

Abstract
Locust bean gum (LBG) was blended with a cellulose/methacrylate-based interpolyelectrolyte complex (IPEC) to assess the hydro-erosive influence of addition of a polysaccharide on the disposition and drug delivery properties inherent to IPEC matrix. The addition of LBG modulated the drug (levodopa) release characteristics of the IPEC by reducing excessive swelling and preventing bulk erosion. After 8 h in pH 4.5 dissolution medium, gravimetric analysis established that IPEC tablet matrix eroded by 30% of the initial weight due to bulk erosion while LBG-blended IPEC (LBG-b-IPEC) demonstrated surface erosion accounting to 62% of initial weight (596→226.8 mg). Mathematical modeling of the drug release data depicted a transformation from non-Fickian mechanism (IPEC matrices) to zero-order drug release pattern (LBG-b-IPEC matrices) with the linearity of release profile being close to 1 (R2 = 0.99). Physicochemical characterizations employing Fourier transform infrared spectroscopy (FTIR) and differential scanning calorimetry (DSC) explicated that LBG interacted with IPEC by its hydrophilic groups associating with the existing water-holding bodies of IPEC to produce compact matrices. The lattice atomistic modeling elucidated that LBG acted as a linker with the formation of intra- and intermolecular hydrogen bonds generating a highly stabilized polysaccharide-polyelectrolytic structure which influenced the improved properties observed.
KEY WORDS: controlled release, interpolyelectrolyte complex, levodopa, locust bean gum, oral drug delivery, swelling and erosion
INTRODUCTION
Macromolecular carbohydrate polymers are widely employed as drug carriers, additives, and excipients in order to achieve the desired drug delivery attributes. The use and manipulation of such polymers have led to design and development of novel drug delivery systems which have enhanced delivery, bioavailability, and targeting of drugs. Hence, polymers are employed to achieve different goals such as controlling the duration of action of a drug and its level in the human body; to target drugs to particular organs, tissues, or cells in the body; and to overcome certain tissue and cellular barriers (1).
In general, the performance of the polymers utilized for each drug delivery system is dependent on their physicochemical properties such as constituents, chain length, and functional groups. A polymer is selected based on the understanding of its surface and bulk properties which are explored to give the desired chemical, interfacial, mechanical, and biological functions (2). The surface properties which determine interfacial interactions include hydrophobicity or hydrophilicity, lubricity, smoothness, and surface energy (3). These properties influence adsorption, adhesion, and biocompatibility, and properties such as water adsorption influence mechanical strength, creep resistance, and durability (3). The bulk properties include permeability, rates of diffusion, and degradation which influence the duration of application of the polymer. These properties are in turn influenced by polymer architecture, molecular weights, and solubility among others.
To modify, improve, or optimize a polymer for a specific drug delivery or achieve tailor-made properties, polymers have been altered by conjugation, grafting, crosslinking, functionalization, depolymerization and polymerization, and polymer blending (4,5). Polymer blending is a physical process of mixing two or more polymers enhanced under mechanical or temperature field. When a chemical reaction is involved, the rates of chemical reactions are influenced by blending components, temperature gradients, and diffusion of reagents and products (6). Polymer blending is one of the commonly employed methods for modification of polymers to improve their physicochemical characteristics. Synthesis and characterization of new polymers may be time-consuming, and all the desired properties may still not be achieved by synthesizing a polymer. However, blending of two polymers with their individual properties may result in optimum characteristics for the desired drug delivery system. In polymer blending, the polymer compositions can be manipulated and controlled in conformity to experimental requirements (7). It is a convenient and less time-consuming alternative to copolymerization and other techniques and also produces new materials with superior properties over the individual polymers utilized (8,9). In addition, polymer blending is cheap, simple to perform, versatile, and effective (10,11).
Blending of polymers exhibits synergism of properties leading to such enhancements as easy fabrication of devices, manipulation of device properties (hydration, degradation rate, and mechanical strength), drug loading, and utilization of the dispersed phase domains as micro-reservoirs for enhanced release properties (12). For instance, hydrophilicity of polycaprolactone has been enhanced by blending with a polysaccharide, chitosan (13). Polymer blending has been employed to increase rate of degradation, control water content thereby manipulating permeability (8,14); control and extend drug release (15,16); achieve zero-order kinetics (17) moderate swelling (15,18), modify protein delivery (19) and modulate porosity (20). The popular techniques of polymer blending include melting and aqueous blending (21). These techniques are usually used for natural polymer blends due to their hydrophilicity. However, the properties obtained after blending is dependent on the compatibility and miscibility of the polymers. The possibility of two or more polymers mixing depends on the polymers, their compositions, and miscibility (22). A polymer blend is said to be miscible when two polymers produce a blend with properties comparable to those of a single-phase material and can be said to be compatible (23). Furthermore, a polymer blend is said to be immiscible and non-compatible when it shows signs of phase separation or opacity (23).
This study was undertaken to explore the possibilities of improving the drug release characteristics of an interpolyelectrolyte complex (IPEC) by the addition of a natural polysaccharide, locust bean gum (LBG). LBG is a neutral polysaccharide and classified as a galactomannan. It has been shown to exhibit synergistic interactions with other polysaccharides such as xanthan, k-carrageenan, guar gum, and karaya thereby modulating release mechanisms (24,25). It has also been previously demonstrated that a cationic methacrylate copolymer (Eudragit E100) interacts with sodium carboxymethylcellulose (NaCMC) to form an IPEC (26). NaCMC, a traditional hydrogel forming carbohydrate derivative, has a tendency to lose its shape in acidic medium exhibiting a first-order drug release profile. Methacrylate copolymer such as Eudragit E100, mainly used as a film-coating agent, was observed to solubilize with ease in an acidic medium and so cannot be used for controlled release delivery. However, a series of studies undertaken in our laboratories indicated that the IPEC formed between the methacrylate copolymer and NaCMC has the potential to form a hydrogel maintaining its three-dimensional shape in acidic medium exhibiting a linear in vitro drug release over 24 h (26–29). However, it underwent excessive swelling and bulk erosion. In an attempt to modulate swelling and erosion, LBG was added and a comparative study between the IPEC and the polymer blend (LBG-b-IPEC) was undertaken. Modulating the release mechanism may enhance the delivery of the model drug, levodopa, at a constant rate over a prolonged period of time. Thermal and structural analyses, swellability and erosion studies, in vitro drug release studies, and molecular mechanistic simulations were undertaken to determine the hydro-erosive influence of the polysaccharide on the performance of drug-loaded IPEC matrices.
MATERIALS AND METHODS
Materials
Methacrylate copolymer based on dimethylaminoethyl methacrylate, butyl methacrylate, and methyl methacrylate with a ratio of 2:1:1 (Eudragit E100, Evonik Röhm GmbH & Co. KG, Darmstadt, Germany); sodium carboxymethylcellulose (NaCMC, Fluka Biochemika, Medium viscosity, Sigma-Aldrich Chemie GmbH, Buchs, Switzerland); locust bean from Ceratonia siliqua seeds (Sigma-Aldrich Inc, Steinheim, Germany); 3-(3,4-dihydroxyphenyl)-l-alanine (levodopa, Sigma-Aldrich, Steinheim, Germany); acetic acid glacial (Rochelle Chemicals, South Africa); hydrochloric acid (Rochelle Chemicals, South Africa); potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, Steinheim, Germany); potassium chloride (KCl) (Saarchem, Krugersdorp, South Africa); and ortho-phosphoric acid (BDH Chemicals, Poole, England).
Synthesis of Interpolyelectrolyte Complex
Methacrylate copolymer (Eudragit E100) beads were milled and 0.84 g was dissolved in various concentration of acetic acid −0.1, 0.2, 0.4, 0.6, 0.8, and 1.0 N. A quantity 1.68 g of sodium carboxymethylcellulose (NaCMC) was dissolved in deionized water under agitation. Thereafter, transparent solution of methacrylate copolymer was added to NaCMC and stirred vigorously for 2 to 3 h. The interpolyelectrolyte complex so formed was collected, lyophilized for 48 h, and pulverized (26,28).
Formation of A Polymer Blend (LBG-b-IPEC)
Interpolyelectrolyte complex (IPEC) of E100 and NaCMC was synthesized as described above in the same quantities and various concentration of acetic acid. After the complex formation, 1.68 g of locust bean was added to each IPEC with different concentrations of acetic acid under continuous agitation for 15 min. The polymer blend was lyophilized for 48 h and milled.
Fourier Transform Infrared (FTIR) Spectroscopy of the IPEC and LBG-b-IPEC
For the elucidation of the interactions and structural modification of IPEC from the native polymers, FTIR spectra of milled E100, NaCMC, the IPEC and LBG-b-IPEC were obtained with a PerkinElmer spectrometer (PerkinElmer Spectrum 100, Beaconsfield, United Kingdom) over a range of 4000–650 cm−1.
Differential Scanning Calorimetry of the IPEC and LBG-b-IPEC
To compare the thermal properties of the IPEC and LBG-b-IPEC, differential scanning calorimetry (DSC 1 STARe system, Mettler Toledo, Schwerzenbach, Switzerland) was undertaken. IPEC and LBG-b-IPEC were subjected to a heating gradient ranging from −10 to 270°C at a rate of 5°C per minute under a nitrogen atmosphere. While temperature-modulated differential scanning calorimetry (TMDSC) was undertaken at a heating gradient ranging from −10 to 130°C and 130 to 250°C with a heating rate of 1°C and amplitude of 0.3°C at a period of 0.8 min. The samples of IPEC analyzed were based on the normalities of acetic acid in the range of 0.1–1.0 N while mass of the samples were in the range of 10.3–10.8 mg for DSC and 5.0–5.5 mg for TMDSC.
Direct Compression of the IPEC and LBG-b-IPEC
A quantity of the model drug 3-(3,4-dihydroxyphenyl)-l-alanine (levodopa) 100 mg per matrix was blended with 500 mg of IPEC powder and directly compressed using a Carver Hydraulic Press (Carver Industries Inc., Wabash, IN) at 3 tons. This was also applied to the LBG-b-IPEC thereby producing drug-loaded IPEC and LBG-b-IPEC matrices. The matrices were utilized for swelling and in vitro drug release comparative studies.
Swellability and Erosion Studies of Directly Compressed Matrices
The swelling tests for the IPEC and LBG-b-IPEC matrices were undertaken in pH 1.5 and 4.5. Before submerging the matrices into pH 1.5 (100 mL HCl/KCl standard buffer solution), the matrices were weighed and placed in mesh baskets. Thereafter, the submerged mesh baskets were placed in a shaker bath (Orbital Shaker incubator, LM-530, Laboratory & Scientific Equipment Co, South Africa) at 37°C over 24 h. Increase in weights was determined gravimetrically at 2-h intervals, and swellability was obtained mathematically using Eq. 1:
| 1 |
where Wt is the weight of the hydrogel at time t and W0 is the weight of hydrogel at time zero.
Mesh basket was not employed for swellability and erosion in pH 4.5 due to erosion of matrices. However, the ring mesh assembly was used to ensure the release of the eroded particles into the medium for more accurate determination of erosion and swelling. Swelling and erosion studies were undertaken in a dissolution system (Erweka DT 700, Erweka GmbH, Heusenstamm, Germany) at 37 ± 0.5°C and 50 rpm over 8 h. The matrices were weighed at 2-h intervals and at the end of eighth hour; the samples were lyophilized to determine the final weight after erosion. Variation in weight was determined gravimetrically while the degrees of swelling and erosion were graphically represented.
In Vitro Drug Release Studies and Data Analysis
To obtain drug release profiles for the IPEC and LBG-b-IPEC matrices, dissolution testing was undertaken employing a USP Apparatus II dissolution system (Erweka DT 700, Erweka GmbH, Heusenstamm, Germany) with the temperature and stirring rate kept at 37 ± 0.5°C and 50 rpm, respectively. The dissolution media (900 mL) comprised buffer of pH 1.5 and 4.5. Samples (5 mL) were withdrawn at intervals over 24 h, and the quantity of levodopa released per time was assessed by UV spectroscopy (LAMBDA 25 UV/Vis spectrophotometer, PerkinElmer, MA, USA). The drug release profiles of the IPEC and LBG-b-IPEC matrices were compared. The drug release profiles were analyzed via dissolution efficiency, first-order and zero-order kinetics, as well as the Higuchi and Korsmeyer-Peppas relationships (26) using DDSolver software.
Static Lattice Atomistic Simulations
All modeling procedures and calculations were performed using the HyperChemTM 8.0.8 Molecular Modeling System (Hypercube Inc., Gainesville, Florida, USA) and ChemBio3D Ultra 11.0 (CambridgeSoft Corporation, Cambridge, UK). The E100 polymer was drawn using ChemBio3D Ultra in its syndiotactic stereochemistry as a 3D model whereas the structures of CMC (four saccharide units) and locust bean gum (LBG) were built from standard bond lengths and angles using sugar builder module on HyperChem 8.0.8. The models were energy-minimized using a progressive-convergence strategy where initially the MM+ force field was used followed by energy minimization using the Amber 3 (Assisted Model Building and Energy Refinements) force field. The conformer having the lowest energy was used to create the polymer-polymer and polymer-solvent complexes. A complex of one polymer molecule with another was assembled by disposing the molecules in a parallel way, and the same procedure of energy-minimization was repeated to generate the final models: CMC-LBG, E100-LBG, and CMC-E100-LBG. Full geometry optimization was carried out in vacuum employing the Polak-Ribiere conjugate gradient algorithm until an RMS gradient of 0.001 kcal/mol was reached. For molecular mechanics calculations in vacuum, the force fields were utilized with a distance-dependent dielectric constant scaled by a factor of 1. The 1–4 scale factors were electrostatic 0.5 and van der Waals 0.5 (30).
RESULTS AND DISCUSSION
Synthesis of IPEC and Formation of LBG-b-IPEC
The stoichiometric ratio for a homogenous interaction between E100 and NaCMC is 0.5:1, respectively. The association interactions are achieved by hydrogen bonding, hydrophilic interactions, and ionic interactions enhanced in the presence of acetic acid, and these interactions were ascertained by the atomic simulations. LBG-b-IPEC was light brown due to the color of locust bean. There was an observed thickening of the blend on addition of locust bean, and this was expected due to the nature of locust bean. However, the blending was undertaken under ambient temperature and so locust bean did not swell excessively.
FTIR Spectroscopy of the IPEC and LBG-b-IPEC
The structural differences of the IPEC and LBG-b-IPEC in comparison to the native polymers are shown in Fig. 1. As observed from the figure, the characteristic peaks of methacrylate copolymers at 2877.05, 1452.94, 1342.09, 1060.48, 961.33, 842.31, and 747.27 cm−1 are absent in the IPEC as well as in the LBG-b-IPEC. However, its characteristic distinct peak at 1722.46 cm−1 shifted to 1725.04 cm−1 in both the IPEC and LBG-b-IPEC while two other peaks, 1271.15 and 1239.97 cm−1, shifted to 1266.47 and 1241.50 cm−1, respectively, in both the IPEC and LBG-b-IPEC. However, the last two peaks are more pronounced in IPEC.
Fig. 1.
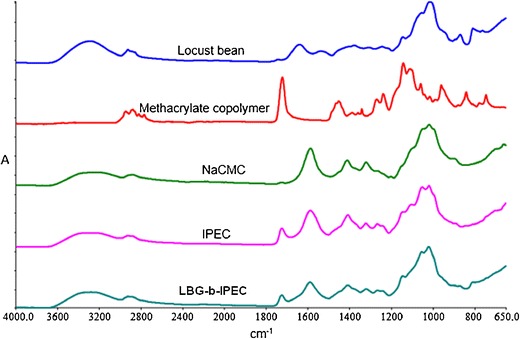
FTIR spectra of native polymers—locust bean, methacrylate copolymer, and NaCMC; the IPEC and LBG-b-IPEC
Not much difference was seen between the spectra of the IPEC and LBG-b-IPEC apart from a slight difference in shift of some peaks. Peak at frequency 1019.53 cm−1 of IPEC shifted to 1021.12 cm−1 in LBG-b-IPEC and is more distinct. Broader peak of LBG-b-IPEC at 1590.79 cm−1 as compared to the IPEC may be due to the superimposing peak 1639.22 cm−1 of locust bean. The peak at frequency 1104.16 cm−1 of IPEC is not seen in ELC while peak 810.75 cm−1 of LBG-b-IPEC is not seen in the IPEC which may be due to the fact that the peak is peculiar to locust bean. In summary, there is no characteristic difference between the IPEC and LBG-b-IPEC which may be an indication that the addition of locust bean to the IPEC did not necessarily generate a chemical change. This suggests that the interactions between LBG and IPEC may be physical.
Differential Scanning Calorimetry of the IPEC and LBG-b-IPEC
The thermograms of the three native polymers, E100, NaCMC, and locust bean, are shown in Fig. 2a. The characteristic melting peak of methacrylate polymer is found at ~58°C while NaCMC and LBG have the characteristic polysaccharide broad endotherms at ~110 and ~90°C, respectively, which is attributed to the loss of water. The broad endotherms of DSC thermograms for NaCMC and LBG collaborate the broad peaks (between 3600 and 3000 cm−1) of FTIR attributed to the presence of hydroxyl groups. As the temperature increases to 210°C, decomposition of the polymers occurs as shown by the upward movement of the thermograms. Comparing Fig. 2b, c, there seemed not to be much difference in the thermograms except perhaps in the depth of the endotherms. IPEC thermograms have shallow endotherms while those of LBG-b-IPEC have deep endotherms. Furthermore, the presence of locust bean in the blend slightly lowered the temperature of the endotherms. For instance, IPEC 0.1 N’s endotherm occurred at ~85°C while that of LBG-b-IPEC 0.1 N occurred at ~80°C. The thermograms for IPEC and LBG-b-IPEC were homogenous indicating the complete interactions and miscibility and so each thermogram appeared as a thermogram for one substance/material.
Fig. 2.
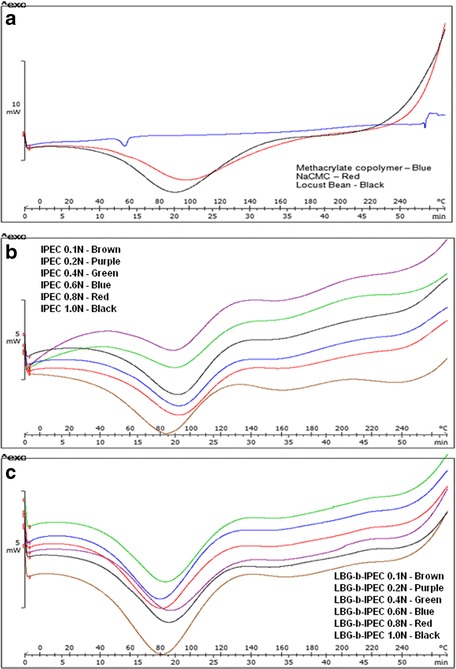
Differential scanning calorimetric curves of a native polymers—methacrylate copolymer, NaCMC, and locust bean; b IPEC synthesized at different normalities of acetic acid; and c LBG-b-IPEC synthesized with different normalities of acetic acid
However, to further compare the thermal transitions of IPEC and LBG-b-IPEC, ADSC was undertaken. The crystallization and melting endotherms were observed in both IPEC and LBG-b-IPEC (data not shown). However, while IPEC exhibited a sinusoidal modulation only, LBG-b-IPEC exhibited a combination of sinusoidal and meander-type of modulation. Although NaCMC had combined modulations, IPEC did not, possibly due to the fact that IPEC is a chemical modification behaving as one material with its own thermal properties. Furthermore, the thermograms of IPEC differed from the thermograms of E100 confirming the observation that it is a new material with its own thermal properties. Introduction of locust bean into IPEC influenced the thermal behavior exhibited by LBG-b-IPEC. Locust bean at −10 to 130°C exhibited a meander-type of modulation only, and therefore, LBG-b-IPEC was exhibiting that aspect of locust bean. In the DSC thermogram, other than the deep endotherm, locust bean transitions were not seen in LBG-b-IPEC thermograms. However, ADSC elucidated the thermal transitions of IPEC and locust bean in ELC clarifying that the blend between IPEC and locust bean may be a physically miscible polymer blend. This, however, was further clarified by molecular mechanics simulations.
Swellability and Erosion Studies of Directly Compressed Matrices
The degree of swelling of both IPEC and LBG-b-IPEC differed in different media (Figs. 3 and 4). Swelling of IPEC in pH 4.5 is about twice the swelling in pH 1.5. Incorporation of locust bean into IPEC decreased the degree of swelling in both media. This is attributed to the fact that the hydrophilic groups of locust bean associated with the existing water bodies of IPEC thereby decreasing the water-holding capacity of the hydrophilic pockets of IPEC. The degree of swelling was dependent on pH and the normality of acetic acid used to synthesize IPEC. In general, as the normality of acetic acid increased from 0.1 to 1.0 N, the swelling decreased in both media. In pH 1.5, IPEC and LBG-b-IPEC matrices swelled but retained their three-dimensional shape (Fig. 4a, b). Decrease in swelling due to the addition of LBG is corroborated by a study in which the inclusion of a polysaccharide, chitosan/alginate, reduced the swelling of IPEC tablets (31). However, in pH 4.5 as the matrix swelled, the polymer begun to dissociate with evidence of surface erosion. As observed with IPEC matrices, rate of swelling was much higher than rate of surface erosion. As swelling continued, bulk erosion occurred from the 16th hour as observed during dissolution studies. IPEC’s process of degradation is a combination of surface and bulk erosion. Initially, the degradation process was faster than diffusion leading to surface erosion. However, as there was continuous ingress of the medium into the matrix, diffusion overtook degradation leading to a burst of the matrix. However, in order to determine final weight after erosion, the swelling and erosion studies were set to end at the eighth hour. In the presence of a locust bean, the rate of surface erosion exhibited by the LBG-b-IPEC was more than the rate of swelling and bulk erosion did not occur at any point. The interactions that occurred during the blending of locust bean with IPEC engineered a constant process of degradation of the LBG-b-IPEC. The decrease in the water-holding capacity of IPEC due to the interactions with locust bean also would decrease the rate of diffusion by the reason of reduced ingress of the medium into the matrix. The interactions which led to surface over bulk erosion may be hydrogen bonding or ionic interactions. The degree of erosion is shown in Table I indicating the final weight after lyophilization. LBG-b-IPEC eroded more than IPEC in pH 4.5 for the different normalities of acetic acid.
Fig. 3.
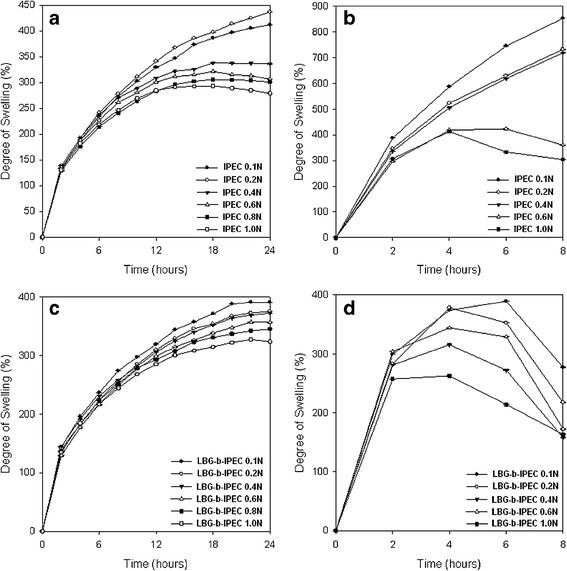
The degree of swelling profiles of a IPEC matrices (pH 1.5), b IPEC matrices (pH 4.5), c LBG-b-IPEC matrices (pH 1.5), and d IPEC-LBG polymeric blend matrices (pH 4.5)
Fig. 4.

Digital images of swollen matrices in pH 1.5 after 24 h: a IPEC matrix and b LBG-b-IPEC blend matrix
Table I.
The degree of erosion (at the eighth hour) by gravimetric method
| Polymer composition | Normality of acetic acid | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 N | 0.2 N | 0.4 N | 0.6 N | 1.0 N | ||||||
| Initial | Final | Initial | Final | Initial | Final | Initial | Final | Initial | Final | |
| IPEC (mg) | 573.2 | 400.7 | 570.9 | 351.2 | 548.6 | 336.7 | 569.7 | 199.0 | 554.4 | 159.9 |
| Polymer blend (mg) | 596.8 | 226.8 | 588.3 | 180.8 | 590.3 | 173.0 | 571.2 | 162.0 | 582.3 | 105.7 |
Swellability studies were undertaken due to its influence on drug release. As the polymer matrix hydrates and swells, the drug diffuses out into the medium. Generally, an increase in rate of swelling increases the rate of drug release. This is achieved by the creation of pores by the ingress of solvent into the matrix through which the drug diffuses out after dissolving. Hence, the more the matrix is hydrated, the more the drug dissolves and diffuses out. However, the IPEC does not swell to its maximum in a few hours, thus continued to swell throughout the 24-h study in pH 1.5 which is indicative that it has the potential to offer a controlled release delivery of drugs and hence reduce frequency of dosage.
The blocking of the pylorus is not envisaged due to the inherent ability of the IPEC to dissociate with time. The presence of food increases the pH of the stomach, and since the IPEC responds to pH, it will dissociate, experience surface erosion, and as observed, undergo bulk erosion at some point. The LBG-b-IPEC is also not expected to block the pylorus as it degrades by surface erosion over time and so will not be retained permanently in the stomach.
In Vitro Drug Release Studies
Slight differences in drug release profiles (Fig. 5a–d) were observed with the matrices prepared with the IPEC and LBG-b-IPEC synthesized at different normalities of acetic acid. Rate of release was slightly increased as the normality increased. Both the IPEC and LBG-b-IPEC released levodopa over 24 h thereby providing a controlled delivery. The linearity of some of the profiles indicated the possible release of levodopa at a constant rate thereby achieving a constant concentration of the drug over a prolonged period. The release mechanism of the IPEC and LBG-b-IPEC in pH 1.5 was observed to be a diffusion-controlled process which comprised hydration and swelling of the matrix, and then dissolution and diffusion of the drug out of the matrix. However, in pH 4.5, the release mechanisms of the IPEC matrices were dependent on the surface and bulk erosion of matrix as well as diffusion-controlled mechanism while that of LBG-b-IPEC was dependent on the surface erosion of matrix and diffusion-controlled mechanism.
Fig. 5.
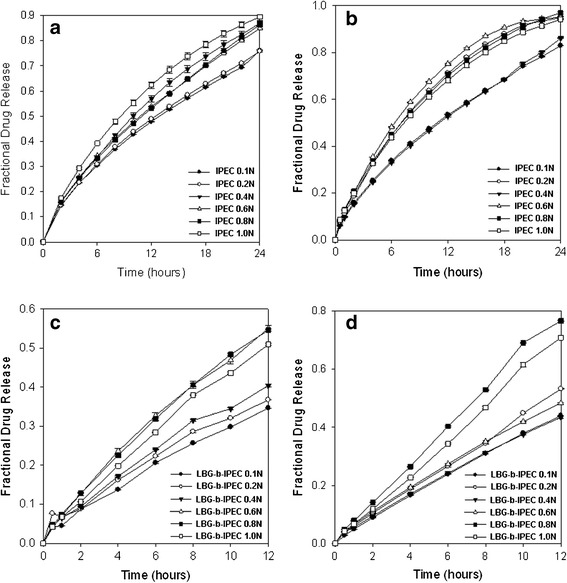
Drug release profiles of a IPEC formulations (pH 1.5), b LBG-b-IPEC formulations (pH 1.5), c IPEC formulations (pH 4.5), and d LBG-b-IPEC formulations (pH 4.5)
To analyze and compare the rates of drug release from the IPEC and LBG-b-IPEC, mathematical models were employed. The drug release profiles were fitted into zero-order and first-order kinetics and the data is shown in Table II. Both the IPEC and LBG-b-IPEC formulations fitted into zero- and first-order kinetics; however, the fitness differed in the two dissolution media. In pH 1.5, the best fit model for the formulation was first order with the exceptions of IPEC (IPEC 0.8 N) and polymeric blend (LBG-b-IPEC 0.4 N) whose best fit was zero order. In pH 4.5, the best fit for most of IPEC formulations was first order while the best fit for most of LBG-b-IPEC formulations was zero order. Therefore, addition of locust bean to the IPEC enhanced IPEC delivery to follow zero-order release kinetics. This is corroborated by a study indicating that polymer blending can modulate drug release to follow zero-order kinetics (17).
Table II.
Mathematical modeling of drug release profiles of IPEC and polymeric blend
| Drug device | Zero order | Zero order | First order | First order | DE (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| pH 1.5 | pH 4.5 | pH 1.5 | pH 4.5 | ||||||
| k o | r 2 | k o | r 2 | k 1 | r 2 | k 1 | r 2 | ||
| EC 0.1 N | 0.0287 | 0.9688 | 0.0281 | 0.9904 | −0.0233 | 0.9879 | −0.0147 | 0.9980 | 59.37 |
| EC 0.2 N | 0.0291 | 0.9665 | 0.0289 | 0.9777 | −0.0238 | 0.9948 | −0.0152 | 0.9926 | 60.71 |
| EC 0.4 N | 0.0344 | 0.9687 | 0.0328 | 0.9867 | −0.0356 | 0.9865 | −0.0178 | 0.9959 | 59.01 |
| EC 0.6 N | 0.0328 | 0.9724 | 0.0454 | 0.9898 | −0.0320 | 0.9776 | −0.0281 | 0.9986 | 60.01 |
| EC 0.8 N | 0.0335 | 0.9786 | 0.0454 | 0.9924 | −0.0338 | 0.9689 | −0.0282 | 0.9993 | 59.07 |
| EC 1.0 N | 0.0351 | 0.9487 | 0.0421 | 0.9946 | −0.0398 | 0.9921 | −0.0252 | 0.9976 | 64.10 |
| ELC 0.1 N | 0.0331 | 0.9737 | 0.0365 | 0.9979 | −0.0294 | 0.9887 | −0.0207 | 0.9986 | 60.58 |
| ELC 0.2 N | 0.0401 | 0.9365 | 0.0430 | 0.9983 | −0.0540 | 0.9899 | −0.0262 | 0.9845 | 67.56 |
| ELC 0.4 N | 0.0341 | 0.9818 | 0.0356 | 0.9960 | −0.0316 | 0.9696 | −0.0200 | 0.9990 | 58.33 |
| ELC 0.6 N | 0.0402 | 0.9155 | 0.0396 | 0.9949 | −0.0570 | 0.9938 | −0.0232 | 0.9990 | 70.25 |
| ELC 0.8 N | 0.0397 | 0.9462 | 0.0650 | 0.9974 | −0.0571 | 0.9604 | −0.0524 | 0.9705 | 65.92 |
| ELC 1.0 N | 0.0386 | 0.9452 | 0.0591 | 0.9981 | −0.0485 | 0.9869 | −0.0437 | 0.9680 | 66.17 |
Dissolution efficiency (DE) is referred to as the area under dissolution time curve within a time frame (t1-t2) and is expressed as percentage of dissolution time curve at maximum dissolution y100 over the same time frame (32). Dissolution efficiency can be employed to determine bioequivalence. The test and reference products can be said to be bioequivalent if the difference between their dissolution efficiencies is within appropriate limits and ±10% is usually used (32). In this study, IPEC formulation was used as reference and it was found out that though polymeric blend had slightly higher dissolution efficiencies, the IPEC and LBG-b-IPEC formulations could be said to be bioequivalent, and based on rate of drug release (on ± 10% limits), they can be used interchangeably. It is worthy to note that the degree of swelling was higher with the IPEC formulation which is directly proportional to the rate of drug release. Therefore, the DEs of LBG-b-IPEC formulations were slightly higher than those of IPEC formulations indicating the role of other factors rather than swelling.
To further analyze the drug release mechanisms, the data obtained were fitted into Higuchi and Korsmeyer-Peppas equations and the overall best fit model was identified for each formulation. Korsmeyer-Peppas is employed to determine the drug release pattern from polymeric systems when the mechanism of release is more than one or not well known (33). The outcomes of the modeling of the drug release data are shown in Table III while the best fit model for each device is shown in Table IV.
Table III.
Mathematical modeling of drug release profiles of the IPEC and polymeric blend
| Drug device | Higuchi | Higuchi | Korsmeyer-Peppas | Korsmeyer-Peppas | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 1.5 | pH 4.5 | pH 1.5 | pH 4.5 | |||||||
| K H | r 2 | K H | r 2 | K KP | r 2 | n | K KP | r 2 | n | |
| EC 0.1 N | 14.266 | 0.9759 | 8.844 | 0.9405 | 9.521 | 0.9993 | 0.65 | 5.028 | 0.9972 | 0.78 |
| EC 0.2 N | 14.514 | 0.9779 | 9.687 | 0.9556 | 9.848 | 0.9997 | 0.64 | 6.630 | 0.9860 | 0.69 |
| EC 0.4 N | 16.810 | 0.9697 | 10.457 | 0.9466 | 10.655 | 0.9984 | 0.67 | 6.274 | 0.9959 | 0.75 |
| EC 0.6 N | 16.126 | 0.9729 | 14.004 | 0.9361 | 10.371 | 0.9999 | 0.66 | 7.706 | 0.9990 | 0.79 |
| EC 0.8 N | 16.191 | 0.9653 | 13.995 | 0.9339 | 9.669 | 0.9999 | 0.69 | 7.527 | 0.9995 | 0.80 |
| EC 1.0 N | 17.971 | 0.9828 | 12.784 | 0.9230 | 13.404 | 0.9965 | 0.61 | 6.395 | 0.9986 | 0.84 |
| ELC 0.1 N | 15.813 | 0.9767 | 10.815 | 0.9095 | 10.412 | 0.9998 | 0.65 | 4.877 | 0.9999 | 0.89 |
| ELC 0.2 N | 19.894 | 0.9796 | 12.633 | 0.8969 | 15.479 | 0.9896 | 0.59 | 5.162 | 0.9976 | 0.93 |
| ELC 0.4 N | 15.917 | 0.9679 | 10.870 | 0.9254 | 9.513 | 0.9996 | 0.69 | 5.473 | 0.9993 | 0.83 |
| ELC 0.6 N | 20.570 | 0.9800 | 12.175 | 0.9308 | 17.660 | 0.9841 | 0.56 | 6.367 | 0.9997 | 0.82 |
| ELC 0.8 N | 19.862 | 0.9864 | 18.759 | 0.8892 | 15.665 | 0.9954 | 0.59 | 7.407 | 0.9976 | 0.95 |
| ELC 1.0 N | 19.331 | 0.9860 | 16.729 | 0.8718 | 15.283 | 0.9948 | 0.59 | 5.715 | 0.9982 | 1.02 |
Table IV.
The best fit models for the drug devices in the two dissolution media
| Drug device | Best fit model | Implication of n | Best fit model | Implication of n |
|---|---|---|---|---|
| pH 1.5 | pH 1.5 | pH 4.5 | pH 1.5 | |
| EC 0.1 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | First order | Non-Fickian/anomalous |
| EC 0.2 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | First order | Non-Fickian/anomalous |
| EC 0.4 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Non-Fickian/anomalous |
| EC 0.6 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Non-Fickian/anomalous |
| EC 0.8 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Non-Fickian/anomalous |
| EC 1.0 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Non-Fickian/anomalous |
| ELC 0.1 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Case II/zero order |
| ELC 0.2 N | First order | Non-Fickian/Anomalous | Zero order | Super case II |
| ELC 0.4 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Non-Fickian/anomalous |
| ELC 0.6 N | First order | Non-Fickian/Anomalous | Korsmeyer-Peppas | Non-Fickian/anomalous |
| ELC 0.8 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Super case II |
| ELC 1.0 N | Korsmeyer-Peppas | Non-Fickian/Anomalous | Korsmeyer-Peppas | Super case II |
The analysis of drug release mechanism by Korsmeyer-Peppas model was further explicated by the diffusional constant, n. For a cylindrical tablet matrix, where n = 0.5, the release mechanism is Fickian diffusion, non-Fickian release or anomalous transport if 0.45 < n < 0.89, case II transport or zero-order release if n = 0.89, and super case II transport if greater than 0.89 (33,34). The values for n are shown in Table III and the implications for each device are shown in Table IV.
As shown in Table IV, most of the devices/formulations fitted into Korsmeyer-Peppas model in both media—pH values of 1.5 and 4.5. Furthermore, the release exponent imply that the mechanism of release for most of the formulations involved a combination of mechanisms which may be envisaged to be hydration, swelling, dissolution, and diffusion in pH 1.5 and hydration, swelling, dissolution, diffusion, and bulk/surface erosion in pH 4.5. Addition of locust bean (a natural polysaccharide) improved the mechanism of release of the IPEC from the non-Fickian mechanism of release to zero order or case II or super case II in pH 4.5. This may be due to the observation that addition of locust bean decreased rate of swelling and enhanced surface erosion. Surface erosion occurs at constant velocity producing reproducible kinetics of erosion and drug release in zero-order pattern (2,35–37).
Molecular Mechanics Assisted Model Building and Energy Refinements
A molecular mechanics conformational searching procedure was employed to acquire the data employed in the statistical mechanics analysis and to obtain differential binding energies of a Polak-Ribiere algorithm and to potentially permit application to polymer composite assemblies. MM+ is a HyperChem modification and extension of Norman Allinger’s Molecular Mechanics program MM2 (38) whereas AMBER is a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics, and free-energy calculations to simulate the structural and energetic properties of molecules (39).
MMER Analysis
Molecular mechanics energy relationship (MMER), a method for analytico-mathematical representation of potential energy surfaces, was used to provide information about the contributions of valence terms, non-covalent Coulombic terms, and non-covalent van der Waals interactions involved in the formation of the polymeric matrix. The MMER model for potential energy factor in various molecular complexes can be written as follows:
| 2 |
where V∑ is related to total steric energy for an optimized structure, Vb corresponds to bond stretching contributions (reference values were assigned to all of a structure’s bond lengths), Vθ denotes bond angle contributions (reference values were assigned to all of a structure’s bond angles), Vφ represents torsional contribution arising from deviations from optimum dihedral angles, Vij incorporates van der Waals interactions due to non-bonded interatomic distances, Vhb symbolizes hydrogen bond energy function, and Vel stands for electrostatic energy.
In the present static lattice atomistic simulations (SLAS) study, a set of three compounds was used to build the energy models for the polysaccharide-polyelectrolytic structure (PsPeS) (Figs. 6 and 7). The synthesis and stability properties of the IPEC entities that may vary with the addition of a polysaccharide were studied using the static system. The global energy relationships for the various complexes derived after assisted model building and energy refinements are as follows:
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
Fig. 6.
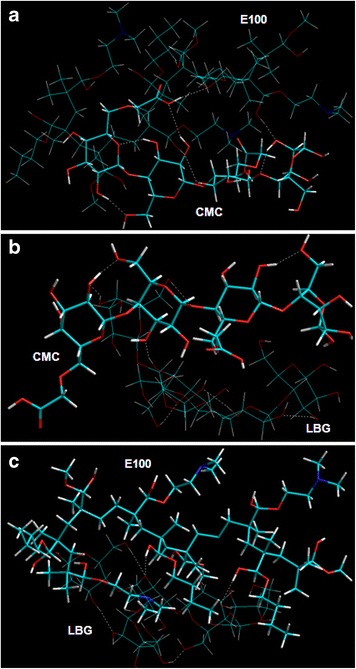
Visualization of geometrical preference of a CMC in complexation with E100; b CMC in complexation with LBG; and c E100 in complexation with LBG, after molecular mechanics simulations. Color codes: C (cyan), O (red), N (blue), and H (white)
Fig. 7.
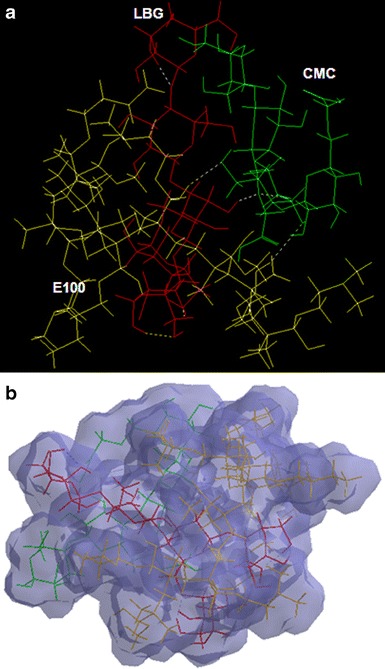
Visualization of geometrical preference of a CMC-E100-LBG tripolymeric complex and b connolly molecular electrostatic potential surfaces in translucent display mode showcasing the matrix compactness, after molecular mechanics simulations. Color codes: CMC (green), LBG (red), and E100 (yellow)
In addition, the total potential energy deviation, ΔEtotal, was calculated as the difference between the total potential energy of the complex system and the sum of the potential energies of isolated individual molecules as follows:
| 10 |
The molecular stability can then be estimated by comparing the total potential energies of the isolated and complexed systems. If the total potential energy of complex is smaller than the sum of the potential energies of isolated individual molecules in the same conformation, the complexed form is more stable and its formation is favored (40).
3D-Computational Modeling for Polymer-Polymer Complexes
The alteration in the performance of the IPEC after addition of a polysaccharide, locust bean gum, led to modeling of the PsPeS based on conformational analysis. We hereby hypothesize that these changes might have been brought upon by the presence of a “structurally defined macromolecular complex.” The measured differences in terms of swelling, erosion, and drug release studies of the polymeric matrices may depend on confound geometry and the extent of network innervations and may require geometrically proportional networks for all simulations to systematically control and isolate the effects of a given parameter. Therefore, to carry out the static lattice atomistic simulations for the polymer-polymer complexes in vacuum, we used the three representative networks shown in Figs. 6 and 7 for our measurements and randomly disposed CMC, LBG, and E100 around each other to form interaction complexes viz. CMC/E100, CMC/LBG, E100/LBG, and CMC/E100/LBG to fully understand the effect of LBG’s interaction. It is evident from Eqs. 2–10 that the formation of CMC-E100, CMC-LBG, E100-LBG, and CMC-E100-LBG (in vacuum) was accompanied by energy stabilization of −24.753, −55.119, −49.984, and −88.208 kcal/mol, respectively. Molecular modeling studies can account for specific interactions between polymer segments and may provide an estimate of whether two polymers will form a compatible blend, and a necessary condition for the miscibility of a mixture of two polymers is a negative free energy of mixing (41). This negative free energy of blending of IPEC (CMC-E100) with LBG confirms the results observed from thermal transitions (Fig. 2) where it was proposed that the blend may be miscible.
This confirms the compatibility of the polymers and stability of the hydrogel in a dried state. Furthermore, the energy data displays the involvement of both bonding and non-bonding interactions (Table V). The formation of CMC-E100 has been described (27), and here, we only explain the influence of the addition of LBG. The introduction of LBG provided additional interaction sites in the matrix system. CMC-LBG, stabilized by all energies except angle contributions, displayed –O…H…O…H…O– hydrogen bonding interactions. However, E100-LBG, stabilized by all constituent energies except torsional contributions, relied mainly on O–H…O–H bonding conformations. Interestingly, the addition of LBG to CMC-E100 formed a highly stabilized structure with constituent H-bonding interaction of all bi-polymeric complexes. A closer look at the tripolymeric matrix system conveys the ability of LBG to act as a linker between E100 and CMC with intermolecular hydrogen bondings occurring between E100 side chain and LBG oxygen atoms and between oxygen atoms belonging to two polysaccharide polymers. LBG side chains are in an intermediate disposition between the E100 and CMC stretched arrangement moving away the polysaccharide symmetry to improve the intermolecular interactions. In addition, some intramolecular hydrogen bonds are also maintained. These underlying chemical interactions may cause torsional and geometrical changes responsible for compactness and resilient characteristics of the composites. The energy minimizations were inherent from rotation of monosaccharide residues (LBG) producing strain due to steric interactions (angle in CMC-LBG and torsional in E100-LBG and tripolymer) which in turn are relieved by the inclusion of non-bonding adjustments. These steric adjustments lead to the formation of H-bonds and hence a dense polymeric matrix showing reduced swelling at either pHs as described in the experimental section. This compactness may further lead to a centralized matrix hardness making the surface more vulnerable to aqueous media as compared to the core (Fig. 7b). This may further lead to two-dimensional surface erosion instead of three-dimensional bulk erosion as discussed in the experimental section. Although the polymeric complexes are stabilized by non-bonding interactions, the inherent bonding interaction in the form of slightly higher torsional energies in CMC-E100-LBG (polysaccharide-polyelectrolytic structure) may induce degradation of the polymeric matrix in a quest to attain further energy stabilization on hydration. This makes the IPB complexes vulnerable to early release of drug due to diffusion of water molecules inside the torsional restraints. These observations are in line with the experimental results wherein slight increased release rates were observed from LBG-b-IPEC (Fig. 5).
Table V.
Computational differential energy attributes calculated for the simulated IPEC/LBG system in a molecular mechanics’ force field setup performed using HyperChem™ 8.0.8 (Hypercube Inc., Gainesville, FL)
| Name | ΔE Total* | ΔE Bond | ΔE Angle | ΔE Dihedral | ΔE Vdw | ΔE H-bond | ΔE Elec |
|---|---|---|---|---|---|---|---|
| CMC-E100 | −24.753 | −0.858 | −1.096 | −0.558 | −25.575 | −2.154 | 5.488 |
| CMC-LBG | −55.119 | −0.244 | 0.339 | −1.494 | −8.257 | −4.796 | −40.668 |
| E100-LBG | −49.984 | −1.088 | −9.293 | 3.108 | −24.559 | −1.479 | −16.673 |
| CMC-E100-LBG | −88.208 | −1.085 | −6.899 | 1.005 | −43.571 | −4.697 | −32.961 |
*ΔE calculated with in comparison to the energy values of the constituent molecules.
CONCLUSIONS
This study explored the incorporation of locust bean into IPEC and its influence on IPEC’s physicochemical properties. The FTIR and DSC analysis indicate there was no characteristic change in the structure of the IPEC and suggest that the LBG-b-IPEC was a physical mixture that may be miscible and appeared as a single-phase material. This attribute was also corroborated by molecular mechanics simulations. Locust bean acted as a linker between E100 and carboxylmethylcellulose with intermolecular hydrogen bonding generating a highly stabilized structure while maintaining some intermolecular hydrogen bonds. The presence of locust bean modified interpolyelectrolyte complex by reducing its excessive swelling by the hydrophilic groups of locust bean associating themselves to the water-holding bodies of IPEC. In addition, incorporation of locust bean prevented the degradation of IPEC by bulk erosion but enhanced surface erosion instead. The steric interactions within the tripolymeric blend led to the formation of hydrogen bonds thereby enhancing compactness which produced a centralized matrix hardness making the surface more vulnerable to erosion than the core. Furthermore, it slightly improved its dissolution efficiency and slightly upgraded the linearity of the drug release profile close to 1 (0.99), thus producing a zero-order release. Computational modeling methods employed for the prediction of preferred molecular conformations polymer-polymer complexes using force field minimizations and modes of interaction corroborated the experimental results.
Acknowledgments
This research was funded by the National Research Foundation (NRF) of South Africa.
Conflict of Interest
The authors confirm that there are no conflicts of interest.
References
- 1.Langer R, Peppas NA. Advances in biomaterials, drug delivery, and bionanotechnology. AIChE J. 2003;49:2990–3006. doi: 10.1002/aic.690491202. [DOI] [Google Scholar]
- 2.Pillai O, Panchagnula R. Polymers in drug delivery. Curr Opin Chem Biol. 2001;5:447–451. doi: 10.1016/S1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- 3.Angelova N, Hunkeler D. Rationalizing the design of polymeric biomaterials. Trends Biotechnol. 1999;17:409–421. doi: 10.1016/S0167-7799(99)01356-6. [DOI] [PubMed] [Google Scholar]
- 4.Ngwuluka NC, Ochekpe NA, Aruoma OI. Naturapolyceutics: the science of utilizing natural polymers for drug delivery. Polymers. 2014;6:1312–1332. doi: 10.3390/polym6051312. [DOI] [Google Scholar]
- 5.Goddard JM, Hotchkiss JH. Polymer surface modification for the attachment of bioactive compounds. Prog Polym Sci. 2007;32:698–725. doi: 10.1016/j.progpolymsci.2007.04.002. [DOI] [Google Scholar]
- 6.Prut EV, Zelenetskii AN. Chemical modification and blending of polymers in an extruder reactor. Russ Chem Rev. 2001;70:65–79. doi: 10.1070/RC2001v070n01ABEH000624. [DOI] [Google Scholar]
- 7.Li J, Barrow D, Howell H, Kalachandra S. In vitro drug release study of methacrylate polymer blend system: effect of polymer blend composition, drug loading and solubilizing surfactants on drug release. J Mater Sci Mater Med. 2010;21:583–588. doi: 10.1007/s10856-009-3899-6. [DOI] [PubMed] [Google Scholar]
- 8.Edlund U, Albertsson AC. Microspheres from poly(D, L-lactide)/poly(1,5-dioxepan-2-one) miscible blends for controlled drug delivery. J Bioact Compatible Polym. 2000;15:214–229. doi: 10.1106/ME8A-MUBN-4LFH-P6XQ. [DOI] [Google Scholar]
- 9.Ramesh Babu V, Sairam M, Hosamani KM, Aminabhavi TM. Preparation of sodium alginate–methylcellulose blend microspheres for controlled release of nifedipine. Carbohydr Polym. 2007;69:241–250. doi: 10.1016/j.carbpol.2006.09.027. [DOI] [Google Scholar]
- 10.Peesan M, Supaphol P, Rujiravanit R. Preparation and characterization of hexanoyl chitosan/polylactide blend films. Carbohydr Polym. 2005;60:343–350. doi: 10.1016/j.carbpol.2005.01.008. [DOI] [Google Scholar]
- 11.Wang Q, Hu X, Du Y, Kennedy JF. Alginate/starch blend fibers and their properties for drug controlled release. Carbohydr Polym. 2010;82:842–847. doi: 10.1016/j.carbpol.2010.06.004. [DOI] [Google Scholar]
- 12.Bajpai AK, Shukla SK, Bhanu S, Kankane S. Responsive polymers in controlled drug delivery. Prog Polym Sci. 2008;33:1088–1118. doi: 10.1016/j.progpolymsci.2008.07.005. [DOI] [Google Scholar]
- 13.Sahoo S, Sasmal A, Sahoo D, Nayak P. Synthesis and characterization of chitosan-polycaprolactone blended with organoclay for control release of doxycycline. J Appl Polym Sci. 2010;118:3167–3175. doi: 10.1002/app.32474. [DOI] [Google Scholar]
- 14.Pitt GG, Cha Y, Shah SS, Zhu KJ. Blends of PVA and PGLA: control of the permeability and degradability of hydrogels by blending. J Controlled Release. 1992;19:189–199. doi: 10.1016/0168-3659(92)90076-4. [DOI] [Google Scholar]
- 15.Huang Y, Yu H, Xiao C. pH-sensitive cationic guar gum/poly (acrylic acid) polyelectrolyte hydrogels: swelling and in vitro drug release. Carbohydr Polym. 2007;69:774–783. doi: 10.1016/j.carbpol.2007.02.016. [DOI] [Google Scholar]
- 16.Mundargi RC, Patil SA, Aminabhavi TM. Evaluation of acrylamide-grafted-xanthan gum copolymer matrix tablets for oral controlled delivery of antihypertensive drugs. Carbohydr Polym. 2007;69:130–141. doi: 10.1016/j.carbpol.2006.09.007. [DOI] [Google Scholar]
- 17.Nanaki S, Karavas E, Kalantzi L, Bikiaris D. Miscibility study of carrageenan blends and evaluation of their effectiveness as sustained release carriers. Carbohydr Polym. 2010;79:1157–1167. doi: 10.1016/j.carbpol.2009.10.067. [DOI] [Google Scholar]
- 18.Jian H, Zhu L, Zhang W, Sun D, Jiang J. Galactomannan (from Gleditsia sinensis Lam.) and xanthan gum matrix tablets for controlled delivery of theophylline: in vitro drug release and swelling behavior. Carbohydr Polym. 2012;87:2176–2182. doi: 10.1016/j.carbpol.2011.10.043. [DOI] [Google Scholar]
- 19.Mi F, Shyu S, Lin Y, Wu Y, Peng C, Tsai Y. Chitin/PLGA blend microspheres as a biodegradable drug delivery system: a new delivery system for protein. Biomaterials. 2003;24:5023–5036. doi: 10.1016/S0142-9612(03)00413-7. [DOI] [PubMed] [Google Scholar]
- 20.Thomas PA, Padmaja T, Kulkarni MG. Polyanhydride blend microspheres: novel carriers for the controlled release of macromolecular drugs. J Controlled Release. 1997;43:273–281. doi: 10.1016/S0168-3659(96)01497-6. [DOI] [Google Scholar]
- 21.Yu L, Dean K, Li L. Polymer blends and composites from renewable resources. Prog Polym Sci. 2006;31:576–602. doi: 10.1016/j.progpolymsci.2006.03.002. [DOI] [Google Scholar]
- 22.Aroguz AZ, Baysal BM. Miscibility studies on blends of poly(phenylene oxide)/brominated polystyrene by viscometry. Eur Polym J. 2006;42:311–315. doi: 10.1016/j.eurpolymj.2005.07.012. [DOI] [Google Scholar]
- 23.Edlund U, Albertsson A. Copolymerization and polymer blending of trimethylene carbonate and adipic anhydride for tailored drug delivery. J Appl Polym Sci. 1999;72:227–239. doi: 10.1002/(SICI)1097-4628(19990411)72:2<227::AID-APP8>3.0.CO;2-Z. [DOI] [Google Scholar]
- 24.Prajapati VD, Jani GK, Moradiya NG, Randeria NP, Nagar BJ. Locust bean gum: a versatile biopolymer. Carbohydr Polym. 2013;94:814–821. doi: 10.1016/j.carbpol.2013.01.086. [DOI] [PubMed] [Google Scholar]
- 25.Dionísio M, Grenha A. Locust bean gum: exploring its potential for biopharmaceutical applications. J Pharm Bioallied Sci. 2012;4:175. doi: 10.4103/0975-7406.99013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ngwuluka NC, Choonara YE, Modi G, du Toit LC, Kumar P, Ndesendo VM, et al. Design of an interpolyelectrolyte gastroretentive matrix for the site-specific zero-order delivery of levodopa in Parkinson’s disease. AAPS PharmSciTech. 2013;14:605–619. doi: 10.1208/s12249-013-9945-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ngwuluka NC, Choonara YE, Kumar P, Modi G, Toit LC, Pillay V. A hybrid methacrylate-sodium carboxymethylcellulose interpolyelectrolyte complex: rheometry and in silico disposition for controlled drug release. Materials. 2013;6:4284–4308. doi: 10.3390/ma6104284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ngwuluka NC, Choonara YE, Kumar P, du Toit LC, Khan RA, Pillay V. A novel pH-responsive interpolyelectrolyte hydrogel complex for the oral delivery of levodopa. I. IPEC modeling and synthesis. J Biomed Mater Res Part A. 2015;103A:1077–1084. doi: 10.1002/jbm.a.35259. [DOI] [PubMed] [Google Scholar]
- 29.Ngwuluka NC, Choonara YE, Kumar P, du Toit LC, Khan RA, Pillay V. A novel pH‐responsive interpolyelectrolyte hydrogel complex for the oral delivery of levodopa. Part II: characterization and formulation of an IPEC‐based tablet matrix. J Biomed Mater Res Part A. 2015;103A:1085–1094. doi: 10.1002/jbm.a.35258. [DOI] [PubMed] [Google Scholar]
- 30.Kumar P, Pillay V, Choonara YE, Modi G, Naidoo D, Du Toit LC. In silico theoretical molecular modeling for Alzheimer’s disease: the nicotine-curcumin paradigm in neuroprotection and neurotherapy. Int J Mol Sci. 2011;12:694–724. [DOI] [PMC free article] [PubMed]
- 31.Pendekal MS, Tegginamat PK. Hybrid drug delivery system for oropharyngeal, cervical and colorectal cancer—in vitro and in vivo evaluation. Saudi Pharm J. 2013;21:177–186. doi: 10.1016/j.jsps.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson NH, Bauer M, Boussac N, Khan-Malek R, Munden P, Sardaro M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J Pharm Biomed Anal. 1998;17:811–822. doi: 10.1016/S0731-7085(98)00011-9. [DOI] [PubMed] [Google Scholar]
- 33.Sriamornsak P, Thirawong N, Weerapol Y, Nunthanid J, Sungthongjeen S. Swelling and erosion of pectin matrix tablets and their impact on drug release behavior. Eur J Pharm Biopharm. 2007;67:211–219. doi: 10.1016/j.ejpb.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 34.Singh R, Maity S, Sa B. Effect of ionic crosslink on the release of metronidazole from partially carboxymethylated guar gum tablet. Carbohydr Polym. 2014;106:414–421. doi: 10.1016/j.carbpol.2014.01.033. [DOI] [PubMed] [Google Scholar]
- 35.Fv B, Schedl L, Göpferich A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials. 2002;23:4221–4231. doi: 10.1016/S0142-9612(02)00170-9. [DOI] [PubMed] [Google Scholar]
- 36.Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new, simple mathematical model quantifying drug release. Eur J Pharm Sci. 2002;15:355–366. doi: 10.1016/S0928-0987(02)00023-4. [DOI] [PubMed] [Google Scholar]
- 37.Siepmann J, Göpferich A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv Drug Deliv Rev. 2001;48:229–247. doi: 10.1016/S0169-409X(01)00116-8. [DOI] [PubMed] [Google Scholar]
- 38.Warhurst D, Craig J, Adagu I, Meyer D, Lee S. The relationship of physico-chemical properties and structure to the differential antiplasmodial activity of the cinchona alkaloids. Malar J. 2003;2:26. doi: 10.1186/1475-2875-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, III, DeBolt S, et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. doi: 10.1016/0010-4655(95)00041-D. [DOI] [Google Scholar]
- 40.Yu BY, Chung JW, Kwak S. Reduced migration from flexible poly(vinyl chloride) of a plasticizer containing beta-cyclodextrin derivative. Environ Sci Technol. 2008;42:7522–7. [DOI] [PubMed]
- 41.Tiller AR, Gorella B. Estimation of polymer compatibility from molecular mechanics calculations. Polymer. 1994;35:3251–3259. doi: 10.1016/0032-3861(94)90130-9. [DOI] [Google Scholar]