

Abstract
Background
Osteogenesis imperfecta (OI) is a congenital disorder characterized by increased bone fragility and low bone mass.
Methods
The presence of COL1A1 or COL1A2 mutation was investigated by direct sequencing in 72 patients with OI type I, III, or IV (27 males and 45 females; age range 0.2-62 years) from 37 unrelated families. The clinical features of these patients were also recorded.
Results
Thirty-seven COL1A1 and COL1A2 mutations were identified, including 28 COL1A1 mutations and 9 COL1A2 mutations. Fifteen (41 %) were novel mutations, and twelve (32 %) were familial mutations. A review of their medical records revealed that the 72 patients could be classified into OI type I (n = 42), III (n = 5), and IV (n = 25). Twenty-nine patients had helical mutations (caused by the substitution of a glycine within the Gly-X-Y triplet domain of the triple helix), and 42 had haploinsufficiency mutations (caused by frameshift, nonsense, and splice-site mutations). Compared with haploinsufficiency, the patients with helical mutations had more severely impaired skeletal phenotypes, including shorter height, lower bone mineral density, poorer walking ability, more frequent manifestations of dentinogenesis imperfecta and scoliosis (p < 0.05).
Conclusions
Genotype and phenotype databases are expected to promote better genetic counseling and medical care of patients with OI.
Electronic supplementary material
The online version of this article (doi:10.1186/s13023-015-0370-2) contains supplementary material, which is available to authorized users.
Keywords: Bone mineral density, Genotype, Height, Osteogenesis imperfecta, Phenotype
Background
Osteogenesis imperfecta (OI) (MIM# 166200, 166210, 259420, and 166220) is a hereditary disease characterized by increased bone fragility, low bone mass, short stature, and other connective tissue manifestations, with a reported incidence of 1:15,000-1:25,000 [1, 2]. Additional extra-skeletal features manifest to a variable degree, including blue sclera, dentinogenesis imperfecta (DI), and hearing loss [3, 4]. In Western populations, OI is generally inherited in an autosomal dominant manner, however, a small number of families inherit their OI in an autosomal recessive pattern [3]. The most commonly used classification is that proposed by Sillence et al. [5], but as modified and expanded by the International Nomenclature Committee for Constitutional Disorders of the skeleton as reported by van Dijk and Sillence 2014 [3] which classifies OI into five major groups of disorders as types 1–5. Type I is usually the mildest form, with normal or near-normal growth and minimal bone deformities. Type II is lethal in the perinatal period, with multiple intrauterine fractures and bone deformities of the extremities. Type III is the most severe form in children who survive the neonatal period, with fractures often presenting at birth. These children have extremely short stature with progressive limb and spine deformities secondary to multiple fractures. Type IV is the most phenotypically heterogeneous group, with mild to moderate bone deformities and variable short stature. In 2012, van Dijk et al. [6] reporting for the committee on best practice guidelines for diagnosis and investigation noted 8 types of OI based on clinical characteristics and molecular genetic defects. However, OMIM now records 17 OI types and a further 3 syndromic forms of OI have been defined at the molecular level, 2 types with craniosynostosis/skull deformity and one type with significant eye involvement.
To date, more than 1,000 different COL1A1/COL1A2 mutations have been identified in patients with OI (https://oi.gene.le.ac.uk, accessed July 20, 2015). There are two general categories of type I collagen mutations giving rise to OI types I to IV. The first are haploinsufficiency mutations caused by frameshift, nonsense, and splice-site mutations, which lead to failure to synthesize the products of one COL1A1 allele. The second involves the synthesis of collagen molecules with structural abnormalities, most frequently caused by the substitution of glycine by another amino acid in the Gly-X-Y triplet domain of the triple helix. Previous studies have reported that OI patients with COL1A1 haploinsufficiency mutations have milder bone fragility and damage than those with COL1A1/COL1A2 helical glycine mutations [7, 8]. Both COL1A1 and COL1A2 genes are very large, and they have rarely been analyzed systematically in Taiwan. The aim of this study was to characterize the correlations of genotype and phenotype for Taiwanese OI patients, with the hope that this may aid in the clinical diagnosis, genetic counseling and prenatal diagnosis of this disease.
Patients and methods
Study population
Seventy-two Taiwanese patients (27 males and 45 females; age range at last follow-up, 0.2-62 years) from 37 unrelated families were diagnosed with OI during the study period (January 1996 through December 2014) at Mackay Memorial Hospital, Taipei, Taiwan. None of the patients belonged to a consanguineous family. Molecular analysis was performed to investigate the presence of COL1A1 or COL1A2 mutations. This is an analysis of only those patients found to have a molecular genetic finding of a mutation in one of the two type I collagen genes. Clinical manifestations and the results of physical examinations and imaging studies of these patients were also recorded. The hospital’s Ethics Committee approved the study protocol, and all of the participants or their parents provided written informed consent.
Clinical assessment
Diagnosis and classification were based on clinical and radiological characteristics according to the Sillence classification system [5]. In this study, every patient was evaluated at our clinic by one author, in person, to minimize inter-observer variation. For each patient, we recorded their height, weight, and bone mineral density (BMD) standard deviation scores (SDSs), walking ability and family history, and the occurrence of blue sclera, DI, hearing loss, bone deformity, and scoliosis. Height and weight were transformed to SDSs on the basis of a standard growth table for Taiwanese children and adolescents [9]. The BMD of the lumbar spine (L1–L4) was assessed using dual energy X-ray absorptiometry (DEXA) with a Hologic QDR 4500 system (Hologic, Bedford, MA, USA) [4]. The BMD results were converted to age- and gender-specific SDSs based on the normative reference data for BMD in Taiwanese children and adults [10, 11]. Pamidronate therapy has been reported to increase BMD, decrease the fracture rate, and substantially improve functional status for OI patients [12]. However, none of our patients had received pamidronate treatment at the time of assessment.
Mutation analysis for type I collagen
Total genomic DNA was isolated from peripheral blood using standard extraction methods. DNA sequencing of all 51 polymerase chain reaction-amplified exons of the COL1A1 gene and 52 exons of the COL1A2 gene, including the intron-exon boundaries, was performed using a BigDye Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). The nucleotide sequence was determined using an Applied Biosystems 3100 DNA sequencer. Sequence traces were aligned with the GenBank reference sequences of COL1A1 genomic DNA (AF017178.2) and cDNA (NM_000088.3), and COL1A2 genomic DNA (AF004877.1) and cDNA (NM_000089.3). DNA mutation numbering was based on the cDNA sequence using the A of the ATG translation initiation start site as nucleotide +1. Novel mutations were identified by their absence from the Osteogenesis Imperfecta Variant Database (https://oi.gene.le.ac.uk/home.php). In addition, identified nucleotide changes were re-examined in 100 control alleles. Polymorphisms were considered if the same nucleotide changes were detected in the control group.
Statistical analysis
Relationships between gender, age, BMD SDS, DI and the existence of each clinical feature in the OI patients were tested using Pearson correlation, and significance was tested using Fisher r-z transformations. The clinical and radiological data were compared between patients with helical mutations versus those with haploinsufficiency mutations using the Student’s t-test for continuous variable, and Pearson’s chi-squared test and Fisher’s exact test for categorical variables. Two-tailed p-values were computed. SPSS version 11.5 (SPSS Inc., Chicago, IL) was used for calculations and differences were considered to be statistically significant when the p value was less than 0.05.
Results
Clinical characteristics
A review of the patients’ medical records revealed that among the 72 OI patients, 42 (58 %) were classified as OI type I, 5 (7 %) as type III, and 25 (35 %) as type IV. None of the patients had OI type II. The SDSs for height, weight, and BMD for all patients were–1.61 ± 2.57,–0.84 ± 1.56, and–2.06 ± 1.62, respectively. Furthermore, when dividing the patients into groups by OI type, the height SDSs were–0.49 ± 1.37,–6.42 ± 5.54, and–2.51 ± 1.85, respectively; and the BMD SDSs were–1.57 ± 1.15,–3.32 ± 1.86, and − 2.69 ± 1.96, respectively, for type I, III, and IV. A triangular face, blue sclera, DI, hearing loss, bone deformity, scoliosis, and walking without assistance were recorded in 18, 89, 43, 19, 58, 36 and 90 % of the patients, respectively. An older age was associated with hearing loss (p < 0.01). The BMD SDS was positively correlated with the ability to walk without assistance (p < 0.01). Patients with DI appeared to be prone to developing bone deformities (p < 0.01) and scoliosis (p < 0.01). Girls with OI had a slightly higher BMD SDS than boys with the same mutation type (p < 0.05) (Additional file 1: Table S1 and Additional file 2: Table S2).
COL1A1 and COL1A2 mutations
Thirty-seven COL1A1 and COL1A2 mutations were identified in the 72 patients, including 28 COL1A1 mutations and 9 COL1A2 mutations. None of the COL1A1 or COL1A2 mutations were the same among these 37 unrelated families, and 15 (41 %) were novel mutations. Among the 28 COL1A1 mutations, 7 were missense mutations, 4 were nonsense mutations, 6 were splicing mutations, and 11 were frameshift mutations. Eight familial mutations were identified. All 9 COL1A2 mutations were missense mutations, 4 of which were from familial inheritance (Tables 1 and 2). Among the 37 causative COL1A1 and COL1A2 mutations, 15 (41 %) were caused by the substitution of a glycine within the Gly-X-Y triplet domain of the triple helix. There were 7 glycine mutations in COL1A1 and 8 in COL1A2. Among the 15 glycine mutations, aspartic acid substitutions were the most common type (n = 6, 40 %), followed by serine (n = 4, 27 %), arginine (n = 2, 13 %), cysteine (n = 2, 13 %), and glutamic acid (n = 1, 7 %) substitutions. Of the 21 haploinsufficiency mutations in COL1A1, 11 were frameshift mutations, 6 were splicing mutations, and 4 were nonsense mutations.
Table 1.
Genetic findings of 28 OI probands with mutations in COL1A1
| Family No. | Type of OI | Exon or Intron | Nucleotide change (DNA level) | Predicted amino acid change (protein level) | Mutation type | Helical mutation or haploinsufficiency | Novel mutation | Familial/Sporadic |
|---|---|---|---|---|---|---|---|---|
| F1 | I | Exon 4 | c.333-9A > G | Splicing | Haploinsufficiency | Yes | S | |
| F2 | I | Exon 5 | c.441delC | Frameshift | Haploinsufficiency | S | ||
| F3 | I | Exon 5 | c.386_387insC | Frameshift | Haploinsufficiency | Yes | F | |
| F4 | IV | Exon 5 | c.391C > T | p. Arg131X | Nonsense | Haploinsufficiency | S | |
| F5 | IV | Exon 6 | c.477_478 insT | Frameshift | Haploinsufficiency | Yes | S | |
| F6 | I | Exon 7 | c.579delT | p. Pro193Profs*72 | Frameshift | Haploinsufficiency | F | |
| F7 | IV | Exon 8 | c.642 + 1G > A | Splicing | Haploinsufficiency | S | ||
| F8 | I | Exon 8 | c.590G > A | p. Gly197Asp | Missense | Helical | S | |
| F9 | I | Exon 9 | c.658C > T | p. Arg220X | Nonsense | Haploinsufficiency | F | |
| F10 | IV | Exon 11 | c.769G > A | p. Gly257Arg | Missense | Helical | F | |
| F11 | IV | Intron 12 | c.858 + 24G > A | Splicing | Haploinsufficiency | Yes | S | |
| F12 | III | Exon 13 | c.878G > A | p. Gly293Asp | Missense | Helical | Yes | S |
| F13 | III | Exon 16 | c.1021G > C | p. Gly341Arg | Missense | Helical | Yes | S |
| F14 | IV | Intron 17 | c.1155 + 3_1155 + 6del | c.1155 + 3_6delAAGT | Splicing | Haploinsufficiency | S | |
| F15 | I | Intron 20 | c.1354-12G > A | Splicing | Haploinsufficiency | F | ||
| F16 | I | Exon 21 | c.1380delT | Frameshift | Haploinsufficiency | F | ||
| F17 | IV | Exon 24 | c.1667delC | Frameshift | Haploinsufficiency | Yes | S | |
| F18 | I | Exon 24 | c.1615-1G > T | Splicing | Haploinsufficiency | Yes | S | |
| F19 | IV | Exon 35 | c.2384-2394 del 11 mers | Frameshift | Haploinsufficiency | Yes | S | |
| F20 | IV | Exon 36 | c.2461G > A | p. Gly821Ser | Missense | Helical | S | |
| F21 | I | Exon 37 | c.2523delT | Frameshift | Haploinsufficiency | S | ||
| F22 | I | Exon 38 | c.2644C > T | p. Arg882X | Nonsense | Haploinsufficiency | F | |
| F23 | I | Exon 40 | c.2775delT | Frameshift | Haploinsufficiency | Yes | S | |
| F24 | III | Exon 42 | c.3064G > A | p. Gly1022Ser | Missense | Helical | S | |
| F25 | I | Exon 42 | c.3076C > T | p. Arg1026X | Nonsense | Haploinsufficiency | F | |
| F26 | IV | Exon 44 | c.3124_3134del11 | Frameshift | Haploinsufficiency | Yes | S | |
| F27 | IV | Exon 47 | c.3505G > A | p. Gly1169Ser | Missense | Helical | S | |
| F28 | III | Exon 52 | c.4308_4309insA | Frameshift | Haploinsufficiency | S |
OI osteogenesis imperfecta
Table 2.
Genetic findings of 9 OI probands with mutations in COL1A2
| Family No. | Type of OI | Exon or Intron | Nucleotide change (DNA level) | Predicted amino acid change (protein level) | Mutation type | Helical mutation or haploinsufficiency | Novel mutation | Familial/Sporadic |
|---|---|---|---|---|---|---|---|---|
| F29 | I | Exon 8 | c.335G > A | p. Gly112Asp | Missense | Helical | Yes | F |
| F30 | IV | Exon 24 | c.1378G > A | p. Gly460Ser | Missense | Helical | S | |
| F31 | IV | Exon 29 | c.1666G > T | p. Gly556Cys | Missense | Helical | Yes | S |
| F32 | IV | Exon 33 | c.2018G > A | p. Gly673Asp | Missense | Helical | S | |
| F33 | I | Exon 37 | c.2197G > T | p. Gly733Cys | Missense | Helical | F | |
| F34 | IV | Exon 37 | c.2279G > A | p. Gly760Glu | Missense | Helical | F | |
| F35 | III | Exon 37 | c.2288G > A | p. Gly763Asp | Missense | Helical | S | |
| F36 | I | Exon 40 | c.2531G > A | p. Gly844Asp | Missense | Helical | Yes | F |
| F37 | IV | Exon 51 | c.3815G > C | p. Cys1272Ser | Missense | - | Yes | S |
OI osteogenesis imperfecta
Genotype and phenotype analysis
The OI type was correlated with mutated genes and mutation types. Compared with COL1A2 mutations, COL1A1 mutations were more frequent in the patients with OI type I (46 % vs. 33 %) and III (14 % vs. 11 %), and less frequent in the patients with OI type IV (39 % vs. 56 %). Helical mutations were more frequent in the patients with OI type III (27 % vs. 5 %) and IV (47 % vs. 38 %), and less frequent in the patients with OI type I (27 % vs. 57 %) compared to those with haploinsufficiency mutations (Table 3). Among the 72 patients, 51 had a COL1A1 mutation and 21 had a COL1A2 mutation. Twenty-nine patients had helical mutations and 42 had haploinsufficiency mutations. Compared with haploinsufficiency, the patients with helical mutations had more severely damaged skeletal phenotypes, including shorter height, lower weight and BMD, poorer walking ability, and more frequent manifestations of DI and scoliosis (p < 0.05) (Table 4, Fig: 1a and b).
Table 3.
The relationship between OI type and mutated genes and mutation types
| Mutated genes and mutation types | OI type | ||
|---|---|---|---|
| I | III | IV | |
| COL1A1 (n = 28) | 13 (46 %) | 4 (14 %) | 11 (39 %) |
| COL1A2 (n = 9) | 3 (33 %) | 1 (11 %) | 5 (56 %) |
| Helical mutation (n = 15) | 4 (27 %) | 4 (27 %) | 7 (47 %) |
| Haploinsufficiency (n = 21) | 12 (57 %) | 1 (5 %) | 8 (38 %) |
OI osteogenesis imperfecta
Table 4.
Relationship between clinical characteristics and different mutation types (helical mutation vs. haploinsufficiency) in COL1A1 and COL1A2 of 71 patients with OI at the time of bone densitometry analysis
| Helical mutation (n = 29) | Haploinsufficiency (n = 42) | p value | |
|---|---|---|---|
| OI Type (I/III/IV) | 9/4/16 | 33/1/8 | <0.001 |
| COL1A1/COL1A2 mutation | 9/20 | 42/0 | <0.001 |
| Gender (M/F) | 12/17 | 15/27 | 0.635 |
| Age (years) | 21.8 ± 17.1 | 21.4 ± 17.0 | 0.941 |
| Height SDS | −2.93 ± 3.04 | −0.61 ± 1.61 | <0.001 |
| Weight SDS | −1.29 ± 1.67 | −0.55 ± 1.44 | <0.05 |
| BMD SDS | −2.73 ± 1.96 (n = 24) | −1.70 ± 1.25 (n = 40) | <0.05 |
| Triangular face | 21 % | 17 % | 0.672 |
| Blue sclera | 83 % | 95 % | 0.085 |
| Dentinogenesis imperfecta | 62 % | 31 % | <0.01 |
| Hearing loss | 17 % | 21 % | 0.668 |
| Fracture at birth | 14 % | 5 % | 0.184 |
| Bone deformity | 69 % | 50 % | 0.115 |
| Scoliosis | 52 % | 26 % | <0.05 |
| Walking without assistance | 76 % (n = 25) | 98 % | <0.01 |
OI osteogenesis imperfecta, SDS standard deviation score, BMD bone mineral density
Fig. 1.
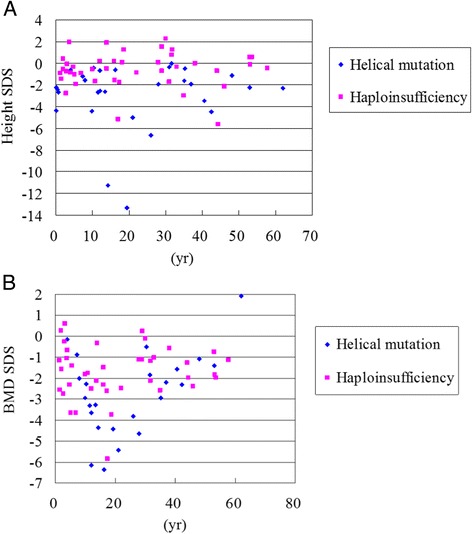
Relationships between age and a height SDS (n = 71), b BMD SDS (n = 64) of different mutation types [COL1A1/COL1A2 helical glycine mutation (blue dot) vs. COL1A1 haploinsufficiency mutation (pink dot)] in patients with osteogenesis imperfecta. SDS, standard deviation score; BMD, bone mineral density
Discussion
A number of studies have described the clinical and molecular findings for Western patients with OI [13–17]; however, only a few studies have been conducted in Asian patients [8, 18, 19]. To the best of our knowledge, this is the first large-scale study to analyze the genotypes and phenotypes of Taiwanese patients with OI. We identified 37 different COL1A1/COL1A2 mutations in 72 patients from 37 unrelated families, including 28 COL1A1 mutations and 9 COL1A2 mutations. Among these 37 COL1A1/COL1A2 mutations, 16 (43 %) were missense mutations, 11 (30 %) were frameshift mutations, 6 (16 %) were splicing mutations, and 4 (11 %) were nonsense mutations. These mutations were unique and were not repeated between the families, meaning that there was no hot-spot COL1A1/COL1A2 mutation in the study population. Fifteen (41 %) of the mutations were novel and 12 (32 %) were familial. In Zhang et al’s study [8], 19 of 58 (33 %) Chinese patients had familial OI, and Venturi et al. [17] reported that 7 of 22 (32 %) Italian OI patients were familial cases, both of which are consistent with our results. However, Lee et al. [18] described a higher incidence of familial OI with 18 of 34 (53 %) Korean patients. Further studies are needed to elucidate whether this is an artifactual difference related to a limitation in sample size or whether there are ethnic differences in the genetic defects underlying OI. Similar to previous population studies on OI in other countries [8, 14–19], most of our Taiwanese OI patients were categorized into type I (58 %), followed by type IV (35 %) and type III (7 %). In addition, none of the patients had OI type II in this study.
Growth retardation is a notable symptom of OI, and it is extremely severe in patients with OI type III. Moriwake and Seino [20] reported a national survey of Japanese OI patients with height SDSs of −3.36 ± 3.59,−7.83 ± 3.54, and − 4.63 ± 3.13 in patients with OI type I, III and IV, respectively, which is consistent with our findings.
Patients with OI have a significantly lower BMD. DEXA can detect a low BMD that may be missed on plain radiographs, even in patients with the milder forms of OI [21]. It may therefore aid in establishing the diagnosis, assessing the prognosis, and possibly monitoring the response to medical treatment. Among the 65 patients with available DEXA data of BMD in the present study, 94 % had a reduced BMD, 48 % with BMD SDS < −2, and 31 % with BMD SDS < −1 and ≧ − 2.
Blue sclera is a distinctive feature of unknown etiology in OI, and most patients with OI type I have blue sclera throughout their lives. In OI type III and IV, the sclera may also be blue at birth and during infancy, however the blue color fades with time during childhood [22]. The majority of our patients (89 %) had blue sclera.
DI is another principle manifestation of OI, and is associated with an abnormal type I collagen molecule. Discoloration and pulpal obliteration are the major characteristics. Lukinmaa et al. [23] reported that DI is frequently observed in OI type III and IV, but not commonly in type I. Our results were consistent with this finding, and 80 % of the OI type III patients had DI, followed by 56 % of the type IV patients and 31 % of the type I patients. We also found that the patients with DI appeared to be prone to developing bone deformities (p < 0.01) and scoliosis (p < 0.01).
Progressive hearing loss is an important symptom of OI, with the most common age at onset being in the second to fourth decades of life [24]. In our study, 19 % of the patients had hearing loss at a mean age of 39.1 years compared to 17.5 years without hearing loss, and an increase in age was associated with the presence of hearing loss (p < 0.01).
In patients with OI, the causes of bone deformities include an imperfect healing process after a fracture, and weight bearing itself without an apparent fracture. Bone deformities are frequently observed in the long bones, and scoliosis, spinal deformities, and compression fractures are also commonly seen [20]. In our study, 58 % of the patients presented with bone deformities.
The pathology of scoliosis is based on vertebral fragility, which progressively deteriorates with age [25]. Karbowski et al. [26] performed a nationwide cross-sectional study on German patients with OI, and reported that scoliosis was observed in 74.5 % (76/102) of their patients with an average age of 24.6 years. In our patients, 36 % (26/72) had scoliosis with a mean age of 27.3 years.
Rauch et al. [7] reported that girls with OI have a slightly higher BMD than boys with the same mutation type. Their histomorphometric observations also showed that bone formation and turnover rates were lower in girls than in boys. Lower bone turnover is expected to result in a slightly higher bone mass. The same results were also observed in our study.
There is a tendency to give more weighting to severity in phenotypic grouping whereas recent reviews stress that there is variability in severity encompassing mild to moderate phenotypes within families in patients with both OI type 1 and type 4 [3]. The persistence of deep blue-gray scleral hue should be given more weighting in favor of OI type 1 in families although scleral hue is difficult to assess in small children with OI type 4 who may have quite marked blue-gray sclera in infancy which subsequently fade to white in later childhood [22].
In our study cohort of Taiwanese OI patients, COL1A1 mutations occurred 3.1 times (28:9) more frequently than COL1A2 mutations, and the replacement of glycine residues by other residues within the Gly-X-Y triplet domain of the triple helix in both COL1A1 and COL1A2 was common. Our results are consistent with those of a previous study on Chinese patients [8]. Of the 15 glycine mutations, 6 (40 %) had glycine-to-aspartic acid substitutions, which is higher than previously reported in a mutation database (7.4 % in COL1A1 and 15.8 % in COL1A2) [27]. The mutation database also reported that serine substitutions were the most common type of mutation in both COL1A1 (38.9 %) and COL1A2 (44 %) [27], however, only 4 (27 %) serine substitutions were identified in our sample.
We found that the OI type was correlated with mutated genes and mutation types. Rauch et al. [7] and Zhang et al. [8] both reported that compared with COL1A2 mutations, COL1A1 mutations were more frequent in patients with OI type I and less frequent in those with OI type IV. Compared with haploinsufficiency mutations, helical mutations occurred more commonly in the patients with OI type III and IV, and less commonly in the patients with OI type I. Although the Sillence classification is based exclusively on phenotypic criteria and is inevitably used inconsistently among different OI investigators, our results were still similar to previous reports [7, 8].
COL1A1 haploinsufficiency mutations result in a quantitative defect, with synthesis of structurally normal type I procollagen at about half of the normal amount. They initiate nonsense-mediated decay of the mRNA derived from that allele leading to mild bone fragility. COL1A1/COL1A2 helical glycine mutations cause the synthesis of collagen molecules with structural abnormalities, which can result in a clinical severity from mild OI type I to lethal OI type II. Previous studies have reported that OI patients with COL1A1 haploinsufficiency mutations have milder bone fragility and damage than those with COL1A1/COL1A2 helical glycine mutations [7, 8, 27], and this is consistent with our results.
Limitations
Due to the limited sample size in this single-center retrospective study, substitutions by many amino acid residues as well as the position of a mutation occurred too infrequently to draw any conclusions about their phenotypic effects. In addition, our results were limited to BMD of the lumbar spine, which may not be representative of other skeletal involvement, especially the appendicular skeleton. The small sample size also reflects the rare nature of this genetic disorder, and both the age range and degree of disease severity varied widely. Therefore, further multicenter studies with larger cohorts and a longer follow-up period are warranted.
Conclusion
Thirty-seven mutations were identified in the COL1A1/COL1A2 genes and were associated with OI type I, III and IV by direct sequencing in our Taiwanese patients, including 28 COL1A1 mutations and 9 COL1A2 mutations. Among them, 15 (41 %) were novel mutations, 12 (32 %) were familial mutations, and 15 (41 %) were caused by the substitution of a glycine within the Gly-X-Y triplet domain of the triple helix. Patients with COL1A1/COL1A2 helical glycine mutations had more severely damaged skeletal phenotypes than those with COL1A1 haploinsufficiency mutations. Genotype and phenotype databases are expected to promote better genetic counseling and medical care of patients with OI.
Ethics approval
Mackay Memorial Hospital IRB.
Acknowledgments
This study was supported by research grants from the National Science Council, Taiwan (NSC-102-2314-B-195-006, NSC-104-2314-B-195-019, and NSC-102-2314-B-195-017-MY3) and Mackay Memorial Hospital (MMH-103-092, MMH-101-111 and MMH-I-S-600). The authors would like to express their thanks to Ms. Tsai-Feng Ho for her professional assistance in biostatistics.
Funding
This study was supported by research grants from the National Science Council, Taiwan (NSC-102-2314-B-195-006, NSC-104-2314-B-195-019, and NSC-102-2314-B-195-017-MY3) and Mackay Memorial Hospital (MMH-103-092, MMH-101-111 and MMH-I-S-600).
Abbreviations
- BMD
bone mineral density
- DEXA
dual energy X-ray absorptiometry
- DI
dentinogenesis imperfecta
- OI
osteogenesis imperfecta
- SDS
standard deviation score
Additional files
Clinical findings of 51 OI patients with mutations in COL1A1 (DOC 119 kb)
Clinical findings of 21 OI patients with mutations in COL1A2 (DOC 63 kb)
Footnotes
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
HYL was responsible for developing the protocol, patient screening and enrollment, outcome assessment, preliminary data analysis, and writing the manuscript. CKC and YNS participated in the development of the protocol and the analytical framework, and contributed to writing the manuscript. MRC, HCC and DMN were responsible for patient screening. Together with SPL, they also supervised the study design and execution, performed the final data analyses, and contributed to writing the manuscript. All authors read and approved the final manuscript.
References
- 1.Orioli IM, Castilla EE, Barbosa-Neto JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet. 1986;23:328–332. doi: 10.1136/jmg.23.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stoll C, Dott B, Roth MP, Alembik Y. Birth prevalence rates of skeletal dysplasias. Clin Genet. 1989;35:88–92. doi: 10.1111/j.1399-0004.1989.tb02912.x. [DOI] [PubMed] [Google Scholar]
- 3.Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014;164A:1470–1481. doi: 10.1002/ajmg.a.36545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin HY, Lin SP, Chuang CK, Chen MR, Chang CY, Niu DM. Clinical features of osteogenesis imperfecta in Taiwan. J Formos Med Assoc. 2009;108:570–576. doi: 10.1016/S0929-6646(09)60375-2. [DOI] [PubMed] [Google Scholar]
- 5.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Dijk FS, Byers PH, Dalgleish R, Malfait F, Maugeri A, Rohrbach M, et al. EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta. Eur J Hum Genet. 2012;20:11–19. doi: 10.1038/ejhg.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rauch F, Lalic L, Roughley P, Glorieux FH. Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res. 2010;25:1367–1374. doi: 10.1359/jbmr.091109. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZL, Zhang H, Ke YH, Yue H, Xiao WJ, Yu JB, et al. The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J Bone Miner Metab. 2012;30:69–77. doi: 10.1007/s00774-011-0284-6. [DOI] [PubMed] [Google Scholar]
- 9.Chen W, Chang MH. New growth charts for Taiwanese children and adolescents based on World Health Organization standards and health-related physical fitness. Pediatr Neonatol. 2010;51:69–79. doi: 10.1016/S1875-9572(10)60014-9. [DOI] [PubMed] [Google Scholar]
- 10.Shu SG. Bone mineral density and correlation factor analysis in normal Taiwanese children. Acta Paediatr Taiwan. 2007;48:323–327. [PubMed] [Google Scholar]
- 11.Yeh LR, Chen CK, Lai PH. Normal bone mineral density in anteroposterior, lateral spine and hip of Chinese men in Taiwan: effect of age change, body weight and height. J Chin Med Assoc. 2004;67:287–295. [PubMed] [Google Scholar]
- 12.Lin HY, Lin SP, Chuang CK, Chen MR, Chang CY. Intravenous pamidronate therapy in Taiwanese patients with osteogenesis imperfecta. Pediatr Neonatol. 2008;49:161–165. doi: 10.1016/S1875-9572(09)60002-4. [DOI] [PubMed] [Google Scholar]
- 13.Ward LM, Lalic L, Roughley PJ, Glorieux FH. Thirty-three novel COL1A1 and COL1A2 mutations in patients with osteogenesis imperfecta types I-IV. Hum Mutat. 2001;17:434. doi: 10.1002/humu.1124. [DOI] [PubMed] [Google Scholar]
- 14.Ries-Levavi L, Ish-Shalom T, Frydman M, Lev D, Cohen S, Barkai G, et al. Genetic and biochemical analyses of Israeli osteogenesis imperfecta patients. Hum Mutat. 2004;23:399–400. doi: 10.1002/humu.9230. [DOI] [PubMed] [Google Scholar]
- 15.Reis FC, Alexandrino F, Steiner CE, Norato DY, Cavalcanti DP, Sartorato EL. Molecular findings in Brazilian patients with osteogenesis imperfecta. J Appl Genet. 2005;46:105–108. [PubMed] [Google Scholar]
- 16.Pollitt R, McMahon R, Nunn J, Bamford R, Afifi A, Bishop N, et al. Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with osteogenesis imperfecta type I-IV. Hum Mutat. 2006;27:716. doi: 10.1002/humu.9430. [DOI] [PubMed] [Google Scholar]
- 17.Venturi G, Tedeschi E, Mottes M, Valli M, Camilot M, Viglio S, et al. Osteogenesis imperfecta: clinical, biochemical and molecular findings. Clin Genet. 2006;70:131–139. doi: 10.1111/j.1399-0004.2006.00646.x. [DOI] [PubMed] [Google Scholar]
- 18.Lee KS, Song HR, Cho TJ, Kim HJ, Lee TM, Jin HS, et al. Mutational spectrum of type I collagen genes in Korean patients with osteogenesis imperfecta. Hum Mutat. 2006;27:599. doi: 10.1002/humu.9423. [DOI] [PubMed] [Google Scholar]
- 19.Kataoka K, Ogura E, Hasegawa K, Inoue M, Seino Y, Morishima T, et al. Mutations in type I collagen genes in Japanese osteogenesis imperfecta patients. Pediatr Int. 2007;49:564–569. doi: 10.1111/j.1442-200X.2007.02422.x. [DOI] [PubMed] [Google Scholar]
- 20.Moriwake T, Seino Y. Recent progress in diagnosis and treatment of osteogenesis imperfecta. Acta Paediatr Jpn. 1997;39:521–527. doi: 10.1111/j.1442-200X.1997.tb03631.x. [DOI] [PubMed] [Google Scholar]
- 21.Moore MS, Minch CM, Kruse RW, Harcke HT, Jacobson L, Taylor A. The role of dual energy x-ray absorptiometry in aiding the diagnosis of pediatric osteogenesis imperfecta. Am J Orthop. 1998;27:797–801. [PubMed] [Google Scholar]
- 22.Sillence D, Butler B, Latham M, Barlow K. Natural history of blue sclerae in osteogenesis imperfecta. Am J Med Genet. 1993;45:183–186. doi: 10.1002/ajmg.1320450207. [DOI] [PubMed] [Google Scholar]
- 23.Lukinmaa PL, Ranta H, Ranta K, Kaitila I. Dental findings in osteogenesis imperfecta: I. Occurrence and expression of type I dentinogenesis imperfecta. J Craniofac Genet Dev Biol. 1987;7:115–125. [PubMed] [Google Scholar]
- 24.Kuurila K, Kaitila I, Johansson R, Grénman R. Hearing loss in Finnish adults with osteogenesis imperfecta: a nationwide survey. Ann Otol Rhinol Laryngol. 2002;111:939–946. doi: 10.1177/000348940211101014. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe G, Kawaguchi S, Matsuyama T, Yamashita T. Correlation of scoliotic curvature with Z-score bone mineral density and body mass index in patients with osteogenesis imperfecta. Spine (Phila Pa 1976) 2007;32:E488–E494. doi: 10.1097/BRS.0b013e31811ec2d9. [DOI] [PubMed] [Google Scholar]
- 26.Karbowski A, Schwitalle M, Eckardt A. Scoliosis in patients with osteogenesis imperfecta: a federal nation-wide cross-sectional study. Z Orthop Ihre Grenzgeb. 1999;137:219–222. doi: 10.1055/s-2008-1037397. [DOI] [PubMed] [Google Scholar]
- 27.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]