

K11- and K48-linked polyubiquitin constitute alternative destruction signals on substrates of the APC/C. Substrates targeted late in mitotic exit, including Aurora kinases, Polo-like kinase, and KIFC1, all depend on K11 linkages for degradation. APC/C coactivator Cdh1 directs K11 linkage assembly via UBE2S in a substrate-specific manner.
Abstract
The ubiquitin proteasome system (UPS) directs programmed destruction of key cellular regulators via posttranslational modification of its targets with polyubiquitin chains. These commonly contain Lys-48 (K48)–directed ubiquitin linkages, but chains containing atypical Lys-11 (K11) linkages also target substrates to the proteasome—for example, to regulate cell cycle progression. The ubiquitin ligase called the anaphase-promoting complex/cyclosome (APC/C) controls mitotic exit. In higher eukaryotes, the APC/C works with the E2 enzyme UBE2S to assemble K11 linkages in cells released from mitotic arrest, and these are proposed to constitute an improved proteolytic signal during exit from mitosis. We tested this idea by correlating quantitative measures of in vivo K11-specific ubiquitination of individual substrates, including Aurora kinases, with their degradation kinetics tracked at the single-cell level. All anaphase substrates tested by this methodology are stabilized by depletion of K11 linkages via UBE2S knockdown, even if the same substrates are significantly modified with K48-linked polyubiquitin. Specific examination of substrates depending on the APC/C coactivator Cdh1 for their degradation revealed Cdh1-dependent enrichment of K11 chains on these substrates, whereas other ubiquitin linkages on the same substrates added during mitotic exit were Cdh1-independent. Therefore we show that K11 linkages provide the APC/C with a means to regulate the rate of substrate degradation in a coactivator-specified manner.
INTRODUCTION
The specificity of substrate targeting by the ubiquitin-proteasome system (UPS) enables precise remodeling of the protein landscape in many cellular processes. One such process is mitotic exit, triggered by destruction of mitotic cyclin at metaphase and accompanied by degradation of many other mitotic regulators, each with distinct timing and kinetics, as cells progress through anaphase and return to G1 phase (Min and Lindon, 2012). All of these degradation events are mediated by the anaphase-promoting complex/cyclosome (APC/C; Pines, 2011). A longstanding question concerns how this single ubiquitin ligase specifies temporal degradation for a large number of substrates. Part of the answer lies with the temporal specificity of APC/C’s coactivators, with Cdc20 active from prometaphase until its degradation during mitotic exit and Cdh1 from anaphase onward (Pines, 2011). The coactivation mechanism is proposed to function partly via recruitment of substrates and partly through enhancement of ubiquitination activity mediated by the APC/C (Barford, 2011), with recent studies attributing the latter role to enhanced E2 efficiency and stabilization of E2–APC/C interaction in the presence of coactivator (Brown et al., 2014; Chang et al., 2014; Kelly et al., 2014; Van Voorhis and Morgan, 2014).
In mammalian cells, APC/C is believed to act via the concerted activity of two different E2s. UBE2C adds the first ubiquitin or short ubiquitin chain to a substrate (“priming”), and UBE2S attaches ubiquitin to the already attached ubiquitin molecules, elongating polyubiquitin chains in a Lys-11 (K11) linkage-specific manner (Williamson et al., 2009; Matsumoto et al., 2010; Wu et al., 2010; Wickliffe et al., 2011a). Linkage specificity of UBE2S accounts for an abrupt increase in K11 linkages seen in cells released from mitotic arrest, when the APC/C reaches its most active state (Matsumoto et al., 2010). It is therefore widely accepted that regulation of mitotic exit is one of the major roles of K11 chains (Bremm and Komander, 2011; Wickliffe et al., 2011b), even though UBE2S is not required for destruction of cyclin B1 in unchallenged mitotic exit (Garnett et al., 2009).
A recent study identified Nek2A as a Ube2S-dependent substrate of the APC/C in prometaphase (Meyer and Rape, 2014). Of interest, that study showed that Nek2A was modified with a variety of short chains by UBE2C, with UBE2S elongating short chains in a K11 linkage–specific manner to generate branched chains. Such heterotypic chains were shown to constitute an improved degradation signal in prometaphase, when the APC/C is partially inhibited by the spindle assembly checkpoint (SAC; Meyer and Rape, 2014). In mitotic exit, however, once the SAC is silenced, there is a dramatic increase in K11-containing polyubiquitin conjugates. It is not known how these K11 linkages contribute to degradation of specific substrates as cells exit mitosis.
In this study, we combine a cell-based ubiquitination assay that allows quantitative interrogation of ubiquitin conjugates on individual substrates (Min et al., 2013; Lee et al., 2014) with in vivo degradation assays that allow tracking of their degradation kinetics at the single-cell level (Clute and Pines, 1999) to advance our understanding of the role and regulation of K11 linkages in mitotic exit.
RESULTS
Abundance of K11 chains increases sharply in mitotic exit
The abrupt increase in K11 linkages in cells released from mitotic arrest suggests that K11 linkages could contribute to mitotic exit. However, whereas UBE2S is required for efficient mitotic progression after chronic drug-induced SAC activation, it is dispensable for mitotic exit in unchallenged mitosis (Garnett et al., 2009), raising the question of whether the boost of K11 linkages at mitotic exit also occurs during unperturbed mitosis. To test this, we synchronized U2OS cells at the G1/S phase boundary by double thymidine block and collected samples after release into fresh medium over a time course that followed progress through mitosis. The mitotic peak appeared 10 h after release, as indicated by phosphorylation of Histone H3 (Figure 1A). The decreasing level of Aurora A at 12 h confirmed that mitotic exit had begun at this time and was correlated with a sharp increase in the intensity of linkages detected using an antibody specific for K11-linked ubiquitin conjugates (Matsumoto et al., 2010; Figure 1A). All material detected with this antibody in mitotic exit extracts was dependent on UBE2S (Figure 1B and Supplemental Figure S1). We concluded that a dramatic increase in abundance of K11 chains accompanies mitotic exit, whether or not cells previously underwent mitotic arrest.
FIGURE 1:
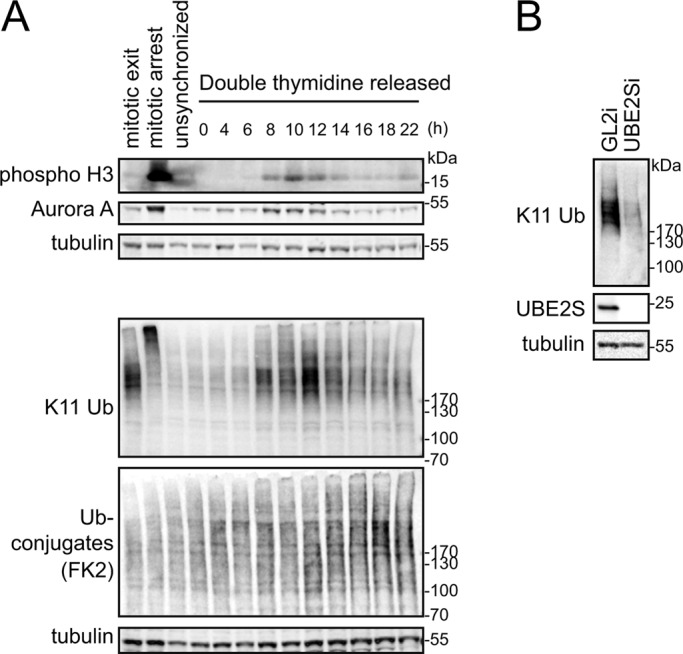
Abundance of K11 linkages increases strongly in unperturbed mitotic exit. (A) U2OS cells were synchronized at G1/S boundary using double thymidine block and then released into fresh medium. Samples taken at the indicated time points were blotted and probed with K11 linkage–specific antibody and other markers. For mitotic control samples, cells from double thymidine block were released into fresh medium containing 10 μM STLC for 16 h. SAC-arrested cells were collected by mitotic shake-off, as “mitotic arrest” sample. AurB inhibitor ZM447439 was used at 10 μM to silence the SAC for 80 min to obtain “mitotic exit” sample. (B) U2OS cells treated with siRNA against GL2 (control) or UBE2S were synchronized at mitotic exit and harvested 44 h after transfection for immunoblotting with antibodies as indicated.
APC/C substrates are modified with K11 ubiquitin linkages during mitotic exit
To investigate the function of K11 linkages during unperturbed mitotic exit, we sought to identify K11 acceptors by testing whether specific known anaphase substrates of APC/C-dependent proteolysis are modified with K11 linkages.
We recently described a sensitive and robust cell-based assay for measuring ubiquitinated fractions of exogenously expressed green fluorescent protein (GFP)–tagged substrates in cells synchronized at mitotic exit, using drug-release protocols that generate a sharp boost in K11 linkages (Supplemental Figure S1). We applied this assay to two well-known anaphase substrates, mitotic kinases Aurora A and Aurora B (AurA and AurB). After purification of AurA- and AurB-Venus from mitotic exit cells, we interrogated samples with linkage-specific K11 antibody and with GFP antibody. K11 linkages were clearly present on both Aurora kinases and abrogated by small interfering RNA (siRNA)–mediated depletion of UBE2S (Ube2Si; Figure 2, A and B). Total ubiquitination on Aurora kinases was only partially affected, with approximately half of polyubiquitin conjugates remaining after Ube2Si (Figure 2, A and C). Ubiquitin conjugates detected on these substrates were specific to mitotic exit (Supplemental Figure S2). We obtained quantitatively comparable results from stable, inducible cell lines (Floyd et al., 2013) and from cells transiently expressing AurA-Venus and AurB-Venus (Figure 2, B and C). Quantifying the total ubiquitin signal as a function of molecular weight as shown in Figure 2D, we observed no apparent decrease in size of substrate–ubiquitin conjugates after Ube2Si. Therefore depletion of UBE2S did not appear to abolish general ubiquitin conjugation in mitotic exit.
FIGURE 2:

K11 chains are built on Aurora kinases at mitotic exit in a UBE2S-dependent manner. U2OS-AurA-Venus cells and U2OS-AurB-Venus cells were transfected with siRNA oligos against GL2 (control) or UBE2S for 44 h. Transfected cells were induced for expression of Venus-tagged substrates for 16 h and synchronized to mitotic exit before harvesting for in vivo ubiquitination assays. (A) K11-specific antibody was used to signal K11 linkage and FK2 antibody for total ubiquitin linkages. GFP antibody recognizes the Venus tag. (B, C) Quantifications of ubiquitin-modified to -unmodified Aurora-Venus measured from A and a further two experiments carried out with cells either inducibly or transiently expressing Aurora-Venus. In each case, the ratio of modified to unmodified substrate was normalized to the corresponding control sample (GL2i). The bar chart shows the mean ± SD of the three independent experiments, showing ratios of K11-specific linkages (B) and total linkages (C). (D) Molecular weight distribution of ubiquitinated Aurora-Venus in control or UBE2S-depleted cells in the blots shown in A. (E) AurA-Venus purified as in A was subjected to DUB “restriction” analysis. For this assay, AurA-Venus on beads was incubated with buffer alone, 1.5 μM USP21, 0.5 μM Cezanne, or 15 μM OTUB1 at 37°C for 30 min as previously described (Mevissen et al., 2013). Arrowheads indicate unspecific staining of USP21 and OTUB1 by the K11 ubiquitin antibody. Quantifications show ubiquitin-modified to -unmodified Aurora-Venus.
We considered the possibility that there might exist compensatory mechanisms of ubiquitination to accommodate cells to UBE2S siRNA treatment—for example, an increase in multiubiquitin versus polyubiquitin conjugates (Dimova et al., 2012), which would mask the loss of UBE2S-dependent K11 linkages. To test this idea, we used ubiquitin chain restriction (UbiCRest) analysis on AurA-Venus purified from untreated cells (Mevissen et al., 2013). Digesting with the non–linkage-specific deubiquitinase (DUB) USP21 abolished all polyubiquitin conjugated to purified AurA-Venus (Figure 2E). Digesting with the K11-specific DUB Cezanne (Bremm et al., 2010) depleted the polyubiquitin fraction in a manner that resembled UBE2Si treatment, with all K11 linkages lost and total ubiquitin staining partially reduced across the entire molecular weight range (Figure 2E). Treatment with Lys-48 (K48)-specific DUB OTUB1 removed a significant fraction of linkages recognized by a K48-specific antibody while having little effect on the amount of K11 linkage detected. For Nek2A-like ’branched’ ubiquitination (Meyer and Rape, 2014), OTUB1 treatment should have decreased the quantity of K11 linkages detected. However, OTUB1 did not decrease K11 chains on AurA, suggesting that such chains are either of a distinct architecture or exist as unbranched chains, although we cannot exclude that OTUB1 fails to hydrolyze K11-modified K48 chains efficiently. In any event, knocking down UBE2S expression faithfully reflects specific removal of K11 linkages from AurA and is a valid route to interfere with K11 linkage formation in mitotic exit cells.
K11 ubiquitin linkages promote substrate degradation in mitotic exit
Next we investigated degradation of individual mitotic exit substrates under conditions of Ube2Si, using live-cell imaging (Clute and Pines, 1999). We chose five substrates that are targeted by APC/C for proteolysis from anaphase onward. These include substrates whose degradation exclusively depends on the coactivator Cdh1, namely AurA, AurB, and Cdc6, as well as substrates that can be targeted by either Cdh1 or Cdc20, namely Plk1 and KIFC1 (Mailand and Diffley, 2005; Floyd et al., 2008; Clijsters et al., 2013; Min et al., 2014).
We expressed fluorescent protein–tagged substrates and traced their levels in single cells in asynchronously proliferating cultures. We then normalized fluorescence levels to anaphase onset and in silico–synchronized cell traces. Knockdown of Ube2S did not alter the timing of anaphase onset (Supplemental Figure S3) or, for each of the substrates analyzed, the timing of degradation with respect to anaphase onset (Figure 3, A and B). However, degradation rates for all substrates were decreased under these conditions (Figure 3, A and B), suggesting that K11 linkages are important in efficient degradation of mitotic exit substrates. Moreover, in the absence of UBE2S, all substrates underwent degradation with similar kinetics despite distinct degradation kinetics under control conditions (Figure 3, C and D), indicating that K11 chains specify accelerated rates of degradation in a substrate-dependent manner.
FIGURE 3:
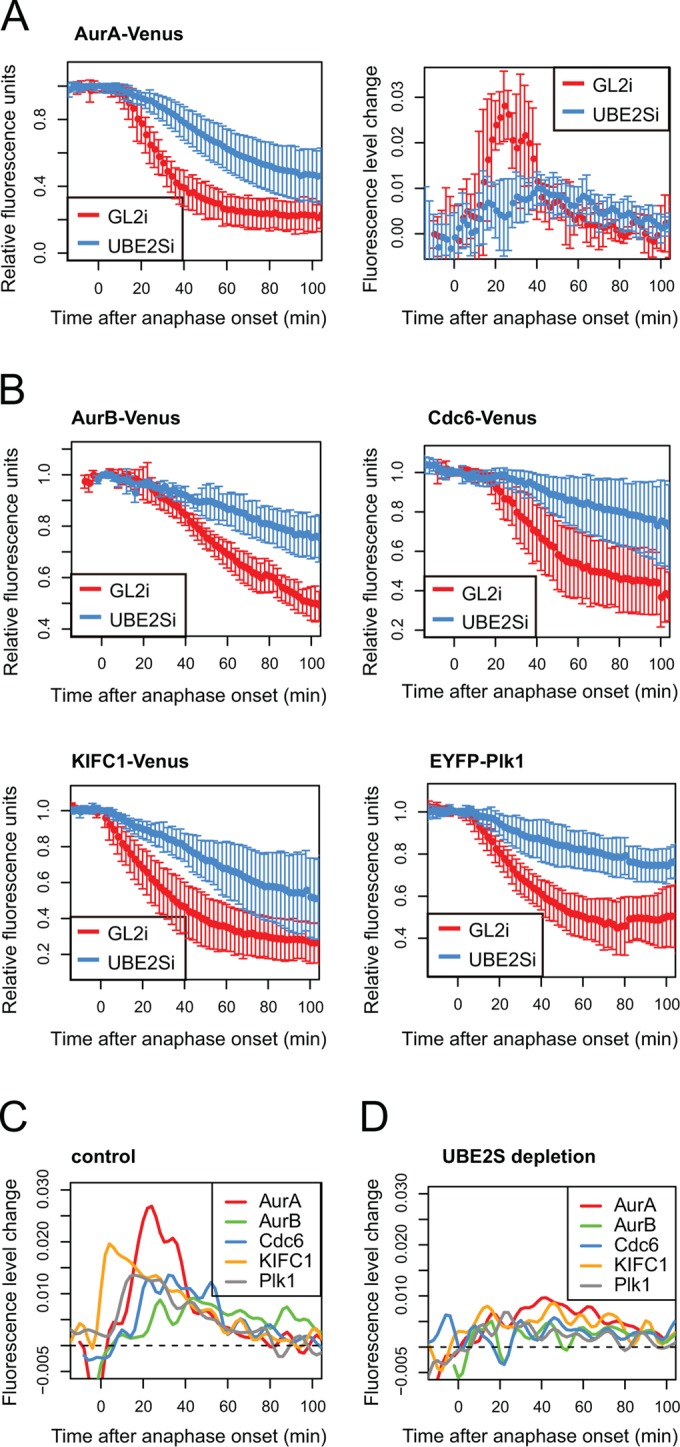
Efficient degradation of mitotic exit substrates requires UBE2S. U2OS cells were transfected with indicated constructs together with control or UBE2S siRNA and imaged by fluorescence time-lapse microscopy from 24 to 48 h after transfection. Fluorescence intensity of Venus over time, in individual mitotic cells, was quantified and plotted as a function of anaphase onset. (A) In vivo degradation curve (left) shows averaged intensities normalized to anaphase ± SD (n ≥ 5); degradation rate curve (right) shows the change in rate over time, derived from degradation curves as described in Min et al. (2013). (B) Degradation curves for indicated substrates under conditions of control (GL2i) or Ube2S knockdown (Ube2Si). (C) Degradation rate curves for mitotic exit substrates under control conditions. (D) Degradation rate curves for mitotic exit substrates after UBE2Si.
Regulation of K11 chain formation by APC/C-Cdh1
Given that the subset of mitotic exit substrates of cellular proteolysis could be major contributors to the K11-linked polyubiquitin detected in Figure 1A, we hypothesized that Cdh1 activity accounted for the dramatic increase in K11 chain assembly at mitotic exit. Indeed, we observed a 60% reduction in K11 linkage after siRNA-mediated Cdh1 depletion (Cdh1i; Figure 4A), supporting the idea that K11 linkages could be the critical output of Cdh1 specificity in substrate degradation. Therefore we interrogated ubiquitin conjugates on individual substrates after Cdh1i. We found that, on the exclusively Cdh1-dependent substrates AurA and AurB (Floyd et al., 2008; Supplemental Figure S4, A–D), Cdh1i treatment abolished K11 chains as effectively as Ube2Si (Figure 4, B–C). By contrast, however, K11 chains on non–Cdh1-dependent substrate KIFC1 (Min et al., 2014; Supplemental Figure S4, E and F) were not largely affected by Cdh1i (Figure 4D).
FIGURE 4:

Regulation of K11 chain assembly by APC/C-Cdh1. (A) U2OS-bioUb cells transfected with siRNA sequence against GL2 (control), Cdh1, or UBE2S were synchronized to mitotic exit. Whole-cell lysate was interrogated by K11 linkage–specific antibody and by biotin antibody to show total ubiquitin conjugates. (B–D) U2OS-bioUb cells were transfected with indicated Venus-tagged constructs together with control (GL2) or Cdh1 or UBE2S siRNA sequence, induced for bioUb expression for 44 h, and synchronized to mitotic exit. Cellular ubiquitination assays were carried out to detect total ubiquitin, K11 linkage, and K48 linkage on each substrate. Bar plot shows mean measurements from three repeats for AurA and AurB and two repeats for KIFC1, with SD plotted where three repeats are available. In vivo degradation assays were carried out in parallel (shown in Supplemental Figure S4), together with representative blots to validate depletions. (E) U2OS-AurA-Venus cells were transfected with indicated siRNAs and synchronized to prometaphase using a sequential thymidine and STLC block. Cells were then released into mitotic exit by treating with 300 nM CDK I/II inhibitor for 45 min before harvesting. Whole-cell lysates (left) or AurA-Venus pull downs (middle) were probed for ubiquitin conjugates. Whole-cell lysates were also tested for knockdown efficacies with indicated antibodies (right).
The coactivation mechanism is proposed to function partly via recruitment of substrates to the APC/C via specific motifs (Barford, 2011) and partly through enhancement of ubiquitination activity mediated by the APC/C, with recent studies attributing the latter role to enhanced E2 efficiency and stabilization of E2-APC/C interactions in the presence of coactivator (Brown et al., 2014; Chang et al., 2014; Kelly et al., 2014; Van Voorhis and Morgan, 2014). Our results confirm that substrate recruitment does not explain the role for Cdh1 in Aurora kinase degradation, since total polyubiquitin associated with Aurora kinases was only reduced by ∼50%, with K48 chains still assembled on these substrates, in the absence of Cdh1 (Figure 4, B and C). Therefore our results indicate that assembly of K11 chains, but not other linkages, correlates with substrate specificity in degradation conferred by Cdh1.
A recent study of coactivator function showed that Cdc20 plays a role in UBE2S activation by stabilizing the interaction between APC/C and UBE2S (Kelly et al., 2014). Cdh1 also binds UBE2S (Williamson et al., 2009), and our findings suggest that Cdh1 may act in a similar way. In the Kelly et al. (2014) study, it was reported that mitotic exit APC/C was less active in stimulating UBE2S activity than APC/CCdc20, based on measurement of UBE2S activity in the presence of Cdh1 and APC/C purified from synchronized cell extracts using APC3 antibody. APC3 is a core APC/C subunit required for cyclin B degradation in metaphase and has been proposed to interact with Cdh1 (Kraft et al., 2005; Chang et al., 2014). Strikingly, we found that depleting APC3 does not abolish assembly of K11 linkage in mitotic exit in the whole-cell lysate or on APC/C substrate AurA (Figure 4E). Therefore APC3 is not required for building K11 chains in mitotic exit. Although unexpected, this result can reconcile the finding of Kelly et al. (2014) that APC3-bound APC/C purified from mitotic exit cells is less active in building K11 linkages than that purified from mitotic-arrested cells with observations that K11 linkages are severalfold more abundant in mitotic exit cells (Figure 1; Matsumoto et al., 2010).
DISCUSSION
In this study, we investigated the function of K11 ubiquitin chains in mitotic exit. We confirmed that K11 ubiquitin linkages dramatically increase in both unperturbed and pharmacologically synchronized mitotic exit in a UBE2S-dependent manner, consistent with the idea that a main cellular function of K11 chain in higher eukaryotes is to regulate mitotic exit. Using UBE2S depletion to specifically interfere with the assembly of K11 chain, we found that it slowed down degradation of all mitotic exit substrates we examined and diminished the kinetic differences in degradation that we observe among different substrates. This correlation is also present in the budding yeast Saccharomyces cerevisiae, in which APC/C and its E2s Ubc4 and Ubc1 assemble K48 chains only (Rodrigo-Brenni and Morgan, 2007) and degradation of a variety of substrates was shown in a recent study to proceed at identical rates (Lu et al., 2014).
From an evolutionary viewpoint, K11 chains in higher eukaryotes may serve to specialize targeting of substrates to allow for more complex regulation of their proteolysis. For example, only one Aurora kinase is present in yeast, whereas there are three in mammals, of which at least two are degraded during mitotic exit. Degradation of AurA starts soon after anaphase onset, and both its degradation and activity are required for assembly of a robust spindle midzone (Floyd et al., 2008; Lioutas and Vernos, 2013; Reboutier et al., 2013). Beyond this point in mitotic exit, AurB activity is retained at the midbody to surveil abscission (Steigemann et al., 2009) but otherwise needs to be degraded to promote respreading of the cell after mitosis (Floyd et al., 2013). These distinct roles in mitotic exit require that AurA and AurB disappear from the cell at different times. K11 linkages could therefore contribute to the temporal organization of substrate degradation by specifying different degradation kinetics.
Our investigation of the role of Cdh1 in K11 chain assembly revealed a correlation between assembly of K11 chains and substrate specificity in degradation conferred by Cdh1, suggesting that K11 linkages may play a role in passaging information from APC/C coactivators to substrate degradation. How would the process of stimulating K11 chain assembly couple with the substrate selectivity of Cdh1? K48 chains can still be assembled on Aurora kinases upon Cdh1 depletion, implying that these substrates are able to receive K48-linked ubiquitin chains from Cdc20-activated APC/C. Indeed, Cdc20 and Cdh1 can both bind to the canonical D-box and KEN-box motifs (Barford, 2011). In this case, binding of substrate to the APC/C per se may not be the only step conferring substrate selectivity: it was recently shown that binding of coactivator increases the affinity of APC/C for an E2 (Chang et al., 2014) and, of importance, that in the case of the S. cerevisiae E2 Ubc4, Cdh1-bound substrate further enhances E2 activity (Van Voorhis and Morgan, 2014). Therefore substrate-specific stimulation may promote coactivator-dependent recruitment of UBE2S. The study from Chang et al. (2014) lends support to this idea by showing that Cdh1 binding induces displacement and flexibility of the APC/C catalytic module APC2-APC11. By this account, coactivator specificity in substrate degradation, mediated by K11 linkages, would arise from coactivator-dependent positioning of the K11-specific machinery at the right proximity to substrates.
We also showed that, unexpectedly, APC3—an APC/C core subunit proposed to recruit coactivator Cdh1 and Cdc20 (Kraft et al., 2005)—plays a less critical role in K11 chain assembly in mitotic exit than Cdh1. APC/C is a highly dynamic enzyme that is itself intensively regulated by phosphorylation-dependent interactions. For example, APC3 is required for Cdc20 binding in metaphase but not prometaphase (Izawa and Pines, 2011). A recent high-resolution structural study indicated that whereas the IR tail of Cdh1 binds to the tetratricopeptide repeat domain (TPR) of APC3, the Cdh1 N-terminus interacts with APC8, APC6, and APC1 (Chang et al., 2014). This multivalent interaction may constitute a state space within which different combinations of binary interactions define distinct functional outputs (e.g., interaction with APC3 is not required for building K11 chains). Alternatively, because the TPR lobe of APC/C contains eight TPRs, including APC3, the high density of these replicate motifs provides a possibility that Cdh1 can plug into alternative positions on the core APC/C. Resolving these ideas will require biochemical studies from highly synchronized cell populations. Nevertheless, our result suggests that in mitotic exit, binding to APC3 is not required for Cdh1 to support K11 chain assembly.
Our studies demonstrate the importance of investigating ubiquitination in a cellular context. Although in vitro studies have generated extensive knowledge of the versatile enzyme activities in the UPS, tracking their activity in cells can reveal unexpected details on how they contribute to cellular functions.
MATERIALS AND METHODS
Cell culture and synchronization
U2OS-bioUb cells and U2OS parental cells were described in Min et al. (2014) and U2OS-AurB-Venus in Floyd et al. (2013). A tetracycline-regulated U2OS-AurA-Venus cell line was created from the same parental line as the U2OS-AurB-Venus cells, using pTRE-AurA-Venus plasmid and established procedures (Floyd et al., 2013). All U2OS cells were cultured in high-glucose DMEM (Life Technologies, Carlsbad, CA). Cell culture medium was supplemented with fetal bovine serum (10%), penicillin-streptomycin, amphotericin B, 500 μg/ml Geneticin (all from PAA Laboratories, Pasching, Austria) and 1 μg/ml tetracycline hydrochloride (Calbiochem, San Diego, CA). Tetracycline was removed from the medium to induce expression of the corresponding construct. Synchronization at mitotic exit was achieved using sequential treatments with thymidine (20 h, 2.5 mM) and KIF11/Eg5 inhibitor S-trityl-l-cysteine (STLC; 16 h, 10 μM) after 3-h release from thymidine. Prometaphase cells were collected by mitotic shake-off and forced into mitotic exit by silencing the SAC using AurB inhibitor ZM447439 (10 μM) for 70 min, unless specified otherwise. For the double thymidine synchronization experiment, cells were first treated with 2.5 mM thymidine for 20 h and then released into fresh medium for 12 h. Then 2.5 mM thymidine was added into the culture for 12 h before release again.
Plasmids, siRNA, and transfection
pVenus-AurB (Floyd et al., 2013), pEYFP-Plk1 (Lindon and Pines, 2004), pVenus-AurA, and pVenus-KIFC1 (Min et al., 2014) were as described before. Cloning details for pTRE-AurA-Venus are available on request. pVenus-Cdc6 was a kind gift from Rob Wolthuis (VUmc Medical Faculty, Amsterdam, The Netherlands; Clijsters et al., 2013). siRNA sequences targeting APC3, Cdh1, and UBE2S were as previously described (Floyd et al., 2008; Garnett et al., 2009; Izawa and Pines, 2011). Plasmids and siRNAs were electroporated into cells using the Neon transfection system (Life Technologies) using a program with voltage 1150 V, width 30 ms, and two pulses. Each transfection was carried out with 2 × 106 to 1 × 107 cells with 5 μg of plasmid per 100 μl of transfection or 1 μM siRNA oligo in the transfection suspension, respectively.
Cellular ubiquitination assay
To prepare one GFP-Trap pull-down sample for immunoblotting, ∼5 × 106 cells expressing a Venus-tagged substrate were lysed in 100 μl of lysis buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% [vol/vol] Triton, 1× protease inhibitor cocktail [Roche Diagnostics GmbH, Mannheim, Germany], 1 mM NaF, 1 mM Na3VO4, and 50 mM N-ethylmaleimide). After dilution with 900 μl of dilution buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.5 mM EDTA, 1× protease inhibitor cocktail, 1 mM NaF, 1 mM Na3VO4, and 50 mM N-ethylmaleimide), the lysate was cleared by centrifugation, applied to 5 μl of prewashed GFP-Trap_A beads (Chromotek GmbH, Martinsried, Germany), and incubated for 2 h at room temperature. The beads were then washed with 8 M urea, 1% (wt/vol) SDS for 1 min, 10 mM Tris-HCl, pH 7.6, 1 M NaCl, 0.5 mM EDTA, 1% (vol/vol) Triton for 3 × 5 min on a roller, and briefly rinsed in 1% (wt/vol) SDS. The beads were then boiled in 5 μl of 4× sample buffer (Life Technologies) containing 100 mM dithiothreitol for 10 min at 95°C, and the eluted sample was separated from the beads by centrifugation at 14,000 × g. Blots were probed with GFP antibody (with an IRDye fluorescent secondary antibody [LI-COR Biosciences Ltd., Cambridge, UK]) and indicated ubiquitin antibodies (with a horseradish peroxidase–linked secondary antibody) for quantitative detection of unmodified and ubiquitinated Venus-tagged substrate, respectively, using a Li-Cor Odyssey Fc. Ubiquitination levels are indicated by the ratio of ubiquitin antibody intensity to GFP antibody intensity.
Ubiquitin smear distribution profiling
The indicated ubiquitin blots were exported from Li-Cor Image studio as a nonsaturated 8-bit image. For each lane, three line scans (plot profiles) were measured in ImageJ (National Institutes of Health, Bethesda, MD). The averaged profiles were generated in R using an in-house script.
In vivo degradation assay
Imaging and analysis were performed as described before (Min et al., 2013, 2014).
Antibodies
Table 1 shows the antibodies used in this study.
Table 1:
Antibodies used in this study.
| Antibody | Source | Dilution for GFP-Trap purified protein | Dilution for whole-cell lysate |
|---|---|---|---|
| Actin | Sigma-Aldrich (A 3853) | 1:1000 | |
| APC3 | Abcam (ab10538) | 1:1000 | |
| Aurora A | BD Biosciences (610939) | 1:1000 | |
| Biotin | Cell Signaling (7075) | 1:100 | 1:1000 |
| Cdh1 | Tim Hunt (Clare Hall Laboratories, London Research Institute, South Mimms, United Kingdom) | 1:50 | |
| FK2 | Enzo (PW0150) | 1:600 | 1:2000 |
| GFP | Abcam (ab290) | 1:2000 | |
| GFP | Roche (11814460001) | 1:500 | |
| K11 | Millipore (MABS107-I) | 1:100–1:500 | 1:1000 |
| K48 | Abcam (ab140601) | 1:500 | |
| Phospho–Histone H3 | Cell Signaling | 1:1000 | |
| Tubulin | Abcam | 1:2000 | |
| UBE2S | Abnova (PAB1701) | 1:1000 |
Supplementary Material
Acknowledgments
We thank Chiara Marcozzi and Ben Kinnersley for help in setting up Aurora B degradation assays and Rob Wolthuis for Cdc6-Venus. Yu Ye and Rhys Grant contributed valuable discussions during the course of this study. Work in C.L.’s lab was funded by the Medical Research Council (G120/892), Cancer Research UK (C3/A10239), and the Department of Genetics. Work in D.K.’s lab was funded by the Medical Research Council (U105192732), the European Research Council (309756), and the Lister Institute for Preventive Medicine. M.M. was supported by the Great Britain China Centre Educational Trust and the Henry Lester Trust. T.M. was funded by the Marie Curie Initial Training Network UPStream.
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- DUB
deubiquitinase
- SAC
spindle assembly checkpoint
- UPS
ubiquitin-proteasome system.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-02-0102) on October 7, 2015.
REFERENCES
- Barford D. Structure, function and mechanism of the anaphase promoting complex (APC/C) Q Rev Biophys. 2011;44:153–190. doi: 10.1017/S0033583510000259. [DOI] [PubMed] [Google Scholar]
- Bremm A, Freund SM, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol. 2010;17:939–947. doi: 10.1038/nsmb.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremm A, Komander D. Emerging roles for Lys11-linked polyubiquitin in cellular regulation. Trends Biochem Sci. 2011;36:355–363. doi: 10.1016/j.tibs.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Brown NG, Watson ER, Weissmann F, Jarvis MA, VanderLinden R, Grace CR, Frye JJ, Qiao R, Dube P, Petzold G, et al. Mechanism of polyubiquitination by human anaphase-promoting complex: RING repurposing for ubiquitin chain assembly. Mol Cell. 2014;56:246–260. doi: 10.1016/j.molcel.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LF, Zhang ZG, Yang J, McLaughlin SH, Barford D. Molecular architecture and mechanism of the anaphase-promoting complex. Nature. 2014;513:388–393. doi: 10.1038/nature13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clijsters L, Ogink J, Wolthuis R. The spindle checkpoint, APC/CCdc20, and APC/CCdh1 play distinct roles in connecting mitosis to S phase. J Cell Biol. 2013;201:1013–1026. doi: 10.1083/jcb.201211019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- Dimova NV, Hathaway NA, Lee BH, Kirkpatrick DS, Berkowitz ML, Gygi SP, Finley D, King RW. APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat Cell Biol. 2012;14:168–176. doi: 10.1038/ncb2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd S, Pines J, Lindon C. APC/C Cdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr Biol. 2008;18:1649–1658. doi: 10.1016/j.cub.2008.09.058. [DOI] [PubMed] [Google Scholar]
- Floyd S, Whiffin N, Gavilan MP, Kutscheidt S, De Luca M, Marcozzi C, Min M, Watkins J, Chung K, Fackler OT, Lindon C. Spatiotemporal organization of Aurora-B by APC/CCdh1 after mitosis coordinates cell spreading through FHOD1. J Cell Sci. 2013;126:2845–2856. doi: 10.1242/jcs.123232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, Pines J, Venkitaraman AR. UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol. 2009;11:1363–1369. doi: 10.1038/ncb1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa D, Pines J. How APC/C-Cdc20 changes its substrate specificity in mitosis. Nat Cell Biol. 2011;13:223–233. doi: 10.1038/ncb2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Wickliffe, Katherine E, Song L, Fedrigo I, Rape M. Ubiquitin chain elongation requires E3-dependent tracking of the emerging conjugate. Mol Cell. 2014;56:232–245. doi: 10.1016/j.molcel.2014.09.010. [DOI] [PubMed] [Google Scholar]
- Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, Peters J-M. The \WD40\ propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol Cell. 2005;18:543–553. doi: 10.1016/j.molcel.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Lee S, Ramirez J, Franco M, Lectez B, Gonzalez M, Barrio R, Mayor U. Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell Mol Life Sci. 2014;71:2747–2758. doi: 10.1007/s00018-013-1526-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon C, Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol. 2004;164:233–241. doi: 10.1083/jcb.200309035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioutas A, Vernos I. Aurora A kinase and its substrate TACC3 are required for central spindle assembly. EMBO Rep. 2013;14:829–836. doi: 10.1038/embor.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Hsiao JY, Davey NE, Van Voorhis VA, Foster SA, Tang C, Morgan DO. Multiple mechanisms determine the order of APC/C substrate degradation in mitosis. J Cell Biol. 2014;207:23–39. doi: 10.1083/jcb.201402041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122:915–926. doi: 10.1016/j.cell.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D, Phu L, Kirkpatrick DS, Hymowitz SG, Rape M, et al. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell. 2010;39:477–484. doi: 10.1016/j.molcel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Mevissen TET, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H-J, Rape M. Enhanced protein degradation by branched ubiquitin chains. Cell. 2014;157:910–921. doi: 10.1016/j.cell.2014.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, Lindon C. Substrate targeting by the ubiquitin-proteasome system in mitosis. Semin Cell Dev Biol. 2012;23:482–491. doi: 10.1016/j.semcdb.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Min M, Mayor U, Dittmar G, Lindon C. Using in vivo biotinylated ubiquitin to describe a mitotic exit ubiquitome from human cells. Mol Cell Proteomics. 2014;13:2411–2425. doi: 10.1074/mcp.M113.033498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, Mayor U, Lindon C. Ubiquitination site preferences in anaphase promoting complex/cyclosome (APC/C) substrates. Open Biol. 2013:3, 130097. doi: 10.1098/rsob.130097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol. 2011;12:427–438. doi: 10.1038/nrm3132. [DOI] [PubMed] [Google Scholar]
- Reboutier D, Troadec M-B, Cremet J-Y, Chauvin L, Guen V, Salaun P, Prigent C. Aurora A is involved in central spindle assembly through phosphorylation of Ser 19 in P150Glued. J Cell Biol. 2013;201:65–79. doi: 10.1083/jcb.201210060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo-Brenni MC, Morgan DO. Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell. 2007;130:127–139. doi: 10.1016/j.cell.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MHA, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Van Voorhis Vanessa A, Morgan David O. Activation of the APC/C ubiquitin ligase by enhanced E2 efficiency. Curr Biol. 2014;24:1556–1562. doi: 10.1016/j.cub.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell. 2011a;144:769–781. doi: 10.1016/j.cell.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Williamson A, Meyer HJ, Kelly A, Rape M. K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol. 2011b;21:656–663. doi: 10.1016/j.tcb.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, Rape M. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci USA. 2009;106:18213–18218. doi: 10.1073/pnas.0907887106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Merbl Y, Huo Y, Gallop JL, Tzur A, Kirschner MW. UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc Natl Acad Sci USA. 2010;107:1355–1360. doi: 10.1073/pnas.0912802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.