

Abstract
Hepatocellular carcinoma (HCC) is characterized by the aberrant expression of a number of genes that govern crucial signaling pathways. The insulin-like growth factor (IGF) axis is important in this context, and the precise regulation of expression of members of this axis is known to be lost in HCC. miR-155 is a well-established oncogene in numerous types of cancer. However, to the best of our knowledge, its effect on the regulation of the IGF axis has not been investigated to date. The present study aimed to elucidate the interactions between miR-155 and key components of the IGF axis, in addition to examining its effect on HCC development. Reverse transcription-quantitative polymerase chain reaction was used to measure the expression of miR-155 in HCC and cirrhotic tissues, in addition to HCC cell lines. Furthermore, the effect of the induction of miR-155 expression on the expression of three members of the IGF axis [IGF II, IGF type-1 receptor (IGF-1R) and IGF-binding protein 3 (IGFBP-3)], was analyzed. Finally, the effect of miR-155 on HCC cell proliferation, migration and clonogenicity was also examined. Quantification of the expression of miR-155 demonstrated that it is upregulated in HCC. Induction of the expression of miR-155 in HCC cell lines led to the upregulation of IGF-II and IGF-IR, and the downregulation of IGFBP-3. In addition, the proliferation, migration and clonogenicity of HCC was increased following induction of miR-155 expression. miR-155 is an oncomiR, which upregulates the oncogenes, IGF-II and IGF-IR, and downregulates the tumor suppressor, IGFBP-3, thereby resulting in increased HCC cell carcinogenicity. Therefore, miR-155 may be a therapeutic target in HCC.
Keywords: microRNA-155, insulin-like growth factor II, insulin-like growth factor type-1 receptor, insulin-like growth factor-binding protein 3, hepatocellular carcinoma
Introduction
microRNAs (miRs) are known to be involved in the development of hepatocellular carcinoma (HCC). They are non-coding microregulators of ~22 nucleotides in length, which are involved in the regulation of hepatocarcinogenesis (1). miRs may function as oncogenes or as tumor suppressors. An example of an oncogenic miR, or oncomiR, is miR-221. HCC cells were shown to overexpress miR-221, which, in turn, enhances HCC cell proliferation, migration and invasion by enhancing AKT signaling (2–4). By contrast, miR-122, a tumor suppressor, is the most abundantly expressed miR in the healthy liver, while it is downregulated in HCC (5). Inducing the expression of miR-122 has been shown to induce apoptosis and suppress proliferation in HepG2 and Hep3B cells, which is in part due to direct targeting of Cyclin G1 (6,7).
miR-155 has been demonstrated to be involved in the development of a number of types of malignancies, such as B cell malignancies (8), breast cancer (9) and colon cancer (10), in addition to hepatocellular carcinoma (11), which was the focus of the present study. It has been reported that miR-155 is an oncomiR that is frequently upregulated in HCC (12). miR-155 was found to be involved in the initiation of hepatocarcinogenesis, in which it provided a link between inflammation and HCC. Zhang et al (13) reported that the inflammation-related transcription factor, nuclear factor κB, induces upregulation of miR-155, which in turn promotes tumorigenesis by activating Wnt signaling. Similarly, loss of function studies demonstrated that silencing miR-155 leads to G0/G1 cell cycle arrest and increased cell death (13). This is in accordance with other observations, where the upregulation of miR-155 in HCC has been shown to correlate with the upregulation of oncogenes, such as cyclin D1, c-MYC and matrix metalloproteinase-9 (14), and the downregulation of tumor suppressors, such as phosphatase and tensin homolog and CCAAT/enhancer binding protein β (12). Therefore, miR-155 is hypothesized to be a potential target for the treatment of HCC.
Insulin-like growth factor-II (IGF-II) is a member of a complex system, termed the IGF axis. The IGF axis consists of two ligands, two receptors and six IGF-binding proteins (IGFBPs). Upon binding of the ligands to the tyrosine kinase receptor, IGF type-1 receptor (IGF-1R), activation of the RAF/MEK/ERK and the PI3K/AKT/mTOR mitogenic signaling pathways occurs, which leads to the induction of cell proliferation, differentiation and survival (15). Uncontrolled IGF-II-IGF-1R interaction, and consequent excessive intracellular mitogenic signals, is a common feature of a number of types of cancer. This is ordinarily restricted by the presence of IGFBPs, which sequester IGF-II in the blood and thereby limit its interaction with IGF-1R (16).
Three IGF axis members are known to be involved in the development of HCC. IGF-II has been reported to be upregulated in HCC tissues, and this increase in expression was correlated with cancer cell proliferation and tumor neovascularization (17,18). IGF-1R has also been demonstrated to be upregulated in HCC. Rodriguez-Tarduchy et al (19) reported that the primary tumorigenic effects of IGFs are regulated by IGF-IR. By contrast, the expression of IGFBP-3 has been found to be downregulated in HCC, and appears to exhibit tumor suppressor effects, by neutralizing IGFs and inducing the expression of p53 (20).
Based on preliminary in silico miR-mRNA binding predictions, the current study aimed to investigate the effect of miR-155 on the three IGF axis family members, and on HCC cellular functions.
Patients and methods
Patients
The present study included 23 patients with HCC, who underwent liver transplant surgery in the Kasr El Einy Hospital (Cairo University, Cairo, Egypt). Four samples of cirrhotic tissues were taken from a subsection of these patients with focal HCC lesions. Ten biopsies from healthy livers were obtained. According to the agreement between the ethical review committee of the German University in Cairo and the institutional review board of Cairo University, ethical approval was obtained for the present study. In addition, all participants provided written informed consent. The institutional ethics committees approving this research comply with the principles set forth in the international reports and guidelines of the Helsinki Declaration and the International Ethical Guidelines for Biomedical Research Involving Human Subjects, issued by the Council for International Organizations of Medical Sciences. The majority of the patients (66.6%) had >1 focal lesion, as indicated in the pathology report, and were subjected to clinical assessment, as summarized in Table I.
Table I.
Clinical assessment of 23 patients with HCC.
| Parameter | Value |
|---|---|
| Age (years) | 49±13.5 |
| Aspartate aminotransferase (U/l) | 100.5±65.8 |
| Alanine aminotransferase (U/l) | 85.6±95.6 |
| Alkaline Phosphatase (U/l) | 110.2±60.7 |
| Serum albumin (g/dl) | 4.6±1.5 |
| Serum α fetoprotein (ng/ml) | 155.7±22.3 |
Data are presented as the mean ± standard deviation. Male:female, 2:1. All patients were positive for hepatitis C virus antibody. HCC, hepatocellular carcinoma.
Cell cultures
HuH-7 and HepG2 cells were maintained in Dulbecco's modified Eagle's medium (Lonza, Basal, Switzerland), supplemented with 4.5 g/l glucose, 4 mmol/l L-glutamine, 10% fetal bovine serum and Mycozap (1:500, Lonza) at 37°C in a 5% CO2 atmosphere.
Transfection of miR oligonucleotides
HuH-7 and HepG2 cell lines were transfected with miScript™ miRNA mimics/inhibitors of miR-155, and scrambled miRs (Qiagen, Venlo, The Netherlands). All transfection experiments were conducted at room temperature using HiPerfect Transfection Reagent (Qiagen), according to the manufacturer's instructions. Experiments were performed in triplicate and repeated 3 times. Cells that were exposed only to the transfection reagent were designated mock cells; cells transfected with scrambled miRs were designated as Scr-miR cells; cells transfected with miR-155 mimics were designated miR-155 cells; and cells transfected with the miR-155 inhibitor were designated anti-miR-155 cells. Cells were lysed 48 h posttransfection and total RNA was extracted for further analysis.
mRNA and miR extraction from liver biopsies and HCC cell lines
mRNA and miR were extracted from liver biopsies and the HCC cell lines. Fresh liver samples (HCC, cirrhotic and healthy tissues) were collected during surgery and were immediately snap-frozen in liquid nitrogen. The specimens were manually pulverized in liquid nitrogen and ~100 mg of the resulting tissue powder was used for large and small RNA extraction, using mirVana miRNA Isolation kit (Ambion Life Technologies, Austin, TX, USA), according to the manufacturer's instructions. Cells were harvested 48 h after transfection, according to the HiPerfect Transfection Reagent instructions. For transfection of HuH-7 cell, 150 ng oligonucleotides was used and for HepG2 cells, 300 ng oligonucleotides were used in 6-well plates. The RNA yield was quantified using a V-530 UV-Vis Spectrophotometer (Jasco International Co., Ltd., Tokyo, Japan), and RNA integrity was tested by 18S rRNA bands detection on 1% agarose gel electrophoresis. RNA samples with an Optical Density 260/280 of >2 were excluded.
miR and mRNA quantification
Extracted miRs were reverse transcribed into single stranded cDNA, using a TaqMan® MicroRNA Reverse Transcription kit (Applied Biosystems Life Technologies, Foster City, CA, USA), and specific primers for hsa-miR-155 and RNU6B. IGF-II mRNA was reverse transcribed into cDNA, using the high-capacity cDNA reverse transcription kit (Applied Biosystems Life Technologies), according to the manufacturer's instructions. The relative expression of miR-155 to that of RNU6B, as well as that of IGF-II, IGF-1R and IGFBP-3 to that of β-microglobulin, was quantified with TaqMan Real-Time Q-PCR (ABI Assay IDs: 002623, 001093, Hs01005963_m1, Hs00609566_m1, Hs00365742_g1 and Hs00984230_m1 respectively), using StepOne™ Systems (Applied Biosystems Life Technologies). The polymerase chain reaction (PCR) for miR quantification included 1 µl TaqMan Small RNA Assay (20X) specific for miR-155 or RNU6B and 1.33 µl cDNA from the miR-155 or RNU6B RT reactions, respectively. TaqMan Gene Expression Assay (1.25 µl) specific for each of the three target genes, IGF-II, IGF-1R and IGFBP-3, and 5 µl cDNA from the mRNA reverse transcription reaction were used for quantification. For both reactions, TaqMan Universal PCR Master Mix II (2X) was used for quantifications (Applied Biosystems Life Technologies) and the cycling conditions were as follows: 2 min at 50°C and 10 min at 95°C followed by 40 cycles of 15 sec at 95°C and 60 sec at 60°C. The relative expression was calculated using the 2−∆∆Ct method. All PCR reactions, including controls, were run in duplicate.
Cell proliferation assay (BrdU)
HuH-7 and HepG2 cells were seeded 24 h prior to transfection in black 96-well plates, and transfected with 12.5 ng and 25 ng oligonucleotides, respectively, according to the HiPerfect instructions, with an initial constant cell count of 5×104 cells/well. At 48 h following oligonucleotide transfection, cells were labeled with BrdU labeling reagent for 4 h (final concentration, 100 uM), using the Cell Proliferation ELISA (Roche Applied Science, Penzberg, Germany). Cells were then fixed using FixDenate for 30 min and incubated with Anti-BrdU POD (final concentration, 10 uM) for 90 min. Colorimetric measurements were performed using Wallac 1420 Victor2™ Multilabel Counter (PerkinElmer, Inc., Waltham, MA, USA). All cell proliferation experiments were conducted in triplicate and repeated three times.
Cell scratch wound healing assay
HuH-7 and HepG2 cells were cultured to 80–90% confluence in 6-well plates. At 48 h post-transfection, 5 scratches/well were made in each plate with 200-µl pipette tip. Detached cells were removed using serum-free medium. Medium [Dulbecco's modified Eagle's medium supplemented with 4.5 g/l glucose, 4 mmol/l L-glutamine, 1% fetal bovine serum and Mycozap (1:500)] was then added and culture plates were incubated at 37°C for 48 h. Migration was then documented and wound closure was quantified with Image J software for Windows (version 1.48; http://rsbweb.nih.gov/ij/download.html), by measuring the surface area covered by the cells. All Scratch assays were conducted in duplicate (two wells/test, representing 10 scratches/test) and repeated three times.
Colony-forming assay
HuH-7 cells were seeded at an initial count of 1,000 cells/well and left to adhere overnight. Cells were then transfected with miR-155 mimics, inhibitors or small interfering RNA to IGF-II (siIGF-II). At 24 h post-transfection, cells were detached by trypsinization, and embedded on a gel composed of a soft 0.36% (w/v) agarose bottom layer and a hard 0.76% cell-free media-enriched agarose top layer. Cells were incubated at 37°C and colonized for 2 weeks. Colonies were observed under a light microscope (Axio Observer A1; Zeiss, Oberkochen, Germany) and the number of cells in each well was counted. All colony-forming assays were conducted in duplicate (two wells/test) and repeated three times.
Bioinformatics
Bioinformatics algorithms; microrna.org (www.microrna.org), miRDB (www.mirdb.org/miRDB/), DIANA Lab (www.diana.cslab.ece.ntua.gr/) and Target Scan (www.targetscan.org/) were used to predict 3′-UTR downstream targets for miR-155. 5′-UTR regions were predicted using bibiserv software (Bielefeld University Bioinformatics Server; http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/submission.html).
Statistical analysis
All data are presented as the mean ± standard error of the mean. Statistical significance was analyzed by performing one-way analysis of variance or paired Student's t-tests using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
miR-155 screening in non-metastatic liver cancer tissues and HCC cell lines
miR-155 expression was significantly higher in HCC and cirrhotic tissues, and in HCC cell lines, compared with that in healthy tissues (P=0.0015, P=0.0036 and P<0.0001, respectively; Fig. 1).
Figure 1.
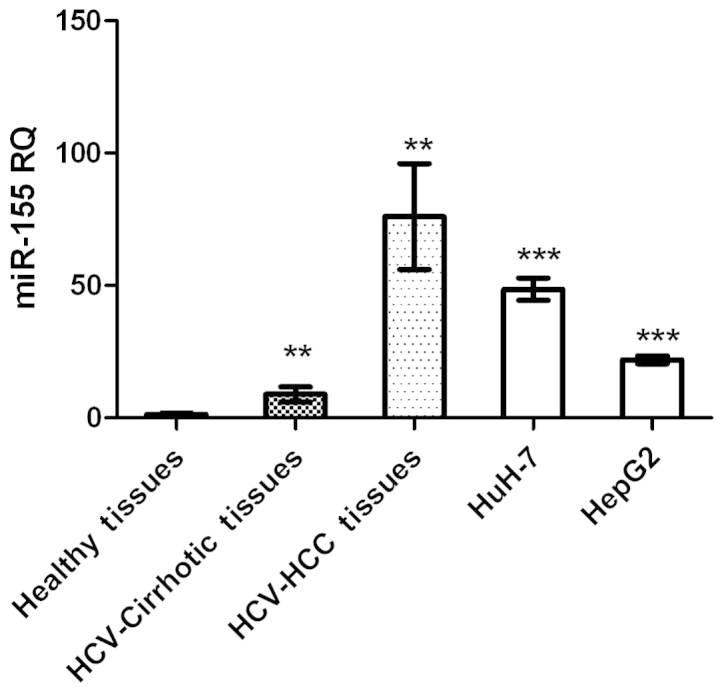
miR-155–5p expression in liver tissues and HCC cell lines. miR-155-5p expression was significantly higher in HCC (n=23) and cirrhotic tissues, as well as in HuH-7 and HepG2 cell lines, compared with healthy tissues (P=0.0015, P=0.0036, P<0.0001 and P<0.0001, respectively). miR, microRNA; HCC, hepatocellular carcinoma; HCV, hepatitis C virus. **P<0.01; ***P<0.001.
Bioinformatics
miR-155 accession numbers and mature sequences were retrieved using the miRBase database (http://www.mirbase.org/). Using different in silico algorithms for predicting binding to the downstream targets at the 3′-UTR and 5′-UTR positions, miR-155 was shown to target IGF-II IGF-1R and IGFBP-3 mRNA. Microrna.org and miRDeep2 computational algorithms, demonstrated that miR-155 targeted the 3′-UTR of the IGF-II ligand and IGF-1R, although not that of IGFBP-3 (Table II). By contrast, miR-155 was predicted to target the 5′-UTR of IGF-II, IGF-1R and IGFBP-3, according to bibiserv software (Table II).
Table II.
Binding sites of miR-155 to IGF-II, IGF-1R and IGFBP-3 UTRs.
| A, 5′-UTR target regions | |
|---|---|
| IGF family member | miR-155 binding position |
| IGF-II | Pos: 658 |
| IGF-1R | Pos: 127 |
| IGFBP-3 | Pos: 114 |
| B, 3′-UTR target regions | |
| IGF family member | miR-155 binding position |
| IGF-II | Pos: 3838 |
| IGF-1R | Pos: 4205 |
| IGFBP-3 | N/A |
Binding site positions: 5′UTR, predicted by bibiserv and 3′UTR, predicted by microrna.org and miRDeep2. UTR, untranslated region; miR, microRNA; IGF-II, insulin-like growth factor II; IGF-1R, IGF type-1 receptor; IGFBP-3, IGF-binding protein 3; N/A, not applicable.
Transfection efficiency of miR-155 oligonucleotides
HuH-7 and HepG2 cells transfected with miR-155 mimics, exhibited an upregulation in miR-155 expression of 20-fold and 17-fold compared with mock HuH-7 and HepG2 cells, respectively (both P<0.0001; Fig. 2).
Figure 2.
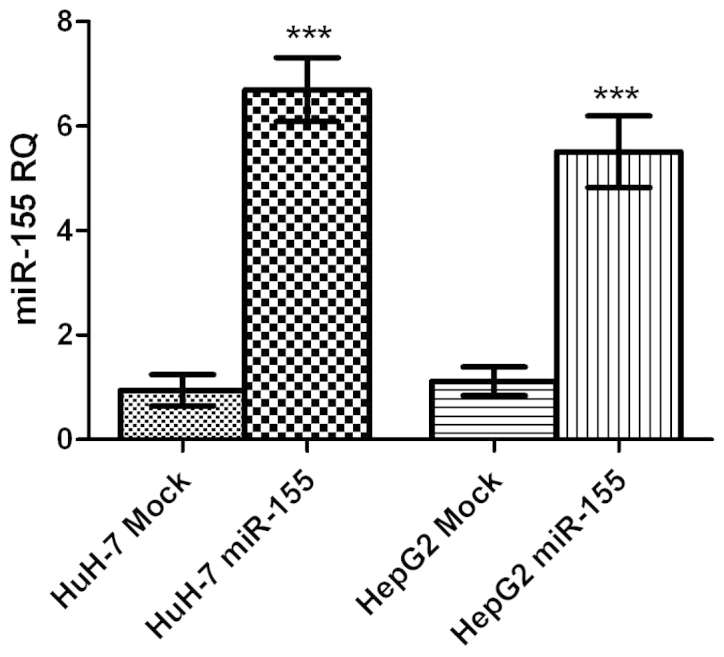
Transfection efficiency of miR-155 oligonucleotides. HuH-7 and HepG2 cells transfected with miR-155 mimics, exhibited upregulation of miR-155 expression by 20-fold and 17-fold, respectively, compared with mock cells (P<0.0001 and P<0.0001). miR-155 expression was normalized to that of RNU6B, as an internal control. miR, microRNA. ***P<0.001.
IGF-II, IGF-1R and IGFBP-3 mRNA expression following induction and inhibition of miR-155 expression
Transfection of HuH-7 with miR-155 mimics resulted in a significant increase in the expression of IGF-II mRNA (P=0.0046) as well as that of IGF-1R mRNA (P=0.0078), compared with control cells. By contrast, a significant downregulation of IGFBP-3 mRNA expression, compared with that of mock cells, was observed (P=0.0458). Inhibition of miR-155, significantly repressed IGF-II and IGF-1R mRNA expression in HuH-7 cells (P=0.0008 and P=0.0006, respectively). By contrast, IGFBP-3 mRNA expression was increased in cells transfected with miR-155, compared with miR-155 mimic-transfected cells, as shown in Fig. 3. The same statistically significant effects were observed in HepG2 cells; for example, a significant increase in IGF-II mRNA expression was observed in HepG2 cells tranfected with miR-155 mimics (P=0.029; data not shown).
Figure 3.
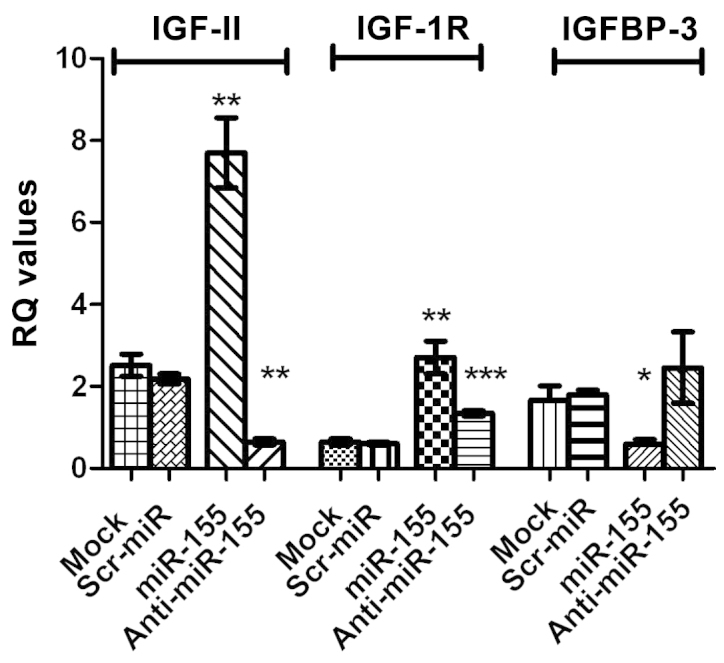
Effect of transfection with miR-155 mimics and inhibitors on IGF-II, IGF-1R and IGFBP-3 mRNA expression. Transfection of HuH-7 cells with miR-155 mimics, resulted in a significant increase in IGF-II and IGF-1R mRNA expression (P=0.0046 and P=0.0078, respectively) compared with that in untransfected mock cells. Transfection of HuH-7 cells with miR-155 mimics resulted in significant downregulation of IGFBP-3 mRNA expression (P=0.0458) compared with that in mock cells. Inhibition of miR-155 significantly repressed IGF-II and IGF-1R mRNA expression in HuH-7 cell lines (P=0.0008 and P=0.0006, respectively), while IGFBP-3 expression was increased compared with cells treated with miR-155 mimics. The same effects were observed in HepG2 cells (data not shown). miR, microRNA; IGF, insulin-like growth factor; IGF-1R, IGF type 1 receptor; IGFBP-3, IGF-binding protein 3. *P<0.05; **P<0.01; ***P<0.001.
Effect of miR-155 inhibition on cellular functions
Cellular proliferation
Transfection with the miR-155 inhibitor, significantly decreased HuH-7 and HepG2 cell proliferation (P=0.0005 and P=0.0009, respectively), compared with that of negative control and mock cells (Fig. 4A). By contrast, transfection with miR-155 mimics significantly enhanced the proliferation HuH-7 and HepG2 cells (P=0.0017 and P=0.0292, respectively Fig. 4A). It is worth noting that cells transfected with siIGF-II were used for confirmation and showed a negative effect on cell proliferation in Huh7 and HepG2 cells (P=0.0005 and P=0.0042, respectively; Fig. 4A), similar to the effect of miR-155 inhibitors.
Figure 4.
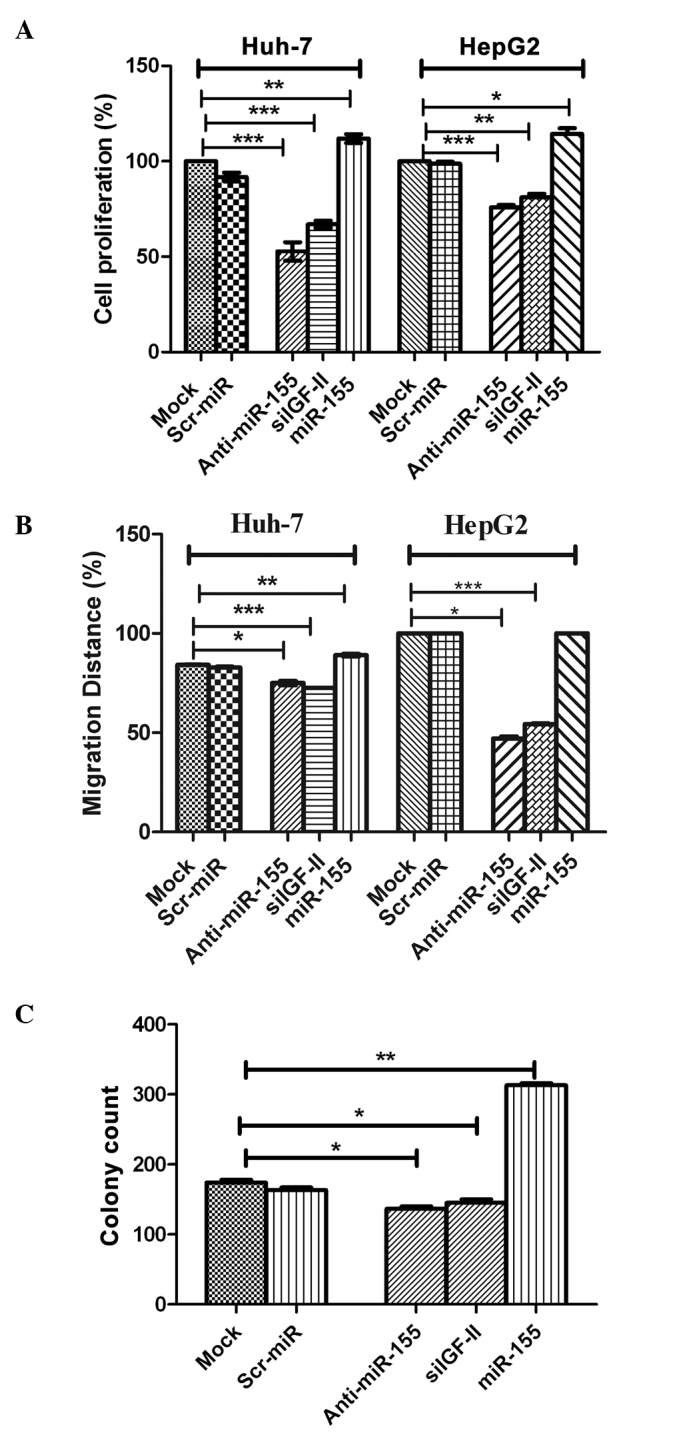
Effect of miR-155 inhibition on cellular functions. (A) Cellular proliferation. Inhibition of miR-155 significantly decreased HuH-7 and HepG2 cell proliferation compared with negative control and mock cells (P=0.0005 and P=0.0009, respectively). This was in contrast to induced miR-155 expression using miR-155 mimics, which significantly enhanced the proliferation of HuH-7 and HepG2 cells (P=0.0017 and P=0.0292, respectively). (B) Cellular migration. Inhibition of miR-155 abrogated the effect of miR-155 on the promotion of migration in HuH-7 and HepG2 cells compared with the controls, with coverage of 48 and 76% (P=0.0002 and P=0.0001, respectively) of the original scratch, respectively. By contrast, transfection of miR-155 mimics resulted in the promotion of tumor cell migration in HuH-7 and HepG2 cells, with coverage of 89 and 100% of the original scratch, compared with mock and Scr-miR, which had an original scratch coverage of 84.2 and 82.7%, and 100 and 100%, in these cell lines, respectively. (C) Cellular clonogenicity. Inhibition of miR-155 suppressed its tumorigenic effect by significantly decreasing the colony-forming ability of HuH-7 cells compared with that of with untransfected mock cells (P=0.0195). Induction of miR-155 overexpression induced high clonogenicity in HuH-7 cells compared with mock cells (P=0.0013). miR, microRNA; Scr-miR, scrambled miR; siIGF, small interfering RNA specific to insulin-like growth factor II. *P<0.05; **P<0.01; ***P<0.001.
Cellular migration
Images for the 2D scratch-migration assay were captured at 5× magnification (21). Migration inhibition by miR-155 antagomirs (inhibitors) abrogated the migratory effect of miR-155 in HuH-7 and HepG2 cells compared with the controls, with coverage of 48 and 76% of the original scratch, respectively (Fig. 4B). By contrast, transfection of miR-155 mimics resulted in the promotion of tumor cell migration in HuH-7 and HepG2 cells, with coverage of 89 and 100% of the original scratch, respectively, compared with mock and Scr-miR, which exhibited original scratch coverage of 84.2 and 82.7%, and 100 and 100%, in the HuH-7 and HepG2 cell lines, respectively (Fig. 4B). siIGF-II also significantly decreased the migration of Huh7 and HepG2 cells compared with mock untransfected cells, with coverage of 72.5 and 54.0% (P=0.0002 and P=0.0001, respectively) of the original scratch, respectively (Fig. 4B).
Cellular clonogenicity
Inhibition of miR-155 suppressed its tumorigenic effect by significantly decreasing the colony-forming capability of HuH-7 cells, compared with that of untransfected mock cells (P=0.0195; Fig. 4C). This was in contrast to the induction of miR-155 overexpression, which induced high clonogenicity in HuH-7 cells, compared with that of mock cells (P=0.0013; Fig. 4C). Furthermore, siIGF-II significantly suppressed colony formation in Huh7 cells compared with mock untransfected cells (P=0.0455; Fig. 4C).
Discussion
One of the pathways that may be involved in the pathogenesis of human HCC, is the IGF axis. Aberrant expression of members of the IGF axis is a hallmark of the initiation, progression and metastasis of HCC (21–25). In addition, miR-155 was recently demonstrated to be involved in HCC proliferation and migration (11,26). Therefore, the present study aimed to investigate whether an IGF axis-miR-155 interaction exists in HCC. In silico analysis demonstrated that miR-155 had putative target sites in the 3′-UTR regions of two IGF axis members, involved in HCC, namely IGF-II and IGF-1R (Table II). The aim of the current study was to examine the interaction between miR-155 and certain IGF axis members, in order to improve the understanding of the mechanisms underlying the development of HCC.
Expression profiling of miR-155 demonstrated higher expression of this molecule in liver biopsies from patients with non-metastatic HCC and cirrhosis, compared with those from healthy controls (Fig. 1). Similarly, when measured in HuH-7 and HepG2 cell lines, miR-155 expression was significantly upregulated compared with healthy controls (Fig. 1). These findings are in accordance with those from other studies, which reported the elevation of miR-155 expression in non-neoplastic tissues compared with that in normal liver tissues (27), and in biopsies from HCV-induced and HBV-induced HCC, compared with healthy tissues (11,13). Since the increase in miR-155 expression occurs at an early stage of hepatocarcinogenesis, even prior to the development of cirrhosis (28), it was hypothesized to act as a oncomir, bridging inflammation with carcinogenesis (13,29). Functional analysis, performed in HuH-7 and HepG2 cell lines, demonstrated that miR-155 promoted cellular proliferation (Fig. 4A), migration (Fig. 4B) and anchorage-independent growth (Fig. 4C). This data is in accordance with previous studies, which demonstrated that miR-155 promotes invasiveness of HCC cells and tumor recurrence, and predicts poor survival in patients with HCC following liver transplantation (14,26). Transfection of HuH-7 cells with inhibitors of miR-155, resulted in a significant reduction in the proliferation, migration and anchorage-independent growth of these cells, compared with mock cells (Fig. 4A–C). This data is in accordance with that from a previous study, which reported reduced cell growth and enhanced cell death in xenograft tumors, following knockdown of miR-155 expression in J7 and Mahlavu hepatoma cells (30). In addition, a separate study showed that miR-155 overexpression leads to the promotion of cell proliferation and tumorigenesis, and the inhibition of hepatocyte apoptosis, through the activation of Wnt signaling, by enhancing β-catenin nuclear accumulation with a simultaneous increase in cyclin D1 and c-myc expression (13).
This indicates that miR-155 may act through alternative mechanisms, to link inflammation with tumorigenesis. Preliminary in silico analysis demonstrated that miR-155 has potential target sites in the 3′-UTR regions of IGF-II and IGF-1R, although not that of IGFBP-3 (Table II). Despite this prediction, IGF-II and IGF-1R mRNA expression was significantly upregulated following ectopic induction of miR-155 expression in HuH-7 and HepG2 cells, compared with that in the respective untransfected mock cells (Fig. 3). This overexpression of the downstream targets was restored to normal levels following transfection of HuH-7 cells with specific antagomirs or miR-155 (Fig. 3). Ordinarily, miRNAs act by base-pairing with 3′-UTR of their target mRNAs, thereby hindering their translation into proteins or enhancing their degradation (31,32). However, the anticipated downregulation by miR-155 of its predicted targets did not occur in the present study. Further analysis using bioinformatic tools, demonstrated that miR-155 has potential target sites in the 5′-UTR regions of IGF-II and IGF-1R (Table II). The present findings were in accordance with those of another study, which proposed a new mechanism of action of miRNAs (33). The authors reported that 5′-UTR binding may lead to the activation of gene expression, through stabilization of the mRNA rather than the induction of its degradation, which may, in part, explain the observed induction of IGF-II and IGF-1R mRNAs by miR-155. This was not the first study to report the enhancement of target mRNA expression by miRNAs as a result of binding to the 5′-UTR region. Other studies have demonstrated that miR-122 and miR-346 may bind to the 5′-UTR of HCV RNA and Receptor-Interacting Protein 140, respectively, resulting in the induction of translation and thereby the upregulation of the expression of these proteins (34–37).
By contrast, the tumor suppressor, IGFBP-3, was predicted to bear a putative miR-155 binding site in its 5′-UTR, although not in its 3′-UTR (Table II). However, it was observed that IGFBP-3 mRNA expression was significantly downregulated in HuH-7 cells transfected with miR-155 mimics, compared with the untransfected mock cells (Fig. 3). The translational repression by miRNA-mediated 5′-UTR binding has been reported for miR-2 in Drosophila (38). The authors reported 6 miRNA binding sites in the 5′-UTR and in the open reading frame, where miR-2 binding inhibits translation initiation in a cap-dependent manner, induces mRNA deadenylation and decrease 80S complex formation, resulting in inhibition of the translation initiation step.
The present results suggest a possible mechanism underlying the involvement of miR-155 in the development of HCC. Following transfection of HuH-7 cells with miR-155 mimics, there was a concomitant increase in the mRNA expression of IGF-II and IGF-1R, which are mitogenic, and a decrease in the mRNA expression of IGFBP-3 mRNA, which is a tumor suppressor. This variability in the expression of the IGF axis family members, potentiates the tumorigenic effect of miR-155 by activating the downstream signaling cascade, eventually leading to tumor formation, progression and metastasis.
In conclusion, the present study demonstrated that miR-155 expression is higher in cirrhotic and non-metastatic HCC tissues, compared with that in healthy controls (Fig. 1), which is in accordance with the results of previous studies, implicating its upregulation in each stage of tumor formation beginning with inflammation, passing through cirrhosis, and culminating with HBV- and HCV-induced HCC (11,27,28). This is in contrast to other metastamiRs, such as miR-17-5p, which was found to be significantly downregulated in non-metastatic HCC, while its expression was upregulated only in metastatic HCC (39,40). The current data indicate that miR-155 is an oncogenic miR that is involved early in the development of HCC. To the best of our knowledge, the present study demonstrated, for the first time, a novel mechanism through which miR-155 regulates multiple oncogenic IGF pathway components in HCC. The current results provide a rationale for the potential use of miR-155 in therapeutic approaches as a ‘one for all’ target, through which multiple oncogenic targets may be inhibited and tumor suppressors induced.
Acknowledgements
The authors would like to thank the patients and healthy volunteers for their participation. This study was funded by the Science and Technology Development Fund.
References
- 1.Sidhu K, Kapoor NR, Pandey V, Kumar V. The ‘macro’ world of microRNAs in hepatocellular carcinoma. Front Oncol. 2015;5:68. doi: 10.3389/fonc.2015.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM, Dejean A. MiR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci USA. 2010;107:264–269. doi: 10.1073/pnas.0907904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan Q, Loya K, Rani B, Möbus S, Balakrishnan A, Lamle J, Cathomen T, Vogel A, Manns MP, Ott M, et al. MicroRNA-221 overexpression accelerates hepatocyte proliferation during liver regeneration. Hepatology. 2013;57:299–310. doi: 10.1002/hep.25984. [DOI] [PubMed] [Google Scholar]
- 4.Jensen MR, Factor VM, Fantozzi A, Helin K, Huh CG, Thorgeirsson SS. Reduced hepatic tumor incidence in cyclin G1-deficient mice. Hepatology. 2003;37:862–870. doi: 10.1053/jhep.2003.50137. [DOI] [PubMed] [Google Scholar]
- 5.Girard M, Jacquemin E, Munnich A, Lyonnet S, Henrion-Caude A. miR-122, a paradigm for the role of microRNAs in the liver. J Hepatol. 2008;48:648–656. doi: 10.1016/j.jhep.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 6.Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 7.Datta J, Kutay H, Nasser MW, Nuovo GJ, Wang B, Majumder S, Liu CG, Volinia S, Croce CM, Schmittgen TD, et al. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008;68:5049–5058. doi: 10.1158/0008-5472.CAN-07-6655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Wang M, Tan LP, Dijkstra MK, van Lom K, Robertus JL, Harms G, Blokzijl T, Kooistra K, van T'veer MB, Rosati S, et al. miRNA analysis in B-cell chronic lymphocytic leukaemia: Proliferation centres characterized by low miR-150 and high BIC/miR-155 expression. J Pathol. 2008;215:13–20. doi: 10.1002/path.2333. [DOI] [PubMed] [Google Scholar]
- 9.Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, Liu MF, Wang ED. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70:3119–3127. doi: 10.1158/0008-5472.CAN-09-4250. [DOI] [PubMed] [Google Scholar]
- 10.Bakirtzi K, Hatziapostolou M, Karagiannides I, Polytarchou C, Jaeger S, Iliopoulos D, Pothoulakis C. Neurotensin signaling activates microRNAs-21 and −155 and Akt, promotes tumor growth in mice and is increased in human colon tumors. Gastroenterology. 2011;141(e1):1749–1761. doi: 10.1053/j.gastro.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Q, Chen X, Lu F, Zhang T, Hao M, Wang Y, Zhao J, McCrae MA, Zhuang H. Aberrant expression of microRNA 155 may accelerate cell proliferation by targeting sex-determining region Y box 6 in hepatocellular carcinoma. Cancer. 2012;118:2431–2442. doi: 10.1002/cncr.26566. [DOI] [PubMed] [Google Scholar]
- 12.Wang B, Majumder S, Nuovo G, Kutay H, Volinia S, Patel T, Schmittgen TD, Croce C, Ghoshal K, Jacob ST. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50:1152–1161. doi: 10.1002/hep.23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Wei W, Cheng N, Wang K, Li B, Jiang X, Sun S. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology. 2012;56:1631–1640. doi: 10.1002/hep.25849. [DOI] [PubMed] [Google Scholar]
- 14.Yan XL, Jia YL, Chen L, Zeng Q, Zhou JN, Fu CJ, Chen HX, Yuan HF, Li ZW, Shi L, et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: Role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology. 2013;57:2274–2286. doi: 10.1002/hep.26257. [DOI] [PubMed] [Google Scholar]
- 15.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 16.Fürstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol. 2002;3:298–302. doi: 10.1016/S1470-2045(02)00731-3. [DOI] [PubMed] [Google Scholar]
- 17.Bae MH, Lee MJ, Bae SK, Lee OH, Lee YM, Park BC, Kim KW. Insulin-like growth factor II (IGF-II) secreted from HepG2 human hepatocellular carcinoma cells shows angiogenic activity. Cancer Lett. 1998;128:41–46. doi: 10.1016/S0304-3835(98)00044-5. [DOI] [PubMed] [Google Scholar]
- 18.Lahm H, Gittner K, Krebs O, Sprague L, Deml E, Oesterle D, Hoeflich A, Wanke R, Wolf E. Diethylnitrosamine induces long-lasting re-expression of insulin-like growth factor II during early stages of liver carcinogenesis in mice. Growth Horm IGF Res. 2002;12:69–79. doi: 10.1054/ghir.2002.0261. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Tarduchy G, Collins MK, García I, López-Rivas A. Insulin-like growth factor I inhibits apoptosis in IL-3-dependent hemopoietic cells. J Immunol. 1992;149:535–540. [PubMed] [Google Scholar]
- 20.Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem. 1997;272:12181–12188. doi: 10.1074/jbc.272.18.12181. [DOI] [PubMed] [Google Scholar]
- 21.Nussbaum T, Samarin J, Ehemann V, Bissinger M, Ryschich E, Khamidjanov A, Yu X, Gretz N, Schirmacher P, Breuhahn K. Autocrine insulin-like growth factor-II stimulation of tumor cell migration is a progression step in human hepatocarcinogenesis. Hepatology. 2008;48:146–156. doi: 10.1002/hep.22297. [DOI] [PubMed] [Google Scholar]
- 22.Gong Y, Cui L, Minuk GY. The expression of insulin-like growth factor binding proteins in human hepatocellular carcinoma. Mol Cell Biochem. 2000;207:101–104. doi: 10.1023/A:1007010818094. [DOI] [PubMed] [Google Scholar]
- 23.Aleem E, Nehrbass D, Klimek F, Mayer D, Bannasch P. Upregulation of the insulin receptor and type I insulin-like growth factor receptor are early events in hepatocarcinogenesis. Toxicol Pathol. 2011;39:524–543. doi: 10.1177/0192623310396905. [DOI] [PubMed] [Google Scholar]
- 24.Khandwala HM, McCutcheon IE, Flyvbjerg A, Friend KE. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr Rev. 2000;21:215–244. doi: 10.1210/edrv.21.3.0399. [DOI] [PubMed] [Google Scholar]
- 25.LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. doi: 10.1016/S0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 26.Han ZB, Chen HY, Fan JW, Wu JY, Tang HM, Peng ZH. Up-regulation of microRNA-155 promotes cancer cell invasion and predicts poor survival of hepatocellular carcinoma following liver transplantation. J Cancer Res Clin Oncol. 2012;138:153–161. doi: 10.1007/s00432-011-1076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoon SO, Chun SM, Han EH, Choi J, Jang SJ, Koh SA, Hwang S, Yu E. Deregulated expression of microRNA-221 with the potential for prognostic biomarkers in surgically resected hepatocellular carcinoma. Hum Pathol. 2011;42:1391–1400. doi: 10.1016/j.humpath.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 28.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tili E, Michaille JJ, Wernicke D, Alder H, Costinean S, Volinia S, Croce CM. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci USA. 2011;108:4908–4913. doi: 10.1073/pnas.1101795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang YH, Lin KH, Chen HC, Chang ML, Hsu CW, Lai MW, Chen TC, Lee WC, Tseng YH, Yeh CT. Identification of postoperative prognostic microRNA predictors in hepatocellular carcinoma. PLoS One. 2012;7:e37188. doi: 10.1371/journal.pone.0037188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattick JS, Makunin IV. Small regulatory RNAs in mammals. Hum Mol Genet. 2005;14:R121–R132. doi: 10.1093/hmg/ddi101. [DOI] [PubMed] [Google Scholar]
- 32.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 33.Henke JI, Goergen D, Zheng J, Song Y, Schüttler CG, Fehr C, Jünemann C, Niepmann M. MicroRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villanueva RA, Jangra RK, Yi M, Pyles R, Bourne N, Lemon SM. MiR-122 does not modulate the elongation phase of hepatitis C virus RNA synthesis in isolated replicase complexes. Antiviral Res. 2010;88:119–123. doi: 10.1016/j.antiviral.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Norman KL, Sarnow P. Modulation of hepatitis C virus RNA abundance and the isoprenoid biosynthesis pathway by microRNA miR-122 involves distinct mechanisms. J Virol. 2010;84:666–670. doi: 10.1128/JVI.01156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jopling CL. Regulation of hepatitis C virus by microRNA-122. Biochem Soc Trans. 2008;36:1220–1223. doi: 10.1042/BST0361220. [DOI] [PubMed] [Google Scholar]
- 37.Tsai NP, Lin YL, Wei LN. MicroRNA mir-346 targets the 5′-untranslated region of receptor-interacting protein 140 (RIP140) mRNA and up-regulates its protein expression. Biochem J. 2009;424:411–418. doi: 10.1042/BJ20090915. [DOI] [PubMed] [Google Scholar]
- 38.Moretti F, Thermann R, Hentze MW. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA. 2010;16:2493–2502. doi: 10.1261/rna.2384610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang F, Yin Y, Wang F, Wang Y, Zhang L, Tang Y, Sun S. miR-17-5p Promotes migration of human hepatocellular carcinoma cells through the p38 mitogen-activated protein kinase-heat shock protein 27 pathway. Hepatology. 2010;51:1614–1623. doi: 10.1002/hep.23566. [DOI] [PubMed] [Google Scholar]
- 40.El Tayebi HM, Omar K, Hegy S, El Maghrabi M, El Brolosy M, Hosny KA, Esmat G, Abdelaziz AI. Repression of miR-17-5p with elevated expression of E2F-1 and c-MYC in non-metastatic hepatocellular carcinoma and enhancement of cell growth upon reversing this expression pattern. Biochem Biophys Res Commun. 2013;434:421–427. doi: 10.1016/j.bbrc.2013.04.003. [DOI] [PubMed] [Google Scholar]