

Abstract
The global incidence of cancer is on the rise, and within the next decade, the disease is expected to become the leading cause of death worldwide. Forthcoming strategies used to treat cancers focus on the design and implementation of multidrug therapies to target complementary cancer specific pathways. A more direct means by which this multitargeted approach can be achieved is by identifying and targeting interpathway regulatory factors. Recent advances in understanding Nek2 (NIMA related kinase 2) biology suggest that the kinase potentially represents a multifaceted therapeutic target. In this regard, pharmacologic modulation of Nek2 with a single agent may effect several mechanisms important for tumor growth, survival, progression, and metastasis. We herein review the development of Nek2 as an oncology target and provide a succinct chronology of drug discovery campaigns focused on targeting Nek2.
In the next two decades, global cancer incidence is expected to double from 12.7 million cases in 2008 to an estimated 21.4 million by 2030.1,2 Many clinical therapies predate the 40+ year war on cancer, and modern therapeutics often have limited efficacy in aggressive drug refractory malignancies. Additional complicating factors, such as aberrant activity of undruggable oncogenic factors, further burden the success of treatment. Personalized medicine has been implemented to more efficiently and effectively diagnose and treat disease. However, the era of personalized medicine is in its infancy, and without the discovery of novel targets and therapeutics the challenges of successfully tackling a surge in cancer incidence are enormous. To meet the projected demand, innovative methods for treating cancers must be investigated.
Current drugs used for the treatment of cancer fall into one of two main classes, chemotherapy or targeted therapy, based on target specificity. Chemotherapy comprises three main drug types: antimetabolites,3 alkylating agents,4 and cell cycle inhibitors.5,6 Though chemotherapy remains the dominant therapy in the clinical setting, design of new chemotherapies represent an outdated approach for identifying more effective anticancer drugs. Moreover, because of the heterogeneous nature of tumors,7 chemotherapies select for and promote drug refractory malignancies.
The enhanced understanding of cancer biology has given rise to the second class of cancer therapies termed targeted therapeutics.8–10 Targeted therapeutics are designed to specifically act on aberrant cancer signaling pathways that are unique to a particular tumor. Multiple generations of inhibitors have achieved profound target specificity effectively limiting off target toxicities. The success of target-based therapeutics was believed to eliminate the need for nonspecific agents; however, to date, this therapeutic goal remains unmet. Akin to chemotherapies, the targeted therapeutic class suffers from transient clinical efficacy, as treatment selects for drug refractory malignancies.11,12
The evolution of drugs for the treatment of cancer has begun to produce more successes in large part due to the combination of both therapeutic classes. On the basis of these successes, a variety of therapeutic combinations were investigated for the treatment of pervasive malignancies.13–16 Drug–drug interaction complications from this treatment strategy can produce undesirable toxicity.17 Toxicity issues from drug–drug interactions have caused a push for the discovery and development of novel therapeutics that can bridge the two main drug classes. The goal of identifying one agent that can produce the benefit of combination therapy without the liability of a drug–drug interaction will require strategic analysis of molecular signaling pathways to identify key interpathway regulatory factors as drug targets.
Recent advances in understanding the biology of Nek2 (NIMA related kinase 2), a serine/threonine kinase, suggest that its pharmacological inhibition has multifaceted therapeutic potential in bridging targeted and nontargeted avenues of chemotherapy. Nek2 is involved in regulating four independent mechanisms of tumor biology: (1) cell-cycle regulation, (2) cell survival, (3) chemosensitization, and (4) metastasis. Many solid tumors overexpress Nek2,18–20 and Nek2 RNAi inhibition results in decreased proliferation in numerous cancer models.21,22 Additionally, overexpression of Nek2 promotes active Akt,23,24 a potent and critical oncogene for a variety of malignancies (reviewed by Vivanco et al.25). By contributing to aberrant Akt activity, Nek2 represents a novel target for blocking a variety of tumor-specific pathways.23,24 Furthermore, recent studies have demonstrated that inhibition of Nek2 has an important role in chemoresensitization of drug refractory tumors by down-regulating the expression of ABC (ATP binding cassette) efflux transporters.24 The ABC family of efflux transporters has been extensively studied, as increased expression of ABCs causes chemoresistance and inhibition results in chemoresensitization (reviewed by Gottesman et al.26). Nek2 has also been implemented in up-regulation Wnt/Wg signaling, altering cell adhesion markers, increasing Jnk (c-Jun N-terminal kinase) activity, and altering β-catenin signaling, which can increase tumor migration, metastasis, and survival.23 Cumulatively, these findings identify the Nek2 kinase as an oncology target that is uniquely positioned in tumor biology such that blocking Nek2 can integrate the effects of both targeted and nontargeted treatment avenues. Herein, we review the development of Nek2 as an oncology target and associated inhibitor discovery campaigns.
Nek2 OVERVIEW
The Nek family of serine/threonine kinases is essential for normal mitosis and cell cycle regulation.27,28 The first characterized member of the Nek family was never in mitosis Aspergillus (NIMA) from the species Aspergillus nidulans, which has shown importance in cell division.29 The closest related mammalian isoform to NIMA is Nek2.30 In mammals, Nek2 is responsible for initiating centrosomal separation at the G2/M stage of the cell cycle.27 To accomplish separation, Nek2 phosphorylates β-catenin,31 ninein-like protein (NIP),32 and centrosomal Nek2-associated protein 1 (C-Nap1).33 Because of the very specific role in cell cycle progression, Nek2 is tightly regulated at the transcriptional and post-translational levels. Specifically, Nek2 is expressed during the S and G2 phases of the cell cycle, and at the post-translational level activity is modulated by protein phosphatase 1 (PP1).24,29,34 Interestingly, PP1 is also a critical regulator of Akt.24,29,34 In malignancies, elevated levels of Nek2 result in centrosomal separation, which can lead to chromosomal instability (CIN), tumor promotion, and progression.35 From a regulatory standpoint, elevated levels of Nek2 overwhelm the endogenous regulatory capacity of PP1, resulting in aberrant activity of Akt.24 Conceptually, overexpressing Nek2 in normal cells can lead to a tumorigenic intracellular environment.
Nek2 AND CANCER
The contributions of aberrant Nek2 activity to the development and maintenance of an oncogenic phenotype have only been resolved in the past decade. The first report of aberrant expression of Nek2 in cancer came from a gene expression array querying over 1700 cancer associated genes expressed in pediatric solid tumors.36 In this study, Nek2 and factors that promote Nek2 activity were found to be co-up-regulated in a rare form of cancer called Ewing’s sarcoma.36 On the basis of the findings, aberrant Nek2 activity was correlated to the development and progression of Ewing’s sarcoma. Further investigation in follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL) identified a significant elevation of Nek2 levels at both the transcriptional and protein level.19 Seminal findings from the study indicate that aberrant expression and activity of Nek2 drive the progression and transformation of lymphomas that ultimately produce a more aggressive disease.19 A hallmark of lymphomas is aneuploidy and chromosomal rearrangement,37 though speculative, aberrant Nek2 activity likely contributes to the defining characteristics of lymphoma. Significantly, Hayward and colleagues later defined a role for Nek2 in driving aneuploidy and centrosome amplification in human breast epithelial cells.20 Gene expression analysis identified elevated levels of Nek2 transcripts in a variety of malignancies that harbor genetic abnormalities such as leukemia, cervical, ovarian, breast, and prostate cancers. In solid tumors, up-regulated Nek2 was identified in both primary and metastatic lesions, suggesting that up-regulation can be an early oncogenic event that leads to promotion and progression of disease.20
Early studies investigating the therapeutic value of targeting Nek2 utilized RNAi in cholangiocarcinoma, a rare inoperable cancer of the liver.38 RNAi was utilized to specifically target and attenuate aberrantly expressed Nek2 in primary cultures of the tumor as well as primary human fibroblasts.38 These studies demonstrated that Nek2 targeted therapies have selectivity for cancer, as suppressed proliferation relative to Nek2 RNAi was only observed in the cholangiocarcinoma.38 In a similar RNAi based study, the efficacy of Nek2 targeted therapy was investigated in xenograft models of breast cancer using triple negative, MDA-MB-231, or estrogen receptor (ER) positive MCF7 breast cancer cell lines.39 Inhibition of Nek2 by RNAi resulted in a statistically significant response in xenografts of both MDA-MB-231 and MCF7.39 Results indicated that targeting Nek2 in breast cancer displays efficacy in reducing tumor growth independent of ER status. Another study identified that Nek2 is important for tumor growth at both primary and secondary breast cancer sites and depletion of Nek2 caused cell death from excessive aneuploidy.40 Further, Nek2 was found to be important for driving the growth of breast cancer metastases in the lung.40 The findings suggest that blocking oncogenic Nek2 in vivo is efficacious at reducing the proliferation of Nek2 mutant tumors and validates Nek2 as an oncogene.
In another study, deliberate overexpression of Nek2 in Drosophila melanogaster led to an up-regulation in Wnt/Wg signaling and an alteration in Rho1, Rac1, and E-cadherin cell migration markers.23 Also, there was an increase in the activation of the Akt and Jnk pathways, which can promote growth and survival.23 β-Catenin levels were found to be reduced at the cell–cell adherens junction complexes with increased signaling around the nucleus, indicative of an invasive phenotype.23 As such, Nek2 was found to be important in the process of tumor migration, metastasis, and survival.23 Nek2 enables tumor progression beyond promoting chromosomal instability (CIN) through the activation of a variety of tumor enabling pathways.23
Nek2 AND DRUG RESISTANCE
Drug efflux is an extensive, complicating factor that limits the therapeutic window for many cancer therapeutics and plays an essential role in increasing drug resistance in a variety of malignancies.1,2,24,41–45 The Nek2 kinase has been identified as a critical player in increasing drug resistance by causing an increase in ABC mediated drug efflux.24 Studies using RNAi to knock down Nek2 transcripts have linked the kinase to ABC expression and chemoresensitization.24,41 Specifically, overexpression of Nek2 in the ARP-1 multiple myeloma cell line causes reduced efficacy of bortezomib, doxorubicin, and etoposide.24 Zhou et al. demonstrated chemoresensitization to chemotherapies using tet-inducible Nek2 RNAi in a xenograft model system.24 Seminal findings demonstrate Nek2 RNAi resensitizes ARP-1 cells to the aforementioned chemotherapies.24 Similarly, knocking down Nek2 by RNAi resensitizes the breast cancer cell lines MDA-MB-231 and MDA-MB-468 to doxorubicin and paclitaxel.41
The proposed mechanism for Nek2 induced drug resistance relies on the deregulation of the phosphatase, PP1. Nek2 contains a binding domain for PP1, which dephosphorylates the activated form of Nek2 to control catalytic activity.46 PP1 is also responsible for regulating Akt by dephosphorylating Akt at Thr-450.47 As levels of Nek2 rise, the regulatory capacity of PP1 is overcome by an excess of activated Nek2, which results in aberrant activation of Akt, leading to an up-regulation in expression of ABC genes (Figure 1). Supporting evidence for the proposed Nek2-PP1-Akt regulatory model comes from Nek2 inhibition studies that show diminished transcripts and protein levels for both multidrug resistance-associated protein 1 (ABCC1) and breast cancer resistant protein (ABCG2).24 The proposed Nek2-PP1-Akt regulatory model implicates the kinase in driving drug resistance. Moreover, evidence presented in these studies suggests that blocking the Nek2 oncogene can restore and enhance chemotherapy efficacy.
Figure 1.

(A) Protein phosphatase 1 (PP1) signaling axis regulates both Nek2 and Akt in a normal cell. (B) In a tumor environment, increased Nek2 expression disrupts the PP1 signaling axis, leading to more active Akt, which drives ABC mediated drug resistance.24
Nek2: THERAPEUTIC IMPACT AND IMPLICATIONS
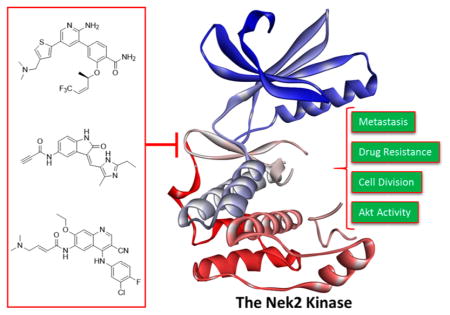
Nek2 has been preliminarily validated as a fruitful drug target for effecting oncogenic pathways that drive cell division, Akt regulation, ABC mediated drug resistance, and metastasis in a diverse set of solid and liquid tumors (Figure 2).20,23,24,36,37–41,48 A majority of all studies have been completed with RNAi knockdown or Nek2 knockin models. RNAi based studies do not necessarily recapitulate the effects observed with small molecule inhibition. Extensive Nek2 target validation utilizing biological models has provided incentive for the development of selective Nek2 small molecules. The small molecules can serve as a tool to further define Nek2 biology and to progress Nek2 as a therapeutic target.
Figure 2.

Aberrant Nek2 activity causes an increase in Akt activity, cell division, drug resistance, and metastasis. Inhibiting the Nek2 oncogene can block Akt, reduce tumor cell proliferation, increase chemotherapy sensitivity, and lower the rate of metastasis. Crystal structure data were obtained from Westood et al.27
Nek2 INHIBITOR DESIGN AND DEVELOPMENT
The concept of targeting Nek2 for chemotherapy treatment is a relatively new idea, since the role of Nek2 in tumor biology has only recently been described.36 Development of the first Nek2 inhibitor relied on the modification of sunitinib (SU11652, 1)49,50 (Figure 3A), which is a pan tyrosine kinase inhibitor with market approval for renal cell carcinomas (RCC) and gastrointestinal stromal tumors (GISTs). Design of the Nek2 inhibitor was geared toward the modification of compound 1 into an irreversible inhibitor through the strategic introduction of an electrophilic moiety (Figure 3).50 Inhibitors were constructed to covalently engage a cysteine residue adjacent to the hinge of the kinase, which dramatically increased inhibitor potency (Figure 3B).50 Only 11 kinases in the kinome are known to have a cysteine residue in a similar location,50 and this suggested a possible fine-tuning of activity to yield highly selective Nek2 inhibitors.
Figure 3.

(A) Evolution from compound 1 to compound 3, a Nek2 selective irreversible inhibitor. (B) (1) Design strategy of a selective Nek2 covalent inhibitor and (2) compound 3 computationally modeled in the Nek2 kinase. The alkyne moiety is in proximity to Cys22 for covalent modification of Nek2. The imidazole moiety provides Nek2 selectivity. Computational binding experiments were completed using AutoDock Vina,58 and the Nek2 crystal structure was obtained from Innocenti et al.55
The most potent Nek2 covalent inhibitor identified, compound 250 (IC50 < 6 nM), had no selectivity for cyclin dependent kinase 1 (Cdk1), and as such, cells treated rapidly exited mitosis.50 The lack of selectivity obscures biological effects, and therefore, further design strategies were employed to mitigate selectivity issues. Compound design was improved by strategically introducing an ethyl and methyl group and changing the alkylating moiety to generate compound 350 that had >25 times selectivity against Cdk1. The selective compound 3 was >130 times less active on Nek2 (IC50 = 770 nM) (Figure 3). Compound 3 was able to acutely inhibit Nek2 in vitro without significantly impacting chromosome congression, the spindle assembly checkpoint, bipolar spindle assembly, and mitotic entry.50 Therefore, compound 3 was selective for Nek2 and did not have substantial activity on other cell-cycle kinases.50 Because of the selectivity profile, compound 3 possesses characteristics that are useful for determining the biological roles of Nek2.50 Identification of small molecule tools with limited off target activity will be required for progressing Nek2 target validation beyond RNAi knockdown experiments.
Noncovalent Nek2 inhibition has been achieved with compound 4,51 a predecessor to the GlaxoSmithKline pololike kinase inhibitor (Plk1) GSK461364 (4a)51 (Figure 4 and Figure 8C).51 The compound displayed excellent activity in colon (COLO205, IC50 = 5 nM), lung (A549, IC50 = 11 nM), breast (MX-1, IC50 = 9 nM), and ovarian (SKOV-3, IC50 = 11 nM) tumor cell lines.51 Although the inhibitor achieved activity on Nek2 (IC50 = 21 nM), inhibition was greater on Plk1 (IC50 = 2 nM).51 Because of the selectivity issue, the amount of efficacy originating from inhibiting Nek2 could not be identified. In a phase I clinical trial, compound 4a caused a high incidence (20%) of venous thromboembolism (VTE), and no phase II data exist on compound 4a.52 Hence, Plk1 activity is likely not well tolerated in patients, despite prolonging stable disease in 15% of individuals on the study.52
Figure 4.

Development of compound 8 from compound 4 and HTS hit 5.
Figure 8.

(A) Proposed four-part Nek2 selective pharmacophore, composed of (1) solubilizing group, (2) hinge warhead, (3) aryl linker, and (4) glycine pocket moiety. (B) Crystal structure of compound 11 bound to the Nek2 kinase.55 Key residues within the ATP active site are highlighted. (C) Computational binding of compound 4 with Nek2. (D) Computational binding of compound 6 with Nek2. (E) Molecular landscape of the Nek2 kinase. Crucial regions have been highlighted to aid in the design of future inhibitors. Computational binding experiments were completed using AutoDock Vina,58 and the Nek2 crystal structure was obtained from Innocenti et al.55
In another example of Nek2 inhibitor design, high throughput screening (HTS) identified a pyrazine-based scaffold 553 with Nek2 inhibitory activity (Figure 4).53 Diversification efforts off the HTS hit commenced, and a nonliner Nek2 structure–activity relationship (SAR) was identified. Further SAR based studies identified the pyrazine moiety as the warhead of the compound. Molecular modeling revealed that the pyrazine forms two hydrogen bonds on the hinge of Nek2.53 Further optimization of the pyrazine scaffold proceeded resulting in compounds that bound to a novel, inactive form of Nek2.53 Specifically, compound 753 promoted a Nek2 conformational change where Tyr70 was recruited to a hydrogen bond network with the carboxylic acid moiety of the compound (Figure 5). Presumably, this unique attribute is related to the flexibility of the compound as other more rigid compounds do not adopt similar binding modalities.53 As such, the conformational change was not identified with compound 953 because of the rigidity of the structure (Figure 5).53
Figure 5.

Induced Nek2 conformational change. Adapted from Whelligan et al.53
Despite the novelty of an induced-fit binding mode, strong geometric constraints at the ATP binding site (Phe148 and Met86) further complicate Nek2 inhibitor design.53 Therefore, the majority of inhibitors based on the pyrazine scaffold had micromolar activity, while low nanomolar activity was never achieved.53 Interestingly, the Nek2 ATP binding site has shared functionality with Plk1, which enables Nek2 chemotypes to have activity on Plk1 creating a problematic situation for selectivity (Figure 6).53
Figure 6.

Nek2 (orange) and Plk1 (blue) 3D structure alignment. Key residues within the ATP binding pocket have been highlighted. Arg136 on Plk1 provides an avenue for the development of Nek2 selective inhibitors. The structure alignment was completed with CLICK: Topology Independent Comparison of Biomolecular 3D Structures. The Nek2 crystal structure was obtained from Innocenti et al.,55 and the Plk1 crystal structure was obtained from Nie et al.59
To identify a potent, Plk1 selective Nek2 inhibitor, work shifted back to compound 4 in an effort to abrogate the Plk1 profile.54 Compound 4 has a thiophene linker bridging the hinge heterocycle with the amide and chiral ether (Figure 4). Remarkably, simply substituting the thiophene linker to a benzene linker significantly reduced activity on Plk1.54 Exclusive Nek2 selectivity was achieved by moving the solubilizing group on the benzimidazole to the 5 position, yielding compound 654 (Figure 4 and Figure 8D).54 The strategic placement of the solubilizing group at the 5 position of benzimidazole creates Nek2 specificity relative to Plk1 because of steric clash with Plk1-Arg136, a bulky amino acid residue not encountered at the same position in Nek2 (Figure 6).54 Therefore, by a change of the thiophene linker to benzene and the benzimidazole substitution to the 5 position, compounds with upward of 150 times selectivity for Nek2 were generated. Unlike the pyrazine-based scaffold, Nek2 inhibitors derived from benzimidazole achieved submicromolar activity and have a high degree of Nek2 selectivity.54
Perhaps the most successful campaign to develop a selective Nek2 inhibitor relied on the combination of the pyrazine scaffold with the benzimidazole scaffold to generate the hybrid compound 855 (Figure 4). Through SAR studies, an aminopyridine warhead was found to be superior to pyrazine, and the phenyl ring was substituted for an alkene to lower the molecular weight of the final compound.55 Therefore, optimal Nek2 potency and selectivity were achieved with a significant optimization in ligand efficiency (IC50 = 22 nM, >100 times Plk1 selectivity). Against a panel of cell cycle kinases and other important kinases, there was good selectivity for Nek2.55 However, compound 8 had only 3 times Nek2 selectivity against GSK3β (glycogen synthase 3β), yet the narrow selectivity window was found to be acceptable because of good global kinase selectivity.55 Like Plk1, GSK3β should be of similar concern for the design and development of future selective Nek2 inhibitors. Potent, selective compounds were progressed into cell-based assays, and the compounds were evaluated for Nek2 inhibition by monitoring the amount of C-Nap1 (centrosomal Nek2-associated protein 1) phosphorylation. Nek2 phosphorylates C-Nap1 to induce centrosome separation,33 and the lead inhibitor blocked Nek2 activity on C-Nap1 with submicromolar activity (IC50 = 0.8 μM).55 The activity further translated into growth inhibition observed in a panel of cancer cell lines: US02 (GI50 = 0.48–2.92 μM), MDA-MB-231 (GI50 = 0.95–7.25 μM), HeLa (GI50 = 0.14–1.20 μM), and MCF7 (GI50 = 0.22–5.42 μM).55 The developed inhibitors were able to preliminarily support RNAi knockdown studies by exhibiting activity in a variety of tumor cell lines. Because of the selectivity and potency of compound 8, the compound was found to serve as an efficient tool to further define Nek2 biology.
In a novel, orthogonal approach to identify a Nek2 inhibitor, epidermal growth factor receptor (EGFR) inhibitors, such as pelitinib (EKB-569, 10)23 and neratinib (HKI-272, 11)23 (Figure 7), were found to have activity on the Nek2 kinase.23 Compound 10 achieved a 661 nM IC50 value on Nek2, while compound 11 achieved a 274 nM IC50.23 On EGFR, compounds 10 and 11 have IC50 values of 38.5 56 and 92 nM,57 respectively. Because Nek2 can promote metastasis by cooperating with mitogen-activated protein kinase (MAPK) and EGFR singling,23 identifying an inhibitor to simultaneously block the EGFR/MAPK cascade as well as Nek2 activity represents an innovative approach to targeting cancers where EGFR and Nek2 are both driving disease progression.23 By use of compound 10, the inhibitor was found to block a Nek2 dependent increase in cytosolic β-catenin (nonmetastatic phenotype) and block Jnk signaling. Compound 10 also partially restored normal function of cell migration markers and reduced cell migratory phenotypes.23 Lapatinib, an EGFR inhibitor without strong Nek2 activity, did not have a significant impact on Nek2 driven phenotypes.23 The study displayed that simultaneous inhibition of Nek2 and EGFR synergize to increase efficacy in Nek2/EGFR driven tumors.23 The full extent of the synergy and dual inhibition on other receptor tyrosine kinases (RTKs) needs to be further investigated to better establish an understanding of Nek2/RTK interplay.
Figure 7.

Novel Nek2/EGFR dual inhibitors.
Considerable development on small molecules that can inhibit Nek2 has been completed. Both kinase selectivity and Nek2 potency were achieved by optimizing regions of compound 4 for Nek2 activity. In general, a selective Nek2 noncovalent pharmacophore is composed of four different regions: (1) solubilizing group, (2) hinge warhead, (3) aryl linker, and (4) glycine pocket moiety (Figure 8A). Also, key areas of the Nek2 kinase can be strategically exploited to develop potent and selective inhibitors (Figure 8B–E). Significant work has been completed to improve Nek2 activity at the biochemical level,53–55 yet thoroughly validating RNAi knockdown studies with selective Nek2 inhibitors still needs much investigation. The inhibitors can be used to further define Nek2 biology and to evaluate Nek2 as a realistic therapeutic target. Also, a Nek2 inhibitor has yet to be evaluated for chemosensitization properties, which has been recently identified as an important aspect to Nek2 biology.24 Because of the oncogene synergy observed between Nek2 and RTK signaling,23 dual inhibition of Nek2/RTK needs further investigation to fully determine therapeutic relevance.
Nek2 FUTURE DIRECTION
The basis for Nek2 as a therapeutic target has been preliminarily evaluated through RNAi knockdown studies and Nek2 knockin models.23,24,38–41 Initial research suggests that the inhibition of Nek2 causes selective cytotoxicity, Akt regulation, reversal of ABC mediated drug resistance, and a decrease in metastasis. Although Nek2 drug discovery and development efforts have been limited, potent and selective inhibitors have been generated.50,53–55 Nek2 inhibitors are in the early stages of development and have primarily been used as selective, molecular probes to further define Nek2 biology. Also, dual Nek2/RTK inhibition represents a novel avenue for therapeutic investigation.23 At present, selective Nek2 inhibitors can be utilized to identify the therapeutic relevance of Akt regulation and the possibility of obstructing ABC mediated drug resistance. Moreover, down-regulating Nek2 demonstrates great potential to elicit inhibitory effects on aberrant Akt signaling; yet, the biological effects of Nek2 inhibitors on the Akt signaling axis remain largely unexplored. As is evident, many important questions surrounding Nek2 biology remain unanswered. With a Nek2 inhibitor, there is a possibility to impact therapeutic outcomes for Nek2 positive cancers and address the forecasted surge in cancer incidence, yet the biology surrounding the kinase needs to be further defined to fully evaluate therapeutic relevance.
ABBREVIATIONS USED
- Nek2
never in mitosis/Aspergillus related kinase 2
- Akt
protein kinase B
- ATP
adenosine triphosphate
- ABC
ATP binding cassette
- MDR
multidrug resistance
- NIMA
never in mitosis/Aspergillus
- NIP
ninein-like protein
- C-Nap1
centrosomal Nek2-associated protein 1
- PP1
protein phosphatase 1
- DLBCL
diffuse large B-cell lymphoma
- FL
follicular lymphoma
- Plk1
polo-like kinase 1
- HFF
human foreskin fibroblast
- ER
estrogen receptor
- ABCC1
a multidrug resistance-associated protein 1
- ABCG2
a breast cancer resistance protein
- PI3K
phosphoinositide 3-kinase
- RCC
renal cell carcinoma
- GIST
gastrointestinal stromal tumor
- Cdk1
cyclin dependent kinase 1
- VTE
venous thromboembolism
- HTS
high throughput screening
- SAR
structure–activity relationship
- Jnk
c-Jun N-terminal kinase
- CIN
chromosomal instability
- RTK
receptor tyrosine kinase
Biographies
Brendan Frett is currently pursuing a Ph.D. degree in the Drug Discovery and Development program at the University of Arizona under the advisement of Professor Hong-yu Li. He has a strong interest in small molecule inhibitors for novel oncology-related targets. His dissertation has focused on identifying a novel clinical candidate and multiple advanced leads. His expertise is in the development and synthesis of druglike compounds, the development of biological assays for high-throughput compound screening, computational modeling, inhibitor design, and the discovery of unique synthetic methodologies.
Robert V. Brown is currently pursuing a Ph.D. in Pharmaceutical Sciences with a minor in Cancer Biology under the tutelage of Professor Lawrence L. Hurley at University of Arizona. His research focuses on the discovery and development of novel therapeutic targets for the treatment of human disease. To these ends, his dissertation focuses on the characterization and molecular targeting of transcriptionally induced, sequence dependent DNA secondary structures known as G-quadruplex(es) and iMotif(s) for modulating aberrant oncogene transcription. His expertise is in the focused characterization and analytics of drug–target interactions and the design/development of low-/mid-/high-throughput biochemical and cell-based assays for screening compounds.
Mingliang Ma is currently an Associate Professor at Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, China, and also a Visiting Scholar in Professor Hong-yu Li’s laboratory in the College of Pharmacy at the University of Arizona. He obtained his Ph.D. degree in physiology from East China Normal University, under the supervision of Professor Ke Wen and Kang Zhao in 2008. His current research interests are in drug discovery and self-assembly in supramolecular chemistry.
Wenhao Hu is currently a Professor and Head of Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, China. He received his Ph.D. from Hong Kong Polytechnic University and was a postdoctoral fellow at the University of Arizona with Professor Michael P. Doyle. He worked as a medicinal chemist at Genesoft Pharmaceutical in South San Francisco and as a process chemist at Bristol-Myers Squibb in New Jersey. His research interests include synthetic methodology, MCRs, medicinal, and process chemistry.
Haiyong Han is currently an Associate Professor in the Clinical Translational Research Division at the Translational Genomics Research Institute (TGen), Phoenix, Arizona. His research involves the identification and characterization of novel genetic alterations in cancers, particularly pancreatic cancer, and the development of new therapeutic agents that target these alterations.
Hong-yu Li is currently an Associate Professor of Medicinal Chemistry at the University of Arizona. He received his Ph.D. degree from the University of Tokyo, Japan, and did his postdoctoral trainings at Columbia University, NY, and Harvard University, MA. He previously worked at Eli Lilly where he focused on oncology drug discovery. His current research interests are in chemical biology and drug discovery, especially for oncology related targets and phenotypes. In his lab at the University of Arizona, a robust oncology pipeline including a clinical candidate and three advanced leads were discovered and currently under development. The clinical candidate is a highly potent (picomolar activity in oncogene-driven cancer cell lines), selective, and orally bioavailable small molecule inhibitor and currently ready for preclinical development and further IND filing.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM, editors. Globocan 2008: Cancer Incidence and Mortality Worldwide. International Agency for Research on Cancer; Lyon, France: 2011. IARC CancerBase No.10. [Google Scholar]
- 3.Kaye SB. New antimetabolites in cancer chemotherapy and their clinical impact. Br J Cancer. 1998;78:1–7. doi: 10.1038/bjc.1998.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Go RS, Adjei AA. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J Clin Oncol. 1999;17:409–422. doi: 10.1200/JCO.1999.17.1.409. [DOI] [PubMed] [Google Scholar]
- 5.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Epothilones, a new class of microtubule-stabilizing agents with a Taxol-like mechanism of action. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 6.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci US A. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108:479–485. doi: 10.1038/bjc.2012.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Druker BJ. STI571 (Gleevec) as a paradigm for cancer therapy. Trends Mol Med. 2002;8:S14–S18. doi: 10.1016/s1471-4914(02)02305-5. [DOI] [PubMed] [Google Scholar]
- 9.Wells SA, Jr, Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA, Skinner M, Krebs A, Vasselli J, Schlumberger M. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28:67–72. doi: 10.1200/JCO.2009.23.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 11.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, Taylor K, Herrmann R, Seymour JF, Arthur C, Joske D, Lynch K, Hughes T. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102:276–283. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 12.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–326. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herbst RS, Sun Y, Eberhardt WE, Germonpré P, Saijo N, Zhou C, Wang J, Li L, Kabbinavar F, Ichinose Y, Qin S, Zhang L, Biesma B, Heymach JV, Langmuir P, Kennedy SJ, Tada H, Johnson BE. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11:619–626. doi: 10.1016/S1470-2045(10)70132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heymach JV, Paz-Ares L, De Braud F, Sebastian M, Stewart DJ, Eberhardt WE, Ranade AA, Cohen G, Trigo JM, Sandler AB, Bonomi PD, Herbst RS, Krebs AD, Vasselli J, Johnson BE. Randomized phase II study of vandetanib alone or with paclitaxel and carboplatin as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:5407–5415. doi: 10.1200/JCO.2008.17.3138. [DOI] [PubMed] [Google Scholar]
- 15.Peters GJ, van der Wilt CL, van Moorsel CJ, Kroep JR, Bergman AM, Ackland SP. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol Ther. 2000;87:227–253. doi: 10.1016/s0163-7258(00)00086-3. [DOI] [PubMed] [Google Scholar]
- 16.Bunn PA, Jr, Kelly K. New chemotherapeutic agents prolong survival and improve quality of life in non-small cell lung cancer: a review of the literature and future directions. Clin Cancer Res. 1998;4:1087–1100. [PubMed] [Google Scholar]
- 17.Beijnen JH, Schellens JH. Drug interactions in oncology. Lancet Oncol. 2004;5:489–496. doi: 10.1016/S1470-2045(04)01528-1. [DOI] [PubMed] [Google Scholar]
- 18.Wai DH, Schaefer KL, Schramm A, Korsching E, Van Valen F, Ozaki T, Boecker W, Schweigerer L, Dockhorn-Dworniczak B, Poremba C. Expression analysis of pediatric solid tumor cell lines using oligonucleotide microarrays. Int J Oncol. 2002;20:441–451. [PubMed] [Google Scholar]
- 19.de Vos S, Hofmann WK, Grogan TM, Krug U, Schrage M, Miller TP, Braun JG, Wachsman W, Koeffler HP, Said JW. Gene expression profile of serial samples of transformed B-cell lymphomas. Lab Invest. 2003;83:271–285. doi: 10.1097/01.lab.0000053913.85892.e9. [DOI] [PubMed] [Google Scholar]
- 20.Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004;64:7370–7376. doi: 10.1158/0008-5472.CAN-04-0960. [DOI] [PubMed] [Google Scholar]
- 21.Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res. 2007;67:9637–9642. doi: 10.1158/0008-5472.CAN-07-1489. [DOI] [PubMed] [Google Scholar]
- 22.Tsunoda N, Kokuryo T, Oda K, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100:111–116. doi: 10.1111/j.1349-7006.2008.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das TK, Dana D, Paroly SS, Perumal SK, Singh S, Jhun H, Pendse J, Cagan RL, Talele TT, Kumar S. Centrosomal kinase Nek2 cooperates with oncogenic pathways to promote metastasis. Oncogenesis. 2013;2:1–14. doi: 10.1038/oncsis.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z, Shi L, Zangari M, Miles R, Bearss D, Tricot G, Zhan F. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23:48–62. doi: 10.1016/j.ccr.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 26.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 27.Westwood I, Cheary DM, Baxter JE, Richards MW, van Montfort RL, Fry AM, Bayliss R. Insights into the conformational variability and regulation of human Nek2 kinase. J Mol Biol. 2009;386:476–485. doi: 10.1016/j.jmb.2008.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fry AM, O’Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci. 2012;125:4423–4433. doi: 10.1242/jcs.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fry AM, Schultz SJ, Bartek J, Nigg EA. Substrate specificity and cell cycle regulation of the Nek2 protein kinase, a potential human homolog of the mitotic regulator NIMA of Aspergillus nidulans. J Biol Chem. 1995;270:12899–12905. doi: 10.1074/jbc.270.21.12899. [DOI] [PubMed] [Google Scholar]
- 30.Reininger L, Tewari R, Fennell C, Holland Z, Goldring D, Ranford-Cartwright L, Billker O, Doerig C. An essential role for the Plasmodium Nek-2 Nima-related protein kinase in the sexual development of malaria parasites. J Biol Chem. 2009;284:20858–20868. doi: 10.1074/jbc.M109.017988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bahmanyar S, Kaplan DD, Deluca JG, Giddings TH, Jr, O’Toole ET, Winey M, Salmon ED, Casey PJ, Nelson WJ, Barth AI. β-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008;22:91–105. doi: 10.1101/gad.1596308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonn S, Jeong Y, Rhee K. Nip2/centrobin may be a substrate of Nek2 that is required for proper spindle assembly during mitosis in early mouse embryos. Mol Reprod Dev. 2009;76:587–592. doi: 10.1002/mrd.20990. [DOI] [PubMed] [Google Scholar]
- 33.Fry AM, Nigg EA. Characterization of mammalian NIMA-related kinases. Methods Enzymol. 1997;283:270–282. doi: 10.1016/s0076-6879(97)83022-4. [DOI] [PubMed] [Google Scholar]
- 34.Helps NR, Luo X, Barker HM, Cohen PT. NIMArelated kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem J. 2000;349:509–518. doi: 10.1042/0264-6021:3490509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meraldi P, Nigg EA. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J Cell Sci. 2001;114:3749–3757. doi: 10.1242/jcs.114.20.3749. [DOI] [PubMed] [Google Scholar]
- 36.Wai DH, Schaefer KL, Schramm A, Korsching E, Van Valen F, Ozaki T, Boecker W, Schweigerer L, Dockhorn-Dworniczak B, Poremba C. Expression analysis of pediatric solid tumor cell lines using oligonucleotide microarrays. Int J Oncol. 2002;20:441–451. [PubMed] [Google Scholar]
- 37.Fukuhara S, Rowley JD, Variakojis D, Golomb HM. Chromosome abnormalities in poorly differentiated lymphocytic lymphoma. Cancer Res. 1979;39:3119–3128. [PubMed] [Google Scholar]
- 38.Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res. 2007;67:9637–9642. doi: 10.1158/0008-5472.CAN-07-1489. [DOI] [PubMed] [Google Scholar]
- 39.Tsunoda N, Kokuryo T, Oda K, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100:111–116. doi: 10.1111/j.1349-7006.2008.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cappello P, Blaser H, Gorrini C, Lin DC, Elia AJ, Wakeham A, Haider S, Boutros PC, Mason JM, Miller NA, Youngson B, Done SJ, Mak TW. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene. 2013:1–10. doi: 10.1038/onc.2013.183. [DOI] [PubMed] [Google Scholar]
- 41.Lee J, Gollahon L. Nek2-targeted ASO or siRNA pretreatment enhances anticancer drug sensitivity in triple-negative breast cancer cells. Int J Oncol. 2013;42:839–847. doi: 10.3892/ijo.2013.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lage H. ABC-transporters: implications on drug resistance from microorganisms to human cancers. Int J Antimicrob Agents. 2003;22:188–199. doi: 10.1016/s0924-8579(03)00203-6. [DOI] [PubMed] [Google Scholar]
- 43.Bell DR, Gerlach JH, Kartner N, Buick RN, Ling V. Detection of P-glycoprotein in ovarian cancer: a molecular marker associated with multidrug resistance. J Clin Oncol. 1985;3:311–315. doi: 10.1200/JCO.1985.3.3.311. [DOI] [PubMed] [Google Scholar]
- 44.Burger H, Foekens JA, Look MP, Meijer-van Gelder ME, Klijn JG, Wiemer EA, Stoter G, Nooter K. RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: correlation with chemotherapeutic response. Clin Cancer Res. 2003;9:827–836. [PubMed] [Google Scholar]
- 45.Ro J, Sahin A, Ro JY, Fritsche H, Hortobagyi G, Blick M. Immunohistochemical analysis of P-glycoprotein expression correlated with chemotherapy resistance in locally advanced breast cancer. Hum Pathol. 1990;21:787–791. doi: 10.1016/0046-8177(90)90046-8. [DOI] [PubMed] [Google Scholar]
- 46.Mi J, Guo C, Brautigan DL, Larner JM. Protein phosphatase-1alpha regulates centrosome splitting through Nek2. Cancer Res. 2007;67:1082–1089. doi: 10.1158/0008-5472.CAN-06-3071. [DOI] [PubMed] [Google Scholar]
- 47.Xiao L, Gong LL, Yuan D, Deng M, Zeng XM, Chen LL, Zhang L, Yan Q, Liu JP, Hu XH, Sun SM, Liu J, Ma HL, Zheng CB, Fu H, Chen PC, Zhao JQ, Xie SS, Zou LJ, Xiao YM, Liu WB, Zhang J, Liu Y, Li DW. Protein phosphatase-1 regulates Akt1 signal transduction pathway to control gene expression, cell survival and differentiation. Cell Death Differ. 2010;17:1448–1462. doi: 10.1038/cdd.2010.16. [DOI] [PubMed] [Google Scholar]
- 48.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 49.Rellos P, Ivins FJ, Baxter JE, Pike A, Nott TJ, Parkinson DM, Das S, Howell S, Fedorov O, Shen QY, Fry AM, Knapp S, Smerdon SJ. Structure and regulation of the human Nek2 centrosomal kinase. J Biol Chem. 2007;282:6833–6842. doi: 10.1074/jbc.M609721200. [DOI] [PubMed] [Google Scholar]
- 50.Henise JC, Taunton J. Irreversible Nek2 kinase inhibitors with cellular activity. J Med Chem. 2011;54:4133–4146. doi: 10.1021/jm200222m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emmitte KA, Adjabeng GM, Andrews CW, Alberti JG, Bambal R, Chamberlain SD, Davis-Ward RG, Dickson HD, Hassler DF, Hornberger KR, Jackson JR, Kuntz KW, Lansing TJ, Mook RA, Jr, Nailor KE, Pobanz MA, Smith SC, Sung CM, Cheung M. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg Med Chem Lett. 2009;19:1694–1697. doi: 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- 52.Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJ, Smith DA, Murray SC, Lampkin TA, Dar MM, Wilson R, de Bono JS, Blagden SP. Phase I study of GSK461364; a specific and competitive polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res. 2011;17:3420–3430. doi: 10.1158/1078-0432.CCR-10-2946. [DOI] [PubMed] [Google Scholar]
- 53.Whelligan DK, Solanki S, Taylor D, Thomson DW, Cheung KM, Boxall K, Mas-Droux C, Barillari C, Burns S, Grummitt CG, Collins I, van Montfort RL, Aherne GW, Bayliss R, Hoelder S. Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization. J Med Chem. 2010;53:7682–7698. doi: 10.1021/jm1008727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solanki S, Innocenti P, Mas-Droux C, Boxall K, Barillari C, van Montfort RL, Aherne GW, Bayliss R, Hoelder S. Benzimidazole inhibitors induce a DFG-out conformation of never in mitosis gene A-related kinase 2 (Nek2) without binding to the back pocket and reveal a nonlinear structure–activity relationship. J Med Chem. 2011;54:1626–1639. doi: 10.1021/jm1011726. [DOI] [PubMed] [Google Scholar]
- 55.Innocenti P, Cheung KM, Solanki S, Mas-Droux C, Rowan F, Yeoh S, Boxall K, Westlake M, Pickard L, Hardy T, Baxter JE, Aherne GW, Bayliss R, Fry AM, Hoelder S. Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: structure–activity relationship, structural biology, and cellular activity. J Med Chem. 2012;55:3228–3241. doi: 10.1021/jm201683b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Torrance CJ, Jackson PE, Montgomery E, Kinzler KW, Vogelstein B, Wissner A, Nunes M, Frost P, Discafani CM. Combinatorial chemoprevention of intestinal neoplasia. Nat Med. 2000;6:1024–1028. doi: 10.1038/79534. [DOI] [PubMed] [Google Scholar]
- 57.Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, Reich MF, Shen R, Shi X, Tsou HR, Wang YF, Wissner A. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 58.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nie Z, Feher V, Natala S, McBride C, Kiryanov A, Jones B, Lam B, Liu Y, Kaldor S, Stafford J, Hikami K, Uchiyama N, Kawamoto T, Hikichi Y, Matsumoto S, Amano N, Zhang L, Hosfield D, Skene R, Zou H, Cao X, Ichikawa T. Discovery of TAK-960: an orally available small molecule inhibitor of polo-like kinase 1 (PLK1) Bioorg Med Chem Lett. 2013;23:3662–3666. doi: 10.1016/j.bmcl.2013.02.083. [DOI] [PubMed] [Google Scholar]