

Abstract
Background
Synergistic action of different enzymes is required to complete the degradation of plant biomass in order to release sugars which are useful for biorefining. However, the use of single strains is often not efficient, as crucial parts of the required enzymatic machinery can be absent. The use of microbial consortia bred on plant biomass is a way to overcome this hurdle. In these, secreted proteins constitute sources of relevant enzyme cocktails. Extensive analyses of the proteins secreted by effective microbial consortia will contribute to a better understanding of the mechanism of lignocellulose degradation.
Results
Here, we report an analysis of the proteins secreted by a microbial consortium (metasecretome) that was grown on either wheat straw (RWS), xylose or xylan as the carbon sources. Liquid chromatography–tandem mass spectrometry was used to analyze the proteins in the supernatants. Totals of 768 (RWS), 477 (xylose) and 103 (xylan) proteins were identified and taxonomically and functionally classified. In RWS, the proteins were mostly affiliated with Sphingobacterium-like consortium members (~50 %). Specific abundant protein clusters were predicted to be involved in polysaccharide transport and/or sensing (TonB-dependent receptors). In addition, proteins predicted to degrade plant biomass, i.e. endo-1,4-beta-xylanases, alpha-l-arabinofuranosidases and alpha-l-fucosidases, were prominent. In the xylose-driven consortium, most secreted proteins were affiliated with those from Enterobacteriales (mostly Klebsiella species), whereas in the xylan-driven one, they were related to Flavobacterium-like ones. Notably, the metasecretomes of the consortia growing on xylose and xylan contained proteins involved in diverse metabolic functions (e.g. membrane proteins, isomerases, dehydrogenases and oxidoreductases).
Conclusions
An analysis of the metasecretomes of microbial consortia originating from the same source consortium and subsequently bred on three different carbon sources indicated that the major active microorganisms in the three final consortia differed. Importantly, diverse glycosyl hydrolases, predicted to be involved in (hemi)cellulose degradation (e.g. of CAZy families GH3, GH10, GH43, GH51, GH67 and GH95), were identified in the RWS metasecretome. Based on these results, we catalogued the RWS consortium as a true microbial enzyme factory that constitute an excellent source for the production of an efficient enzyme cocktail for the pretreatment of plant biomass.
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-015-0387-8) contains supplementary material, which is available to authorized users.
Keywords: Enzyme cocktail, Metasecretome, Microbial consortium, Glycosyl hydrolases, Sphingobacterium, Wheat straw, Xylan, Xylose
Background
Recently, important developments have paved the way for the use of plant biomass (e.g. wheat straw) as a source of sugars, which—in turn—are useful for the production of bioethanol, plastics and pharmaceutical–chemical intermediates [1]. However, the bioconversion of plant biomass to sugars is a prime bottleneck with respect to efficiency. Next to the need for a physicochemical treatment that affects the complex bonds in lignocellulosic matter, the action of effective degradative enzymes is required. In the latter process, synergism between various enzymes (e.g. oxidases, xylanases, arabinofuranosidases, cellobiohydrolases, endoglucanases and beta-glucosidases) seems to be a “conditio-sine-qua-non” for an efficient process [2, 3]. In the light of process parameters, lignocellulose-degradative enzymes are required to work under diverse conditions, including high temperatures, low or high pH and the presence of residual pretreatment chemicals and inhibitors [4]. Given the difficulty of working with high diversities of required enzymes, plant biomass hydrolysis by microbial consortia instead of single strains has been proposed [5–8]. However, a clear disadvantage of this strategy is that the sugars released from the lignocellulose will be immediately consumed by concurring microorganisms. To overcome this hurdle, enzymes that are released by the microbial consortia into the medium may be applied directly on plant biomass or be used as a supplement to commercial cellulolytic enzyme cocktails. Gladden et al. [9] used a secreted protein fraction of a compost-derived microbial consortium to saccharify pretreated switchgrass, demonstrating that this strategy has excellent potential for the development of (hemi)cellulolytic enzyme cocktails. Additionally, Park et al. [10] reported the development of a cellulase cocktail by combining thermophile-secreted endoglucanases produced by a microcrystalline cellulose-degrading microbial consortium with a recombinant cellobiohydrolase and beta-glucosidase.
Metagenomics and metatranscriptomics studies provide extraordinary insight into the metabolic potential and expression of enzymes in lignocellulolytic microbial consortia [11, 12]. Moreover, total microbial community proteomics (metaproteomics) and—more specifically—metasecretomics (analysis of the total surface-bound and secreted proteins that make up the “secretome” of a microbial community) have the potential to provide a high-resolution representation of proteins and their dynamics outside of the microbial cells. This allows the identification of proteins that are involved directly in the deconstruction of plant biomass [13, 14]. Recently, several studies have reported the analysis of secreted proteins in cultures of single microbial strains. For example, Ganoderma lucidum cultivated in sugarcane bagasse [15], Thermobifida fusca bred on different lignocellulosic biomass (e.g. corn stover and wood chips) [16], Aspergillus fumigatus grown in the presence of glucose, avicel and rice straw [17] and Ceriporiopsis subvermispora during growth on aspen wood [18]. However, little is known about the proteins secreted from a wheat straw-degrading microbial consortium across carbon sources with distinct complexities. The presence and abundance of proteins in complex secretomes would guide us in our search for key enzymes that are useful for biorefining. In addition, assessment of the taxonomic affiliation of the secreted proteins enables us to identify the metabolically active microbes, helping to correlate specific functions with the taxa that are truly involved in plant biomass degradation.
Here, we report an analysis of the secreted proteins (metasecretome) from a phylogenetically stable soil-derived microbial consortium [19] that was grown in mineral medium with 1 % of either wheat straw (RWS), xylan or xylose as the unique carbon sources. The selection of untreated instead of pretreated wheat straw was based on the contention that this substrate better selects microbes with high capacity to degrade complex and recalcitrantly bound (hemi)cellulose structures. In addition, the selection of xylan and xylose, as comparators, was based on the composition of the wheat straw. Thus, xylan is a major component of the hemicellulose in wheat straw, xylose being the monomer and unique component of xylan. The metasecretomes were harvested after 11 days of incubation at pH 7.2 under mesophilic aerobic conditions. The analyses indicated that Sphingobacterium-like organisms are major players in the degradation of wheat straw, as they were found to secrete key enzymes such as endo-1,4-beta-xylanases (GH10), alpha-l-arabinofuranosidases (GH51) and alpha-l-fucosidases (GH95). Moreover, pullulanases from Klebsiella-like organisms were detected in the RWS consortial metasecretome. In contrast, all secreted proteins in the xylose-driven consortial metasecretome were tracked to Enterobacteriales, suggesting that the presence of a pentose like xylose increases the prevalence of bacteria belonging to this class. This study, identified the key enzymes that are predicted to be particularly involved in the attack on wheat straw in comparison to xylan and xylose, giving clues as to the design of enzyme cocktails for future applications.
Results
Microbially enriched cultures and metasecretome extraction
Microbially enriched cultures were produced with cells from an RWS-derived source consortium [8, 19]. Flasks containing 25 ml mineral medium and 1 % of wheat straw (RWS), xylan or xylose were inoculated with 25 µl of cell suspension (~5 log bacterial cells/ml in the first enrichment flasks). Bacterial growth was found to differ along the 11 days of incubation (Fig. 1). In the RWS-driven consortium, the densities increased from the inoculum level to around 8.5 log bacterial cells/ml after 5 days and then to 9 log bacterial cells/ml after 11 days. However, for the xylan- and xylose-driven consortia, we observed a maximum peak of growth after 5 days (~7.4 and 7.8 log bacterial cells/ml for xylan and xylose, respectively) and relatively stable communities afterwards (~7.0 and 7.6 log bacterial cells/ml for xylan and xylose, respectively, after 11 days of incubation) (Additional file 1).
Fig. 1.
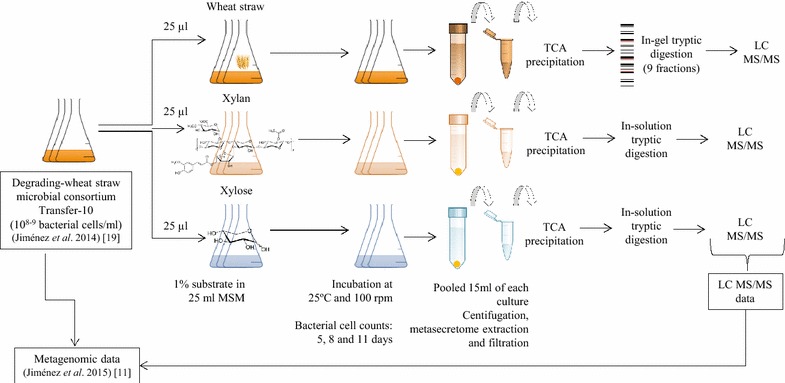
Schematic representation of the methodology used in this study
The secreted proteins (metasecretome) of each treatment were recovered from 15 ml pooled final cultures. Selection of the sampling point (11 days) was based on the assumption that some of the highly recalcitrant compounds in the wheat straw might have been sufficiently degraded, allowing enzyme systems to work on the lignocellulose moieties of the substrate. Proteins in 2 ml of supernatant were precipitated by the trichloroacetic acid (TCA) method. Prior SDS-PAGE analysis of these fractions showed a high complexity of protein bands in the RWS compared with the xylose- and xylan-driven consortia. Given this result, we performed an in-gel tryptic digestion of nine fractions for the RWS treatment, selecting proteins of sizes between 20 and 150 kDa (Additional file 1). Moreover, the secreted proteins from the xylose and xylan treatments were digested with trypsin (in solution) and subsequently used for liquid chromatography–tandem mass spectrometry (LC–MS/MS) (Fig. 1).
Metasecretomes: overview of the data
Totals of 3652, 2402 and 459 peptide sequences, from RWS, xylose and xylan, respectively, were detected and identified from the filtered LC–MS/MS data. Based on database searches (affiliation of the detected peptide sequences to the 388,324 predicted metagenome proteins from the original wheat straw-degrading microbial consortium) [11], 768 (RWS), 477 (xylose) and 103 (xylan) proteins were identified as traceable to the metagenome data. Of these total proteins, only 22 were common between RWS and xylan and five between RWS and xylose. We did not find any common proteins between the xylan and xylose consortial metasecretomes and also not between all three systems (Fig. 2).
Fig. 2.
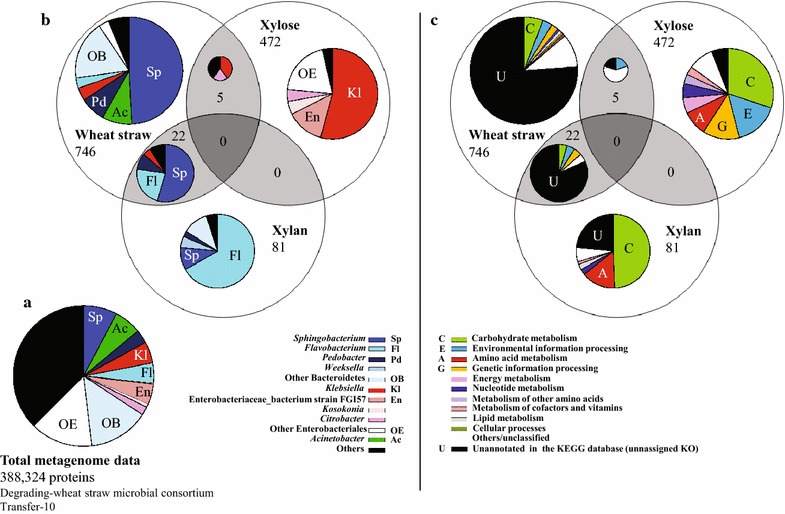
Taxonomic and functional assignment of proteins. a Taxonomic assignment of detected proteins in the metagenome database and b in each metasecretome, c functional assignment of proteins detected in each metasecretome
Taxonomic and functional affiliation of secreted proteins
The secreted proteins, in each system, and the total proteins from the database (metagenome) were analyzed using the KEGG database on the GhostKOALA platform (GHOSTX threshold: 100) [20] (Fig. 2). Overall, 8, 6, 5, 5 and 3 % of the predicted proteins from the metagenome were affiliated to proteins from Sphingobacterium, Acinetobacter, Flavobacterium, Klebsiella and Pedobacter-like organisms, respectively (Fig. 2a). In the RWS consortial metasecretome, most of the proteins were affiliated with proteins detected in Bacteroidetes, specifically 48 % Sphingobacterium, 8 % Pedobacter, 3 % Flavobacterium and 18 % “others”. In addition, 8 and 4 % of the proteins were affiliated with those from Acinetobacter and Klebsiella-like organisms, respectively. In the metasecretome from the xylose-grown consortium, approximately 96 % of the proteins were assigned to predicted proteins encoded by the genomes of different Enterobacteriales (i.e. 54 % Klebsiella, 13 % Enterobacteriaceae bacterium strain FGI57, 5 % Kosakonia, 4 % Citrobacter and 20 % “others”). Finally, the predominance (~95 %) of Bacteroidetes-related proteins was found in the metasecretome of the xylan-driven consortium, among which 56 % were similar to proteins produced by Flavobacterium-like organisms. Thus, a shift in the secreted proteome was found, depending on the carbon source (Fig. 2b).
Regarding the functional annotation, around 86 % of all proteins detected in the RWS metasecretome were unannotated/unclassified based on the KEGG orthology system, thus representing as-yet-undescribed proteins with potential function (Fig. 2c). However, the remaining proteins in this metasecretome were mostly classified within the KEGG functional classes (orthology groups) carbohydrate metabolism (6 %), environmental information (3 %) and genetic information (2 %) processing. In contrast, in the metasecretome of the xylose-driven consortium, 86 % of the proteins were classifiable based on the KEGG orthology system, the most abundant functions being related with carbohydrate metabolism (30 %), environmental information (16 %) and genetic information processing (13 %). In the metasecretome of the xylan-grown consortium, 49 and 15 % of the proteins were classified within the classes carbohydrate and amino acid metabolism, leaving 29 % of unannotated/unclassified proteins plus 7 % low-abundance classes (Fig. 2c).
Analysis of proteins detected in the metasecretomes: abundant functions
The unique proteins detected in the RWS (746), xylose (472) and xylan (81) metasecretomes were—separately—compared to each other, and clustered, at 70 % of amino acid identity. This allowed to infer the “abundant” clusters (clusters containing >2–3 detected proteins) of proteins for each metasecretome. Thus, we could discern 38 (RWS), 38 (xylose) and 11 (xylan) abundant protein clusters (more than 3 protein sequences per cluster in RWS and xylose; and more than 2 proteins per clusters in xylan) (Table 1; Additional files 2, 3). In addition, 408, 243 and 53 singletons (i.e. clusters containing only a single protein sequence) were found in the RWS-, xylose- and xylan-driven metasecretomes, respectively.
Table 1.
Abundant clusters (clusters at 70 % identity with more than 3 sequences) of secreted proteins in RWS
| Cluster | # PS | LRS | Best PSI-BLAST hit vs. NCBI (organism source) | Accession No. | COV (%) | e value | Identity | Domain ID | Domain description |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 7 | 161 | Hemagglutinin (Sphingobacterium) | WP_045753147.1 | 76 | 8E−74 | 90 | pfam01832 | Mannosyl-glycoprotein endo-beta-N-acetylglucosaminidase |
| 2 | 6 | 159 | RNA methyltransferase (Sphingobacterium) | KKX48125.1 | 100 | 3E−110 | 100 | pfam00588 | SpoU rRNA methylase family |
| 3 | 6 | 136 | Flagellin (Enterobacter) | WP_042717930.1 | 100 | 4E−84 | 98 | pfam00669 | Bacterial flagellin |
| 4 | 6 | 155 | Membrane protein (Sphingobacterium) | WP_045755907.1 | 100 | 5E−94 | 90 | pfam14322 | Starch-binding associating with outer membrane (SusD) |
| 5 | 6 | 161 | Pyruvate dehydrogenase (Sphingobacterium) | WP_021189278.1 | 100 | 6E−111 | 100 | pfam02780 | Transketolase |
| 6 | 6 | 83 | Membrane protein (Sphingobacterium) | WP_045753095.1 | 100 | 1E−40 | 94 | TIGR04056 | TonB-linked outer membrane protein, SusC/RagA family |
| 7 | 6 | 153 | Outer membrane lipoprotein (Acinetobacter) | WP_004695452.1 | 96 | 5E−104 | 100 | pfam03548 | Outer membrane lipoprotein carrier protein LolA |
| 8 | 6 | 154 | Molecular chaperone GroEL (Sphingobacterium) | WP_045754355.1 | 100 | 2E−94 | 99 | PRK00013 | Chaperonin GroEL |
| 9 | 5 | 149 | Hypothetical protein (Sphingobacterium) | WP_021190406.1 | 100 | 6E−90 | 94 | TIGR04056 | TonB-linked outer membrane protein, SusC/RagA family |
| 10 | 4 | 153 | Aminopeptidase (Sphingobacterium) | WP_045755761.1 | 100 | 3E−107 | 99 | cd02619 | C1 peptidase family, also referred to as the papain family |
| 11 | 4 | 155 | Hypothetical protein (Sphingobacterium) | WP_038697635.1 | 98 | 9E−95 | 86 | pfam06283 | Trehalose utilization |
| 12 | 4 | 131 | Hypothetical protein (Sphingobacterium) | WP_021189714.1 | 100 | 1E−74 | 85 | cd02968 | Synthesis of cytochrome c oxidase |
| 13 | 4 | 154 | Hypothetical protein (Sphingobacterium) | WP_021188014.1 | 55 | 6E−49 | 98 | pfam11138 | Protein of unknown function (DUF2911) |
| 14 | 4 | 153 | Hypothetical protein (Sphingobacterium) | WP_021192491.1 | 88 | 8E−88 | 98 | pfam13472 | GDSL-like lipase/acylhydrolase family |
| 15 | 3 | 85 | Hypothetical protein (Sphingobacterium) | WP_037526146.1 | 97 | 1E−48 | 99 | ND | ND |
| 16 | 3 | 135 | Alpha-l-arabinofuranosidase (Sphingobacterium) | WP_045756399.1 | 100 | 1E−88 | 99 | pfam06964 | Alpha-l-arabinofuranosidase |
| 17 | 3 | 144 | Hypothetical protein (Sphingobacterium) | KKX50961.1 | 100 | 5E−94 | 100 | TIGR04056 | TonB-linked outer membrane protein, SusC/RagA family |
| 18 | 3 | 153 | Hypothetical protein (Sphingobacterium) | WP_045755931.1 | 62 | 1E−52 | 86 | ND | ND |
| 19 | 3 | 162 | Pyruvate dehydrogenase (Sphingobacterium) | WP_021192396.1 | 100 | 6E−102 | 99 | TIGR01349 | Pyruvate dehydrogenase |
| 20 | 3 | 82 | TonB-linked outer membrane protein (Sphingobacterium) | WP_003012895.1 | 84 | 1E−21 | 70 | TIGR04056 | TonB-linked outer membrane protein, SusC/RagA family |
| 21 | 3 | 137 | Membrane protein (Klebsiella) | WP_032728567.1 | 61 | 1E−48 | 98 | PRK10554 | Outer membrane porin protein C |
| 22 | 3 | 122 | Flagellin (Alicycliphilus) | WP_013723065.1 | 100 | 3E−68 | 95 | pfam00669 | Bacterial flagellin |
| 23 | 3 | 152 | Porin (Serratia) | WP_024529021.1 | 100 | 5E−99 | 96 | pfam02264 | LamB porin, maltoporin |
| 24 | 3 | 157 | Hypothetical protein (Sphingobacterium) | KKX47093.1 | 87 | 2E−83 | 99 | TIGR04056 | TonB-linked outer membrane protein, SusC/RagA family |
| 25 | 3 | 162 | Superoxide dismutase (Flavobacterium) | WP_041515972.1 | 93 | 2E−95 | 92 | pfam00080 | Copper/zinc superoxide dismutase |
| 26 | 3 | 140 | Hypothetical protein (Sphingobacterium) | WP_045754573.1 | 100 | 2E−84 | 99 | COG1729 | Periplasmic TolA-binding protein (function unknown) |
| 27 | 3 | 90 | Flagellar motor protein MotB (Sphingobacterium) | WP_038696344.1 | 82 | 2E−36 | 86 | ND | ND |
| 28 | 3 | 97 | Pyruvate dehydrogenase (Sphingobacterium) | WP_045753967.1 | 100 | 6E−60 | 100 | pfam00198 | 2-oxoacid dehydrogenases acyltransferase |
| 29 | 3 | 71 | Pullulanase (Klebsiella) | EHS90708.1 | 100 | 4E−41 | 96 | TIGR02103 | Alpha-1,6-glucosidases, pullulanase-type |
| 30 | 3 | 162 | Hypothetical protein (Sphingobacterium) | WP_021191620.1 | 100 | 4E−110 | 99 | pfam07980 | SusD family |
| 31 | 3 | 82 | Hypothetical protein (Acinetobacter) | WP_005399834.1 | 100 | 1E−50 | 99 | pfam03724 | META domain; unknown function, some are secreted |
| 32 | 3 | 83 | Hypothetical protein (Acinetobacter) | WP_004981111.1 | 86 | 2E−35 | 86 | ND | ND |
| 33 | 3 | 145 | Hypothetical protein (Sphingobacterium) | KKX46588.1 | 100 | 4E−85 | 93 | TIGR04056 | TonB-linked outer membrane protein, SusC/RagA family |
| 34 | 3 | 151 | Pullulanase (Klebsiella) | EHS90708.1 | 100 | 2E−100 | 95 | TIGR02103 | Alpha-1,6-glucosidases, pullulanase-type |
| 35 | 3 | 156 | PhoH-like ATP-binding protein (Salmonella) | WP_001731743.1 | 100 | 3E−106 | 100 | COG1702 | Phosphate starvation-inducible protein PhoH, predicted ATPase |
| 36 | 3 | 83 | Gliding motility protein RemB (Sphingobacterium) | WP_038696674.1 | 97 | 4E−37 | 81 | ND | ND |
| 37 | 3 | 151 | Aldose 1-epimerase (Sphingobacterium) | KKX49924.1 | 100 | 6E−97 | 95 | cd09019 | Galactose mutarotase_like |
| 38 | 3 | 154 | Membrane protein (Sphingobacterium) | WP_038697956.1 | 100 | 1E−95 | 95 | TIGR04056 | TonB-linked outer membrane protein, SusC/RagA family |
#PS number of protein sequences, LSR length of representative sequence in amino acids, COV query coverage, ND not identified
On the basis of the PSI-BLAST annotation, the representative sequences of the abundant clusters, in the RWS-driven metasecretome, were mostly classified as hypothetical proteins (14 clusters), membrane proteins (5), proteins involved in motility (e.g. flagellin) (4), pyruvate dehydrogenases (3) and proteins involved in polysaccharide deconstruction (e.g. pullulanases and alpha-l-arabinofuranosidases) (3). Moreover, RNA methyltransferases and chaperones like GroEL were found (Table 1). Domain detection using the NCBI conserved domain database revealed that the hypothetical and membrane proteins (19 of 38 abundant clusters: 50 %) might reflect plant polysaccharide sensors/transporters such as TonB-dependent receptors. Interestingly, the most abundant protein cluster was affiliated with a hemagglutinin from Sphingobacterium-like organisms. Such proteins contain a pfam01832 domain, which is related with a mannosyl-glycoprotein endo-beta-N-acetylglucosaminidase (EC:3.2.1.96) (Table 1).
In the xylose-driven metasecretome, we observed high abundances and diversities of proteins involved in several different pathways (Additional file 2). The high-abundance protein clusters (38) were related to functions mostly dealing with sugar metabolism (e.g. fructose-bisphosphate aldolases, xylose isomerases and xylulose kinases) (5 clusters), next to ribosomal proteins (3). In addition, some oxidoreductases, dehydrogenases and ABC transporters were detected. Regarding the xylan-driven metasecretome, the most abundant protein clusters encompassed proteins such as glycine dehydrogenases (cluster 1 with 5 sequences), hydroperoxidases (cluster 2 with 4 sequences) and xylose isomerases (cluster 3 with 3 sequences) from Flavobacterium-like organisms (97 % amino acid identity) (Additional file 3).
A total of 22 proteins was common between the RWS and xylan systems. These were mostly affiliated with Bacteroidetes-like membrane-localized proteins (mainly involved in transport, polysaccharide-binding and motility). In addition, four aldehyde dehydrogenases, two hypothetical proteins and one superoxide dismutase were detected. Moreover, one pullulanase from Klebsiella-like organisms (97 % identity) was common in the RWS- and xylan-driven consortium metasecretomes (Additional file 4).
Overview of carbohydrate-active proteins detected in the three metasecretomes
All proteins detected in the three metasecretomes were also annotated based on the CAZy database [21]. Fifty-nine, thirty-one and one unique proteins from the RWS-, xylose- and xylan-driven metasecretomes, respectively, were homologous (e value threshold: 0.01) to particular carbohydrate-active enzymes (Fig. 3). Based on this annotation, in the RWS consortial metasecretome, proteins of the CAZy families CBM48/GH13/CBM41 (14 %) and CBM50/GH73 (12 %) were the most abundant (proteins with 2 or 3 CAZy modules), followed by AA5 (glyoxal/galactose oxidases) and glycosyl hydrolases (GHs) of CAZy families GH43, GH16, GH3, GH51, GH10, GH20, GH95, GH12 and GH67. In the xylose metasecretome, the proteins were mostly affiliated with those from the CAZy families GH1 (16 %), CBM50 (13 %), GH3 (13 %) and GH37 (10 %). In addition, we observed the presence of glycosyl transferases (GTs) of families GT66, GT2, GT30 and GT4. The one protein identified in the xylan-driven metasecretome was classified as a presumptive GH97 family protein. This protein was affiliated, based on PSI-BLAST, with an alpha-glucosidase from Flavobacterium sp. F52 (WP_008466439—100 % coverage and 98 % identity).
Fig. 3.
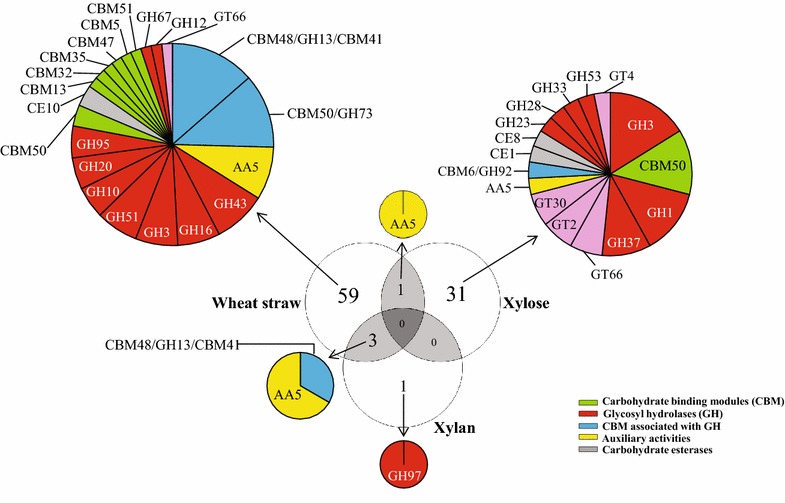
Carbohydrate-active annotation of proteins detected in each metasecretome
Plant biomass-degrading proteins detected in the wheat straw metasecretome
The proteins annotated as carbohydrate-active enzymes in the RWS metasecretome were manually analyzed and re-annotated based on the PSI-BLAST hits (Table 2). Using this analysis, we observed eight proteins (RWS1–8) containing CBM48/GH13/CBM41 modules. These were all affiliated with two pullulanases from Klebsiella-like organisms (identity from 95 to 100 %). In addition, the proteins detected within CAZy families GH43 (5), GH51 (4), GH16 (4), GH3 (4), GH10 (3), GH95 (3), GH12 (1) and GH67 (1) (Fig. 3) were identified as GH43A/D19 precursor (probably a beta-xylosidases) (RWS21–25), alpha-l-arabinofuranosidases (RWS26–29), glycerophosphodiester phosphodiesterases (RWS13–16), putative beta-glucosidases (RWS17–20), endo-1,4-beta-xylanases (RWS9–11), alpha-l-fucosidases (RWS31–33), peptidases (RWS12) and alpha-glucuronidases (RWS30), respectively. All these last 25 proteins were affiliated with Sphingobacterium-like sequences, showing high identity and coverage (>70 %). In addition, proteins affiliated with those of CAZy family AA5 were detected, of which two were tracked to the Gammaproteobacteria, specifically Pseudomonas and Acinetobacter species. The PSI-BLAST analyse had catalogued these as “membrane/flagellar proteins” (RWS34–38) (Table 2).
Table 2.
Plant biomass-degrading enzymes found in the RWS metasecretome
| Protein number | CAZy family | Action on (activity)a | Best PSI-BLAST hit vs. NCBI (organism source) | Accession no. | COV (%) | e value | Identity (%) | COV2 | #P |
|---|---|---|---|---|---|---|---|---|---|
| RWS1 | CBM48|GH13|CBM41 | Alpha-glucan polysaccharides (alpha-amylase/pullulanase) | Pullulanase (Klebsiella) | EHS90708.1 | 100 | 3E−45 | 98 | 43 | 5 |
| RWS2 | CBM48|GH13|CBM41 | Pullulanase (Klebsiella) | WP_017144955.1 | 100 | 8E−48 | 100 | 37 | 3 | |
| RWS3 | CBM48|GH13|CBM41 | Pullulanase (Klebsiella) | EHS90708.1 | 100 | 9E−34 | 97 | 33 | 3 | |
| RWS4 | CBM48|GH13|CBM41 | Pullulanase (Klebsiella) | EHS90708.1 | 100 | 4E−41 | 96 | 28 | 3 | |
| RWS5 | CBM48|GH13|CBM41 | Pullulanase (Klebsiella) | EHS90708.1 | 100 | 2E−64 | 99 | 45 | 3 | |
| RWS6 | CBM48|GH13|CBM41 | Pullulanase (Klebsiella) | EHS90707.1 | 100 | 1E−91 | 98 | 34 | 4 | |
| RWS7 | CBM48|GH13|CBM41 | Pullulanase (Klebsiella) | WP_017144955.1 | 100 | 2E−41 | 97 | 48 | 5 | |
| RWS8 | CBM48|GH13|CBM41 | Pullulanase (Klebsiella) | EHS90708.1 | 100 | 2E−100 | 95 | 30 | 3 | |
| RWS9 | GH10 | Hemicellulose (endoxylanase) | XynB19 precursor (Sphingobacterium) | ACX30647.1 | 100 | 9E−47 | 95 | 57 | 5 |
| RWS10 | GH10 | Endo-1,4-beta-xylanase precursor (Sphingobacterium) | ACX30652.1 | 100 | 4E−67 | 95 | 38 | 3 | |
| RWS11 | GH10 | Endo-1,4-beta-xylanase precursor (Sphingobacterium) | ACX30652.1 | 98 | 1E−101 | 92 | 27 | 5 | |
| RWS12 | GH12 | Cellulose (endoglucanase) | Peptidase M16 (Sphingobacterium) | WP_045752104.1 | 100 | 3E−96 | 97 | 17 | 2 |
| RWS13 | GH16 | (Hemi)cellulose (endoglucanase/endogalactanase) | Glycerophosphodiester phosphodiesterase (Sphingobacterium) | WP_031286917.1 | 100 | 2E−70 | 99 | 55 | 5 |
| RWS14 | GH16 | Glycerophosphoryl diester phosphodiesterase (Sphingobacterium) | KKX48537.1 | 100 | 7E−50 | 100 | 34 | 3 | |
| RWS15 | GH16 | Glycerophosphoryl diester phosphodiesterase (Sphingobacterium) | KKX48537.1 | 100 | 5E−77 | 100 | 24 | 3 | |
| RWS16 | GH16 | Glycerophosphodiester phosphodiesterase (Sphingobacterium) | WP_031286917.1 | 100 | 2E−95 | 99 | 41 | 5 | |
| RWS17 | GH3 | Cellulose (beta-glucosidase) | Beta-glucosidase (Sphingobacterium) | CDS95530.1 | 100 | 7E−106 | 95 | 21 | 3 |
| RWS18 | GH3 | Glycoside hydrolase family 3 (Sphingobacterium) | KKX49060.1 | 81 | 1E−52 | 92 | 27 | 2 | |
| RWS19 | GH3 | Glycoside hydrolase family 3 (Sphingobacterium) | KKX49060.1 | 100 | 9E−94 | 92 | 21 | 3 | |
| RWS20 | GH3 | Glycoside hydrolase family 3 (Sphingobacterium) | KKX49060.1 | 75 | 3E−34 | 96 | 36 | 2 | |
| RWS21 | GH43 | Hemicellulose (beta-xylosidase/alpha-arabinofuranosidase) | Aldose epimerase (Sphingobacterium) | WP_045756400.1 | 100 | 4E−81 | 81 | 11 | 2 |
| RWS22 | GH43 | GH43D19 precursor (Sphingobacterium) | ACX30655.1 | 100 | 3E−83 | 98 | 29 | 3 | |
| RWS23 | GH43 | GH43D19 precursor (Sphingobacterium) | ACX30655.1 | 100 | 1E−51 | 100 | 37 | 2 | |
| RWS24 | GH43 | GH43D19 precursor (Sphingobacterium) | ACX30655.1 | 97 | 8E−40 | 94 | 38 | 2 | |
| RWS25 | GH43 | GH43A19 precursor (Sphingobacterium) | ACX30649.1 | 100 | 3E−88 | 98 | 15 | 2 | |
| RWS26 | GH51 | Hemicellulose (alpha-arabinofuranosidase) | Alpha-l-arabinofuranosidase (Sphingobacterium) | WP_045756399.1 | 100 | 1E−88 | 99 | 27 | 2 |
| RWS27 | GH51 | Alpha-l-arabinofuranosidase (Sphingobacterium) | WP_045756399.1 | 92 | 5E−45 | 99 | 44 | 2 | |
| RWS28 | GH51 | Alpha-l-arabinofuranosidase (Sphingobacterium) | WP_038697950.1 | 100 | 3E−69 | 96 | 33 | 2 | |
| RWS29 | GH51 | Alpha-l-arabinofuranosidase (Sphingobacterium) | WP_045756399.1 | 100 | 3E−52 | 100 | 43 | 2 | |
| RWS30 | GH67 | Hemicellulose (alpha-glucuronidase) | Alpha-glucuronidase (Sphingobacterium) | WP_031288024.1 | 90 | 5E−69 | 83 | 15 | 2 |
| RWS31 | GH95 | Hemicellulose (alpha-fucosidase) | Alpha-l-fucosidase (Sphingobacterium) | WP_045753034.1 | 99 | 8E−72 | 76 | 37 | 4 |
| RWS32 | GH95 | Alpha-l-fucosidase (Sphingobacterium) | WP_045753034.1 | 100 | 1E−44 | 82 | 46 | 3 | |
| RWS33 | GH95 | Alpha-l-fucosidase (Sphingobacterium) | WP_045753034.1 | 100 | 5E−71 | 77 | 37 | 4 | |
| RWS34 | AA5 | Lignin or polysaccharides (glyoxal/galactose oxidases)b | Cell envelope biogenesis protein OmpA (Flavobacterium) | WP_042565523.1 | 100 | 2E−44 | 99 | 18 | 2 |
| RWS35 | AA5 | Outer membrane protein omp38 (Acinetobacter) | WP_004819145.1 | 66 | 7E−53 | 98 | 39 | 5 | |
| RWS36 | AA5 | Flagellar motor protein MotB (Sphingobacterium) | WP_031288562.1 | 100 | 4E−82 | 98 | 24 | 4 | |
| RWS37 | AA5 | Flagellar motor protein MotB (Sphingobacterium) | WP_038696344.1 | 98 | 4E−53 | 97 | 65 | 6 | |
| RWS38 | AA5 | Membrane protein (Pseudomonas) | WP_016485176.1 | 100 | 8E−81 | 100 | 29 | 3 |
Discussion
Complete degradation of plant biomass depends on the synergistic action of different enzymes. Thus, analysis of the proteins that are secreted by lignocellulose-grown microbial consortia can shed light on the complexity of the proteome involved in degradation. Here, we provide an analysis of the metasecretome of microbial consortia grown on raw wheat straw (RWS), xylan and xylose as the sole carbon sources (Fig. 1). The source microbial consortium bred on wheat straw had already revealed beta-galactosidase, beta-xylosidase, beta-mannosidase, cellobiohydrolase and beta-glucosidase extracellular activities [19]. We observed low consortial growth (~107 bacterial cells/ml), next to a low abundance of secreted/detected proteins (103) on xylan, as compared to RWS (~109 cells/ml and 768 proteins) (Additional file 1). Thus, somewhat unexpectedly, the xylan was less palatable than the wheat straw to the consortium used. Wheat straw likely contains diverse high-complexity as well as low-complexity carbon sources, which collectively provide “niches” that spur the growth of the different microbes that had been “primed” for its use given their preselection on this substrate.
Detection of the proteins in the metasecretomes of each treatment was performed with the help of a metagenomic database obtained from the original microbial consortium [11]. This methodology was found to improve the detection of proteins compared with the use of public databases such as NCBI or UniProt [22]. However, a potential caveat in this process should be mentioned: the proteins detected were mostly fragmentary and so only yielded hits with gene fragments. Also, signal peptides were not detected with the proteins, as reported [16]. To quantify the relative abundance of the proteins, the high-throughput isobaric tag (iTRAQ) technique has been proposed [13, 17]. Alternatively, label-free methods and the “exponentially modified protein abundance index” (emPAI) have been applied [16]. In our study, we applied a label-free method, refraining from using the emPAI. The rationale was that this index may suffer from biases, specifically in the spectrum counting, due to the fact that small proteins tend to have fewer peptides identified per protein compared with large proteins [23]. To fill this gap, we performed a “semi-quantitative” analysis of the secreted proteins using an innovative clustering method, in which the detected proteins (in each treatment) were grouped at 70 % of amino acid identity. Then, we considered clusters having more than three sequences to constitute “abundant” types of proteins. Moreover, in our study, the functional annotation of the proteins was supported by the search for conservative domains in the most abundant protein clusters. This methodology can assist in the assignment of hypothetical proteins to a known function, used by us, for example, in the case of the TonB-dependent receptors (Table 1).
Although the RWS-bred microbial consortium contained bacteria as well as fungi, the abundance of bacteria (estimated as ~109 16S rRNA gene copies/ml) was higher than the fungi (~107 ITS1 copies/ml) [8]. The majority (around 70 %) of the predicted protein-encoding genes, in the RWS metagenome data, had homologs on published bacterial genomes, which supported the contention of primary selection of bacterially dominated substrate-degrading microbial consortia. In contrast, metagenomic sequences affiliated with the Eukarya amounted to less than 0.001 % of the totals [11]. Based on these premises and due to the fact that the proteins in the metasecretomes of each treatment were identified with the help of a metagenomic database from the original microbial consortium (RWS), it is logical to assume that fungal proteins were present in low abundance and could not be easily detected.
Based on the taxonomic assignment of abundant proteins (Fig. 2), we inferred that different organisms in our consortia have different roles. Specifically, Sphingobacterium, Pedobacter and Flavobacterium types were hypothesized to act mainly on the complex substrates (wheat straw and xylan), whereas Klebsiella-like organisms were more tuned to utilizing sugars such as xylose. In contrast, in our original consortium, the latter organisms were surmised to degrade (hemi)cellulose through the expression of endoglucanases, xylanases and glucosidases [8, 11, 19]. Moreover, the detection of TonB-dependent receptors in the RWS metasecretome indicated that sensing of plant polysaccharides and transport of sugars might have been dominant in particular Bacteroidetes-like organisms [24–26]. Genes for such proteins have previously been found to be enriched in the metagenome from the source consortium when compared with that from the microbial source (forest soil) [11]. The most abundant protein cluster, in RWS, showed a domain that belongs to mannosyl-glycoprotein endo-beta-N-acetylglucosaminidase from Sphingobacterium-like consortium members (Table 1). Such proteins, called autolysins, may be involved in the degradation of peptidoglycan. They are involved in daughter cell separation during vegetative cell growth, hydrolyzing the septum after cell division [27, 28]. Thus, these proteins might not have a primary role in lignocellulose degradation.
Regarding the carbohydrate-active enzymes detected in the RWS metasecretome (Fig. 3; Table 2), Sphingobacterium-like organisms might be assumed as being the sources of most of the secreted enzymes involved in cellulose (GH3, GH12) and hemicellulose deconstruction (e.g. GH10, GH43, GH51 and GH95). Recently, Jiménez et al. [11] reported an overrepresentation of genes for family GH3, GH43 and GH95 proteins in the metagenome of the source consortium when compared with that of the forest soil inoculum. Together, these data suggest a correlation between the enrichment of genes for these families, their expression and the concomitant protein secretion. We postulate that this type of selection strongly impacts the wheat straw deconstruction rate. The absence of cellobiohydrolases (CBHs) and endoglucanases (specifically of families GH5 and GH9) in the RWS metasecretome could be related to the consortium response to the composition of the substrate or to their low abundance. Other studies have shown similar results. For instance, D’haeseleer et al. [14] reported the absence of CBHs in the metasecretome of a thermophilic bacterial consortium adapted to switchgrass (JP-1 % SG). They reported the presence of only one abundant and overrepresented protein involved in cellulose hydrolysis (a GH5). In contrast, the majority of secreted GHs were related with the deconstruction of hemicellulose or alpha-glucan polysaccharides (GH13 and GH31). Interestingly, comparison of this metasecretome [14] with that of the same consortium that had been perturbed by cultivation with microcrystalline cellulose (McCel) [10] demonstrated that the GHs complement was different for the McCel-degrading microbial consortium. In particular, one CBH and three endoglucanases were detected in McCel that were not detected in the JP-1 % SG. These data, plus our results, indicate that the lignocellulolytic microbial consortia, and their metasecretomes, respond differently as related to the composition of the substrates used. Additionally, we hypothesized that the microbes in RWS could get energy and act efficiently at the expense of mainly the hemicellulose fractions instead of the microcrystalline cellulose.
Moreover, finding proteins affiliated with CAZy family AA5 (oxidative enzyme—glyoxal/galactose oxidase) may pinpoint potential roles for Sphingobacterium, Acinetobacter or Pseudomonas in this activity. Glyoxal oxidases are copper-radical oxidases with a broad specificity that are able to oxidize aldehydes to the corresponding carboxylic acids. Such proteins are considered to be one of the central H2O2-generating enzymes. Also, galactose oxidases (GAO) are copper-containing enzymes that catalyze a reaction comprising two separable half-reactions, i.e. oxidation of a primary alcohol, and reduction of O2 to H2O2. GAO catalyze the oxidation of a wide range of carbohydrates (including galactose) but also primary alcohols to the corresponding aldehydes, with the reduction of O2 into H2O2 [37]. Based on that, we hypothesize that such proteins are involved in the oxidation of polysaccharides or in lignin transformations in the RWS metasecretome, possibly improving the deconstruction of the plant biomass.
The finding of eight proteins with CBM48/GH13/CBM41 (pullulanases—EC 3.2.1.41) modules affiliated with those from Klebsiella-like organisms (Table 2) pinpointed Klebsiella as the source organism for this specific activity. Enzymes of family GH13 (called debranching enzymes) constitute the major glycoside hydrolase family acting on substrates containing alpha-glucoside linkages (alpha-glucan polysaccharides). Pullulanases are a specific class of GH13 glucanases, with type I pullulanases specifically attacking alpha-1,6 linkages and type II pullulanases also alpha-1,4 linkages. Alpha-glucan polysaccharides (e.g. starch, rhamnogalacturonan and (1–6)-alpha-glucomannan) are present in plant cell walls and hence the aforementioned protein may be denoted as plant polysaccharide-degrading enzymes. Pullulanases break down pullulan, an exopolysaccharide produced from starch and other biopolymers [29]. These enzymes are useful in hydrolyzing side chain residues of hemicellulose and other alpha cross-links that could be present in the wheat straw used for breeding [30]. Recently, Adav et al. [16] reported the upregulation of GH13 proteins (alpha-glucosidases or pullulanases) in the secretome of Thermobifida fusca when grown on corn stover, hay and wood chips, suggesting that such proteins might enhance the hydrolysis of plant biomass.
Interestingly, D’haeseleer et al. [14] reported the presence of proteins affiliated with CAZy families GH1, GH2, GH3, GH10 and GH51 in the metasecretome of a thermophilic bacterial consortium growing on switchgrass (JP-1 % SG). In addition, endoglucanases (GH5 and GH9), cellobiohydrolases (GH48), xylanases (GH10) and arabinofuranosidases were found in the metasecretome of microcrystalline cellulose-degrading consortia (McCel) [10]. These last studies reflect the importance of proteins from these families to deconstruct plant biomass. Especially hemicellulolytic enzymes such as endo-1,4-beta-xylanases (GH10), beta-xylosidases (GH43), alpha-l-arabinofuranosidases (GH43 or GH51) and alpha-l-fucosidases (GH95) could be indispensable in the formulation of efficient enzyme cocktails. These could be added to cellulose- and alpha-glucan polysaccharides-degrading enzymes, i.e. endoglucanases (GH5), cellobiohydrolases, beta-glucosidases (GH1or GH3) and pullulanases (GH13). The commercial development of hemicellulases for the enzymatic hydrolysis of plant biomass is not as advanced as that of cellulases because current commercial preparations have been primarily used on pretreated biomass from which the hemicellulose part was partially removed before saccharification [4]. Moreover, current fungus-derived cellulases tend to have only weak hemicellulolytic activity and are not adequate for the complete conversion of hemicellulose from plant biomass. Development of low-cost commercial hemicellulases that work synergistically with cellulases is one of the goals of many current research activities [31]. Based on this premise, the hemicellulases secreted by the here-described RWS consortium could constitute an excellent source for the preparation of enzyme cocktails that can be applied directly or as a supplement in plant biomass pretreatment. Complete genes (with start and final codons) for these secreted hemicellulases can be identified in the assembled metagenome files [11] and then might be synthesized, codon-optimized, cloned and expressed, in order to produce the individual enzymes and combine them in cocktails. Recently, four genes from the GH43 family (Sphingobacterium origin) were already expressed successfully in E. coli from our metagenome data (unpublished observations).
Regarding the xylose consortium metasecretome, the profile of carbohydrate-active enzymes was quite different from that of the RWS one. Clearly, the presence of the xylan monomer xylose repressed the production and release of hemicellulases (e.g. GH43, GH51 and GH95) and pullulanases (GH13). However, oligosaccharide-degrading enzymes (GH1 and GH3) and GTs were detected. Unfortunately, we could not find enzymes directly involved in polysaccharide degradation in the xylan-driven metasecretome, possibly because these were not abundant. However, we did find an alpha-glucosidase (GH97) apparently from a Flavobacterium-like organism as well as enzymes related with the conversion (isomerization) of sugars that are release after degradation of xylan (e.g. xylose isomerases). Such sugar isomerases were present in the metasecretomes of both the xylan- and xylose-driven consortia. In previous work, extracellular xylose isomerases have been found in the supernatant of a thermophilic bacterial consortium adapted to switchgrass [14]. These enzymes may have an extracellular role, transforming monosaccharides produced from hemicellulose, potentially as a means of relieving product inhibition on the primary hemicellulases or as a way to circumvent competition for these monosaccharides from other members of the microbial community.
Conclusions
Analyses of the metasecretomes of lignocellulolytic microbial consortia grown on 1 % of either xylose, xylan or wheat straw revealed several key proteins to become abundant in relation to the substrate used. First, the microbial consortium performed poorly on xylan as compared to xylose and wheat straw. In terms of protein “richness”, the metasecretome of the RWS-driven consortium stood out as the “richest”. In the RWS (as well as xylan) consortium, proteins released by Bacteroidetes (e.g. Sphingobacterium, Flavobacterium and Pedobacter) were dominant. In contrast, proteins secreted by Enterobacteriales abounded in the xylose-driven metasecretome. In this respect, we also found Klebsiella-like organisms to secrete pullulanases in the RWS-driven consortium.
The highly diverse RWS metasecretome revealed an abundance of CAZy-defined GH3, GH10, GH13, GH43, GH51, GH67 and GH95 family enzymes, which are all predicted to be involved in plant biomass breakdown. The fact that these proteins were mainly affiliated with Sphingobacterium-like enzymes suggested sphingobacteria are highly relevant players in the degradation of lignocellulose. Thus, an intricate relationship between the active consortial members was shown, providing evidence of synergistic activities of the different proteins in the deconstruction of plant biomass. We advocate the use of the RWS consortium released proteins as potential enhancers of efficiencies in plant biomass treatments. Future experiments that combine metasecretomes, other hemicellulases (e.g. synthesized from the metagenome) with available commercial cellulases can help to improve the plant biomass degradation rates for biorefining. Finally, an analysis of the secreted proteins during consortium development (time course) posits as very valuable, allowing to observe the differential expression and secretion of the enzymes involved in plant biomass deconstruction.
Methods
Microbial enriched cultures design
The original lignocellulose decomposing consortium was developed with a forest soil as the microbial source inoculum [8]. Briefly, the soil-derived cell suspensions were introduced into triplicate flasks containing 25 ml of mineral salts medium (MSM) with 1 % of “raw” wheat straw (RWS), and incubated at 25 °C with shaking at 100 rpm. A dilution-to-stimulation approach was followed up to the 10th transfer [8, 19]. Then, 25 µl (1 × 108 bacterial cells/ml) were transferred from the 10th transfer flask to three flasks (in triplicate) containing sterile and fresh MSM plus 1 % (w/v) of different carbon sources, i.e. xylose, xylan (from beechwood) and wheat straw. Subsequently, all flasks were incubated at 25 °C with shaking at 100 rpm. The numbers of bacterial cells per milliliter were quantified by microscope cell counting in a Burke-Turk chamber (Blaubrand®) according to the manufacturer’s protocol. After 11 days of incubation, the microbial cells as well as particulate matter were removed by centrifugation (7500g for 30 min at 4 °C). The supernatants (from 15 ml of pooled culture in each treatment) were subsequently filtered by 0.45 µm (Whatman FP30/0.45—cellulose acetate membrane) (in order to remove wheat straw particles) followed by 0.22 µm syringe filters (Whatman FP30/0.22—cellulose acetate membrane) (in order to remove microbial cells) (Fig. 1).
Metasecretome recovery and liquid chromatography–tandem mass spectrometry
A total of 2 ml of filtered supernatant (secreted proteins) for each treatment were precipitated with tri-chloroacetic acid (TCA) (final concentration of 15 %). The samples were incubated on ice for 15 min and then overnight at −20 °C. They were then kept at room temperature and proteins were precipitated by centrifugation at 14,000 rpm for 15 min at 4 °C. Following this, supernatants were gently removed, and the tubes spun briefly to remove remaining liquid. The protein pellets were suspended in 1 ml of 100 % ice-cold acetone by five times vortexing for 30 s, over 1 h at −20 °C. Then, samples were centrifuged at 14,000 rpm for 15 min at 4 °C and the acetone was removed, followed by air-drying the pellets for 10 min. Pellets were resuspended with 2× sample buffer (100 mM DTT, 2 % SDS, 80 mM Tris–HCl, pH 6.8, 0.006 % bromophenol blue, 15 % glycerol), after which they were checked with 10 % SDS-PAGE at 80 V for 1.5 h. This was followed by coomassie staining for 6 h in shaking platform at room temperature, and de-staining (40 % methanol and 10 % glacial acetic acid) for 2 h at room temperature.
The secreted proteins from the RWS system were divided into 9 fractions (ranging from 20 to 150 KDa) from the SDS-PAGE gel. These gel fragments were washed with 25 mM of ammonium bicarbonate and 50 % acetonitrile. The xylose and xylan derived samples were also prepared for in-solution digestion. Proteolytic treatment of all samples was performed using trypsin (Promega Benelux BV, Leiden), dissolved in 100 mM ammonium bicarbonate buffer. In the case of the RWS fractions, the gel pieces were mixed with 20 µl of 10 ng/µl trypsin in 100 mM ammonium bicarbonate buffer and incubated overnight at 37 °C. Peptides were extracted from the gel with 75 % of acetonitrile (in 5 % formic acid), dried and resuspended in 20 μl of 0.1 % of formic acid.
After trypsin digestion, the peptide mix was injected into an Ultimate 3000 nano-LC–MS/MS system (Dionex, Amsterdam, The Netherlands), in line connected to an Q-Exactive-Plus-mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The sample mixture was loaded on a trapping column (Acclaim PepMap; C18; 5-mm length by 300-µm inside diameter; 5-µm particle size; 100-Å porosity; Dionex) and washed. After 3 min, the mixture was separated using a 60-min linear gradient from 5 % of 0.1 % of formic acid in water to 90 % of 0.1 % in acetonitrile at a flow rate of 250 nl/min. The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the eight most abundant multiple charged ions (2, 3, 4, 5 and 6 times). Full-scan MS spectra were acquired from m/z 400 to 1800 in the Q-Exactive-Plus-mass spectrometer at a target value of 3E6 with a resolution of 70,000. Peptides were analyzed in the orbitrap with a resolution of 35,000. The scan range for MS/MS was set to m/z 200–2000.
Peptide identification and data processing
The mass spectrometry datasets produced for each system were analyzed for peptide and protein identification using PEAKS software v7.0 [32]. Spectra for the RWS metasecretomes were pooled (9 spectra). Database searches were carried out against a total metagenome dataset (MG-RAST ID 4547280.3: 388,324 proteins detected by FragGeneScan software) [11]. The following search parameters were applied: (i) trypsin was chosen as the protein-digesting enzyme, (ii) false discovery rate (FDR), 0.1 %, (iii) fragment mass error tolerance 0.02 Da, (iv) peptides with >10 ppm mass error were discarded, (v) proteins with at least two unique peptide matchings were retained, and two missed cleavages were tolerated, (vi) fixed modifications (carbamidomethylation, 57), variable modifications (oxidation—methionine—16). The total metagenome dataset and the detected proteins in each system were taxonomically and functionally classified by the GhostKOALA annotation tool [33] using a cutoff GHOSTX score of 100. Based on this threshold, we predicted the medium–high confidence in the taxonomic affiliation [20]. In order to obtain “semi-quantitative” data, we clustered (threshold of 70 % amino acid identity) the detected proteins using the CD-hit software [34]. Clusters composed of more than three protein sequences (“abundant” proteins) were retrieved. A representative and randomly picked protein sequence for each cluster was manually annotated by PSI-BLAST [35] and inspected for conserved domains using the NCBI conserved domain database. Additionally, the proteins detected in each metasecretome were affiliated with carbohydrate-active enzymes using the CAZymes analysis toolkit (CAT) platform [36] (e value threshold 0.01), and obtained hits were manually inspected by PSI-BLAST.
Authors’ contributions
DJJ helped to design the experiments, performed the bioinformatic analyses and drafted the manuscript. MM carried out the microbial enrichment procedure, participated in the liquid chromatography–tandem mass spectrometry and helped to revise the manuscript. JDvE conceived the study, and participated in its design, coordination and drafting of the manuscript. All authors read and approved of the final manuscript.
Acknowledgements
We thank Harald Ruijssenaars and Richard van Kranenburg (CORBION) for support. Further thanks to Margot Jeronimus-Stratingh for liquid chromatography–tandem mass spectrometry experimental support. This work was supported by the Netherlands Ministry of Economic Affairs and the BE-Basic partner organizations (http://www.be-basic.nl).
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- AA
auxiliary activities
- CAZy
carbohydrate-active enzymes database
- CAT
cazymes analysis toolkit
- CBM
carbohydrate-binding modules
- emPAI
exponentially modified protein abundance index
- FDR
false discovery rate
- GHs
glycosyl hydrolases
- GTs
glycosyl transferases
- iTRAQ
isobaric tags for relative and absolute quantitation
- KEGG
Kyoto encyclopedia of genes and genomes
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- PSI-BLAST
position-specific iterated-BLAST
- RWS
wheat straw consortium
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
Additional files
10.1186/s13068-015-0387-8 Bacterial cell counts in the enriched cultures and SDS-PAGE of protein secretomes. a Bacterial cell numbers (log10 cells per milliliter of culture) in xylose, xylan and wheat straw after 5, 8 and 11 days of incubation, b SDS-PAGE of the secreted proteins by the microbial consortium after 11 days of incubation in xylose, xylan and wheat straw.
10.1186/s13068-015-0387-8 Abundant clusters (clusters at 70 % identity with more than 3 sequences) of secreted proteins in xylose.
10.1186/s13068-015-0387-8 Abundant clusters (clusters at 70 % identity with more than 2 sequences) of secreted proteins in xylan.
10.1186/s13068-015-0387-8 Common proteins between RWS and xylan metasecretomes.
Footnotes
D. J. Jiménez and M. Maruthamuthu contributed equally to this work
Contributor Information
Diego Javier Jiménez, Email: d.j.jimenez.avella@rug.nl.
Mukil Maruthamuthu, Email: m.marutha.muthu@rug.nl.
Jan Dirk van Elsas, Email: j.d.van.elsas@rug.nl.
References
- 1.Liguori R, Amore A, Faraco V. Waste valorization by biotechnological conversion into added value products. Appl Microbiol Biotechnol. 2013;97(14):6129–6147. doi: 10.1007/s00253-013-5014-7. [DOI] [PubMed] [Google Scholar]
- 2.Hasunuma T, Okazaki F, Okai N, Hara KY, Ishii J, Kondo A. A review of enzymes and microbes for lignocellulosic biorefinery and the possibility of their application to consolidated bioprocessing technology. Bioresour Technol. 2013;135:513–522. doi: 10.1016/j.biortech.2012.10.047. [DOI] [PubMed] [Google Scholar]
- 3.Rodrigo de Souza W. Microbial degradation of lignocellulosic biomass. In: Chandel A, Da Silva S, editors. Sustainable degradation of lignocellulosic biomass—techniques, applications and commercialization. Brazil: InTech; 2013. p. 208–209. doi:10.5772/54325.
- 4.Meyer AS, Rosgaard L, Sørensen HR. The minimal enzyme cocktail concept for biomass processing. J Cereal Sci. 2009;50:337–344. doi: 10.1016/j.jcs.2009.01.010. [DOI] [Google Scholar]
- 5.Yang H, Wu H, Wang X, Cui Z, Li Y. Selection and characteristics of a switchgrass-colonizing microbial community to produce extracellular cellulases and xylanases. Bioresour Technol. 2011;102:3546–3550. doi: 10.1016/j.biortech.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Cheng JR, Zhu MJ. A novel co-culture strategy for lignocellulosic bioenergy production: a systematic review. Int J Mod Biol Med. 2012;1(3):166–193. [Google Scholar]
- 7.Hui W, Jiajia L, Yucai L, Peng G, Xiaofen W, Kazuhiro M, et al. Bioconversion of un-pretreated lignocellulosic materials by a microbial consortium XDC-2. Bioresour Technol. 2013;136:481–487. doi: 10.1016/j.biortech.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Jiménez DJ, Korenblum E, van Elsas JD. Novel multispecies microbial consortia involved in lignocellulose and 5-hydroxymethylfurfural bioconversion. Appl Microbiol Biotechnol. 2014;98:2789–2803. doi: 10.1007/s00253-013-5253-7. [DOI] [PubMed] [Google Scholar]
- 9.Gladden JM, Allgaier M, Miller CS, Hazen TC, VanderGheynst JS, Hugenholtz P, et al. Glycoside hydrolase activities of thermophilic bacterial consortia adapted to switchgrass. Appl Environ Microbiol. 2011;77(16):5804–5812. doi: 10.1128/AEM.00032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park JI, Steen EJ, Burd H, Evans SS, Redding-Johnson AM, Batth T, et al. A thermophilic ionic liquid-tolerant cellulase cocktail for the production of cellulosic biofuels. PLoS One. 2012;7(5):e37010. doi: 10.1371/journal.pone.0037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiménez DJ, Chaves-Moreno D, van Elsas JD. Unveiling the metabolic potential of two soil-derived microbial consortia selected on wheat straw. Sci Rep. 2015;5:13845. doi: 10.1038/srep13845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simmons CW, Reddy AP, D’haeseleer P, Khudyakov J, Billis K, Pati A, et al. Metatranscriptomic analysis of lignocellulolytic microbial communities involved in high-solids decomposition of rice straw. Biotechnol Biofuels. 2014;7(1):495. doi: 10.1186/s13068-014-0180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adav SS, Ravindran A, Cheow ES, Sze SK. Quantitative proteomic analysis of secretome of microbial consortium during saw dust utilization. J Proteom. 2012;75(18):5590–5603. doi: 10.1016/j.jprot.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 14.D’haeseleer P, Gladden JM, Allgaier M, Chain PS, Tringe SG, Malfatti SA, et al. Proteogenomic analysis of a thermophilic bacterial consortium adapted to deconstruct switchgrass. PLoS One 2013;8(7):e68465. [DOI] [PMC free article] [PubMed]
- 15.Manavalan T, Manavalan A, Thangavelu KP, Heese K. Secretome analysis of Ganoderma lucidum cultivated in sugarcane bagasse. J Proteom. 2012;77:298–309. doi: 10.1016/j.jprot.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Adav SS, Cheow ES, Ravindran A, Dutta B, Sze SK. Label free quantitative proteomic analysis of secretome by Thermobifida fusca on different lignocellulosic biomass. J Proteom. 2012;75(12):3694–3706. doi: 10.1016/j.jprot.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 17.Liu D, Li J, Zhao S, Zhang R, Wang M, Miao Y, et al. Secretome diversity and quantitative analysis of cellulolytic Aspergillus fumigatus Z5 in the presence of different carbon sources. Biotechnol Biofuels. 2013;6(1):149. doi: 10.1186/1754-6834-6-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hori C, Gaskell J, Igarashi K, Kersten P, Mozuch M, Samejima M, et al. Temporal alterations in the secretome of the selective ligninolytic fungus Ceriporiopsis subvermispora during growth on aspen wood reveal this organism’s strategy for degrading lignocellulose. Appl Environ Microbiol. 2014;80(7):2062–2070. doi: 10.1128/AEM.03652-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiménez DJ, Dini-Andreote F, van Elsas JD. Metataxonomic profiling and prediction of functional behaviour of wheat straw degrading microbial consortia. Biotechnol Biofuels. 2014;7:92. doi: 10.1186/1754-6834-7-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki S, Kakuta M, Ishida T, Akiyama Y. GHOSTX: an improved sequence homology search algorithm using a query suffix array and a database suffix array. PLoS One. 2014;9(8):e103833. doi: 10.1371/journal.pone.0103833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(database issue):D490–5. [DOI] [PMC free article] [PubMed]
- 22.Heyer R, Kohrs F, Reichl U, Benndorf D. Metaproteomics of complex microbial communities in biogas plants. Microb Biotechnol. 2015 doi: 10.1111/1751-7915.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Florens L, Carozza MJ, Swanson SK, Fournier M, Coleman MK, Workman JL, et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods. 2006;40(4):303–311. doi: 10.1016/j.ymeth.2006.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denancé N, et al. Plant carbohydrate scavenging through tonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One. 2007;2(2):e224. doi: 10.1371/journal.pone.0000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dodd D, Mackie RI, Cann IK. Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol. 2011;79(2):292–304. doi: 10.1111/j.1365-2958.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernández-Gómez B, Richter M, Schüler M, Pinhassi J, Acinas SG, González JM, et al. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 2013;7(5):1026–1037. doi: 10.1038/ismej.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto W, Ochiai A, Momma K, Itoh T, Mikami B, Maruyama Y, et al. Crystal structure of the glycosidase family 73 peptidoglycan hydrolase FlgJ. Biochem Biophys Res Commun. 2009;381(1):16–21. doi: 10.1016/j.bbrc.2009.01.186. [DOI] [PubMed] [Google Scholar]
- 28.Lipski A, Hervé M, Lombard V, Nurizzo D, Mengin-Lecreulx D, Bourne Y, et al. Structural and biochemical characterization of the β-N-acetylglucosaminidase from Thermotoga maritima: toward rationalization of mechanistic knowledge in the GH73 family. Glycobiology. 2015;25(3):319–330. doi: 10.1093/glycob/cwu113. [DOI] [PubMed] [Google Scholar]
- 29.Singh RS, Saini GK, Kennedy JF. Pullulan: microbial sources, production and applications. Carbohydr Polym. 2008;73(4):515–531. doi: 10.1016/j.carbpol.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Domań-Pytka M, Bardowski J. Pullulan degrading enzymes of bacterial origin. Crit Rev Microbiol. 2004;30(2):107–121. doi: 10.1080/10408410490435115. [DOI] [PubMed] [Google Scholar]
- 31.Mohanram S, Amat D, Choudhary J, Arora A, Nain L. Novel perspectives for evolving enzyme cocktails for lignocellulose hydrolysis in biorefineries. Sustain Chem Process. 2013;1:15. doi: 10.1186/2043-7129-1-15. [DOI] [Google Scholar]
- 32.Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, et al. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics 2012;11(4):M111.010587. [DOI] [PMC free article] [PubMed]
- 33.GhostKOALA annotation tool. http://www.kegg.jp/ghostkoala/. Accessed 8 May 2015.
- 34.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26(5):680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC. CAZymes analysis toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology. 2010;20(12):1574–1584. doi: 10.1093/glycob/cwq106. [DOI] [PubMed] [Google Scholar]
- 37.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6(1):41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]