

Abstract
Objective:
This study aims to compare the pharmacokinetics and oral bioavailability of a capsule combining 17β-estradiol and progesterone in a non–peanut oil–containing formulation with those of widely used and approved separate formulations of estradiol and progesterone coadministered to healthy postmenopausal women.
Methods:
This was an open-label, balanced, randomized, single-dose, two-treatment, three-period, three-sequence, cross-over, partial-replicate, reference-scaled study. Postmenopausal women (aged 40-65 y) were randomly assigned to one of three dosing sequences of test and reference products (TRR, RTR, or RRT, where T is the test drug and R is the coadministered reference product), with each of the three periods separated by a 14-day washout. The primary pharmacokinetic endpoints were Cmax, AUC(0-t), and AUC(0-inf) for the test and reference products, assessed for bioequivalence using the scaled average bioequivalence or unscaled average bioequivalence method. Safety was assessed by clinical observation, participant-reported adverse events, and laboratory data, including blood levels of hormones.
Results:
Sixty-six women were randomly assigned, and 62 women (94.0%) completed all three study periods. All AUC and Cmax parameters met bioequivalence criteria for all analytes (estradiol, progesterone, and estrone), except Cmax for total estrone. The extent of estradiol and progesterone absorption was similar between the test product and the reference products. Four adverse events—all considered mild and unrelated to the study drugs—were reported.
Conclusions:
The combination 17β-estradiol/progesterone product demonstrates bioavailability similar to those of the respective reference products of estradiol and progesterone. If regulatory approval is obtained, this new hormone therapy would be the first treatment of menopause symptoms to combine progesterone with 17β-estradiol in an oral formulation.
Keywords: Bioavailability, Estradiol, Bioidentical hormone therapy, Menopause, Progesterone, Combination
The ability to combine the natural hormones 17β-estradiol (estradiol) and progesterone in a single-dose form to treat menopause symptoms while providing endometrial protection would be clinically useful. A comprehensive search suggests that no single drug combining these two hormones has been approved by the US Food and Drug Administration (FDA). A product that combines 17β-estradiol with progesterone and offers good bioavailability of both hormones is difficult to achieve biochemically. Progesterone has poor bioavailability—it is highly lipophilic and undergoes a complex metabolic process, making it difficult to administer orally or transdermally.1 Administering clinically effective oral doses of estradiol and progesterone together in a formulation that does not compromise the bioavailability of either hormone is challenging because of differences in their structure and solubility.
Many compounding pharmacies manufacture products that combine estrogen and progesterone. Considering the poor bioavailability of oral and transdermal progesterone and the difficulty in determining the appropriate ratio of progesterone to estradiol, compounded hormone products should be viewed with caution.1,2 Pharmacokinetic studies of products manufactured by compounding pharmacies (with the aim to ensure adequate bioavailability) are rarely performed, and few clinical trials have appropriately evaluated the safety and efficacy of compounded hormones.3 For example, despite the widespread use of custom-compounded progesterone gels and creams, no evidence exists to show that any of them opposes estradiol sufficiently to protect against endometrial hyperplasia.
Although some individuals have a legitimate need for compounded hormone therapy (HT),4,5 use of compounded HT greatly exceeds the scope of need. Determining the full extent to which these unregulated, largely untested compounded HT combinations are being used in the United States is difficult because sales are not tracked and some may be obtained without a prescription. However, compounded hormones are believed to make up a large and growing share of estrogen-progestogen therapy (EPT) for menopausal symptoms,4,5 suggesting an unmet need.
An investigational oral drug that combines 2 mg of solubilized 17β-estradiol with 200 mg of progesterone in a gelatin capsule (TX-001HR; TherapeuticsMD Inc, Boca Raton, FL) has been developed. The estradiol and progesterone ingredients used in the combination are plant-derived and chemically identical to human hormones of ovarian origin. Consistent with FDA guidelines that call for comparing the bioequivalence of any new product with the market standard, a randomized, open-label, three-period, three-sequence, two-treatment, partial-replicate, cross-over study was conducted to characterize the drug's pharmacokinetic and safety profiles in healthy postmenopausal women. The objective was to show that the bioavailability of the estradiol and progesterone compounds used in TX-001HR was equivalent to the bioavailability of the same doses of commercially available, separate formulations of oral estradiol and progesterone coadministered under fed conditions.
METHODS
Participant selection
Participants eligible for this randomized study were healthy postmenopausal women aged 40 to 65 years with a body mass index between 18.5 and 30 kg/m2. “Postmenopausal” was defined as a plasma estradiol level lower than 50 ng/L, a plasma follicle-stimulating hormone level higher than 30 IU/L, and no vaginal bleeding for at least 3 years. At screening, women were required to provide detailed medical history and to undergo physical and gynecologic examinations. Laboratory testing was performed to determine baseline hormone levels and to evaluate other hematological safety parameters.
Exclusion criteria included pregnancy or breast-feeding, allergy or hypersensitivity to estradiol or progesterone, underlying disease or recent major illness, history of substance abuse, and use of menopausal therapy in the 6 months before dosing. Current smokers or women who had smoked in the past 3 months were also excluded. In relation to the study check-in date, other prohibited substances were as follows: prescription medications or hormonal agents in the past 14 days, over-the-counter medications (including herbal products) in the past 7 days, grapefruit juice or poppy-containing foods in the past 48 hours, and caffeine in the past 24 hours. Participants were also instructed to abstain from taking prescription, over-the-counter, or herbal medications; grapefruit juice; and poppy-containing foods for the duration of the study.
The study protocol was approved by an independent ethics committee, and a written informed consent form was obtained from each participant before screening and after enrollment. All procedures followed the ethical guidelines of Good Clinical Practices and the Declaration of Helsinki.
Study design
In this randomized open-label study, the bioavailability of TX-001HR (a softgel capsule containing 2 mg of solubilized estradiol and 200 mg of progesterone) was compared with the bioavailability of separate oral formulations of estradiol (Estrace; estradiol USP tablets 2 mg; Teva Pharmaceuticals, Sellersville, PA) and progesterone (Prometrium; progesterone softgel capsule 200 mg; Catalent Pharma Solutions, St Petersburg, FL) administered together (reference products). The doses of estradiol and progesterone were selected in accordance with the doses recommended in the most recent FDA guidance for bioequivalence studies of oral estradiol and oral progesterone in postmenopausal women under fed conditions.6,7
Progesterone and estradiol are highly variable drugs.8 Thus, the scaled average bioequivalence (SABE) method recommended by the FDA for bioequivalence studies of highly variable drugs was applied.9 We used a replicate, three-sequence, three-period, cross-over design, in which participants served as their own controls and received the coadministered reference products twice.
We used SAS version 9.2 (SAS Institute Inc, Cary, NC) to generate a randomization schedule. Based on the randomized schedule, participants were assigned, in equal numbers, to one of three dosing sequences (TRR, RTR, or RRT, where T is the test drug and R is the coadministered reference product). In each sequence, participants received a single dose of TX-001HR in one study period and a single dose of estradiol plus a single dose of progesterone in each of the remaining two periods. The same doses of the test and reference products were used for all three study periods (Fig. 1). The dose in each of the three study periods was separated by a 14-day washout to eliminate drug carryover effects. Study randomization was balanced, and access to the randomization code was controlled.
FIG. 1.
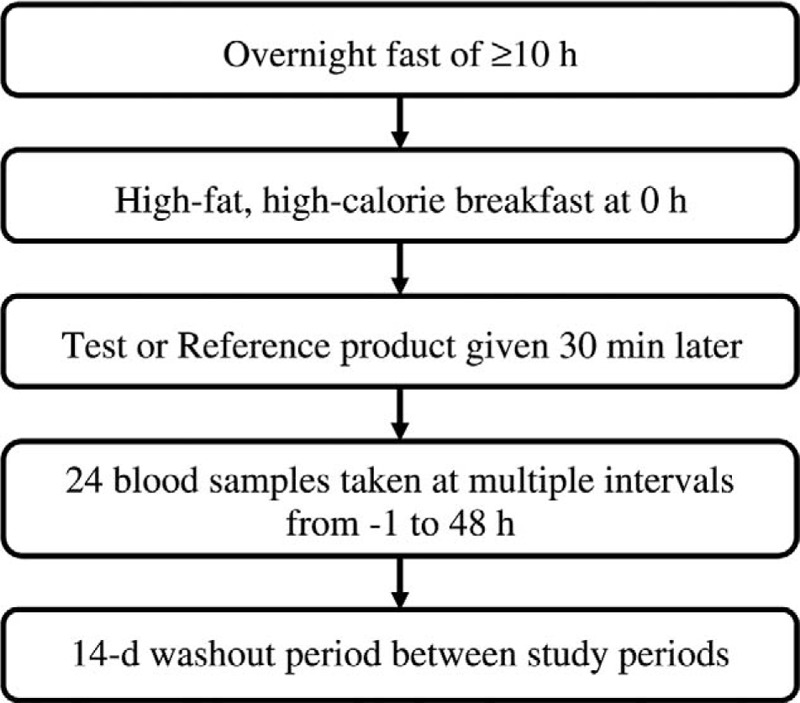
Flow chart outlining the dosing procedure for each study period.
At the start of each study period and on days 1, 15, and 29, women checked into the clinical facility at least 11 hours before dosing, where they remained for at least 24 hours after dosing. Women fasted overnight and consumed a high-fat high-calorie breakfast in the morning, after which they were given a single dose of the assigned medication and were asked to sit upright and to fast for the next 4 hours. In the day before dosing and during the 3 days after dosing, women's well-being was assessed. They underwent complete physical examination, which included measurement of vital signs, a urine pregnancy test, and a drug screen. Adverse events were also recorded. At the end of period III of the study (48 h after the final dose), all women underwent poststudy safety assessment.
During each period, 24 blood samples (6-8 mL) were drawn: 3 blood samples were collected in the 75 minutes before dosing (−01.00, 00.50, and 00.00 h), and 21 blood samples were collected in the 48 hours after dosing (00.25, 00.50, 00.67, 00.83, 01.00, 01.33, 01.67, 02.00, 02.50, 03.00, 04.00, 05.00, 06.00, 07.00, 08.00, 10.00, 12.00, 18.00, 24.00, 36.00, and 48.00 h). The samples were collected in Vacutainer tubes containing K2EDTA and centrifuged at 4,000 rpm for 10 minutes at 4°C. The plasma samples were divided into two aliquots (one for measuring progesterone and unconjugated estrone and one for quantifying unconjugated estradiol and total estrone) and stored at −70°C pending analyses. Pharmacokinetic evaluation was performed at Micro Therapeutic Research Laboratories Private Ltd (Chennai, India) using WinNonlin software version 5.3 (Pharsight/Certara, St Louis, MO). The analysts responsible for determining drug concentrations in the blood samples were blinded to the randomization code throughout the study.
Analytical methods
Plasma samples were analyzed for progesterone, estradiol, estrone, and total estrone—by three methods using high-performance liquid chromatography (HPLC)–tandem mass spectrometry—within the concentration ranges shown in Table 1. The HPLC–tandem mass spectrometry device was equipped with a turbo ion spray and operated in positive ion mode. All three methods used multiple reaction monitoring for detection and quantification of progesterone, estradiol, estrone, and total estrone. Progesterone, estradiol, estrone, and estrone metabolites are endogenous steroid compounds. Calibration standards were prepared in charcoal-stripped human plasma, and quality control (QC) standards were prepared in unstripped human plasma matrix. Baseline endogenous concentrations of estradiol, estrone, and total estrone were measured and added to the spiked QC concentrations of the analytes. This corrected concentration was used as the nominal QC concentration for each analyte to backcalculate with calibration curve standards prepared in charcoal-stripped plasma. For estradiol, estrone, and total estrone, matrix stability experiments (eg, benchtop, freeze-thaw, and long-term stability) were conducted in charcoal-stripped and unstripped human plasma in the presence of coanalytes, including progesterone.
TABLE 1.
Concentration ranges for liquid chromatography–tandem mass spectrum assay
| Parameter | Concentration range |
| Progesterone, ng/mL | 0.4-121.8 |
| Unconjugated estradiol, pg/mL | 25.3-5,034.4 |
| Unconjugated estrone, pg/mL | 5.0-1,004.0 |
| Total estrone, ng/mL | 0.1-100.1 |
Progesterone
Progesterone was extracted from 500 μL of human plasma using a liquid-liquid extraction method. The validated concentration range for progesterone was 0.4 to 122.5 ng/mL. Separation was achieved by reverse-phase HPLC column (Symmetry C18, 100 Å, 5.0 μm, 50 × 4.6 mm; Waters) using 19-norethindrone as internal standard.
The intra-assay and interassay coefficients of variation for accuracy and precision for progesterone were within the FDA-allowable limits (20% for the lower limit of quantification [LLOQ] and 15% for all other QC samples). For intra-assay precision, the coefficient of variation for the limit of quantification QC (LOQQC) sample was less than 6.2%, and the coefficient of variation for the other QC samples was less than 12.1%. For interassay precision, the coefficient of variation for all QC samples was less than 8.8%. The nominal percentage of progesterone for 2 × and 4× dilution samples was 95.6% and 98.3%, respectively. The specificity and selectivity of the method were also verified against endogenous matrix components. Recovery of progesterone from plasma was 68.7%, and recovery of internal standard was 61.7%. The stability of progesterone in plasma samples was established at room temperature (25°C) for 10 hours; freeze-thaw stability was validated for four cycles (at mean [SD] storage temperatures of −70°C [20°C] and −30°C [10°C]); and stability after long-term storage was established in the presence of coanalytes estradiol, estrone, and estrone metabolites at −70°C and −30°C for 126 days. The stability of extracted progesterone samples was at least 78 hours in an autosampler maintained at a temperature of 10°C.
Estradiol and estrone
Estradiol and estrone were extracted from 500-μL aliquots of human plasma initially using solid-phase extraction followed by derivatization with dansyl chloride and a liquid-liquid extraction procedure. The validated concentration range for estradiol was 25.3 to 5,034.4 pg/mL, and the validated concentration range for estrone was 5.0 to 1,004.0 pg/mL. Separation was achieved by reverse-phase HPLC column (Synergi 2.5 μm Fusion-RP, 100 Å, 100 × 2.0 mm; Phenomenex) using estrone-d4 as internal standard.
Estradiol
The intra-assay and interassay coefficients of variation for accuracy and precision for estradiol were within the allowable limits (20% for LLOQ and 15% for all other QC samples). For intra-assay precision, the coefficient of variation for the LOQQC sample was less than 19.1%, and the coefficient of variation for all other QC samples was less than 12.8%. For interassay precision, the coefficient of variation for all QC samples was less than 12.2%. The nominal percentage of estradiol for 2× and 4× dilution samples was 91.2% and 97.7%, respectively. The specificity and selectivity of the method were also verified against endogenous matrix components. The average recovery of estradiol from plasma was 77.1%, and the average recovery of internal standard was 77.6%. The stability of estradiol in plasma samples was established at room temperature (25°C) for 7 hours; freeze-thaw stability was validated for 4 cycles (at mean [SD] storage temperatures of −70°C [20°C] and −30°C [10°C]); and stability after long-term storage was established in the presence of coanalytes progesterone, estrone, and estrone metabolites at −70°C and −30°C for 109 days. The stability of derivatized estradiol in postextracted samples was at least 42 hours in an autosampler maintained at a temperature of 5°C.
Estrone
The intra-assay and interassay coefficients of variation for accuracy and precision for estrone were within the allowable limits (20% for LLOQ and 15% for all other QC samples). For intra-assay precision, the coefficient of variation for the LOQQC sample was less than 8.8%, and the coefficient of variation for all other QC samples was less than 13.4%. For interassay precision, the coefficient of variation for all QC samples was less than 7.9%. The nominal percentage of estrone for 2× and 4× dilution samples was 91.1% and 95.2%, respectively. The specificity and selectivity of the method were also verified against endogenous matrix components. The average recovery of estrone from plasma was 75%, and the average recovery of internal standard was 77.6%. The stability of estrone in plasma samples was established at room temperature (25°C) for 7 hours; freeze-thaw stability was validated for 4 cycles (at mean [SD] storage temperatures of −70°C [20°C] and −30°C [10°C]); and stability after long-term storage was validated in the presence of coanalytes estradiol, progesterone, estrone, and estrone metabolites at −70°C and −30°C for 109 days. The stability of the derivatized estrone in postextracted samples was at least 42 hours in an autosampler maintained at a temperature of 5°C.
Total estrone
To estimate total estrone (combined quantification of free estrone plus estrone obtained from hydrolysis of estrone 3-sulfate and estrone 3-[β-d-glucuronide]), we initially extracted samples using solid-phase extraction followed by enzymatic hydrolysis with β-glucuronidase and sulfatase; samples were backextracted using hexane and dried under nitrogen. The residue obtained was further derivatized with dansyl chloride, followed by liquid-liquid extraction to isolate total estrone. The validated concentration range for total estrone was 0.1 to 100.2 ng/mL. Separation was achieved by reverse-phase HPLC column (Synergi 2.5 μm Fusion-RP, 100 Å, 100 × 2.0 mm; Phenomenex) using estrone-d4 as internal standard.
The intra-assay and interassay coefficients of variation for accuracy and precision for total estrone were within the allowable limits (20% for LLOQ and 15% for all other QC samples). For intra-assay precision, the coefficient of variation for the LOQQC sample was less than 15.2%, and the coefficient of variation for all other QC samples was less than 10.5%. For interassay precision, the coefficient of variation for all QC samples was less than 12.7%. The nominal percentage of total estrone for 2× and 4× dilution samples was 97.0% and 99.1%, respectively. The specificity and selectivity of the method were also verified against endogenous matrix components. The average recovery of total estrone from plasma was 80.8%, and the average recovery of internal standard was 83.8%. The stability of total estrone in plasma samples was established at room temperature (25°C) for 12 hours; freeze-thaw stability was validated for 4 cycles (at mean [SD] storage temperatures of −70°C [20°C] and −30°C [10°C]); and stability after long-term storage was validated in the presence of coanalytes estradiol and progesterone at −70°C and −30°C for 112 days. The stability of the derivatized total estrone in postextracted samples was at least 37 hours in an autosampler maintained at a temperature of 10°C.
Study endpoints
The FDA guidance for establishing the bioequivalence of oral estradiol calls for providing pharmacokinetic parameters for baseline-adjusted serum levels of total estrone, unconjugated estradiol, and unconjugated estrone.6 Total estrone consists of estrone, estrone sulfates, and estrone glucuronides.10 After estradiol is orally ingested, it is rapidly converted into estrone (primarily estrone sulfate) via first-pass metabolism.10,11 This reservoir of circulating estrone continuously undergoes reconversion into estradiol. Thus, total estrone levels are an important measure of estradiol bioavailability.10,11 In evaluating the bioequivalence of oral progesterone, the FDA requires pharmacokinetic parameters for baseline-adjusted serum levels of progesterone.7
In this study, the primary pharmacokinetic endpoints were Cmax, AUC(0-t), and AUC(0-inf) for progesterone, unconjugated estradiol, and total and unconjugated estrone in plasma after treatment with TX-001HR versus the reference products (Table 2). Secondary endpoints were tmax, Kel, t1/2, and AUC%extrapobs. Pharmacokinetic parameters for the specified analytes were determined for each participant during each period by noncompartmental analyses using baseline-adjusted concentrations.
TABLE 2.
Primary and secondary pharmacokinetic endpoints
| Pharmacokinetic endpoint | Definition |
| Primary endpoints | |
| AUC(0-t) | AUC from time 0 to the last sampling time |
| AUC(0-inf) | AUC from time 0 to infinity |
| Cmax | Maximal concentration |
| Secondary endpoints | |
| AUC% extrap obs | Percentage of AUC extrapolated to infinity from Tlast to infinity based on observed value for concentration at Tlast |
| Kel | Elimination rate constant |
| t1/2 | Terminal elimination half-life |
| tmax | Time to reach peak plasma concentration |
AUC, area under the plasma concentration–time curve.
The SABE method for highly variable drugs was used to compare TX-001HR with the coadministered reference products in cases where the within-subject coefficient of variation for the reference product was 30% or more. A pharmacokinetic endpoint for an analyte was identified as bioequivalent when the 95% upper confidence bound on linearized SABE statistic was 0 or less. The unscaled average bioequivalence method was used to evaluate bioequivalence in cases where the within-subject coefficient of variation was less than 30%. A pharmacokinetic endpoint for an analyte was identified as bioequivalent when the 90% CI on the test-to-reference ratio fell between 0.80 and 1.25. Bioequivalence criteria had to be met for all three primary parameters (Cmax, AUC(0-t), and AUC(0-inf)) to establish an analyte as bioequivalent.
Safety was assessed by clinical observation, laboratory data, and plasma levels of the study drugs at the beginning and end of the study. Poststudy assessments were performed at the conclusion of period III, 48 hours after administration of the final dose.
Statistical analysis
Statistical analyses were performed using SAS software version 9.2. For pharmacokinetic and safety analyses, data from participants who completed all three study periods were included. Descriptive statistics were used to evaluate all primary and secondary pharmacokinetic parameters for hormones. PROC GLM was used for all bioequivalence calculations, except for those in which the coefficient of variation was less than 30%. In those cases, PROC MIXED was used to obtain the 90% CI.
RESULTS
Participant disposition and baseline characteristics
Healthy postmenopausal women (N = 66) were randomly assigned to one of the three treatment sequences described in “Methods.” Women had a mean (SD) age of 49.5 (5.6) years (range, 40-64 y) and a mean (SD) body mass index of 24.8 (3.1) kg/m2 (range, 18.7-29.9 kg/m2). The mean (SD) height was 150.6 (5.4) cm (range, 138.0-162.0 cm), and the mean (SD) weight was 56.1 (7.0) kg (range, 42.0-75.0 kg).
Overall, 94% of participants (62 of 66) completed all three study periods (Fig. 2). Four women did not report to the facility for period II and/or period III. Analyses for total estrone were conducted using data for 61 participants; data for one woman who had a predose estrone level higher than 5% of Cmax were excluded, in keeping with FDA guidance for bioequivalence studies.9
FIG. 2.
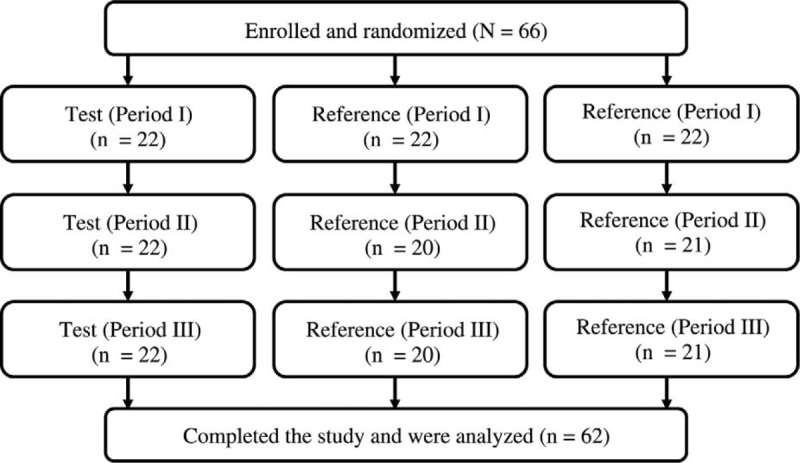
Disposition of the participants.
Pharmacokinetic results
Plasma concentrations of progesterone, unconjugated estradiol, unconjugated estrone, and total estrone were determined for the 62 women who completed the study. Table 3 shows the primary and secondary pharmacokinetic parameters (untransformed data; mean [SD]) for each hormone measured across all three periods for TX-001HR and the reference products.
TABLE 3.
Pharmacokinetic parameters for TX-001HR and reference products
| Pharmacokinetic parameter | Untransformed data | ||
| TX-001HRa | Reference 1b,c | Reference 2b,d | |
| Progesterone (n = 62) | |||
| Cmax, ng/mL | 89.2 (149.7) | 72.7 (101.9) | 69.7 (87.1) |
| AUC(0-t), ng h/mL | 120.1 (164.1) | 125.9 (152.3) | 111.6 (113.3) |
| AUC(0-inf), ng h/mL | 131.4 (172.5) | 142.1 (160.5) | 126.6 (117.3) |
| tmax, mean (range), h | 3.0 (0.8-10.0) | 3.0 (1.0-12.0) | 4.0 (0.7-18.0) |
| Kel, h−1 | 0.31 (0.24) | 0.27 (0.19) | 0.28 (0.25) |
| t1/2, he | 4.6 (4.5) | 5.2 (5.0) | 5.0 (4.6) |
| AUC% extrap obs | 4.3 (2.5) | 4.8 (3.8) | 5.2 (4.1) |
| Unconjugated estradiol (n = 62) | |||
| Cmax, pg/mL | 64.8 (51.0) | 69.1 (33.1) | 73.4 (43.4) |
| AUC(0-t), pg h/mL | 1,403.7 (763.8) | 1,508.2 (876.7) | 1,658.3 (976.6) |
| AUC(0-inf), pg h/mL | 2,459.4 (4,498.3) | 2,842.9 (4,582.7) | 2,111.0 (1,175.4) |
| tmax, mean (range), h | 9.0 (0.5-36.0) | 10.0 (0.5-35.1) | 10.0 (0.3-36.6) |
| Kel, h−1 | 0.04 (0.02) | 0.05 (0.04) | 0.05 (0.03) |
| t1/2, he | 31.9 (96.0) | 25.1 (28.8) | 20.9 (12.1) |
| AUC% extrap obs | 22.8 (16.7) | 25.5 (20.3) | 25.0 (16.5) |
| Unconjugated estrone (n = 62) | |||
| Cmax, pg/mL | 426.6 (179.3) | 455.5 (189.5) | 467.2 (207.4) |
| AUC(0-t), pg h/mL | 9,096.1 (4,377.3) | 10,156.0 (5,141.6) | 10,507.4 (5,183.1) |
| AUC(0-inf), pg h/mL | 11,995 (6,679) | 13,446 (8,699) | 14,066.2 (7,563.2) |
| tmax, mean (range), h | 5.5 (0.8-36.0) | 8.0 (1.7-18.0) | 10.0 (1.7-18.0) |
| Kel, h−1 | 0.04 (0.01) | 0.04 (0.02) | 0.04 (0.02) |
| t1/2, he | 20.3 (9.4) | 19.5 (9.8) | 20.8 (9.4) |
| AUC% extrap obs | 21.3 (11.2) | 20.4 (11.1) | 21.9 (11.9) |
| Total estrone (n = 61) | |||
| Cmax, ng/mL | 35.4 (17.1) | 19.9 (7.4) | 19.9 (8.0) |
| AUC(0-t), ng h/mL | 201.8 (94.2) | 182.8 (88.8) | 199.7 (94.4) |
| AUC(0-inf), ng h/mL | 213.2 (104.6) | 193.7 (100.5) | 203.1 (81.5) |
| tmax, mean (range), h | 2.5 (0.7-7.0) | 4.0 (1.3-18.0) | 4.0 (1.3-10.0) |
| Kel, h−1 | 0.08 (0.04) | 0.08 (0.04) | 0.07 (0.02) |
| t1/2, he | 10.4 (4.0) | 9.9 (3.1) | 10.8 (3.7) |
| AUC% extrap obs | 4.5 (3.7) | 4.6 (3.5) | 5.4 (4.0) |
Data are presented as mean (SD) unless otherwise stated.
aThe test product is TX-001HR, which combines 2 mg of 17β-estradiol and 200 mg of progesterone in a single capsule (TherapeuticsMD Inc, Boca Raton, FL).
bThe reference product consists of separate formulations of estradiol (Estrace; estradiol USP tablets 2 mg; Teva Pharmaceuticals, Sellersville, PA) and progesterone (Prometrium; progesterone softgel capsule 200 mg; Catalent Pharma Solutions, St Petersburg, FL) administered together.
cReference 1 refers to the first time the reference product was received.
dReference 2 refers to the second time the reference product was received.
eFor these data, t1/2 reflects estimated half-life based on terminal log-linear data points.
Table 4 presents bioequivalence data for each analyte. Progesterone and unconjugated estradiol had a within-subject coefficient of variation of 30% or more for all three primary pharmacokinetic parameters, and total estrone had a within-subject coefficient of variation of 30% or more for Cmax. Thus, the SABE method was used to test these analytes for bioequivalence. Unconjugated estrone had a within-subject coefficient of variation lower than 30% for all three primary pharmacokinetic parameters, and total estrone had a within-subject coefficient of variation lower than 30% for AUC(0-t) and AUC(0-inf). Thus, the unscaled average bioequivalence method was used for these endpoints.
TABLE 4.
Bioequivalence analyses for each analyte
| Analyte/parameter | Test-to-reference ratio | Coefficient of variation % | 95% Upper confidence bound | Meets bioequivalence criteriaa |
| Progesterone | ||||
| AUC(0-t) | 1.1 | 122.2 | −0.54 | Yes |
| AUC(0-inf) | 0.9 | 116.4 | −0.49 | Yes |
| Cmax | 1.2 | 173.7 | −0.79 | Yes |
| Unconjugated estradiol | ||||
| AUC(0-t) | 0.9 | 42.6 | −0.09 | Yes |
| AUC(0-inf) | 0.8 | 47.4 | −0.06 | Yes |
| Cmax | 0.9 | 35.4 | −0.04 | Yes |
| Analyte/parameter | Test-to-reference ratio | Coefficient of variation % | 90% CI | Meets bioequivalence criteriab |
| Unconjugated estrone | ||||
| AUC(0-t) | 0.9 | 18.0 | 0.85-0.94 | Yes |
| AUC(0-inf) | 0.9 | 26.3 | 0.83-0.93 | Yes |
| Cmax | 0.9 | 23.3 | 0.87-0.99 | Yes |
| Total estrone | ||||
| AUC(0-t) | 1.1 | 29.7 | 0.98-1.12 | Yes |
| AUC(0-inf) | 1.1 | 29.7 | 0.99-1.11 | Yes |
| Cmax | 1.8 | 35.9 | 0.34c | No |
aScaled average bioequivalence requires a test-to-reference ratio of between 0.80 and 1.25 and a 95% upper confidence bound on linearized statistic of 0 or less.
bUnscaled average bioequivalence requires that the 90% CI on the test-to-reference ratio be between 0.80 and 1.25.
cA 95% upper confidence bound on scaled average bioequivalence statistic.
AUC(0-t), AUC(0-inf), and Cmax met the respective bioequivalence criteria for all analytes, with the exception of Cmax for total estrone. The extent of estradiol and progesterone absorption and the rate of progesterone absorption were similar between TX-001HR and the reference products. The rate of estradiol absorption (tmax) was slightly faster with TX-001HR than with the reference formulation of estradiol. Semilogarithmic plots of drug concentrations across time for each analyte are presented in Figure 3.
FIG. 3.
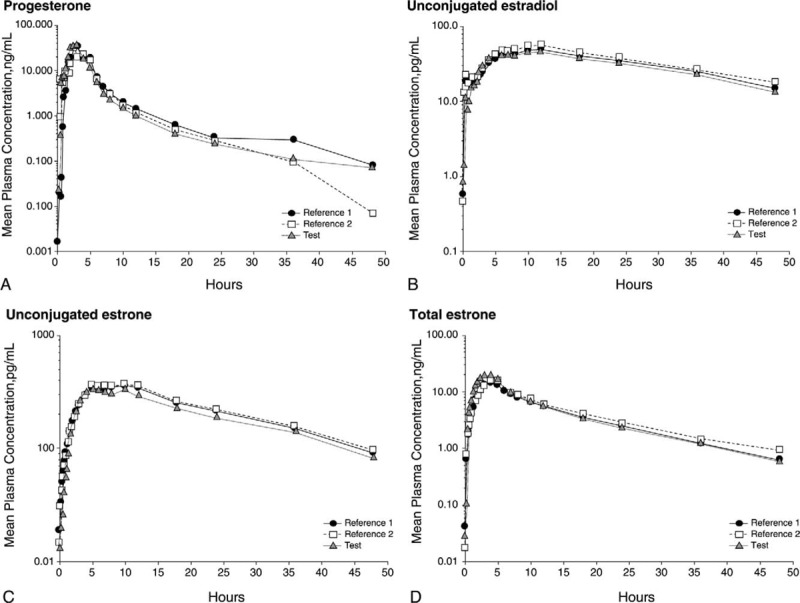
Semilogarithmic plot of area under the curve across time for mean plasma levels of (A) progesterone (corrected; n = 62), (B) unconjugated estradiol (corrected; n = 62), (C) unconjugated estrone (n = 62), and (D) total estrone (corrected; n = 61) after treatment with the reference products for the first time and the second time versus treatment with the test product.
Safety outcomes
Four adverse events were identified during poststudy assessment. All were mild in intensity and considered unrelated to TX-001HR or the reference products. Laboratory test results showed that three women had anemia (considered unrelated to the study), for which they were advised to take supplemental iron, and one woman had an elevated glucose level. Poststudy assessments showed no other clinically significant changes in laboratory values for any of the women. No serious adverse events were reported, and no deaths occurred.
DISCUSSION
The data indicate that the 17β-estradiol and progesterone used in TX-001HR have bioavailability similar to those of the respective reference products of estradiol (Estrace) and progesterone (Prometrium) administered together under fed conditions. In this study, TX-001HR met bioequivalence for all analytes and all parameters, except for Cmax for total estrone. Based on these bioavailability findings, TX-001HR is expected to have a safety profile similar to that of the combined reference products. No treatment-related adverse events occurred with either drug during the study.
Our study looked at progesterone levels under fed conditions, which differ from levels under fasting conditions. A cross-over study showed that taking micronized progesterone with food significantly enhances its bioavailability.12,13 In that study by Simon et al,13 postmenopausal women (n = 15) received a 200-mg dose of oral micronized progesterone in the morning after a typical breakfast and while fasting, with a 7-day washout between doses. The mean (SD) maximal serum levels of progesterone on day 1 were almost fourfold higher when the drug was taken immediately after breakfast than when the drug was taken while fasting (81.6 [113.4] vs 19.4 [17.9] ng/mL, respectively; P = 0.05).12 The maximal progesterone levels observed under fed conditions in that study were similar to the maximal levels reported in this study, which ranged from 69.7 to 89.2 ng/mL. That study also found that AUC values almost doubled when progesterone was taken with food.12
The maximal estradiol levels observed with the test and reference products in this study are consistent with a pharmacokinetic study of oral 17β-estradiol by Lobo and Cassidenti.14 They reported a mean Cmax of 65 pg/mL after oral administration of a single 2-mg dose of micronized estradiol to nonsmoking women.14
Ensuring that the progestogen and estrogen components used in combination HT have sufficient bioavailability is critical because improperly formulated EPT combinations could have serious health consequences for users. For example, data from multiple studies indicated that a sufficient level of progestagen is required to protect the endometrium from estrogenic stimulation, thus preventing endometrial hyperplasia, a precursor of endometrial cancer.1,15-18 For this reason, the FDA requires drug manufacturers seeking approval of investigational HT to provide high-quality evidence that the drug is safe and effective from pharmacokinetic studies and from clinical trials (in which endometrial safety is evaluated).19
In contrast, compounded HT is not reviewed and approved by the FDA, and makers of compounded HT are not required to conduct the costly trials needed to prove the safety and efficacy of the medications they create.20 Compounded hormones that have inadequate bioavailability or do not contain the correct amounts of estrogens and progestogen may fail to protect the uterus. Several studies that evaluated compounded transdermal progesterone creams and gels in postmenopausal women observed posttreatment serum levels of progesterone far below the luteal level of 5 ng/mL.21-26 Wren et al15 reported serum levels of progesterone similar to baseline levels among 21 women treated with a 100-μg estradiol patch plus up to 64 mg of progesterone cream daily for three 28-day cycles. In addition, a biopsy after cycle 3 showed that 13 women developed endometrial proliferation during the study.15 Vashisht et al25,26 treated postmenopausal women (N = 54) with a compounded cream delivering progesterone 40 mg/day and with a compounded gel delivering estradiol 1 mg/day for 48 weeks. Biopsy results at the study's conclusion showed that the progesterone had provided insufficient endometrial protection for 32% of participants: 27% had endometrial proliferation and 5% had complex hyperplasia.25,26
Many US women turned to compounded HT after findings from the Women's Health Initiative indicated that the risks of HT (used to prevent chronic diseases) outweighed its benefits.5,27,28 Surveys suggested that some women have been persuaded by marketing claims from compounding pharmacies that compounded hormones—also sometimes referred to as bioidentical hormones—are safer than FDA-approved hormones, such as those used in the Women's Health Initiative.29,30 Iftikhar et al29 surveyed 184 women who were consulting a clinician at the Mayo Clinic for menopausal symptoms and reported that 63 were using, or were familiar with, custom-compounded HT. Of those women, 42 (67%) believed that compounded HT was safer than FDA-approved HT.29 However, clinical evidence from high-quality randomized trials in support of such claims does not exist.5,20
Spark et al30,31 surveyed 366 Australian women who were taking compounded progesterone and found that 89% started using compounded progesterone because they viewed it as natural. Women's desire for EPT formulations that contain bioidentical hormones, coupled with the need for a formulation that has proven safety and efficacy, was the impetus behind the development of TX-001HR. The active ingredients in TX-001HR are chemically and biologically identical to endogenous estradiol and progesterone. If approved, the non–peanut oil–containing TX-001HR would be the first FDA-approved EPT option for postmenopausal women with nut allergy who prefer oral progesterone to progestins. It would be a safer alternative to unregulated compounded EPT for symptomatic postmenopausal women with intact uterus who prefer an oral HT regimen that combines estradiol with progesterone.
Healthy postmenopausal women (aged 40-65 y) with intact uterus (N = 1,750) are being recruited for the phase 3 randomized placebo-controlled REPLENISH trial (NCT01942668), which aims to evaluate the safety and efficacy of TX-001HR. Eligible participants will be randomly assigned to one of several doses of this novel estradiol/progesterone combination or to placebo for 1 year. The primary endpoint is the incidence of endometrial hyperplasia at 12 months in the overall population and the improvement in climactic symptoms at 12 weeks in a subset of women experiencing more severe vasomotor symptoms. To our knowledge, REPLENISH is the first phase 3 randomized controlled trial of an oral estradiol-progesterone combination designed to treat menopausal symptoms.
CONCLUSIONS
Results of this pharmacokinetic study show that the investigational drug TX-001HR, which combines natural progesterone and 17β-estradiol in a single oral capsule, is bioequivalent to an FDA-approved formulation of oral 17β-estradiol administered concurrently with an FDA-approved formulation of oral progesterone. Bioequivalence data from this early-stage trial suggest that the safety of TX-001HR would be similar to that of the respective reference products. The ongoing phase 3 REPLENISH trial is evaluating the safety and efficacy of TX-001HR in a large population of postmenopausal women. Should the FDA approve TX-001HR, it would be another FDA-approved alternative to compounded EPT for women who prefer to treat their menopausal symptoms with a progesterone-based HT regimen.
Acknowledgments
We acknowledge the medical writing support provided by Christin Melton, ELS, CMPP, and Laura Ninger, ELS, of Precise Publications LLC, which was funded by TherapeuticsMD Inc.
Footnotes
Funding/support: The study was funded by TherapeuticsMD Inc. Medical writing support and editorial assistance were provided by Christin Melton, ELS, CMPP, and Laura Ninger, ELS, of Precise Publications and were funded by TherapeuticsMD Inc.
Financial disclosure/conflicts of interest: J.H.P. was formerly an employee of Wyeth Research; has received consultant fees from Wyeth/Pfizer, Besins Healthcare, Shionogi Inc, Metagenics, and TherapeuticsMD Inc; and holds stock options with TherapeuticsMD Inc. C.B. is a statistical consultant to TherapeuticsMD Inc. B.B., S.M., and J.M.A. are employees of TherapeuticsMD Inc.
REFERENCES
- 1.Du JY, Sanchez P, Kim L, Azen CG, Zava DT, Stanczyk FZ. Percutaneous progesterone delivery via cream or gel application in postmenopausal women: a randomized cross-over study of progesterone levels in serum, whole blood, saliva, and capillary blood. Menopause 2013; 20:1169–1175. [DOI] [PubMed] [Google Scholar]
- 2.McAuley JW, Kroboth FJ, Kroboth PD. Oral administration of micronized progesterone: a review and more experience. Pharmacotherapy 1996; 16:453–457. [PubMed] [Google Scholar]
- 3.Bhavnani BR, Stanczyk FZ. Misconception and concerns about bioidentical hormones used for custom-compounded hormone therapy. J Clin Endocrinol Metab 2012; 97:756–759. [DOI] [PubMed] [Google Scholar]
- 4.The North American Menopause Society The 2012 hormone therapy position statement of: The North American Menopause Society. Menopause 2012; 19:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Committee on Gynecologic Practice and the American Society for Reproductive Medicine Practice Committee Committee opinion no. 532: compounded bioidentical menopausal hormone therapy. Obstet Gynecol 2012; 120:411–415. [DOI] [PubMed] [Google Scholar]
- 6.US Food and Drug Administration. Draft guidance on estradiol. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM238055.pdf Accessed August 29, 2014. [Google Scholar]
- 7.US Food and Drug Administration. Draft guidance on progesterone. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM209294.pdf Accessed August 29, 2014. [Google Scholar]
- 8.Kuhl H. Pharmacokinetics of oestrogens and progestogens. Maturitas 1990; 12:171–197. [DOI] [PubMed] [Google Scholar]
- 9.US Food and Drug Administration. Guidance for industry. Bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM377465.pdf Accessed September 2, 2014. [Google Scholar]
- 10.Kuhl H. Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric 2005; 8 suppl 1:3–63. [DOI] [PubMed] [Google Scholar]
- 11.Stanczyk FZ, Archer DF, Bhavnani BR. Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception 2013; 87:706–727. [DOI] [PubMed] [Google Scholar]
- 12.US Food and Drug Administration. Center for Drug Evaluation and Research application number: NDA 19-781. Clinical Pharmacology and Biopharmaceutics Review(s) for Prometrium. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/98/19781-clinpharm.pdf Accessed September 3, 2014. [Google Scholar]
- 13.Simon JA, Robinson DE, Andrews MC, et al. The absorption of oral micronized progesterone: the effect of food, dose proportionality, and comparison with intramuscular progesterone. Fertil Steril 1993; 60:26–33. [PubMed] [Google Scholar]
- 14.Lobo RA, Cassidenti DL. Pharmacokinetics of oral 17β-estradiol. J Reprod Med 1992; 37:77–84. [PubMed] [Google Scholar]
- 15.Wren BG, McFarland K, Edwards L, et al. Effect of sequential transdermal progesterone cream on endometrium, bleeding pattern, and plasma progesterone and salivary progesterone levels in postmenopausal women. Climacteric 2000; 3:155–160. [DOI] [PubMed] [Google Scholar]
- 16.Effects of hormone replacement therapy on endometrial histology in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. The Writing Group for the PEPI Trial. JAMA 1996; 275:370–375. [DOI] [PubMed] [Google Scholar]
- 17.Pickar JH, Yeh I, Wheeler JE, Cunane MF, Speroff L. Endometrial effects of lower doses of conjugated equine estrogens and medroxyprogesterone acetate. Fertil Steril 2001; 76:25–31. [DOI] [PubMed] [Google Scholar]
- 18.Heiss G, Wallace R, Anderson GL, et al. Health risks and benefits 3 years after stopping randomized treatment with estrogen and progestin. JAMA 2008; 299:1036–1045. [DOI] [PubMed] [Google Scholar]
- 19.US Department of Health and Human Services, Food and Drug Administration, and Center for Drug Evaluation and Research. Guidance for industry. Estrogen and estrogen/progestin drug products to treat vasomotor symptoms and vulvar and vaginal atrophy symptoms—recommendations for clinical evaluation. Available at: http://www.fda.gov/downloads/Drugs/DrugSafety/informationbyDrugClass/UCM135338.pdf Accessed June 20, 2014. [Google Scholar]
- 20.De Villiers TJ, Pines A, Panay N, et al. Updated 2013 International Menopause Society recommendations on menopausal hormone therapy and preventive strategies for midlife health. Climacteric 2013; 16:316–337. [DOI] [PubMed] [Google Scholar]
- 21.Cooper A, Spencer C, Whitehead MI, Ross D, Barnard GJ, Collins WP. Systemic absorption of progesterone from Progest cream in postmenopausal women. Lancet 1998; 351:1255–1256. [DOI] [PubMed] [Google Scholar]
- 22.Carey BJ, Carey AH, Patel S, Carter G, Studd JW. A study to evaluate serum and urinary hormone levels following short and long term administration of two regimens of progesterone cream in postmenopausal women. BJOG 2000; 107:722–726. [DOI] [PubMed] [Google Scholar]
- 23.Zargar-Shoshtari S, Wahhabaghei H, Mehrsai A, Wen J, Alany R. Transdermal delivery of bioidentical progesterone using dutasteride (A 5α-reductase inhibitor): a pilot study. J Pharm Pharm Sci 2010; 13:626–636. [DOI] [PubMed] [Google Scholar]
- 24.Stanczyk FZ, Paulson RJ, Roy S. Percutaneous administration of progesterone: blood levels and endometrial protection. Menopause 2005; 12:232–237. [DOI] [PubMed] [Google Scholar]
- 25.Vashisht A, Wadsworth F, Carey A, Carey B, Studd J. A study to look at hormonal absorption of progesterone cream used in conjunction with transdermal estrogen. Gynecol Endocrinol 2005; 21:101–105. [DOI] [PubMed] [Google Scholar]
- 26.Vashisht A, Wadsworth F, Carey A, Carey B, Studd J. Bleeding profiles and effects on the endometrium for women using a novel combination of transdermal oestradiol and natural progesterone cream as part of a continuous combined hormone replacement regime. BJOG 2005; 112:1402–1406. [DOI] [PubMed] [Google Scholar]
- 27.MacLennan AH, Gill TK, Broadbent JL, Taylor AW. Continuing decline in hormone therapy use: population trends over 17 years. Climacteric 2009; 12:122–130. [DOI] [PubMed] [Google Scholar]
- 28.Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA 2002; 288:321–333. [DOI] [PubMed] [Google Scholar]
- 29.Iftikhar S, Shuster LT, Johnson RE, Jenkins SM, Wahner-Roedler DL. Use of bioidentical compounded hormones for menopausal concerns: cross-sectional survey in an academic menopause center. J Womens Health (Larchmt) 2011; 20:559–565. [DOI] [PubMed] [Google Scholar]
- 30.Spark MJ, Willis J, Byrne G. Compounded progesterone: why is it acceptable to Australian women? Maturitas 2012; 73:318–324. [DOI] [PubMed] [Google Scholar]
- 31.Spark MJ, Willis J, Byrne G, Iacono T. The multifaceted nature of access to compounded progesterone: a cross-sectional study from Australia. Maturitas 2014; 77:155–162. [DOI] [PubMed] [Google Scholar]