyceraldehyde 3-phosphate dehydrogenase step in GNG from PEP consumed 1 RE, which was offset when lactate (lactate dehydrogenase produces 1 RE), but not pyruvate/alanine, was used as a gluconeogenic precursor. The sum of these fluxes was used to determine net RE production using the following equation:
RE = 5.75 × TCA + 3.5 × ACAC + 2.5 × BHB + GNGglycerol – 0.1 × GNGPEP
(Equation 1)
where TCA is flux through the TCA cycle measured by 13C isotopomer analysis of perfusate or plasma glucose, ACAC is acetoacetate production measured in effluent perfusate or by 13C-acetoacetate turnover, and BHB is β-hydroxybutyrate production measured in effluent perfusate or by 13C–β-hydroxybutyrate turnover. GNGglycerol is GNG from glycerol and GNGpep is GNG from TCA cycle intermediates measured by 2H analysis of perfusate or plasma glucose. The equation assumes (a) that all acetyl-CoA originates from palmitate; should acetyl-CoA originate from longer fatty acids or lactate, then the factor of 5.75 would very slightly underestimate RE from β-oxidation (e.g., oleate changes the factor to 5.77 and lactate changes the factor to 6); and (b) that substrates for GNGpep are 90% lactate and 10% pyruvate/alanine based on the accepted hepatocellular redox state; should pyruvate/alanine contribute more, then RE would be overestimated, and if glutamine contributed, the RE would be underestimated.
Theoretical oxygen consumption was calculated based on ½ O2 + NADH + H+ → H2O + NAD+, as in the following equation:
Theoretical MVO2 = RE/2
(Equation 2)
This equation underestimates hepatic oxygen consumption to the extent that (a) not all sources of RE production can be measured; (b) sources of O2 consumption that do not consume RE (e.g., CYP450) have not been measured.
Metabolic modeling
The effect of incomplete backward scrambling on the analysis of glucose isotopomers after [U-13C]lactate/[U-13C]pyruvate or [U-13C]propionate metabolism was examined using the programs tcaSIM and tcaCALC (www.utsouthwestern.edu/education/medical-school/departments/airc/tools-references/software-downloads/running-programs.html). Experimentally determined fluxes (relative to TCA cycle flux) of anaplerosis (2.7), pyruvate cycling (1.5), and GNG (1.2) (Figure 2) were assigned in tcaSim (ypc = 2.6, ys = 0.1, pk = 1.5). Lactate/pyruvate enrichment was assigned to Lac123 = 0.1 or 0. Propionate enrichment was assigned to AS123 = 1 or 0. Backward scrambling (rof) was arrayed from 0.8 to 1. The simulated 13C multiplets formed in glucose were used to recalculate fluxes using the simple equations (64). Backward scrambling was confirmed using glucose C1, C2, and C3 isotopomers formed during [U-13C]lactate/[U-13C]pyruvate + propionate or [U-13C]propionate + lactate/pyruvate perfusions as inputs into tcaCALC and fit to a model of ypc, ys, pk, and rof.
Assessment of tissue redox state and energy charge
Liver mitochondrial NAD+/NADH was estimated from the plasma acetoacetate/β-hydroxybutyrate ratio (29) in WT and knockdown mice following tracer infusions. Plasma was immediately treated with NaBD4 to preserve acetoacetate as deuterium-labeled β-hydroxybutyrate, and the sample was analyzed by LC-MS (63). After correction for natural abundance, the M+1/M ratio was taken as the acetoacetate/β-hydroxybutyrate ratio. The NAD+/NADH ratio was estimated from the β-hydroxybutyrate dehydrogenase equilibrium (i.e., NAD+/NADH = [acetoacetate]/[β-hydroxybutyrate] × 1/KβHBDH; where KβHBDH =4.92 × 10–2 ) (32).
Snap-frozen liver samples were collected from a subset of WT and knockdown mice on the HFD. Organic acid concentrations were measured by gas chromatography–MS (GC-MS) as previously described (66). The Q/QH2 ratio was estimated from the succinate dehydrogenase equilibrium (i.e., Q/QH2 = [Fum]/[succ] × 1/KSDH; where KSDH = 172) (31). The ΔG for combined complexes I and II was calculated as previously described (31). The NADP+/NADPH ratio was estimated from the malic enzyme equilibrium (i.e., NADP+/NADPH = [pyr]/[mal] × [CO2] × 1/KME, where [CO2] = 1.2 × 10–3 and KME = 3.4 × 10–2) (32).
Liver ATP, ADP, and AMP were measured using an HPLC method we previously described (67) that was modified for MS detection. Briefly, the frozen liver samples were spiked with [13C10,15N5]ATP and [13C10,15N5]AMP (Sigma-Aldrich) internal standards before extraction. Analysis was performed on an API 3200 triple quadrupole LC-MS/MS mass spectrometer (Applied Biosystems/Sciex Instruments) in positive electrospray ionization mode. A reverse-phase C18 column (Waters xBridge, 150 × 2.1 mm, 3 μm) and a gradient elution consisting of water/methanol (5:95, v/v) with 4 mM dibutylamine acetate (eluent A) and acetonitrile with 4 mM dibutylamine acetate (eluent B) was used to achieve separation. Positive-ion–mode ESI and multiple reaction monitoring (MRM) were used to quantify nucleotides in standard solutions and biological samples.
Determination of hepatic insulin sensitivity
Overnight-fasted (~16 hours) mice were infused with insulin (Humulin R, Eli Lilly) at 5 mU–1kg–1min–1. Dextrose (30%) mixed with [U-13C6]glucose (~50 μmoles/ml) was infused at variable rates to maintain euglycemia (~120 mg/dl) for 120 minutes. Glucose concentration was detected in tail blood using a glucometer. Glucose infusion rates and plasma glucose enrichment during the last 60 minutes were used to estimate glucose turnover. Plasma [U-13C6] glucose enrichment was measured by LC-MS/MS under negative ion electrospray ionization with MRM, using 179 → 119 (unlabeled glucose) and 192 → 128 ([U-13C6] glucose) transitions. Clamped glucose production was compared with basal glucose production in a subgroup of mice used to determine basal fluxes (see above), but under otherwise identical conditions.
Insulin action was determined using a previously reported approach (59) with some modifications. Briefly, overnight-fasted WT and knockdown mice were anesthetized and laparotomy was performed to obtain a small (~50 mg) biopsy of liver. Insulin (1 IU/kg) was injected into the portal vein, and 3 minutes later, another portion of liver was obtained. Western blotting was used to determine the AKT-p/AKT ratios in these samples, and the fold induction of the ratio following insulin injection was taken as insulin action.
Histology
H&E staining of liver in overnight-fasted mice was performed as we previously described (13). Inflammation was assessed in 8 WT and 5 knockdown livers by a blinded colleague who counted the occurrence of infiltrate clusters in 15 fields of view per liver.
Staining of ROS was performed using DHE staining. Livers from overnight-fasted mice were isolated and immediately frozen in a tissue-freezing medium (O.C.T. compound, Tissue-Tek) and stored at –80°C until staining. Samples were equilibrated to –21°C and sectioned at 8-μm thickness on a Leica CM3050S cryotome. Resulting sections were affixed to silanated-adhesive slide glass (Superfrost-Plus, Fisher Scientific), protected from light, and briefly air dried prior to staining. Serial section companion slides were prepared for immediate DHE staining and for short-term storage at –80°C prior to DHE staining. Thawed sections were incubated (37°C) under coverslips with 2 μM DHE Na-Hepes-Krebs buffer for 30 minutes. Sections were immediately reviewed and imaged using Cy3 single-bandpass fluorescence microscopy (510–560 nm excitation, 572–648 nm emission) on a Leica DM2000 photomicroscope equipped with an Optronics Microfire CCD camera. Control diet panels were 592 μm wide, and HFD panels were 1184 μm wide.
In vitro analysis of oxidative stress
H4IIE cells (ATCC, CRL-1548) were cultured in 100 mm2 sterile cell culture dishes in high-glucose DMEM (Sigma Aldrich, D6429) plus 10% FBS media at 37°C in 5% CO2 to a confluency of approximately 80%. The medium was changed overnight to low-glucose DMEM (Sigma-Aldrich, D6046) supplemented with penicillin-streptomycin. The cells were treated in triplicate for 5 hours with 0.2 mM or 0.8 mM BSA–palmitic acid in low-glucose DMEM with or without 100 μM mercaptopicolinate (PEPCK inhibitor). Cells were subsequently incubated in 8 ml of low-glucose DMEM containing 15 μM 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (Invitrogen, catalog C-400) at 37°C for 1 hour in the dark. The medium was removed, and cells were scraped and pelleted by centrifugation twice in 1 ml of cold PBS. The pellet was subsequently sonicated in 400 μl of PBS and centrifuged at 9,500 g for 10 minutes at 4°C. The supernatant was diluted with 600 μl of PBS and analyzed by fluorescence spectrophotometry with excitation at 485 nm and an emission scan from 500 to 560 nm. The fluorescent intensity at 526 nm per mg protein was reported as the fold change from the untreated controls (BSA only, ± mercaptopicolinate).
Gene-expression analysis
qPCR was performed as previously reported (13). Analysis was performed in triplicate with 25 ng cDNA, 150 nmol/l of each primer, and 5 μl SYBR Green PCR master mix (Applied Biosystems) on an Applied Biosystems Prism 7900HT sequence detection system. The comparative threshold method was used to determine relative mRNA levels with cyclophilin b (Ppib) as the internal control. PCR array of oxidative stress was performed using the oxidative stress RT Profiler (catalog PAMM-065Z, QIAGEN).
Western blot analysis
Approximately 50 mg of frozen liver tissue was homogenized in RIPA buffer (Cell Signaling) with complete protease inhibitors (Roche) followed by two 10-second sonications at 10% amplitude (Branson). The following antibodies were obtained from Cell Signaling Technology: Akt (catalog 9272), p-Akt (S473; catalog 4060), NF-κB (catalog 8242), and pNF-κB (S536; catalog 3033).
Other assays and hormone measurements
Plasma ketone body and NEFA concentrations were determined using commercial assays (Wako Chemicals Inc.). Plasma insulin was measured by enzyme-linked immunoassay using the Mouse Insulin ELISA Kit (CrystalChem). Hepatic triglyceride concentrations were determined using an analytical kit (Sigma-Aldrich). Oxidative damage indicated by malondialdehyde levels was assayed using the thiobarbituric acid–reactivity assay as we previously described (13).
Human biopsies
Biopsies were obtained from nondiabetic African-Americans and Hispanics with metabolic syndrome and elevated liver enzymes as part of a larger study (7, 68). Subjects underwent an evaluation to ensure there were no viral, heritable, or environmental (ethanol) reasons for elevations in serum aminotransferases and were offered liver biopsy (n = 16). A total of 8 subjects underwent ultrasound-guided percutaneous liver biopsy by a hepatologist. All samples were read and scored by an independent hepatopathologist using standards established by the NASH Clinical Research Network (34).
Statistics
Data are reported as mean ± SEM. Significant differences (P < 0.05) were determined using 1-way ANOVA, 2-tailed t test or 1-tailed t test, as indicated in figure legends, and with correction for multiple comparisons where appropriate. Outliers were detected by a Grubb’s test. Significant correlations were determined using 2-tailed (or 1-tailed where designated) Pearson correlation calculations. All statistics and graph preparations were performed using Prism 6 (Graph Pad Software Inc.).
Study approval
All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. All human subjects gave written informed consent prior to participation, and the study was approved by the Institutional Review Board at UT Southwestern Medical Center (approval no. 062007-025).
Author contributions
ADS, CRM, IC, MAM, EJP, and SCB designed experiments; SS, BK, JAGD, JAF, LR, NES, TH, LAN, KL, EJP, and XF performed experiments; SS, XF, JAGD, MEM, JMS, and JL acquired data; SS, BK, JAGD, NES, XF, MEM, JMS, JL, and SCB analyzed data; JDB performed liver biopsies; MAM provided reagents; and JDB and SCB wrote the manuscript.
Supplementary Material
Acknowledgments
Support for this work was provided by NIH RO1DK078184 (to S.C. Burgess), P01DK058398 (to S.C. Burgess), DK087977 (to J.D. Browning and S.C. Burgess), RL1DK081187 (to E.J. Parks), UL1DE019584 (to J.D. Browning, E.J. Parks, and S.C. Burgess), P41 EB015908 (to C.R. Malloy), UL1-RR024982 (Clinical Translational Science Award), and the Robert A. Welch Foundation I-1804 (to S.C. Burgess). J.A.G. Duarte was supported by an educational grant from the Portuguese Foundation for Science and Technology (SFRH/BD/44294/2008). The authors thank Thomas E. Rogers for assessment of human liver biopsies. Celeste Thomas provided graphic art support.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(12):4447–4462. doi:10.1172/JCI82204.
References
- 1.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277(34):30409–30412. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 2.Basu R, et al. Pathogenesis of prediabetes: role of the liver in isolated fasting hyperglycemia and combined fasting and postprandial hyperglycemia. J Clin Endocrinol Metab. 2013;98(3):E409–E417. doi: 10.1210/jc.2012-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beylot M, Soloviev MV, David F, Landau BR, Brunengraber H. Tracing hepatic gluconeogenesis relative to citric acid cycle activity in vitro and in vivo. Comparisons in the use of [3-13C]lactate, [2-13C]acetate, and α-keto[3-13C]isocaproate. J Biol Chem. 1995;270(4):1509–1514. doi: 10.1074/jbc.270.4.1509. [DOI] [PubMed] [Google Scholar]
- 4.Walter P, Paetkau V, Lardy HA. Paths of carbon in gluconeogenesis and lipogenesis. 3. The role and regulation of mitochondrial processes involved in supplying precursors of phosphoenolpyruvate. J Biol Chem. 1966;241(11):2523–2532. [PubMed] [Google Scholar]
- 5.Chakravarty K, Cassuto H, Reshef L, Hanson RW, Cox MM. Factors that control the tissue-specific transcription of the gene for phosphoenolpyruvate carboxykinase-C. Crit Rev Biochem Mol Biol. 2005;40(3):129–154. doi: 10.1080/10409230590935479. [DOI] [PubMed] [Google Scholar]
- 6.Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- 7.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14(6):804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hakimi P, et al. Phosphoenolpyruvate carboxykinase and the critical role of cataplerosis in the control of hepatic metabolism. Nutr Metab (Lond) 2005;2: doi: 10.1186/1743-7075-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adina-Zada A, Zeczycki TN, St Maurice M, Jitrapakdee S, Cleland WW, Attwood PV. Allosteric regulation of the biotin-dependent enzyme pyruvate carboxylase by acetyl-CoA. Biochem Soc Trans. 2012;40(3):567–572. doi: 10.1042/BST20120041. [DOI] [PubMed] [Google Scholar]
- 10.Williamson JR, Scholz R, Browning ET. Control mechanisms of gluconeogenesis and ketogenesis. II. Interactions between fatty acid oxidation and the citric acid cycle in perfused rat liver. J Biol Chem. 1969;244(17):4617–4627. [PubMed] [Google Scholar]
- 11.Williamson JR, Cooper RH. Regulation of the citric acid cycle in mammalian systems. FEBS Lett. 1980;117(suppl):K73–K85. doi: 10.1016/0014-5793(80)80572-2. [DOI] [PubMed] [Google Scholar]
- 12.Begriche K, Massart J, Robin M-A, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58(4):1497–1507. doi: 10.1002/hep.26226. [DOI] [PubMed] [Google Scholar]
- 13.Satapati S, et al. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res. 2012;53(6):1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mantena SK, et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J. 2009;417(1):183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rector RS, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52(5):727–736. doi: 10.1016/j.jhep.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landau BR, Schumann WC, Chandramouli V, Magnusson I, Kumaran K, Wahren J. 14C-labeled propionate metabolism in vivo and estimates of hepatic gluconeogenesis relative to Krebs cycle flux. Am J Physiol. 1993;265(4 pt 1):E636–E647. doi: 10.1152/ajpendo.1993.265.4.E636. [DOI] [PubMed] [Google Scholar]
- 17.Jones JG, Solomon MA, Cole SM, Sherry AD, Malloy CR. An integrated (2)H and (13)C NMR study of gluconeogenesis and TCA cycle flux in humans. Am J Physiol Endocrinol Metab. 2001;281(4):E848–E856. doi: 10.1152/ajpendo.2001.281.4.E848. [DOI] [PubMed] [Google Scholar]
- 18.Jin ES, Jones JG, Merritt M, Burgess SC, Malloy CR, Sherry AD. Glucose production, gluconeogenesis, and hepatic tricarboxylic acid cycle fluxes measured by nuclear magnetic resonance analysis of a single glucose derivative. Anal Biochem. 2004;327(2):149–155. doi: 10.1016/j.ab.2003.12.036. [DOI] [PubMed] [Google Scholar]
- 19.Magnusson I, et al. Noninvasive tracing of Krebs cycle metabolism in liver. J Biol Chem. 1991;266(11):6975–6984. [PubMed] [Google Scholar]
- 20.Di Donato L, Des Rosiers C, Montgomery JA, David F, Garneau M, Brunengraber H. Rates of gluconeogenesis and citric acid cycle in perfused livers, assessed from the mass spectrometric assay of the 13C labeling pattern of glutamate. J Biol Chem. 1993;268(6):4170–4180. [PubMed] [Google Scholar]
- 21.Befroy DE, et al. Direct assessment of hepatic mitochondrial oxidative and anaplerotic fluxes in humans using dynamic 13C magnetic resonance spectroscopy. Nat Med. 2014;20(1):98–102. doi: 10.1038/nm.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasenour CM, et al. Mass spectrometry-based microassay of 2H and 13C plasma glucose labeling to quantify liver metabolic fluxes in vivo. Am J Physiol Endocrinol Metab. 2015;309(2):E191–E203. doi: 10.1152/ajpendo.00003.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen KF, Blair JB, Shulman GI. Triiodothyronine treatment increases substrate cycling between pyruvate carboxylase and malic enzyme in perfused rat liver. Metabolism. 1995;44(11):1380–1383. doi: 10.1016/0026-0495(95)90133-7. [DOI] [PubMed] [Google Scholar]
- 24.Roden M, et al. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes. 2000;49(5):701–707. doi: 10.2337/diabetes.49.5.701. [DOI] [PubMed] [Google Scholar]
- 25.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egnatchik RA, Leamy AK, Jacobson DA, Shiota M, Young JD. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol Metab. 2014;3(5):544–553. doi: 10.1016/j.molmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.She P, Shiota M, Shelton KD, Chalkley R, Postic C, Magnuson MA. Phosphoenolpyruvate carboxykinase is necessary for the integration of hepatic energy metabolism. Mol Cell Biol. 2000;20(17):6508–6517. doi: 10.1128/MCB.20.17.6508-6517.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burgess SC, et al. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007;5(4):313–320. doi: 10.1016/j.cmet.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berry MN, Williamson DH, Wilson MB. Concentrations of acetoacetate and d-(--)-3-hydroxybutyrate in rat liver and blood. Biochem J. 1965;94:17C–19C. doi: 10.1042/bj0940017C. [DOI] [PubMed] [Google Scholar]
- 30.Bergman C, Kashiwaya Y, Veech RL. The effect of pH and free Mg2+ on ATP linked enzymes and the calculation of Gibbs free energy of ATP hydrolysis. J Phys Chem B. 2010;114(49):16137–16146. doi: 10.1021/jp105723r. [DOI] [PubMed] [Google Scholar]
- 31.Sato K, et al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995;9(8):651–658. doi: 10.1096/fasebj.9.8.7768357. [DOI] [PubMed] [Google Scholar]
- 32.Krebs HA, Veech RL. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv Enzyme Regul. 1969;7:397–413. doi: 10.1016/0065-2571(69)90030-2. [DOI] [PubMed] [Google Scholar]
- 33.Pernicova I, Korbonits M. Metformin — mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 34.Kleiner DE, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 35.Ishak K, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22(6):696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 36.Cheng Z, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15(11):1307–1311. doi: 10.1038/nm.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14(2):193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koliaki C, Roden M. Hepatic energy metabolism in human diabetes mellitus, obesity and non-alcoholic fatty liver disease. Mol Cell Endocrinol. 2013;379(1–2):35–42. doi: 10.1016/j.mce.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Felig P, Wahren J, Hendler R, Brundin T. Splanchnic glucose and amino acid metabolism in obesity. J Clin Invest. 1974;53(2):582–590. doi: 10.1172/JCI107593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koliaki C, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21(5):739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 41.Rector RS, et al. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology. 2013;57(6):2213–2223. doi: 10.1002/hep.26285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satapati S, et al. Partial resistance to peroxisome proliferator-activated receptor-alpha agonists in ZDF rats is associated with defective hepatic mitochondrial metabolism. Diabetes. 2008;57(8):2012–2021. doi: 10.2337/db08-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cotter DG, et al. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J Clin Invest. 2014;124(12):5175–5190. doi: 10.1172/JCI76388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iozzo P, et al. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology. 2010;139(3):846–856.e846. doi: 10.1053/j.gastro.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 45.Burgess SC, et al. Impaired tricarboxylic acid cycle activity in mouse livers lacking cytosolic phosphoenolpyruvate carboxykinase. J Biol Chem. 2004;279(47):48941–48949. doi: 10.1074/jbc.M407120200. [DOI] [PubMed] [Google Scholar]
- 46.Owen MR, Halestrap AP. The mechanisms by which mild respiratory chain inhibitors inhibit hepatic gluconeogenesis. Biochim Biophys Acta. 1993;1142(1–2):11–22. doi: 10.1016/0005-2728(93)90079-u. [DOI] [PubMed] [Google Scholar]
- 47.Hasenour CM, Berglund ED, Wasserman DH. Emerging role of AMP-activated protein kinase in endocrine control of metabolism in the liver. Mol Cell Endocrinol. 2013;366(2):152–162. doi: 10.1016/j.mce.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moreno-Sanchez R, et al. Reactive oxygen species are generated by the respiratory complex II — evidence for lack of contribution of the reverse electron flow in complex I. FEBS J. 2013;280(3):927–938. doi: 10.1111/febs.12086. [DOI] [PubMed] [Google Scholar]
- 49.Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014;289(12):8312–8325. doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao D, et al. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J Biol Chem. 2010;285(39):29965–29973. doi: 10.1074/jbc.M110.128694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakamura S, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284(22):14809–14818. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Egnatchik RA, Leamy AK, Noguchi Y, Shiota M, Young JD. Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism. 2014;63(2):283–295. doi: 10.1016/j.metabol.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab. 2012;23(3):142–153. doi: 10.1016/j.tem.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I) J Biol Chem. 2011;286(31):27103–27110. doi: 10.1074/jbc.M111.252502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28(12):551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Xu Z, Chen L, Leung L, Yen TS, Lee C, Chan JY. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci U S A. 2005;102(11):4120–4125. doi: 10.1073/pnas.0500660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vomhof-Dekrey EE, Picklo MJ., Sr The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J Nutr Biochem. 2012;23(10):1201–1206. doi: 10.1016/j.jnutbio.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Huang LJ, Chuang IC, Dong HP, Yang RC. Hypoxia-inducible factor 1alpha regulates the expression of the mitochondrial ATPase inhibitor protein (IF1) in rat liver. Shock. 2011;36(1):90–96. doi: 10.1097/SHK.0b013e318219ff2a. [DOI] [PubMed] [Google Scholar]
- 59.Gomez-Valades AG, et al. Overcoming diabetes-induced hyperglycemia through inhibition of hepatic phosphoenolpyruvate carboxykinase (GTP) with RNAi. Mol Ther. 2006;13(2):401–410. doi: 10.1016/j.ymthe.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 60.Sun Y, et al. Phosphoenolpyruvate carboxykinase overexpression selectively attenuates insulin signaling and hepatic insulin sensitivity in transgenic mice. J Biol Chem. 2002;277(26):23301–23307. doi: 10.1074/jbc.M200964200. [DOI] [PubMed] [Google Scholar]
- 61.Chawla A, Nguyen KD, Goh YPS. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11(11):738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burgess SC, et al. Effect of murine strain on metabolic pathways of glucose production after brief or prolonged fasting. Am J Physiol Endocrinol Metab. 2005;289(1):E53–E61. doi: 10.1152/ajpendo.00601.2004. [DOI] [PubMed] [Google Scholar]
- 63.Sunny NE, et al. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am J Physiol Endocrinol Metab. 2010;298(6):E1226–E1235. doi: 10.1152/ajpendo.00033.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jones JG, Naidoo R, Sherry AD, Jeffrey FMH, Cottam GL, Malloy CR. Measurement of gluconeogensis and pyruvate recycling in the rat liver: a simple analysis of glucose and glutamate isotopomers during metabolism of [1,2,3-13C3]propionate. FEBS Lett. 1997;412(1):131–137. doi: 10.1016/S0014-5793(97)00764-3. [DOI] [PubMed] [Google Scholar]
- 65.Malloy CR, Jones JG, Jeffrey FM, Jessen ME, Sherry AD. Contribution of various substrates to total citric acid cycle flux and anaplerosis as determined by 13C isotopomer analysis and O2 consumption in the heart. MAGMA. 1996;4(1):35–46. doi: 10.1007/BF01759778. [DOI] [PubMed] [Google Scholar]
- 66.Desrosiers C, Fernandez CA, David F, Brunengraber H. Reversibility of the mitochondrial isocitrate dehydrogenase reaction in the perfused rat liver. J Biol Chem. 1994;269(44):27179–27182. [PubMed] [Google Scholar]
- 67.Burgess SC, et al. Diminished hepatic gluconeogenesis via defects in tricarboxylic acid cycle flux in peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α)-deficient mice. J Biol Chem. 2006;281(28):19000–19008. doi: 10.1074/jbc.M600050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.