

Abstract
To achieve the potent therapeutic effects of human immunoglobulin G (IgG), highly concentrated formulations are required. However, the stabilization for highly concentrated human IgG is laborious work. In the present study, to investigate the potentials of polypseudorotaxane (PPRX) hydrogels consisting of polyethylene glycol (PEG) and α- or γ-cyclodextrin (α- or γ-CyD) as pharmaceutical materials for highly concentrated human IgG, we designed the PPRX hydrogels including human IgG and evaluated their pharmaceutical properties. The α- and γ-CyDs formed PPRX hydrogels with PEG (M.W. 20,000) even in the presence of highly concentrated human IgG (>100 mg/mL). According to the results of 1H-NMR, powder X-ray diffraction, and Raman microscopy, the formation of human IgG/CyD PPRX hydrogels was based on physical cross-linking arising from their columnar structures. The release profiles of human IgG from the hydrogels were in accordance with the non-Fickian diffusion model. Importantly, the stabilities of human IgG included into the hydrogels against thermal and shaking stresses were markedly improved. These findings suggest that PEG/CyD PPRX hydrogels are useful to prepare the formulation for highly concentrated human IgG.
KEY WORDS: cyclodextrin, human IgG, hydrogel, polypseudorotaxane, stability
INTRODUCTION
Currently, therapeutic antibodies have by far become the largest growth area in the pharmaceutical industry (1). Immunoglobulin G (IgGs), as one of the classes of therapeutic antibodies, are characterized among various medicines by their high binding specificity to their targets with minimum side effects (2). IgG replacement is a lifesaving treatment for patients with primary immunodeficiency disease (3). For most patients who require IgG replacement, the subcutaneous administration of IgG offers advantages over the intravenous one such as independence from hospital-based infusion settings, alternative for patients with poor venous access, better tolerability in those patients who are not able to tolerate the intravenous administration, flexibility of dosing, ease of administration, less cost to administer, and lower side effect profile (3). Because of the lower administration capacity of the subcutaneous formulation than the intravenous one, concentrated antibody formulations having the concentration of more than 100 mg/mL are increasingly needed for subcutaneous administration of high protein doses. The latter represents particular formulation challenges because of their higher aggregation tendency, higher viscosity, and analytical challenges (4–7).
Protein aggregation is considered as one of the most challenging problems in protein formulations as it can occur at almost any stage of production, processing, storage, shipping, and administration to patients. Especially, proteins easily form aggregates at high concentration, even though a highly concentrated protein formulation is useful in the clinical field (4). For instance, 50 mg/mL of IgG solution forms more than 30% of soluble and insoluble aggregates after storing for 3 months at 37°C, whereas 5 mg/mL of IgG solution forms c.a. 15% of soluble aggregates (8). Regretfully, protein aggregation can severely influence the pharmacokinetics as well as the safety of the protein drug, since the phenomenon of protein aggregation can result in the loss of biological activity, increased immunogenicity, or toxicity (9,10).
An important way of inhibiting protein aggregation in formulations is the addition of appropriate excipients such as polyols, surfactants, or amino acids (11–14). However, many of the commonly employed excipients suffer from certain shortcomings. For example, polyols, such as sugars, are used in relatively high concentrations to stabilize proteins during quiescent storage and thermal stress (9,15). In addition, they cannot stabilize proteins against interfacial damage and during mechanical agitation (14,16). Regretfully, some additives such as glucose, sorbitol, and leucine have negligible effect on the thermal stability of highly concentrated IgG solution (50 mg/mL) at physiologically acceptable concentration (17). Meanwhile, small amounts of surfactants, such as polysorbates, are effective in preventing aggregation under various stresses including stirring (18), freeze-thawing (19), or shaking (18,20), but they have the negative effect during long-term storage due to the increased tendency of protein oxidation (14,21,22). Because of the drawback, there is a need for alternatives, either a different class of excipients or a new delivery system for proteins.
Cyclodextrins (CyDs), cyclic oligosaccharides, can form inclusion complexes with various molecules not only in aqueous solution but also in the solid state. Therefore, CyDs can improve the solubility, dissolution rate, and bioavailability of the drugs, so the widespread use of CyDs is well known in the pharmaceutical field (23). Recently, many supramolecular assemblies based on CyDs have attracted great attention since the target guest molecules for CyDs are widely expanding from low molecular weight compounds through macromolecules such as proteins and synthetic polymers (24). The supramolecular assembly of polyethylene glycol (PEG) and α-CyD was firstly reported by Harada et al. because a number of cyclic molecules are spontaneously threaded on the polymer chain (25–27). After threading on a PEG chain, hydroxyl groups of α-CyD and neighbor α-CyD form hydrogen bonds, resulting in the formation of an insoluble supramolecular assembly (25–27). Subsequently, the formation of a supramolecular inclusion complex consisting of γ-CyD and two PEG chains was reported (28). These complexes are called polypseudorotaxanes (PPRXs) and can be utilized as drug carriers (29–33). PPRXs have the following advantages: (1) preparation by a very simple method, (2) dethreading from the polymer chain by dilution, (3) high safety profile of the components, and (4) formation of hydrogels when high molecular weight PEGs (>2000 Da) are used (34).
We previously reported the potential use of PPRX hydrogels consisting of PEG (M.W. 20,000) and α- or γ-CyD as a sustained-release system for low concentrations (c.a. 1 mg/mL) of protein drugs such as insulin and lysozyme (24,35). However, no studies have been published examining the potential use of PEG/CyD PPRX hydrogels for highly concentrated human IgG (c.a. 100 mg/mL). In addition, the inhibitory effects of PEG/CyD PPRX hydrogels on the aggregation of antibodies are still unclear. Furthermore, there are few reports regarding stabilization of highly concentrated human IgG using hydrogels. In this study, we prepared a highly concentrated human IgG formulation using PEG/CyDs PPRX hydrogels and characterized the structures of the hydrogels. Moreover, we evaluated the release profile and the release mechanism of human IgG from the hydrogels. Finally, the physicochemical stabilities of human IgG encapsulated into PEG/CyDs PPRX hydrogels against thermal and shaking stresses were investigated.
MATERIALS AND METHODS
Materials
Human IgG was purchased from Equitech-Bio (Kerrville, TX). PEG (M.W. 20 kDa) was purchased from Katayama Chemical Co. (Osaka, Japan). CyDs were supplied by Nihon Shokuhin Kako (Tokyo, Japan). All other chemicals and solvents were of analytical reagent grade, and deionized double-distilled water was used throughout the study.
Hydrogel Formation
α-CyD (145 mg/mL) or γ-CyD (232 mg/mL) and human IgG (130 mg/mL) were dissolved in phosphate buffer (pH 7.4) including 0.94 g/L of sodium hydrogen phosphate anhydrous, 2.87 g/L of sodium dihydrogen phosphate dehydrate, 5.37 g/L of l-arginine hydrochloride, 5.84 g/L of sodium chloride, and 10.0 g/L of purified sucrose. Next, 800 μL of either human IgG/α-CyD solution or human IgG/γ-CyD solution was added to 100 or 200 μL of PEG solution (100 mg/mL), respectively, and mixed by gentle pipetting. The solutions were kept in a refrigerator at 4°C for 12 h to obtain a viscous gel.
1H-NMR Spectroscopic Studies
1H-NMR spectra were taken at 25°C on a JEOL JNM-ECP500 spectrometer (Tokyo, Japan) operating at 500 MHz. Dried samples of α- and γ-CyD PPRX hydrogels (1 mg) were dissolved in 0.6 mL of deuterated DMSO (DMSO-d6). The DMSO signal was used as an internal reference for 1H-NMR. Chemical shifts were expressed in parts per million (ppm) relative to that of the DMSO signal (2.49 ppm). To calculate the stoichiometry of the CyD PPRXs, the integral values of the anomeric protons (4.8 ppm) of CyDs and those of the ethylene protons (3.5 ppm) of PEG were compared.
Powder X-ray Diffraction Studies
Powder X-ray diffraction patterns of the dried samples of PEG/α- and PEG/γ-CyD PPRX hydrogels and their individual components were measured by a Rigaku Ultima IV X-ray diffractometer (Tokyo, Japan) under the following conditions: Ni-filtered CuKα radiation, a voltage of 40 kV, a current of 40 mV, a time constant of 2 s, and a scanning speed of 5°/min.
Raman Microscopy
PEG/α- and PEG/γ-CyD PPRX hydrogels and their individual components were measured using a RENISHAW inVia Raman microscope (New Mills, UK). Measurement conditions were as follows: excitation wavelength 532 nm, laser power 100% (45 mW/1 line), exposure time 5 s, spectral acquisition 7979, cumulated number 1, and spatial resolution 1 μm.
In Vitro Release Studies
The in vitro release rates of human IgG from PEG/α- and PEG/γ-CyD PPRX hydrogels were measured by the modified dispersed amount method (36). CyD PPRX hydrogels containing 104 μg of human IgG were diluted with 4 mL of phosphate buffer (pH 7.4). The samples were incubated at 37°C with mild shaking at 150 rpm. At certain time intervals, aliquots of 200 μL of the dissolution medium were withdrawn and replaced with the same volume of the fresh phosphate buffer. Then, the samples were centrifuged at 12,000 rpm for 10 min and the released amounts of human IgG from PEG/CyD PPRX hydrogels were measured spectrophotometrically at 280 nm using a Hitachi U-2000 (Tokyo, Japan). The in vitro release profiles of human IgG from PEG/CyD solutions were also performed in comparison with those of the CyD PPRX hydrogels.
Kinetic Analysis of Human IgG Release Data
The data of in vitro human IgG release from the prepared hydrogels were analyzed according to Higuchi’s and Korsmeyer-Peppas’s model.
Higuchi’s model (Eq. 1):
| 1 |
where Qt is the amount of drug released in time t, and kH is Higuchi’s release rate constant.
Korsmeyer-Peppas’s model (Eq. 2):
| 2 |
where Mt/M∞ is the fraction of drug released at time t, kP is the release rate constant incorporating structural and geometric characteristics of the hydrogel, and n is the release exponent.
Thermal Stability
Five hundred microliters of human IgG/CyD PPRX hydrogels was placed at 60°C for 30 min. Then, 30 mL of phosphate buffer (pH 7.4) was added and shaken for 3 h with a shaking rate of 60 rpm at 37°C to dissolve the hydrogels. To remove the aggregated human IgG, the samples were centrifuged at 12,000 rpm for 10 min and the amount of human IgG in the supernatant was measured spectrophotometrically at 280 nm using a Hitachi U-2000 (Tokyo, Japan).
Shaking Stability
One hundred microliters of human IgG/CyD PPRX hydrogels was shaken for 1 week at 500 rpm at room temperature. Next, 800 μL of phosphate buffer (pH 7.4) was added and the mixture was shaken for 12 h with a shaking rate of 150 rpm at 37°C. Finally, the samples were centrifuged at 12,000 rpm for 10 min, and then the amount of human IgG in the supernatant was determined spectrophotometrically at 280 nm using a Hitachi U-2000 (Tokyo, Japan).
Statistical Analysis
Data are given as the mean ± SE. Statistical significance of means for the studies was determined by analysis of variance followed by Scheffe’s test. p values for significance were set at 0.05.
RESULTS
Formation of CyD PPRX Hydrogels
Figure 1 shows the pictures of the resulting hydrogels after mixing the human IgG/CyD solutions and PEG solution and standing for 12 h at 4°C. As described above, highly concentrated antibody formulations (>100 mg/mL) are required in the clinical field. Therefore, the final concentrations of human IgG were set at 116 and 104 mg/mL in the α- and γ-CyD systems, respectively. When α- and γ-CyD solutions containing human IgG were added to the PEG solution, the solutions became opaque within several minutes and finally changed to gel. In addition, the induction time for gelation in the α-CyD system was shorter than that in the γ-CyD system. On the other hand, the β-CyD solution did not form any hydrogels (data not shown). Thereby, these results suggest the formation of the α- and γ-CyD PPRX hydrogels including highly concentrated human IgG.
Fig. 1.

Photographs of PEG/α-CyD (a) and PEG/γ-CyD (b) PPRX hydrogels including human IgG
1H-NMR Spectroscopy
The stoichiometry of the PPRX was determined by measuring peak areas of the anomeric protons of CyDs and the ethylene protons of PEG in the CyD PPRX hydrogels including human IgG in 1H-NMR spectra after dissolving the solid PPRXs in DMSO-d6. Figure 2 demonstrates the peaks of CyDs and PEG in both the systems where the number of moles of α- and γ-CyDs involved in the PPRX formation were 113.3 and 94.8, respectively (Table I). Furthermore, the coverage of the PEG chain by α- and γ-CyDs was 51.5% and 43.1%, respectively, when it is assumed that two (ethylene glycol) repeat units are included in one CyD cavity (25,28).
Fig. 2.
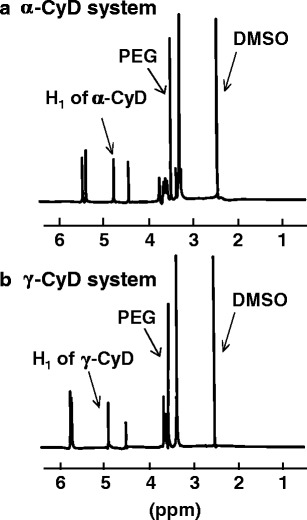
1H-NMR spectra of PEG/α-CyD (a) and PEG/γ-CyD (b) PPRX hydrogels including human IgG in DMSO-d 6 at 25°C
Table I.
Compositions of PEG/CyD PPRX Hydrogels Including Human IgG
| System | Number of CyDa | Coverageb |
|---|---|---|
| PEG/α-CyD | 113.3 ± 13.0 | 51.5 ± 5.9 |
| PEG/γ-CyD | 94.8 ± 3.4 | 43.1 ± 1.6 |
Each value represents the mean ± SE of three experiments
aNumber of total CyD units in the PPRX
bCoverage = 2 (CyD per PEG)/(PEG repeat units), assuming two PEG repeat units per CyD
Powder X-ray Diffraction Patterns
The formation of CyD PPRXs with PEG and their crystal packing structures were confirmed by powder X-ray diffraction patterns (Fig. 3). The X-ray diffraction patterns of human IgG/CyD PPRX hydrogels were markedly different from those of physical mixtures, but the same as those of plain CyD PPRX hydrogels (37). In particular, the pattern of α-CyD PPRX hydrogels gave diffraction peaks at 2θ = 7.5°, 13.0°, 19.8°, and 22.5° and that of γ-CyD PPRX hydrogels gave diffraction peaks at 2θ = 7.4°, 14.8°, 16.6°, and 22.1°. These data resembled the diffraction patterns of the hexagonal and tetragonal columnar channels of the linearly aligned α- and γ-CyD cavities in the crystalline phase (38–40). Therefore, the diffraction patterns of α- and γ-CyD PPRX hydrogels including human IgG were indexed on the basis of the two-dimensional hexagonal and tetragonal unit cells with dimensions a = b = 27.11 Å and a = b = 23.98 Å, respectively, as shown in Table II. The d-spacings of the hkl (200) reflection were used to calculate the unit cell dimensions. The calculated d-spacings (dcal) were in excellent agreement with those observed (dobs), confirming that α- and γ-CyD PPRX hydrogels including human IgG formed the hexagonal or tetragonal columnar necklace-like inclusion complex, respectively.
Fig. 3.
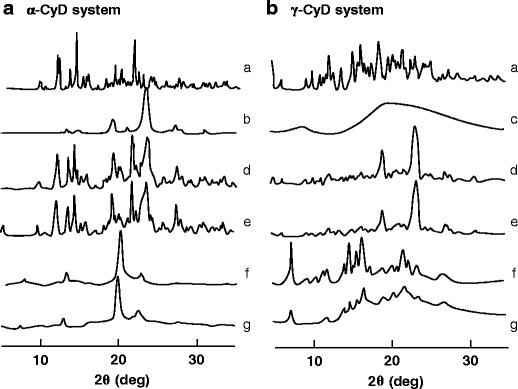
Powder X-ray diffraction patterns of a PEG/α-CyD and b PEG/γ-CyD PPRX hydrogels. a CyD alone, b PEG alone, c IgG alone, d PEG/CyD physical mixture, e PEG/CyD/IgG physical mixture, f plain PEG/CyD PPRX hydrogels, and g PEG/CyD PPRX hydrogels including IgG
Table II.
Crystallographic Characteristics of PEG/CyD PPRX Hydrogels Including Human IgG
| 2θ (deg) | (hkl) | d obs (Å) | d cal a (Å) |
|---|---|---|---|
| α-CyD system | |||
| 7.52 | (200) | 11.74 | 11.74 |
| 13.00 | (220) | 6.80 | 6.78 |
| 19.82 | (420) | 4.47 | 4.44 |
| 22.53 | (600) | 3.94 | 3.91 |
| γ-CyD system | |||
| 7.36 | (200) | 11.99 | 11.99 |
| 14.79 | (400) | 5.98 | 6.00 |
| 16.55 | (420) | 5.35 | 5.36 |
| 22.05 | (600) | 4.03 | 4.00 |
aCalculated assuming a hexagonal unit cell with a = b = 27.11 Å, and a tetragonal unit cell with a = b = 23.98 Å, respectively
Figure 4 shows schematic proposed structures of α- and γ-CyD PPRX hydrogels including human IgG. α- and γ-CyDs formed PPRX hydrogels with one PEG chain and two PEG chains, respectively, even in the presence of human IgG. In addition, α- and γ-CyD PPRXs form columnar channel structures, resulting in physical cross-linking points to form the hydrogels.
Fig. 4.
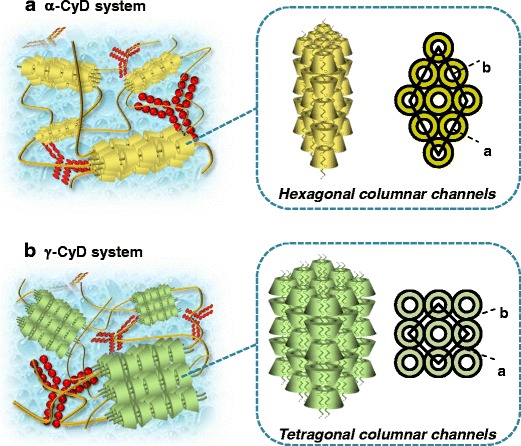
Proposed scheme for PEG/α-CyD (a) and PEG/γ-CyD (b) PPRX hydrogels including human IgG
Raman Microscopy
To examine the distribution of human IgG and CyDs into the hydrogels, both compounds were visualized by the Raman microscope, which non-invasively provides the imaging data obtained from Raman spectroscopy (Fig. 5). The imaging data were obtained from the peaks observed at 950 cm−1 deriving from C–H of CyDs and at 1000 cm−1 deriving from phenylalanine of human IgG. As a result, both α- and γ-CyDs were occasional in the hydrogels. On the other hand, human IgG was homogeneously dispersed in the hydrogels, compared to CyDs. These results suggest the preparation of CyD PPRX hydrogels homogeneously including highly concentrated human IgG.
Fig. 5.
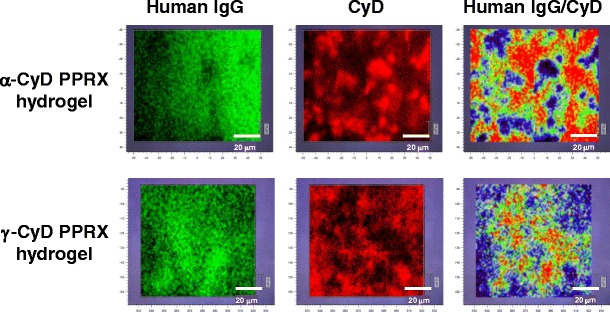
Raman imaging of PEG/CyD PPRX hydrogels including human IgG
In Vitro Release Studies
Figure 6a shows the release profile of human IgG from CyD PPRX hydrogels in phosphate buffer (pH 7.4) at 37°C. The release of human IgG from α- and γ-CyD PPRX hydrogels reached 80 and 90% by 6 h, respectively. In addition, there was a remarkable difference between α- and γ-CyD PPRX hydrogels by the first 3 h. After that, there was no notable change in the release behavior of α- and γ-CyD PPRX hydrogels, although almost all human IgG was released from both systems by the end of the experiment. These results suggest that the PPRX supramolecular hydrogels can act as a sustained-release system for human IgG.
Fig. 6.
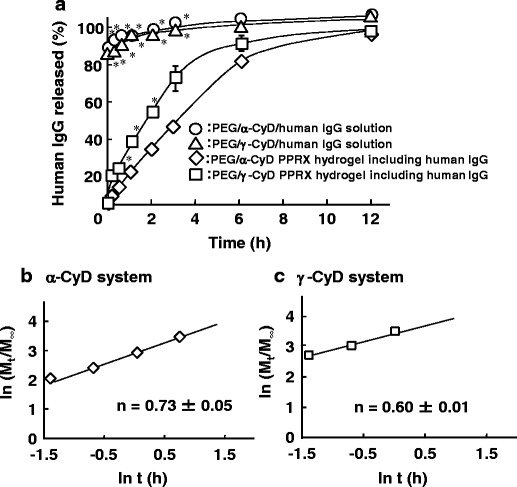
a In vitro release profile of human IgG from CyD PPRX hydrogels in phosphate buffer (pH 7.4) at 37°C. b, c Plots of released human IgG from PEG/CyD PPRX hydrogels based on Korsmeyer-Peppas equations. Each point represents the mean ± SE of three experiments. *p < 0.05 versus PEG/γ-CyD PPRX system
To estimate the release mechanism of human IgG from the CyD PPRX hydrogels, the release kinetic profiles were applied to kinetic models such as the Higuchi and Korsmeyer-Peppas equations. Table III summarizes the release kinetic parameters and correlation coefficients (r) calculated for α- and γ-CyD PPRX hydrogels. The releases of human IgG from α- and γ-CyD PPRX hydrogels were fitted to the diffusion controlled mechanism (Higuchi model) with r values of 0.93 and 0.98, respectively. In addition, to determine the diffusion mechanism of human IgG from the hydrogels, the release profiles were evaluated by the Korsmeyer-Peppas model (Fig. 6b, c). The α- and γ-CyD PPRX hydrogels showed a fair linearity with r values of 0.98 and 0.97, respectively (Table III). In addition, the exponent values (n) in the α- and γ-CyD systems were 0.73 and 0.60, respectively, indicating that the release mechanism of human IgG from the CyD PPRX hydrogels obeyed the non-Fickian diffusion. These results suggest that the CyD PPRX hydrogels release human IgG via coupled diffusion and erosion mechanisms.
Table III.
Release Constants and r Values of PEG/CyD PPRX Hydrogels Including Human IgG Calculated from Higuchi and Korsmeyer-Peppas Equations
| Release model | Parameter | α-CyD system | γ-CyD system |
|---|---|---|---|
| Higuchi | k H (%/h1/2) | 20.54 ± 0.56* | 32.68 ± 0.97 |
| r | 0.93 ± 0.00 | 0.98 ± 0.01 | |
| Korsmeyer- Peppas | k P (%/hn) | 19.23 ± 0.90* | 33.24 ± 1.32 |
| r | 0.98 ± 0.02 | 0.97 ± 0.01 |
Each value represents the mean ± SE of three experiments
*p < 0.05 versus γ-CyD system
Stress-Stability Studies
Figure 7 shows the effects of CyDs, PEG, and PEG/CyD PPRX hydrogels on the thermal and shaking stability of highly concentrated human IgG in the α-CyD system (116 mg/mL) and γ-CyD system (104 mg/mL). Human IgG in α- and γ-CyD PPRX hydrogels negligibly formed aggregates after the incubation at 60°C for 30 min (Fig. 7a). On the other hand, CyDs and PEG did not show the inhibitory effects on the aggregation of human IgG under the present experimental conditions, although these samples were in the solution state. Likewise, both α- and γ-CyD PPRX hydrogels improved the shaking stability of human IgG, but not CyDs or PEG (Fig. 7b). These results suggest that the physicochemical stabilities of highly concentrated human IgG are markedly improved by encapsulation into the CyD PPRX hydrogels.
Fig. 7.
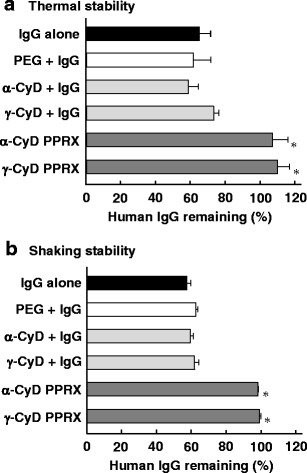
Effects of CyDs, PEG, and PEG/CyD PPRX hydrogels on the thermal and shaking stability of human IgG in phosphate buffer (pH 7.4). Each value represents the mean ± S.E. of three to six experiments. *p < 0.05 versus IgG alone
DISCUSSION
Currently, numerous antibodies are utilized in the clinical field to treat rheumatoid arthritis, multiple sclerosis, cancer, etc. However, highly concentrated antibodies easily form aggregates, resulting in the loss of biological activity and increased immunogenicity or toxicity (9,10). Some additives such as sugars and amino acids have been used to improve the stabilities of highly concentrated antibodies (50 mg/mL); however, they provide negligible stabilizing effects (17). In addition, the inhibitory effects of hydroxypropyl-β-CyD (HP-β-CyD) on aggregation of highly concentrated IgG (100 mg/mL) are also investigated (41). However, HP-β-CyD did not elicit the stabilizing effect against thermal stability of IgG, although it improved the shaking stability of IgG. Thus, the stabilization for highly concentrated human IgG is laborious work. In the present study, we first reported the utility of CyD PPRX hydrogels to improve the stability of highly concentrated human IgG.
α- or γ-CyD formed the PEG PPRX hydrogel including highly concentrated human IgG (Fig. 1), but β-CyD did not (data not shown). Li et al. (34) reported that PEG chains with a molecular weight higher than 2000 Da formed the PPRX hydrogel with α-CyD. In addition, double-stranded PEG chains are known to be threaded through the γ-CyD cavity to form PPRX (28). Meanwhile, a β-CyD cavity is too wide and too narrow to form PPRX with one and two PEG chains, respectively (37). Therefore, α- and γ-CyD, not β-CyD, are useful as the ring components of the PPRX hydrogels with PEG, when highly concentrated human IgG is still included.
According to the results of powder X-ray diffraction patterns, α- and γ-CyD PPRX hydrogels including human IgG formed the hexagonal and tetragonal columnar structures, respectively (Figs. 3 and 4). However, the samples measured by powder X-ray diffraction are dried solid samples, not gel state. Therefore, we measured the powder X-ray diffraction pattern of the PPRX hydrogels in the gel state (data not shown). As a result, the degree of crystallinity of the PPRX hydrogel decreased in the gel state, compared with solid state. However, the peaks derived from PPRX were observed even in the gel state, indicating that the PPRX structure is included in the hydrogels.
The distribution of α- or γ-CyD and human IgG may exist at different points in both the α- and γ-CyD PPRX hydrogels, respectively (Fig. 5). It is acknowledged that the formation of CyD PPRX hydrogels was based on physical cross-linking, arising from their crystalline columnar structures (34). In addition, CyDs are reported to be thickly packed in these cross-linking points (34). Actually, the similar patterns of CyDs were observed in the PPRX hydrogels (Fig. 4). Hence, it is impossible that human IgG exists at the cross linkage points of the hydrogels because there is no space due to the existence of many CyD molecules. Therefore, it is highly likely that human IgG homogeneously exists in the CyD PPRX hydrogels (Fig. 5), implying the suitable property of CyD PPRX hydrogels as pharmaceutical materials for protein drugs, although thereafter further investigation is required.
In our preliminary result, we clarified that sliding friction values of α- and γ-CyD PPRX hydrogels containing human IgG were between 10 and 20 N (data not shown), indicating that the PPRX hydrogels possess an injectable physicochemical property. Importantly, the sliding friction values were almost the same before and after the storage at 4°C for 1 year. Furthermore, Li et al. (42) reported that α-CyD PPRX hydrogels with PEG (M.W. = 8000–35,000) show thixotropic and reversible properties and are injectable with 18G–27G needles. Thus, CyD PPRX hydrogels are likely to be physicochemically injectable and stable, although further investigations are required.
Recently, there have been a number of reports regarding the utility of CyD PPRX hydrogels as controlled-release carriers for protein drugs such as insulin (35), bovine serum albumin (43), and lysozyme (24,44). In the present study, the release of human IgG from the γ-CyD PPRX hydrogel was faster than that from the α-CyD PPRX hydrogel (Fig. 6a and Table III). This could be attributed to the difference in the crystal packing structure of both types of PPRX hydrogels. Generally, tetragonal packing of γ-CyD molecules tends to facilitate the penetration of water molecules into the crystal structure, compared to the hexagonal packing. In addition, the higher crystallinity of the hexagonal packing structure of α-CyD molecules could contribute to the slower release rate of human IgG from the α-CyD/PPRX hydrogel (40). Therefore, α- and γ-CyD PPRX hydrogels may be relatively adequate for the sustained-release type and fast-release type of human IgG. Importantly, our preliminary data suggested that the biological activity of a commercially available IgG product was approximately 100% after storage for 7 days and then release from PPRX hydrogels (data not shown). Thereafter, the attentive in vitro biological activity, in vivo pharmacokinetics, and pharmacodynamics should be examined.
Proteins subjected to heating undergo conformational changes that can lead to the formation of aggregates, and those to mechanical stresses adsorb to the air-water interface, leading to structural alterations that can initiate aggregation through cavitation, local thermal effects, bubble entrapment, and transportation of the aggregated protein (45–47). In both cases, the conformational and/or structural changes of IgG should be involved in the aggregations. Meanwhile, there were few reports about the stabilizing effects of CyD PPRX hydrogels for protein drugs. In the present study, however, the stability of human IgG included into the hydrogels against both thermal and shaking stresses were markedly improved (Fig. 7). Thereby, it is speculated that the improvements of the stabilities of human IgG in the CyD PPRX hydrogels may be due to the encapsulation of the protein in the pores of the CyD PPRX hydrogels, which might hinder the mobility of the protein with subsequent decrease in the intermolecular interaction of the protein molecules. Thus, CyD PPRX hydrogels have the potential for stable pharmaceutical formulations including highly concentrated human IgG. Hereafter, comparison of not only the release but also the stability between the PPRX hydrogel system and other hydrogel systems such as gelatin hydrogel should be required.
CONCLUSIONS
In this study, we demonstrated the formation of CyD supramolecular hydrogels encapsulating highly concentrated human IgG. The resulting hydrogels were formed by the primary driving force arising from the columnar structures of PPRXs and prolonged the release of human IgG. Importantly, remarkable improvement in the stability of human IgG against thermal and shaking stresses was provided by CyD PPRX hydrogels. Recently, a large number of antibodies have been developed as promising drugs. However, to prepare the formulation which provides the stabilization of highly concentrated antibodies is a laborious work. The results obtained in the present study suggest that PEG/CyD PPRX hydrogels could be a promising delivery system and a stabilizing material for highly concentrated human IgG.
Acknowledgments
The authors thank Nihon Shokuhin Kako Co., Ltd. (Tokyo, Japan) for providing parent CyDs. The authors are grateful to T. Minamoto and N. Watabe, Renishaw Inc., for their assistance on a Raman microscope experiment.
Conflict of Interest
This study was funded by Terumo Corporation (Kanagawa, Japan). S. Koyama, R. Iibuchi, S. Mieda, K. Handa, and T. Kimoto are researchers of Terumo Corporation, R&D Headquarters.
References
- 1.Chames P, Regenmortel MV, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Novaretti MC, Dinardo CL. Immunoglobulin: production, mechanisms of action and formulations. Rev Bras Hematol Hemoter. 2011;33:377–82. doi: 10.5581/1516-8484.20110102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shapiro RS. Why I use subcutaneous immunoglobulin (SCIG) J Clin Immunol. 2013;33(Suppl 2):S95–8. doi: 10.1007/s10875-012-9853-2. [DOI] [PubMed] [Google Scholar]
- 4.Daugherty AL, Mrsny RJ. Formulation and delivery issues for monoclonal antibody therapeutics. Adv Drug Deliv Rev. 2006;58:686–706. doi: 10.1016/j.addr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 5.Harris RJ, Shire SJ, Winter C. Commercial manufacturing scale formulation and analytical characterization of therapeutic recombinant antibodies. Drug Dev Res. 2004;61:137–54. doi: 10.1002/ddr.10344. [DOI] [Google Scholar]
- 6.Kanai S, Liu J, Patapoff TW, Shire SJ. Reversible self-association of a concentrated monoclonal antibody solution mediated by Fab-Fab interaction that impacts solution viscosity. J Pharm Sci. 2008;97:4219–27. doi: 10.1002/jps.21322. [DOI] [PubMed] [Google Scholar]
- 7.Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004;93:1390–402. doi: 10.1002/jps.20079. [DOI] [PubMed] [Google Scholar]
- 8.Saluja A, Badkar AV, Zeng DL, Kalonia DS. Ultrasonic rheology of a monoclonal antibody (IgG2) solution: implications for physical stability of proteins in high concentration formulations. J Pharm Sci. 2007;96:3181–95. doi: 10.1002/jps.20970. [DOI] [PubMed] [Google Scholar]
- 9.Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm. 1999;185:129–88. doi: 10.1016/S0378-5173(99)00152-0. [DOI] [PubMed] [Google Scholar]
- 10.Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm. 2005;289:1–30. doi: 10.1016/j.ijpharm.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Bolli R, Woodtli K, Bartschi M, Hofferer L, Lerch P. L-Proline reduces IgG dimer content and enhances the stability of intravenous immunoglobulin (IVIG) solutions. Biologicals. 2010;38:150–7. doi: 10.1016/j.biologicals.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Charman SA, Mason KL, Charman WN. Techniques for assessing the effects of pharmaceutical excipients on the aggregation of porcine growth hormone. Pharm Res. 1993;10:954–62. doi: 10.1023/A:1018994102218. [DOI] [PubMed] [Google Scholar]
- 13.Ignatova Z, Gierasch LM. Inhibition of protein aggregation in vitro and in vivo by a natural osmoprotectant. Proc Natl Acad Sci U S A. 2006;103:13357–61. doi: 10.1073/pnas.0603772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerwin BA, Heller MC, Levin SH, Randolph TW. Effects of Tween 80 and sucrose on acute short-term stability and long-term storage at -20 degrees C of a recombinant hemoglobin. J Pharm Sci. 1998;87:1062–8. doi: 10.1021/js980140v. [DOI] [PubMed] [Google Scholar]
- 15.Jorgensen L, Hostrup S, Moeller EH, Grohganz H. Recent trends in stabilising peptides and proteins in pharmaceutical formulation—considerations in the choice of excipients. Expert Opin Drug Deliv. 2009;6:1219–30. doi: 10.1517/17425240903199143. [DOI] [PubMed] [Google Scholar]
- 16.Serno T, Carpenter JF, Randolph TW, Winter G. Inhibition of agitation-induced aggregation of an IgG-antibody by hydroxypropyl-β-cyclodextrin. J Pharm Sci. 2010;99:1193–206. doi: 10.1002/jps.21931. [DOI] [PubMed] [Google Scholar]
- 17.Szenczi A, Kardos J, Medgyesi GA, Závodszky P. The effect of solvent environment on the conformation and stability of human polyclonal IgG in solution. Biologicals. 2006;34:5–14. doi: 10.1016/j.biologicals.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Kiese S, Papppenberger A, Friess W, Mahler HC. Shaken, not stirred: mechanical stress testing of an IgG1 antibody. J Pharm Sci. 2008;97:4347–66. doi: 10.1002/jps.21328. [DOI] [PubMed] [Google Scholar]
- 19.Hillgren A, Lindgren J, Alden M. Protection mechanism of Tween 80 during freeze-thawing of a model protein, LDH. Int J Pharm. 2012;237:57–69. doi: 10.1016/S0378-5173(02)00021-2. [DOI] [PubMed] [Google Scholar]
- 20.Mahler HC, Muller R, Friess W, Delille A, Matheus S. Induction and analysis of aggregates in a liquid IgG1-antibody formulation. Eur J Pharm Biopharm. 2005;59:407–17. doi: 10.1016/j.ejpb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Ha E, Wang W, Wang WJ. Peroxide formation in polysorbate 80 and protein stability. J Pharm Sci. 2002;91:2252–64. doi: 10.1002/jps.10216. [DOI] [PubMed] [Google Scholar]
- 22.Kerwin BA. Polysorbates 20 and 80 used in the formulation of protein biotherapeutics: structure and degradation pathways. J Pharm Sci. 2008;97:2924–35. doi: 10.1002/jps.21190. [DOI] [PubMed] [Google Scholar]
- 23.Uekama K, Hirayama F, Irie T. Cyclodextrin drug carrier systems. Chem Rev. 1998;98:2045–76. doi: 10.1021/cr970025p. [DOI] [PubMed] [Google Scholar]
- 24.Higashi T, Tajima A, Motoyama K, Arima H. Cyclodextrin/poly(ethylene glycol) polypseudorotaxane hydrogels as a promising sustained-release system for lysozyme. J Pharm Sci. 2012;101:2891–9. doi: 10.1002/jps.23232. [DOI] [PubMed] [Google Scholar]
- 25.Harada A, Kamachi M. Complex formation between poly(ethylene glycol) and α-cyclodextrin. Macromolecules. 1990;23:2821–3. doi: 10.1021/ma00212a039. [DOI] [Google Scholar]
- 26.Harada A, Li J, Kamachi M. The molecular necklace: a rotaxane containing many threaded α-cyclodextrins. Nature. 1992;356:325–7. doi: 10.1038/356325a0. [DOI] [Google Scholar]
- 27.Harada A, Hashidzume A, Yamaguchi H, Takashima Y. Polymeric rotaxanes. Chem Rev. 2009;109:5974–6023. doi: 10.1021/cr9000622. [DOI] [PubMed] [Google Scholar]
- 28.Harada A, Li J, Kamachi M. Double-strand inclusion complexes of cyclodextrin threaded on poly(ethylene glycol) Nature. 1994;370:126–8. doi: 10.1038/370126a0. [DOI] [Google Scholar]
- 29.Li J. Cyclodextrin inclusion polymers forming hydrogels. Adv Polym Sci. 2009;222:79–112. doi: 10.1007/12_2008_6. [DOI] [Google Scholar]
- 30.Li J. Self-assembled supramolecular hydrogels based on polymer-cyclodextrin inclusion complexes for drug delivery. NPG Asia Mater. 2010;2:112–8. doi: 10.1038/asiamat.2010.84. [DOI] [Google Scholar]
- 31.Li J, Loh XJ. Cyclodextrin-based supramolecular architectures: syntheses, structures, and applications for drug and gene delivery. Adv Drug Deliv Rev. 2008;60:1000–17. doi: 10.1016/j.addr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Li JJ, Zhao F, Li J. Polyrotaxanes for applications in life science and biotechnology. Appl Microbiol Biotechnol. 2011;90:427–43. doi: 10.1007/s00253-010-3037-x. [DOI] [PubMed] [Google Scholar]
- 33.Wenz G, Han BH, Muller A. Cyclodextrin rotaxanes and polyrotaxanes. Chem Rev. 2006;106:782–817. doi: 10.1021/cr970027+. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Harada A, Kamachi M. Sol-gel transition during complex formation between α-cyclodextrin and high molecular weight poly(ethylene glycols) in aqueous solution. Polym J. 1994;26:1019–26. doi: 10.1295/polymj.26.1019. [DOI] [Google Scholar]
- 35.Abu IIH, Higashi T, Anno T, Motoyama K, Abd-ElGawad AE, El-Shabouri MH, et al. Potential use of γ-cyclodextrin polypseudorotaxane hydrogels as an injectable sustained release system for insulin. Int J Pharm. 2010;392:83–91. doi: 10.1016/j.ijpharm.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Higashi T, Hirayama F, Misumi S, Arima H, Uekama K. Design and evaluation of polypseudorotaxanes of pegylated insulin with cyclodextrins as sustained release system. Biomaterials. 2008;29:3866–71. doi: 10.1016/j.biomaterials.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 37.Harada A. Preparation and structures of supramolecules between cyclodextrins and polymers. Coord Chem Rev. 1996;148:115–33. doi: 10.1016/0010-8545(95)01157-9. [DOI] [Google Scholar]
- 38.Takeo K, Kuge T. Complexes of starchy materials with organic compounds. Part IV. X-ray diffraction of γ-cyclodextrin complexes. Agric Biol Chem. 1970;34:568–74. doi: 10.1271/bbb1961.34.568. [DOI] [Google Scholar]
- 39.Topchieva IN, Tonelli AE, Panova IG, Matuchina EV, Kalashnikov FA, Gerasimov VI, et al. Two-phase channel structures based on α-cyclodextrin-polyethylene glycol inclusion complexes. Langmuir. 2004;20:9036–43. doi: 10.1021/la048970d. [DOI] [PubMed] [Google Scholar]
- 40.Toropainen T, Heikkila T, Leppanen J, Matilainen L, Velaga S, Jarho P, et al. Crystal structure changes of γ-cyclodextrin after the SEDS process in supercritical carbon dioxide affect the dissolution rate of complexed budesonide. Pharm Res. 2007;24:1058–66. doi: 10.1007/s11095-006-9227-7. [DOI] [PubMed] [Google Scholar]
- 41.Härtl E, Winter G, Besheer A. Influence of hydroxypropyl-β-cyclodextrin on the stability of dilute and highly concentrated immunoglobulin g formulations. J Pharm Sci. 2013;102:4121–31. doi: 10.1002/jps.23729. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Ni X, Leong KW. Injectable drug-delivery systems based on supramolecular hydrogels formed by poly(ethylene oxide)s and α-cyclodextrin. J Biomed Mater Res A. 2003;65:196–202. doi: 10.1002/jbm.a.10444. [DOI] [PubMed] [Google Scholar]
- 43.Ma D, Tu K, Zhang LM. Bioactive supramolecular hydrogel with controlled dual drug release characteristics. Biomacromolecules. 2010;11:2204–12. doi: 10.1021/bm100676a. [DOI] [PubMed] [Google Scholar]
- 44.Ma D, Zhang LM, Xie X, Liu T, Xie MQ. Tunable supramolecular hydrogel for in situ encapsulation and sustained release of bioactive lysozyme. J Colloid Interface Sci. 2011;359:399–406. doi: 10.1016/j.jcis.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 45.Telikepalli SN, Kumru OS, Kalonia C, Esfandiary R, Joshi SB, Middaugh RC, Volkin DB. Structural characterization of IgG1 mAb aggregates and particles generated under various stress conditions. J. Pharm. Sci. 2014; 103:796–809. [DOI] [PMC free article] [PubMed]
- 46.Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant. 2005;20(Suppl 6):vi3–9. doi: 10.1093/ndt/gfh1092. [DOI] [PubMed] [Google Scholar]
- 47.Uversky VN. A GLYmmer of insight into fibril formation. Structure. 2005;13:1090–2. doi: 10.1016/j.str.2005.07.003. [DOI] [PubMed] [Google Scholar]