

Oligomerization or homotypic clustering diverts Golgi membrane proteins into the canonical GGA1/clathrin-dependent Golgi-to-lysosome pathway revealing the presence of cellular quality control that could be useful for therapies designed to down-regulate specific proteins in the secretory pathway.
Abstract
Manganese protects cells against forms of Shiga toxin by down-regulating the cycling Golgi protein GPP130. Down-regulation occurs when Mn binding causes GPP130 to oligomerize and traffic to lysosomes. To determine how GPP130 is redirected to lysosomes, we tested the role of GGA1 and clathrin, which mediate sorting in the canonical Golgi-to-lysosome pathway. GPP130 oligomerization was induced using either Mn or a self-interacting version of the FKBP domain. Inhibition of GGA1 or clathrin specifically blocked GPP130 redistribution, suggesting recognition of the aggregated GPP130 by the GGA1/clathrin-sorting complex. Unexpectedly, however, GPP130’s cytoplasmic domain was not required, and redistribution also occurred after removal of GPP130 sequences needed for its normal cycling. Therefore, to test whether aggregate recognition might be a general phenomenon rather than one involving a specific GPP130 determinant, we induced homo-oligomerization of two unrelated Golgi-targeted constructs using the FKBP strategy. These were targeted to the cis- and trans-Golgi, respectively, using domains from mannosidase-1 and galactosyltransferase. Significantly, upon oligomerization, each redistributed to peripheral punctae and was degraded. This occurred in the absence of detectable UPR activation. These findings suggest the unexpected presence of quality control in the Golgi that recognizes aggregated Golgi proteins and targets them for degradation in lysosomes.
INTRODUCTION
The Golgi membrane protein GPP130 normally cycles between its steady-state location in the cis-Golgi and endosomes (Puri et al., 2002). An increase in Golgi manganese causes it to leave this itinerary and instead traffic to lysosomes, where it is degraded (Mukhopadhyay et al., 2010). Although the role of GPP130 is unknown, this trafficking switch is fortuitous because the bacterially produced Shiga toxin and Shiga toxin 1 (STx/STx1), which cause severe diarrhea and fatal kidney failure (Beddoe et al., 2010), depend on GPP130 for their invasion of host cells (Natarajan and Linstedt, 2004; Mukhopadhyay and Linstedt, 2012). The toxins bind GPP130 in endosomes and piggyback a ride to the Golgi, which allows them to escape degradation in the lysosome (Mukhopadhyay and Linstedt, 2012; Mukhopadhyay et al., 2013). Indeed, Mn-induced down-regulation of GPP130 prevents STx/STx1 trafficking, and Mn protects cells and mice against Shiga toxicosis (Mukhopadhyay and Linstedt, 2012).
The switch in sorting fate of GPP130 caused by Mn is an instance of an important general regulatory mechanism in which sorting during membrane trafficking is used as a control point. The best-known examples of this type of sorting switch are those induced by ligand binding to cell surface receptors. Ligand binding induces conformational changes and/or posttranslational modifications of cytoplasmically exposed sorting signals. This alters the interaction of these signals with vesicle coat adaptors driving endocytosis (Traub and Bonifacino, 2013; Piper et al., 2014). A related scenario takes place when cholesterol regulates the endoplasmic reticulum (ER) exit of the sterol regulatory element–binding protein (SREBP) cleavage–activating protein (SCAP)/SREBP complex. Cholesterol binding changes SCAP’s conformation, leading to binding of another factor that masks SCAP’s sorting signal for ER exit (Brown and Goldstein, 1997; Sun et al., 2007). There are also disease-related examples. Human immunodeficiency virus type 1 protein Nef alters the sorting of the major histocompatibility complex-1 by mediating the indirect interaction of MHC1 with the AP-1 adaptor protein at the trans-Golgi network (TGN; Le Gall et al., 1998). Mutation of the amyloid precursor protein in Alzheimer’s disease blocks its interaction with the adaptor AP-4 at the TGN, resulting in greater cleavage to the pathogenic amyloid-β peptide (Roeth et al., 2004). Control of sorting is also a critical aspect of metal ion homeostasis, and this may be the proper context in which to place Mn control of GPP130 trafficking. Similar to Mn-induced GPP130 trafficking, elevated copper causes redistribution of the Golgi proteins ATP7A and ATP7B, which are copper-translocating pumps. The pumps move from the TGN to the cell surface, where they expel excess copper (Polishchuk and Lutsenko, 2013).
For each case of regulated sorting, understanding the underlying mechanism responsible for enrichment of the sorted membrane protein is critical to elucidating the switch. As in several of the foregoing examples, local enrichment is typically achieved by direct or adaptor-mediated interactions of the sorted cargo with an assembling vesicle coat complex, resulting in selective export in a budding vesicle or membrane tubule (Bonifacino and Glick, 2004; Traub and Bonifacino, 2013). Alternatively, local enrichment may be driven by accumulation in particular lipid domains (Surma et al., 2012) or interaction with localized domains of assembled cytoskeleton (Irannejad and von Zastrow, 2014).
Significantly, recent work indicates that the switch in GPP130 sorting may involve a novel mechanism for its local enrichment in the plane of the membrane. When Mn is elevated, the GPP130 lumenal stem domain binds Mn, and this binding causes the GPP130 oligomerization/aggregation that is required and sufficient for its altered sorting (Tewari et al., 2014). Here we sought to extend our understanding of this pathway by answering the following questions. Is a TGN-localized vesicle coat complex adaptor involved? If so, are specific sequence elements in GPP130 required, or is this sorting switch something more general such that it acts on any oligomerized/aggregated Golgi protein? Our work implicates γ-adaptin homologous ARF–interacting protein 1 (GGA1) and clathrin in a sorting switch that appears general to Golgi proteins.
RESULTS
Role of the canonical Golgi–lysosome pathway in oligomerization-induced trafficking
Manganese-induced GPP130 trafficking involves redistribution to multivesicular bodies (MVBs), where GPP130 is internalized to intraluminal vesicles and then appearance in LAMP2 positive lysosomes, where its degradation is blocked by leupeptin inhibition of lysosomal hydrolases (Mukhopadhyay et al., 2010). The redistribution is insensitive to a dominant-negative (DN) version of dynamin, microtubule disruption or DN-Rab5 indicating that neither endocytosis nor early endosome trafficking is involved (Mukhopadhyay et al., 2010). In contrast, it is blocked by DN-Rab7-T22N (Mukhopadhyay et al., 2010), which inhibits trafficking from MVBs to lysosomes (Bucci et al., 2000; Vanlandingham and Ceresa, 2009). The canonical route from the Golgi toward lysosomes is mediated by Golgi-associated GGA1 and clathrin (Black and Pelham, 2000; Puertollano et al., 2001b; Zhu et al., 2001). Thus we used a DN version of GGA1 and a small-molecule inhibitor of clathrin to test whether the oligomerization-induced trafficking of GPP130 occurs via this canonical route.
For GGA1, HeLa cells were transfected with either a wild-type version or a version lacking the clathrin-binding domains (GGA1-DN), which is known to block export of lysosome-directed cargo from the TGN (Puertollano et al., 2001b). The constructs were green fluorescent protein (GFP) tagged, and each was Golgi localized upon expression (Figure 1A, rows 1 and 2). Cells were then treated with Mn for 5 h, and GPP130 redistribution was determined. In cells expressing the wild-type GGA1, GPP130 moved from its predominant Golgi localization to peripheral punctae (Figure 1A, row 3), indicating movement to lysosomes, as previously described (Mukhopadhyay et al., 2010). Note that there may have been some interference by the wild-type GGA1 construct. Although the number of punctae arising in Mn-treated untransfected cells (Mukhopadhyay et al., 2010; Tewari et al., 2014) was essentially identical to that in GGA1-transfected cells, loss of GPP130 from the Golgi was slower in GGA1-expressing cells, suggesting that GGA1 overexpression may have affected exit. In contrast, cells expressing GGA1-DN showed little or no GPP130 redistribution (Figure 1A, row 4). (Also note that thresholding of the GPP130 channel [last column] is presented to illustrate the peripheral punctae, which are dim due to GPP130 degradation. Identical parameters were used for thresholding and all other analyses within an experiment to allow direct comparison.)
FIGURE 1:

Dominant-negative GGA1 blocks Mn-induced GPP130 trafficking. (A) Cells were transfected with GFP-tagged GGA1 or GGA1-DN and then either left untreated (Ø) or treated with Mn for 5 h. Images show GFP fluorescence from the transfected proteins and anti-GPP130 staining, with the final panel using uniform thresholding of the GPP130 image to aid visualization of small punctae. Note that GGA1-DN blocked GPP130 appearance in punctae. Bar, 5 μm. (B) Cells expressing GGA1 or GGA1-DN were treated with monensin for 30 min to induce GPP130 trafficking to endosomes. (C) Quantified appearance of GPP130 in peripheral punctae of cells expressing GGA1 or GGA1-DN after Mn or monensin (Mon) treatment (mean ± SEM, n = 3, >10 cells/experiment, *p < 0.05). (D) Total cell fluorescence of GPP130 in cells expressing GGA1 or GGA1-DN before or after Mn (mean ± SEM, n = 3, >10 cells/experiment).
Monensin treatment provided a convenient control to test the specificity of the GGA1 inhibition. Monensin is a proton ionophore that neutralizes acidic compartments (Tartakoff, 1983), causing GPP130 redistribution to endosomes presumably because GPP130 endosome-to-Golgi retrieval is pH dependent (Linstedt et al., 1997; Starr et al., 2007). Thus TGN export of GPP130 upon monensin treatment is expected to occur via a distinct pathway from lysosome-directed cargo and occurs independently of GGA1. Indeed, the marked appearance of GPP130 in peripheral endosomes after monensin addition was unabated by expression of either wild-type or GGA1-DN (Figure 1B).
The specific dependence of Mn-induced GPP130 redistribution on GGA1 was confirmed by image analysis for independent experiments quantifying the number of distinct GPP130 punctae per cell. A significant block was evident for GGA1-DN after Mn treatment but not after monensin treatment (Figure 1C). Further, the loss of total GPP130 fluorescence per cell after Mn treatment, which is a measure of its degradation in lysosomes (Mukhopadhyay et al., 2010), was abrogated in cells expressing GGA1-DN (Figure 1D).
Given these results for Mn-induced GPP130 redistribution, we also tested the role of GGA1 for GPP130-FM, a previously characterized construct (Tewari et al., 2014) in which GPP130 residues 1–247 are followed by three copies of a modified version of the FKBP12 domain termed FM (after its F36M substitution). The FM domain self-interacts in a manner that is inhibited by the drug AP21998 (AP), an analogue of rapamycin. Washout of AP forms large complexes due to multiplexing of the three FM domains, and, upon AP washout, GPP130-FM leaves the Golgi, redistributes to peripheral puncta, and is degraded over time (Tewari et al., 2014). As shown, many of the puncta costain with rab7, a marker of late endosomes (Figure 2, A and B), and degradation, which occurs over ∼5 h (Figure 2, C and D), is blocked by lysosomal inhibitors (Figure 2, E and F). Thus GPP130-FM redistributes to endosomes and lysosomes, where it is degraded. As expected, neither GGA1 nor GGA1-DN altered GPP130-FM localization before AP washout (Figure 3, rows 1 and 2), and redistribution of GPP130-FM was readily apparent in cells expressing wild-type GGA1 (Figure 3, row 3). Of interest, GGA1 partially redistributed along with GPP130-FM, consistent with the possible persistence of a GGA1-based sorting complex associated with GPP130-FM beyond the TGN. In contrast, in cells expressing GGA1-DN the appearance of GPP130-FM in peripheral punctae was significantly reduced (Figure 3, row 4). The inhibitory effect of GGA1-DN was significant (Figure 3B).
FIGURE 2:

Lysosomal redistribution of GPP130-FM. (A) Cells were cotransfected with HA-tagged GPP130-FM and GFP-tagged Rab7 and then treated with a 1-h AP washout. Images compare GFP fluorescence from the transfected proteins and anti-HA staining, with the indicated regions expanded and presented as insets. At the 1-h time point, cells vary in their extent of residual Golgi-localized GPP130-FM. To better illustrate our analysis, the cell shown here has mostly punctae-localized GPP130-FM but equivalent results were obtained in all cells. Arrowheads mark examples of costained punctae. Bars, 10 μm. (B) The percentage of total GPP130-FM punctae counted that were positive for Rab7 is quantified for the indicated time points (mean ± SEM, n = 3, >10 cells/experiment, p < 0.05 for 0 h vs 1 or 5 h). (C, D) Blot and quantification showing level of GPP130-FM in cell extracts prepared at the indicated times after AP washout with tubulin as a loading control (mean ± SEM, n = 3). (E, F) Blot and quantification showing level of GPP130-FM in cell extracts prepared after either 0 or 5 h of AP washout or 24 h of pretreatment with pepstatin and leupeptin (+inh) followed by 5 h of AP washout (mean ± SEM, n = 3).
FIGURE 3:

GGA1-DN blocks GPP130-FM trafficking. (A) Cells were cotransfected with HA-tagged GPP130-FM and either GFP-tagged GGA1 or GGA1-DN and then either left in AP or treated with a 1 h of AP washout. Images show GFP fluorescence from the transfected proteins and anti-HA staining, with the final panel using uniform thresholding of the GPP130-FM image to aid visualization of small punctae. Bar, 5 μm. (B) Quantified appearance of GPP130 in peripheral punctae of cells expressing GGA1 or GGA1-DN after 0 or 1 h of AP washout (mean ± SEM, n = 3, >10 cells/experiment, *p < 0.05).
Dependence on GGA1 implies a role for clathrin. As a test, we used treatment with Pitstop2, a selective clathrin inhibitor. Pitstop2 occupies the clathrin terminal domain groove, preventing this domain’s interaction with clathrin box ligands, which stalls clathrin-coated-pit dynamics (von Kleist et al., 2011). Only short-term treatments were used to minimize indirect and toxic effects of the drug. HeLa cells expressing GPP130-FM were transferred to serum-free medium (serum interferes with Pitstop2) in the continued presence of AP. Then the washout of AP was carried out for 10 min with medium containing either dimethyl sulfoxide (DMSO) alone or DMSO containing Pitstop2. As expected, the 10-min washout yielded only a partial redistribution in control cells, but the peripheral punctae were clearly evident. In contrast, no such redistribution occurred in cells treated with the clathrin inhibitor, and this was confirmed by quantification (Figure 4). Taken together, the data in this section indicate that trafficking of oligomerized GPP130 to lysosomes requires TGN export that is GGA1 and clathrin dependent.
FIGURE 4:

Clathrin inhibitor blocks oligomer-induced GPP130 trafficking. (A) GPP130-FM– transfected cells in the presence of AP were washed into serum-free medium lacking AP and containing either vehicle alone (DMSO) or vehicle containing the clathrin inhibitor Pitstop2. The cells were fixed and analyzed after 10 min to minimize indirect and toxic effects of Pitstop2. Images show GFP fluorescence from GPP130-FM and anti-giantin staining to mark the Golgi, as well as thresholded GPP130 images to mark cytoplasmic punctae. Note that Pitstop2 blocked GPP13-FM appearance in punctae. Bar, 5 μm. (B) Quantified appearance of GPP130-FM in peripheral punctae of control and Pitstop2-treated cells after AP washout (mean ± SEM, n = 3, >10 cells/experiment, *p < 0.05).
Sorting signals for oligomerization-induced GPP130 trafficking?
GGA1 functions as a coat complex adaptor by interacting with cytoplasmic motifs in diverse cargo (Puertollano et al., 2001b; He et al., 2002). Although GPP130 has a complex trafficking itinerary that involves multiple distinct domains, its cytoplasmic domain has not yet been implicated in its targeting. Because it is not needed for Golgi targeting, we could test the role of the cytoplasmic domain in oligomerization-induced trafficking of GPP130. We deleted residues 2–11 of the 12-residue cytoplasmic domain in our FM-domain GPP130 construct to create ∆cyto-GPP130-FM. As expected, the construct was localized to the Golgi in the continued presence of AP, whereas after AP washout, ∆cyto-GPP130-FM redistributed to peripheral punctae in a manner indistinguishable from wild type (Figure 5A). Image analysis confirmed the significant increase in punctae (Figure 5B), as well as the loss of total staining over time, reflecting degradation (Figure 5C). Thus the cytoplasmic domain of GPP130 is not needed for its oligomerization-induced trafficking.
FIGURE 5:

Oligomer-induced GPP130 trafficking is independent of its cytoplasmic domain. (A) Cells were transiently transfected with ∆cyto-GPP130-FM in the presence of AP and then examined at the indicated times after AP washout. Images show GFP fluorescence from ∆cyto-GPP130-FM and anti-giantin staining to mark the Golgi. The final panels present the GPP130 images after uniform thresholding. Note redistribution of ∆cyto-GPP130-FM. Bar, 5 μm. (B) Quantified appearance of ∆cyto-GPP130-FM in peripheral punctae at 0 and 1 h after AP washout (mean ± SEM, n = 3, >10 cells/experiment, *p < 0.01). (C) Determination of total fluorescence per cell after AP washout normalized to average value before AP washout (mean ± SEM, n = 3, >10 cells/experiment, error bars too small to be seen).
Next we turned our attention to the GPP130 lumenal coiled-coil domain, which contains sequence elements that independently mediate Shiga toxin binding and Mn binding, as well as Golgi targeting and endosome cycling (Bachert et al., 2001; Mukhopadhyay et al., 2010; Tewari et al., 2014). The entire coiled-coil stretches from residue 36 to 245. Previous work indicated that a deletion of 88–245 shifts the steady-state location of the protein to the trans-Golgi, blocks its cycling to endosomes, and blocks its Mn responsiveness (Bachert et al., 2001; Mukhopadhyay et al., 2010). Therefore we tested a GPP130-1-87-FM construct to see whether it retained the oligomerization-induced trafficking response. The construct was Golgi localized before AP washout, and, upon washout, it moved into peripheral punctae, ultimately undergoing degradation (Figure 6A). Also shown are the quantified results for both its appearance in peripheral punctae (Figure 6B) and its loss of staining over time (Figure 6C). Thus, even without most of its coiled-coil stem domain, GPP130 underwent oligomerization-induced trafficking and degradation. We reasoned that further deletions might be problematic due to the requirement for Golgi targeting in any construct to be tested. Also, we began to consider the possibility that the response might not be specific to GPP130 but instead part of a general pathway in the Golgi. Therefore we next tested the behavior of cis- and trans-Golgi markers after their forced oligomerization.
FIGURE 6:

Redistribution of truncated GPP130-FM. (A) Cells expressing GPP130-1-87-FM (cycling deficient and Mn insensitive) were incubated in the absence of AP for the indicated times. Images show thresholded GFP fluorescence from GPP130-1-87-FM and anti-giantin staining to mark the Golgi. Note redistribution and then loss of GPP130-1-87-FM. Bar, 5 μm. (B) Quantified appearance of GPP130-1-87-FM in peripheral punctae after AP washout (mean ± SEM, n = 3, >10 cells/experiment). (C) Total GPP130-1-87-FM or giantin fluorescence as a percentage of starting value and at the indicated times of AP washout (mean ± SEM, n = 3, >10 cells/experiment, error bars too small to be seen).
Induced oligomerization causes lysosomal targeting of other Golgi proteins
We first studied the cis-Golgi enzyme mannosidase-1 because a suitable construct (mannosidase-1 [Man1]–FM) had already been characterized in which tandem copies of the FM domain and a hemagglutinin (HA) tag were appended to the C-terminus of a truncated mouse α-1,2 mannosidase-1B composed of its cytoplasmic, transmembrane, and stem domains (Rizzo et al., 2013). The construct shows normal cis-Golgi localization in the presence of AP and migration to the trans-Golgi after a short AP washout (Rizzo et al., 2013).
In our experiments, HeLa cells were transfected with the Man1-FM construct in the presence of AP. After 24 h, the AP was removed, and the construct’s localization was determined over time using anti-HA antibodies. As expected, in the presence of AP, Man1-FM was strongly localized to the Golgi, as indicated by comparing its staining pattern in the same cells with that of the cis-Golgi marker GRASP65 (Figure 7A). Similar to our previous observations after induced oligomerization of GPP130 (by either Mn addition for endogenous GPP130 or AP washout for GPP130-FM), Man1-FM clearly redistributed to endosome-like structures. The peripheral punctae were easily detected by 15 min of AP washout and continued to be present, whereas the overall staining became more and more reduced. GRASP65 in the same cells showed no change.
FIGURE 7:

Mannosidase-1 leaves the Golgi and is degraded upon oligomerization. (A) HeLa cells were transiently transfected with Man1-FM in the presence of AP, and the AP was washed off for the indicated times to induce polymerization. Thresholded staining of Man1-FM detected using anti-HA antibodies is compared with GRASP65-GFP fluorescence in the same cells. Bar, 5 μm. (B) The number of cytoplasmic structures per cell was determined at each time point after AP washout for both Man1-FM and the Golgi control GRASP65 using ImageJ (see Materials and Methods). (C) Total cell fluorescence levels of GRASP65 and Man1-FM were determined using ImageJ and normalized to the level of AP-treated cells before washout (mean ± SEM, n = 3, >10 cells/experiment). (D) Immunoblot to determine Man1-FM levels at the indicated times of AP washout with GPP130 detection as loading control. (E) Levels of Man1-FM were quantified at the indicated AP washout time points and normalized to control levels (mean ± SEM, n = 3, *p < 0.01).
The effect was quantified by determining both the number of punctae per cell (Figure 7B) and the total fluorescence per cell (Figure 7C). An immunoblotting assay was also used to confirm degradation. As expected, Man1-FM recovery in cell extracts was strongly reduced upon AP washout (Figure 7D), yielding a reduction of >90% by 5 h (Figure 7E). Endogenous GPP130 served as a control and showed no change. Similar to Mn-induced GPP130 and GPP130-FM, redistribution of Man1-FM to peripheral punctae occurred in cells expressing GGA1 but not GGA1-DN (Figure 8A), and this was confirmed by quantifying appearance in punctae after 1 h of AP washout (Figure 8B). Again, the wild-type GGA1 construct may have slowed exit somewhat, but the inhibition by GGA1-DN was significantly greater. Further, the peripheral puncta colocalized with Rab7 (Figure 8, C and D). Taken together, these findings suggest that Man1-FM redistribution occurs via the same pathway as GPP130.
FIGURE 8:

GGA1-DN blocks Man1-FM trafficking. (A) Images show GFP fluorescence of GGA1 or GGA1-DN and anti-HA staining of Man1-FM after 1 h of AP washout. Thresholded Man1-FM staining is also shown. Bar, 5 μm. (B) Quantified number of cytoplasmic punctae per cell for Man1-FM at 0 or 1 h after AP washout (mean ± SEM, n = 3, >10 cells/experiment, *p < 0.05). (C) Cells were cotransfected with HA-tagged Man1-FM and GFP-tagged Rab7 and then treated with a 1-h AP washout. Images compare GFP fluorescence and anti-HA staining with arrowheads marking examples of costained punctae. Bar, 10 μm. (D) The percentage of total Man1-FM punctae counted that were positive for Rab7 is quantified for the indicated time points (mean ± SEM, n = 3, >10 cells/experiment, p < 0.05 for 0 h vs 1 or 5 h).
Because both Man1 and GPP130 are predominately in the cis-Golgi at steady state, we next tested whether a trans-Golgi protein would also respond to oligomerization by leaving the Golgi and undergoing degradation. A similar strategy was used in which the cytoplasmic, transmembrane, and stem domains of the trans-Golgi protein galactosyltransferase (GT) were fused to the tandem array of FM domains followed, in this case, by GFP. Before AP washout, the construct was strongly Golgi localized, but after washout, it selectively redistributed to peripheral punctae, and, over time, the total GT-FM levels per cell were reduced (Figure 9A). These changes were marked compared with giantin in the same cells, and both appearance in punctae (Figure 9B) and selective loss (Figure 9C) were confirmed by image analysis. As expected, degradation was apparent by immunoblot analysis (Figure 9, D and E). Cycloheximide treatment showed that all of our constructs (GPP130-FM, Man1-FM, and GT-GM) were stable in the Golgi in their nonoligomerized forms, with redistribution and degradation occurring only upon oligomerization induced by AP washout (Supplemental Figure S1).
FIGURE 9:

Induced oligomerization causes degradation of galactosyltransferase. (A) Images show thresholded GFP fluorescence of GT-FM and anti-giantin antibody staining after the indicated times of AP washout. Bar, 5 μm. (B) Quantified number of cytoplasmic punctae per cell is shown for GT-FM and the giantin control at the indicated time points after AP washout (mean ± SEM, n = 3, >10 cells/experiment). (C) Total cell fluorescence levels of GT-FM and giantin were determined using ImageJ and normalized to the level of AP treated cells before washout (mean ± SEM, n = 3, >10 cells/experiment). (D, E) Blot and quantification showing level of GT-FM in cell extracts prepared at the indicated times after AP washout with GPP130 as a loading control (mean ± SEM, n = 3).
The lysosome-directed trafficking response is not associated with unfolded protein response activation
Given the possibility that the pathway described here is a type of quality control, it seemed important, even if unlikely, to test whether our conditions causing oligomerization-induced trafficking of Golgi proteins also activated the unfolded protein response (UPR). For this purpose, we assayed up-regulation of the ATPase chaperone Bip, which is a standard UPR marker. As a positive control, HeLa cells were treated with tunicamycin to block N-glycosylation and activate the UPR (Oslowski and Urano, 2011). As expected, Bip levels increased (Figure 10). In contrast, treatment with Mn did not alter Bip levels. To test the effect of AP washout, we needed to transfect the cells with GPP130-FM. Transfection frequency was ∼70%, and we noted that transfection alone elevated the level of Bip somewhat. However, AP washout of the transfected cells yielded Bip levels that were identical to those of cells before AP washout or control transfected cells (Figure 10). Therefore, based on Bip level, the UPR was not activated under conditions in which oligomerized Golgi proteins are induced to leave the Golgi and undergo degradation. Taken together, the experiments here reveal a novel type of quality control in the Golgi in which clustered Golgi proteins are routed for degradation.
FIGURE 10:
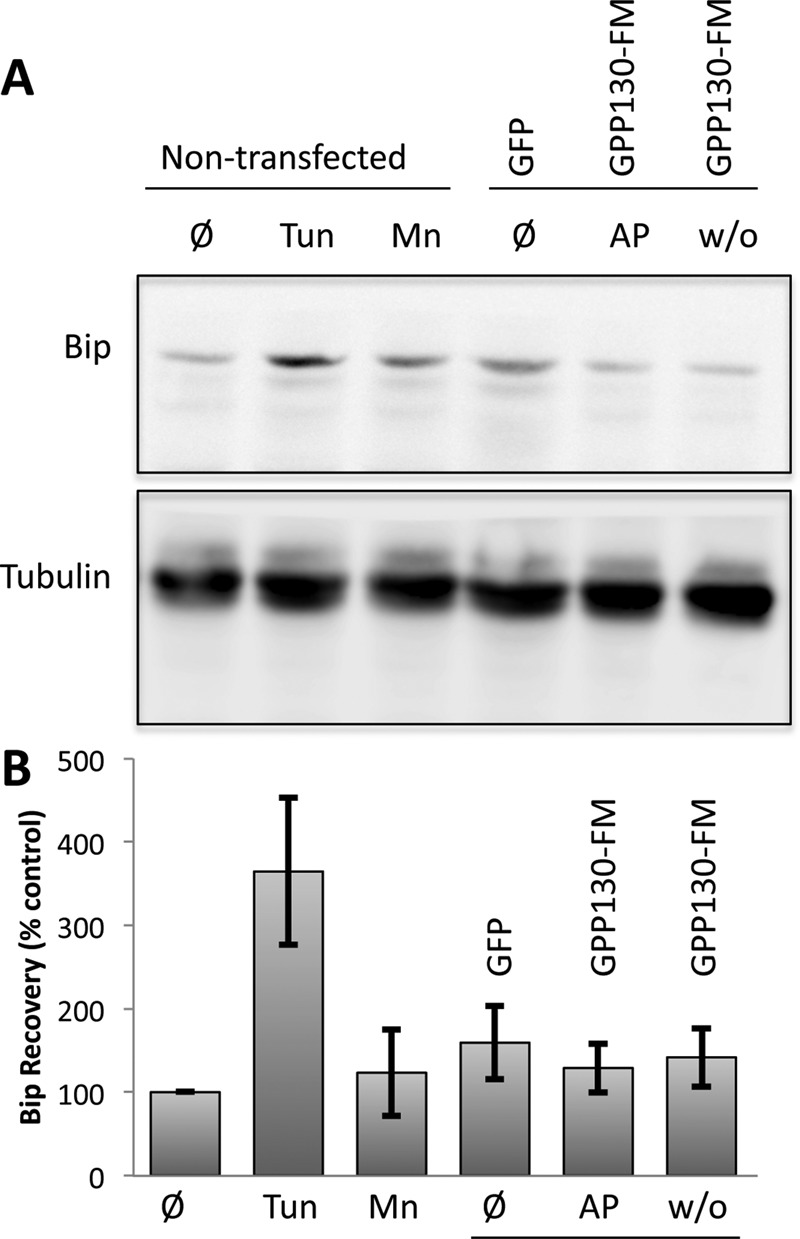
Oligomerization-induced trafficking is not associated with UPR activation. (A) Immunoblots using anti-Bip or anti-tubulin antibodies of extracts from nontransfected cells (lanes 1–3) or cells transfected with GPP130-GFP or GPP130-FM (lanes 4–6). The cells were untreated (Ø), tunicamycin treated (Tun) to induce the UPR, Mn treated (Mn) to cause endogenous GPP130 redistribution, left in AP (AP), or subjected to AP washout (w/o) to cause oligomerization of GPP130-FM. (B) Quantification of Bip recovery as a percentage of Bip levels in untreated cells (mean ± SEM, n = 3).
DISCUSSION
Our interest in the targeting of Golgi proteins to the lysosome started with Mn-induced degradation of GPP130 (Mukhopadhyay et al., 2010). Subsequently, using mutagenesis, cross-linking, fluorescence recovery after photobleaching analysis, and FM-mediated oligomerization, we found that Mn oligomerizes GPP130 and that oligomerization is required and sufficient to alter GPP130 trafficking such that it moves from the Golgi to lysosomes, where it is degraded (Tewari et al., 2014). Here we report the surprising finding that other, unrelated Golgi membrane proteins also left the Golgi and were degraded when their oligomerization was induced. We tested membrane proteins with a type II topology, which is the characteristic topology of Golgi glycosylation enzymes, but proteins of other topologies present in the Golgi will need to be tested. Movement of the complexes in an anterograde direction within the Golgi is most easily explained by their size-based exclusion from retrieval vesicles (Rizzo et al., 2013). Of interest, redistribution and degradation of GPP130 and GPP130-FM are insensitive to nocodazole (our unpublished results; Mukhopadhyay et al., 2010), indicating that an intact Golgi ribbon is not required, which differs from the intra-Golgi transport of large or aggregated soluble cargo (Lavieu et al., 2014). The requirement for GGA1 and clathrin suggests that upon reaching the TGN, the Golgi protein complexes are engaged by the canonical Golgi-to-lysosome pathway. However, based on GPP130, the sorting appears independent of a cytoplasmic sorting signal, raising the possibility that it also involves yet-to-be-described machinery. Elucidating the mechanism of recognition is an important next step, given that it appears to extend to at least several Golgi membrane proteins, and for each, it must distinguish between their normal and oligomerized states. Taken together, these observations suggest the presence of a quality control pathway in the Golgi based on recognition of aggregation state rather than exposure of unfolded domains.
Involvement of the Golgi in quality control, although perhaps underappreciated, is not new. Studies in both yeast and mammalian cells have shown that misfolded proteins may leave the ER before being trafficked back to the ER for dislocation and proteasome-mediated degradation (Hammond and Helenius, 1994; Jenness et al., 1997; Dusseljee et al., 1998; Vashist et al., 2001; Pan et al., 2013; Iannotti et al., 2014), and the UPR up-regulates ER-to-Golgi trafficking factors (Travers et al., 2000; Caldwell et al., 2001). Trafficking of selected substrates to the Golgi may assist in ER quality control, but the mechanistic basis is poorly understood (Caldwell et al., 2001; Arvan et al., 2002). The proteasome is also involved in turnover of cytoplasmically exposed Golgi proteins, at least under certain conditions (Puthenveedu and Linstedt, 2001). Lysosome-mediated degradation of misfolded Golgi proteins has also been observed (Wang et al., 2011). In yeast, there is a Golgi-localized ubiquitin ligase, Tul1p, that promotes sorting of mutated proteins with polar transmembrane domains to MVBs (Reggiori and Pelham, 2002). Further, at a nonpermissive temperature, the mutated yeast plasma membrane H+ATPase Pma1p1-7 is targeted directly from the Golgi to the endosomal system for vacuolar degradation (Chang and Fink, 1995). In mammalian cells, certain mutated forms of human lipoprotein lipase fail to be secreted and are targeted from the Golgi to lysosomes for degradation (Busca et al., 1996), and the TGN glycoprotein furin is somehow aggregated by lysosomal protease inhibitors, causing its transport to lysosomes (Wolins et al., 1997). Finally, under acute ER stress, glycosylphosphatidylinositol-linked, ER-associated degradation substrates traffic through the Golgi to the cell surface before being degraded in lysosomes (Satpute-Krishnan et al., 2014).
In contrast to all of these examples, we assume that the oligomerization/clustering taking place in our experiments is not causing folding defects. The Mn–GPP130 interaction is not yet structurally characterized. Because it is a direct interaction, however, it presumably involves simultaneous coordination of the metal by GPP130 residues on the outer surface of its coiled coil (Tewari et al., 2014). This would cluster the GPP130 dimers without perturbing folding. Similarly, the FM domains we used are positioned well away from the membrane within the Golgi lumen and undergo a well-characterized interaction with one another. Binding via this mechanism is unlikely to cause folding defects, and the UPR was not activated in our experiments. Nevertheless, the FM domain on membrane-anchored proteins creates polymers constrained from entering 40- to 80-nm vesicles and tubules (Rizzo et al., 2013), and it is capable of stapling single cisternae via lumenal interactions in-trans (Lavieu et al., 2014).
How might oligomerized/clustered Golgi proteins be differentially recognized by GGA1/clathrin in the TGN? At this point, we can only speculate. One possibility is that recognition is based on avidity. For the sake of argument, assume that many membrane proteins share a generic determinant that binds weakly to a hypothetical multiadhesive sorting receptor. Under normal conditions, when the cargo is in its native oligomeric state, the weak interaction with its fast off-rate would yield little sorting. If, on the other hand, the cargo becomes clustered, allowing its many copies to interact simultaneously with the multiadhesive sorting receptor, then sorting would occur. Given that sorting of GPP130 is independent of its cytoplasmic domain, we would predict a lumenal cargo/receptor interaction and a cytoplasmic receptor/GGA1 interaction. Among known receptors, both mannose-6-phosphate receptors and sortilins use GGA1 and clathrin to mediate Golgi-to-lysosome trafficking of their cargo (Puertollano et al., 2001a; Takatsu et al., 2001; Canuel et al., 2009). Although it is important to test the possible role of these receptors, they are not known to act on integral membrane cargo, and there is no indication of how they would discriminate between clustered and nonclustered cargo.
Another possibility is differential partitioning of oligomerized cargo in the plane of the membrane. By this view, clusters of transmembrane domains would favor lipid microenvironments that are also recognized by GGA1. Arguably, clustering, if it extends to the transmembrane domains, can impose a rigidity of protein orientation due to mismatch of transmembrane domain length with a given bilayer width. A monomer or dimer with a relatively long transmembrane domain can bury its hydrophobic residues using a skewed orientation, but skewing a clustered set of transmembrane domains exacerbates the mismatch. This effect drives a multipass protein containing relatively long transmembrane domains into thicker sections of bilayer (Lin and London, 2013).
A final possibility is that the clustered proteins are traveling to the lysosome by default. That is, the clustered complexes are left behind after all of the active sorting events take place at the TGN (including retrieval of their nonclustered counterparts), and these residual membranes head to MVB/lysosomes. This contradicts the view that the cell surface is the default pathway in mammalian cells. Of greater importance, it fails to say anything about the role of GGA1 and clathrin or, more generally, how “default” vesicles acquire targeting and fusion factors necessary for delivery to MVB/lysosomes.
We have shown that several Golgi proteins leave the Golgi upon oligomerization and are degraded, suggesting the presence of an ER-independent, Golgi-based quality control system sensitive to oligomeric state. This process seems to provide another level of quality control in the secretory pathway, in addition to the widely known quality control process in the ER. The primary purpose may be to rid cells of clustered or aggregated proteins that accumulate in the Golgi due to defective Golgi-based processing. The pathway may also prove amenable to manipulations, making it therapeutically useful. Oligomerization and the ensuing lysosomal degradation of GPP130 protect against Shiga toxicosis in cultured cells and mice (Mukhopadhyay and Linstedt, 2012). The drug suramin can aggregate the prion protein PrP in a post-ER/Golgi compartment, redirecting it to lysosomes and delaying onset of prion disease (Gilch et al., 2001). Future studies will likely reveal additional components in the oligomer-induced degradation of Golgi proteins and provide new potential targets for therapeutic intervention.
MATERIALS AND METHODS
Antibodies and other reagents
Polyclonal antibodies against GPP130 and giantin and monoclonal antibodies against GPP130 and GFP have been described (Puri et al., 2002; Mukhopadhyay et al., 2010). For immunoblots, monoclonal anti-HA (Sigma-Aldrich, St. Louis, MO) was used at 1:1000, polyclonal anti-Bip (Abcam, Cambridge, MA) was used at 1:500, and anti–γ-tubulin (Sigma-Aldrich) was used at 1:1000. Horseradish peroxidase–conjugated goat anti-rabbit or anti-mouse secondary antibodies were used at 1:3000 (Bio-Rad, Hercules, CA). For immunofluorescence, Alexa Fluor 488 and Alexa Fluor 568 (Invitrogen, Carlsbad, CA) were used at 1:400. AP (now called D/D solubilizer) was purchased from Clontech (Mountain View, CA). Pitstop2 was from Abcam, and tunicamycin was from Sigma-Aldrich. Leupeptin and pepstatin were purchased from Sigma-Aldrich.
Constructs
GPP130-FM, which contains a GFP tag, was previously described (Tewari et al., 2014). A version substituting an HA tag in place of the GFP tag was used for cotransfection with GGA1 constructs. ∆cyto-GPP130-FM was engineered by looping out residues 2–11 from the 12-residue cytoplasmic domain in GPP130-FM using a PCR-based loop-out modification of the QuikChange protocol (Stratagene, La Jolla, CA), as described (Tewari et al., 2014). Similarly, looping out residues 88–247 from GPP130-FM created GPP130-1-87-FM. Subcloning residues 1–75 of galactosyltransferase in-frame before the FM repeats and GFP using Nhe1 and EcoR1 restriction sites generated GT-FM. All constructs were confirmed by restriction analysis and sequencing. GGA1-GFP and GGA1-DN-GFP (Puertollano and Alonso, 1999) were a kind gift from Juan Bonifacino (National Institutes of Health, Bethesda, MD). GPP130-GFP (residues 1–247; Mukhopadhyay et al., 2010), GFP-Rab7 (Mukhopadhyay et al., 2010), and GRASP65-GFP (Sengupta et al., 2009) have been described previously.
Cell culture and transfection
HeLa cells were grown in MEM with 100 IU/ml penicillin-G and 100 μg/ml streptomycin (Fisher Scientific, Hanover Park, IL) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and maintained at 37°C in a 5% CO2 incubator. Manganese treatment was performed by adding freshly prepared MnCl2 to the culture medium at 500 μM for the indicated times. DNA transfections were performed using the JetPEI transfection reagent (PolyPlus, Illkirch, France) according to the manufacturer’s protocol. Cultures were transfected 24 h after plating and, unless noted otherwise, used for experiments 24 h after transfection. For GGA1 coexpression experiments, only cells expressing moderately high levels of GGA1 and GGA1-DN were analyzed to ensure adequate levels for inhibition by GGA1-DN.
Controlled polymerization
Cells were transfected with the FM constructs, and after 16–18 h, they were placed in fresh medium containing 1 μM AP for 24 h. This was followed by another change into medium containing 1 μM AP and 100 μg/ml cyclohexamide for 30 min. Next the cells were incubated in medium containing only 100 μg/ml cycloheximide for various times of AP washout before fixation and imaging. Only cells exhibiting moderate expression levels were analyzed. Note that the control cells contained both cycloheximide and AP throughout the course of the experiment. For the nocodazole experiments, HeLa cells were treated with 1 μg/ml nocodazole and 100 μg/ml cycloheximide for 3 h, followed by either Mn addition for 1 or 5 h or AP removal for 1 h in GPP130-FM–transfected cells. Nocodazole and cycloheximide were present during the entire course of the experiment. For Pitstop2 experiments, 24 h posttransfection, the GPP130-FM transfected cells were serum deprived for 45 min in the presence of AP, followed by AP washout for 10 min in the presence of Pitstop2 (30 μM) in DMSO or DMSO alone. We also confirmed that this period of serum deprivation did not affect GPP130-FM redistribution to endosomal punctae.
Immunoblot analysis
HeLa cells from 60-mm plates were harvested in Tris-SDS lysis buffer (25 mM Tris-Cl, pH 6.7, 1.5% SDS, 0.1% β-mercaptoethanol, and 1 mM phenylmethanesulfonylfluoride), passed through a 25-gauge needle ∼10 times, and clarified by centrifugation at 15,000 rpm in a microfuge for 15 min at 4°C. Protein levels were determined using the Pierce BCA Protein Assay (Thermo Scientific, Waltham, MA), and equal protein amounts of lysate were analyzed by SDS–PAGE and immunoblotting. Immunoblots were quantified using ImageGauge software (Fujifilm, Valhalla, NY). For the GPP130-FM, Man1-FM, and GT-FM blots, the transfected cells were subjected to AP washout for the indicated times before lysis. To inhibit lysosomal hydrolases, GPP130-FM– transfected cells were preincubated with leupeptin (100 μg/ml) and pepstatin (50 μg/ml) for 24 h before AP washout for 5 h in the continued presence of the inhibitors. For Bip assays, cells were treated for 6 h with 3 μM tunicamycin, 500 μM Mn, or a washout of AP from GPP130-FM–transfected cells.
Microscopy and image analysis
Immunofluorescence on 3% paraformaldehyde–fixed cells was performed exactly as described (Tewari et al., 2014) using a spinning-disk confocal microscope and sectioning at 0.3-μm steps. Display images are maximum value projections, except for those that are thresholded to highlight cytoplasmic punctae. For these, a uniform threshold was applied after background subtraction. The images were analyzed using ImageJ (National Institutes of Health, Bethesda, MD) as described (Tewari et al., 2014). To measure fluorescence per cell, background was subtracted from each Z-section, and average value projections were created. Cells were outlined using the Gaussian blur filter, and mean fluorescence was measured using the Measure plug-in of ImageJ. The number of cytoplasmic punctae per cell was determined using the Analyze Particle plug-in of ImageJ (particle range setting of 10–100 pixels) from maximum value projections that were uniformly thresholded after background subtraction. For total fluorescence, the data were normalized using the control values. For Rab7 colocalization, manual examination of maximum value projections was used to determine the total number of anti–HA-stained GPP130-FM punctae and the percentage of these that were positive for Rab7-GFP. Golgi fluorescence was the mean intensity in the Golgi region as marked by giantin staining. For all experiments except supplemental, data are presented as averages of three experiments with at least 10 cells per condition per experiment. Comparisons between two groups were performed using two-tailed Student’s t test, assuming equal variances. In general, p < 0.05 was considered significant, and the determined p values are provided in the figure legends. Asterisks in graphs denote statistically significant differences.
Supplementary Material
Acknowledgments
We thank Tina Lee, Somshuvra Mukhopadhyay (University of Texas at Austin, Austin, TX), and Emily Simon for critical advice on the manuscript and Alberto Luini (Consiglio Nazionale delle Ricerche, Naples, Italy) and Juan Bonifacino (National Institutes of Health, Bethesda, MD) for generous reagent gifts. Funding was provided by National Institutes of Health R01 Grant GM08411101 (to A.D.L.).
Abbreviations used:
- AP
AP21998
- DN
dominant negative
- FM
F36M substitution of FKBP12
- GGA
γ-adaptin homologous ARF–interacting protein
- GT
galactosyltransferase
- Man1
mannosidase-1
- MVB
multivesicular body
- STx
Shiga toxin
- TGN
trans-Golgi network
- UPR
unfolded protein response.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-04-0207 on October 7, 2015.
REFERENCES
- Arvan P, Zhao X, Ramos-Castaneda J, Chang A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic. 2002;3:771–780. doi: 10.1034/j.1600-0854.2002.31102.x. [DOI] [PubMed] [Google Scholar]
- Bachert C, Lee TH, Linstedt AD. Lumenal endosomal and Golgi-retrieval determinants involved in pH-sensitive targeting of an early Golgi protein. Mol Biol Cell. 2001;12:3152–3160. doi: 10.1091/mbc.12.10.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beddoe T, Paton AW, Le Nours J, Rossjohn J, Paton JC. Structure, biological functions and applications of the AB5 toxins. Trends Biochem Sci. 2010;35:411–418. doi: 10.1016/j.tibs.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MW, Pelham HR. A selective transport route from Golgi to late endosomes that requires the yeast GGA proteins. J Cell Biol. 2000;151:587–600. doi: 10.1083/jcb.151.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busca R, Martinez M, Vilella E, Pognonec P, Deeb S, Auwerx J, Reina M, Vilaro S. The mutation Gly142→Glu in human lipoprotein lipase produces a missorted protein that is diverted to lysosomes. J Biol Chem. 1996;271:2139–2146. doi: 10.1074/jbc.271.4.2139. [DOI] [PubMed] [Google Scholar]
- Caldwell SR, Hill KJ, Cooper AA. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J Biol Chem. 2001;276:23296–23303. doi: 10.1074/jbc.M102962200. [DOI] [PubMed] [Google Scholar]
- Canuel M, Libin Y, Morales CR. The interactomics of sortilin: an ancient lysosomal receptor evolving new functions. Histol Histopathol. 2009;24:481–492. doi: 10.14670/HH-24.481. [DOI] [PubMed] [Google Scholar]
- Chang A, Fink GR. Targeting of the yeast plasma membrane [H+]ATPase: a novel gene AST1 prevents mislocalization of mutant ATPase to the vacuole. J Cell Biol. 1995;128:39–49. doi: 10.1083/jcb.128.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusseljee S, Wubbolts R, Verwoerd D, Tulp A, Janssen H, Calafat J, Neefjes J. Removal and degradation of the free MHC class II beta chain in the endoplasmic reticulum requires proteasomes and is accelerated by BFA. J Cell Sci. 1998;111:2217–2226. doi: 10.1242/jcs.111.15.2217. [DOI] [PubMed] [Google Scholar]
- Gilch S, Winklhofer KF, Groschup MH, Nunziante M, Lucassen R, Spielhaupter C, Muranyi W, Riesner D, Tatzelt J, Schatzl HM. Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. EMBO J. 2001;20:3957–3966. doi: 10.1093/emboj/20.15.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Chang WP, Koelsch G, Tang J. Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2. FEBS Lett. 2002;524:183–187. doi: 10.1016/s0014-5793(02)03052-1. [DOI] [PubMed] [Google Scholar]
- Iannotti MJ, Figard L, Sokac AM, Sifers RN. A Golgi-localized mannosidase (MAN1B1) plays a non-enzymatic gatekeeper role in protein biosynthetic quality control. J Biol Chem. 2014;289:11844–11858. doi: 10.1074/jbc.M114.552091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol. 2014;27:109–116. doi: 10.1016/j.ceb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenness DD, Li Y, Tipper C, Spatrick P. Elimination of defective alpha-factor pheromone receptors. Mol Cell Biol. 1997;17:6236–6245. doi: 10.1128/mcb.17.11.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieu G, Dunlop MH, Lerich A, Zheng H, Bottanelli F, Rothman JE. The Golgi ribbon structure facilitates anterograde transport of large cargoes. Mol Biol Cell. 2014;25:3028–3036. doi: 10.1091/mbc.E14-04-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gall S, Erdtmann L, Benichou S, Berlioz-Torrent C, Liu L, Benarous R, Heard JM, Schwartz O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity. 1998;8:483–495. doi: 10.1016/s1074-7613(00)80553-1. [DOI] [PubMed] [Google Scholar]
- Lin Q, London E. Altering hydrophobic sequence lengths shows that hydrophobic mismatch controls affinity for ordered lipid domains (rafts) in the multitransmembrane strand protein perfringolysin O. J Biol Chem. 2013;288:1340–1352. doi: 10.1074/jbc.M112.415596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt AD, Mehta A, Suhan J, Reggio H, Hauri HP. Sequence and overexpression of GPP130/GIMPc: evidence for saturable pH-sensitive targeting of a type II early Golgi membrane protein. Mol Biol Cell. 1997;8:1073–1087. doi: 10.1091/mbc.8.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Bachert C, Smith DR, Linstedt AD. Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol Biol Cell. 2010;21:1282–1292. doi: 10.1091/mbc.E09-11-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science. 2012;335:332–335. doi: 10.1126/science.1215930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Redler B, Linstedt AD. Shiga toxin-binding site for host cell receptor GPP130 reveals unexpected divergence in toxin-trafficking mechanisms. Mol Biol Cell. 2013;24:2311–2318. doi: 10.1091/mbc.E13-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan R, Linstedt AD. A cycling cis-Golgi protein mediates endosome-to-Golgi traffic. Mol Biol Cell. 2004;15:4798–4806. doi: 10.1091/mbc.E04-05-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011;490:71–92. doi: 10.1016/B978-0-12-385114-7.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S, Cheng X, Sifers RN. Golgi-situated endoplasmic reticulum alpha-1, 2-mannosidase contributes to the retrieval of ERAD substrates through a direct interaction with gamma-COP. Mol Biol Cell. 2013;24:1111–1121. doi: 10.1091/mbc.E12-12-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Dikic I, Lukacs GL. Ubiquitin-dependent sorting in endocytosis. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a016808. a016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk R, Lutsenko S. Golgi in copper homeostasis: a view from the membrane trafficking field. Histochem Cell Biol. 2013;140:285–295. doi: 10.1007/s00418-013-1123-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Aguilar RC, Gorshkova I, Crouch RJ, Bonifacino JS. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science. 2001a;292:1712–1716. doi: 10.1126/science.1060750. [DOI] [PubMed] [Google Scholar]
- Puertollano R, Alonso MA. MAL, an integral element of the apical sorting machinery, is an itinerant protein that cycles between the trans-Golgi network and the plasma membrane. Mol Biol Cell. 1999;10:3435–3447. doi: 10.1091/mbc.10.10.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Randazzo PA, Presley JF, Hartnell LM, Bonifacino JS. The GGAs promote ARF-dependent recruitment of clathrin to the TGN. Cell. 2001b;105:93–102. doi: 10.1016/s0092-8674(01)00299-9. [DOI] [PubMed] [Google Scholar]
- Puri S, Bachert C, Fimmel CJ, Linstedt AD. Cycling of early Golgi proteins via the cell surface and endosomes upon lumenal pH disruption. Traffic. 2002;3:641–653. doi: 10.1034/j.1600-0854.2002.30906.x. [DOI] [PubMed] [Google Scholar]
- Puthenveedu MA, Linstedt AD. Evidence that Golgi structure depends on a p115 activity that is independent of the vesicle tether components giantin and GM130. J Cell Biol. 2001;155:227–238. doi: 10.1083/jcb.200105005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Pelham HR. A transmembrane ubiquitin ligase required to sort membrane proteins into multivesicular bodies. Nat Cell Biol. 2002;4:117–123. doi: 10.1038/ncb743. [DOI] [PubMed] [Google Scholar]
- Rizzo R, Parashuraman S, Mirabelli P, Puri C, Lucocq J, Luini A. The dynamics of engineered resident proteins in the mammalian Golgi complex relies on cisternal maturation. J Cell Biol. 2013;201:1027–1036. doi: 10.1083/jcb.201211147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J Cell Biol. 2004;167:903–913. doi: 10.1083/jcb.200407031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpute-Krishnan P, Ajinkya M, Bhat S, Itakura E, Hegde RS, Lippincott-Schwartz J. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell. 2014;158:522–533. doi: 10.1016/j.cell.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D, Truschel S, Bachert C, Linstedt AD. Organelle tethering by a homotypic PDZ interaction underlies formation of the Golgi membrane network. J Cell Biol. 2009;186:41–55. doi: 10.1083/jcb.200902110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T, Forsten-Williams K, Storrie B. Both post-Golgi and intra-Golgi cycling affect the distribution of the Golgi phosphoprotein GPP130. Traffic. 2007;8:1265–1279. doi: 10.1111/j.1600-0854.2007.00607.x. [DOI] [PubMed] [Google Scholar]
- Sun LP, Seemann J, Goldstein JL, Brown MS. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci USA. 2007;104:6519–6526. doi: 10.1073/pnas.0700907104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surma MA, Klose C, Simons K. Lipid-dependent protein sorting at the trans-Golgi network. Biochim Biophys Acta. 2012;1821:1059–1067. doi: 10.1016/j.bbalip.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Takatsu H, Katoh Y, Shiba Y, Nakayama K. Golgi-localizing, gamma-adaptin ear homology domain, ADP-ribosylation factor-binding (GGA) proteins interact with acidic dileucine sequences within the cytoplasmic domains of sorting receptors through their Vps27p/Hrs/STAM (VHS) domains. J Biol Chem. 2001;276:28541–28545. doi: 10.1074/jbc.C100218200. [DOI] [PubMed] [Google Scholar]
- Tartakoff AM. Perturbation of vesicular traffic with the carboxylic ionophore monensin. Cell. 1983;32:1026–1028. doi: 10.1016/0092-8674(83)90286-6. [DOI] [PubMed] [Google Scholar]
- Tewari R, Jarvela T, Linstedt AD. Manganese induces oligomerization to promote down-regulation of the intracellular trafficking receptor used by Shiga toxin. Mol Biol Cell. 2014;25:3049–3058. doi: 10.1091/mbc.E14-05-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub LM, Bonifacino JS. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb Perspect Biol. 2013;5:a016790. doi: 10.1101/cshperspect.a016790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009;284:12110–12124. doi: 10.1074/jbc.M809277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Kim W, Belden WJ, Spear ED, Barlowe C, Ng DT. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J Cell Biol. 2001;155:355–368. doi: 10.1083/jcb.200106123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wang S, Thibault G, Ng DT. Routing misfolded proteins through the multivesicular body (MVB) pathway protects against proteotoxicity. J Biol Chem. 2011;286:29376–29387. doi: 10.1074/jbc.M111.233346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolins N, Bosshart H, Kuster H, Bonifacino JS. Aggregation as a determinant of protein fate in post-Golgi compartments: role of the luminal domain of furin in lysosomal targeting. J Cell Biol. 1997;139:1735–1745. doi: 10.1083/jcb.139.7.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Doray B, Poussu A, Lehto VP, Kornfeld S. Binding of GGA2 to the lysosomal enzyme sorting motif of the mannose 6-phosphate receptor. Science. 2001;292:1716–1718. doi: 10.1126/science.1060896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.