

Abstract
The RAS proteins play a role in cell differentiation, proliferation, and survival. Aberrant RAS signaling has been found to play a role in 30% of all cancers. KRAS, a key member of the RAS protein family, is an attractive cancer target, as frequent point mutations in the KRAS gene render the protein constitutively active. A number of attempts have been made to target aberrant KRAS signaling by identifying small molecule compounds that (1) are synthetic lethal to mutant KRAS, (2) block KRAS/GEF interactions, (3) inhibit downstream KRAS effectors, or (4) inhibit the post-translational processing of RAS proteins. In addition, inhibition of novel targets outside the main KRAS signaling pathway, specifically the cell cycle related kinase PLK1, has been shown have an effect in cells that harbor mutant KRAS. Herein we review the use of various high-throughput screening assays utilized to identify new small-molecule compounds capable of targeting mutant KRAS-driven cancers.
INTRODUCTION
The RAS (rat sarcoma) protein family members are all low-molecular-weight GTP-binding proteins that play a role in regulating cell differentiation, proliferation, and survival.1,2 There are three main members of the RAS family: HRAS, NRAS, and KRAS, all of which have been found to drive cancer formation and progression.1–3 Mutations in RAS are found in approximately 30% of human cancers.2 In the absence of a RAS mutation, increased RAS activity in human tumors has been shown to be the result of gene amplification, overexpression, or increased upstream activation.4 Single point mutations of the RAS gene, affecting residues G12 and G13, abolish GAP-induced GTP hydrolysis through steric hindrance, while mutations of residue Q61 interfere with the coordination of a water molecule necessary for GTP hydrolysis.5 These mutations render the protein constitutively active, and the persistence of active GTP-bound RAS leads to the constant activation of its downstream effector pathways. KRAS is the most frequently mutated RAS isoform, having been shown to be mutated in 90% of pancreatic adenocarcinomas, 45% of colorectal cancers, and 35% of lung adenocarcinomas.2,6 KRAS mutations have been associated with increased tumorigenicity and poor prognosis.4 Additionally, the inhibition of activated RAS has been shown to revert malignant cells to a nonmalignant phenotype and cause tumor regression both in vitro and in vivo.4,7,8 Thus, KRAS is an attractive therapeutic target for a number of cancers.
While the development of a small molecule inhibitor of the constitutively active KRAS protein would be ideal as a cancer therapeutic, 25 years of work on drugs targeting the GTP binding pocket of mutant KRAS have thus far proven to be unsuccessful. To date, no effective therapy that specifically targets mutant KRAS is available. Targeting the constitutively active molecular switch of KRAS is quite difficult because the role of GDP or GTP is to stabilize inactive or active states of the RAS protein.9 This is in contrast to protein kinases in which phosphoryl transfer from ATP to a substrate is a rapid, catalytic process.9 Additionally, because of picomolar affinity between KRAS and GTP, as well as the micromolar concentration of GTP in the cell, a competitive inhibitor is not particularly feasible.9 Furthermore, KRAS activation and signaling are accomplished through protein–protein interactions (PPIs) with guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), and the various KRAS effector proteins.10 PPIs are challenging to target because of the relatively featureless topologies of the surfaces involved.11 In spite of these issues, a number of attempts have been made to target aberrant KRAS signaling at different levels.
KRAS SIGNALING PATHWAY
Gaining a comprehensive understanding of the life cycle of the KRAS protein is essential to mounting a successful drug discovery effort. After RAS proteins are translated, they must undergo a series of post-translational modifications (Figure 1).2,12–14 Initially, the thiol group of the terminal cysteine in KRAS is farnesylated by farnesyltransferase (FTase).15–18 Next, the CAAX (C = cysteine; A = isoleucine; X = serine or methionine)19,20 protease RAS-converting enzyme 1 (RCE1) cleaves the terminal AAX amino acids, and the carboxy group of the cysteine is methylated by isoprenylcysteine carboxylmethyl-transferase 1 (ICMT1).21–23 Finally, palmitoyl transferase (PTase) transfers a palmitoyl moiety to cysteine residues located just upstream of the C-terminus of HRAS, NRAS, and the KRAS-4A isoform.3 The RAS protein then forms a stable interaction with the cell membrane via its farnesyl and palmitoyl groups as well as the positively charged C-terminal lysine residues. Thus, the post-translational modification of KRAS offers several possible drug targets: (1) FTase,24 (2) CAAX endopeptidase, 25,26 (3) methyltransferase,27 and (4) palmitoyl transferase.16
Figure 1.

Post-translational processing of RAS proteins. Farnesyltransferase (FTase) catalyzes the transfer of a farnesyl group to the terminal cysteine of new synthesized RAS proteins. Next, the three C-terminal amino acids are cleaved by the endopeptidase RAS-converting enzyme 1 (RCE1). Carboxymethylation of of the terminal cysteine residue is accomplished by isoprenylcysteine carboxylmethyltransferase 1 (ICMT1). Finally, palmitoyl transferase (PTase) transfers a palmitoyl group to C-terminal cysteine residues of HRAS, NRAS, and the KRAS-4A isoform.
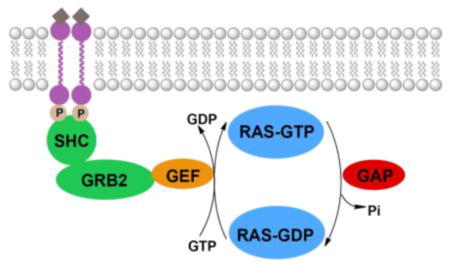
RAS proteins cycle between the inactive GDP-bound form and the active GTP-bound form. This activation state is regulated by GEFs and GAPs (Figure 2).6,28–33 When a growth factor binds to a tyrosine kinase receptor, it forms an active homodimer that undergoes autophosphorylation. This leads to binding of the adaptor proteins SHC and growth factor receptor bound 2 (GRB2). GRB2 then associates with GEFs through its SH3 domain.34 GEFs exchange bound GDP for GTP, thereby activating the RAS protein and initiating a signaling cascade. To terminate signaling, GAPs stimulate the intrinsic GTPase activity of RAS proteins, causing the hydrolysis of GTP to GDP, leading to deactivation of the RAS protein. Thus, upstream targets for KRAS inhibition include (1) tyrosine kinases,35 (2) the GRB2/GEF interaction,36 and (3) GEFs.37
Figure 2.

Upstream signaling of RAS. Activated growth-factor-recptor tyrosine kinases undergo autophosphorylation and interact with the adaptor proteins SHC and GRB2. These proteins associate with various GEFs which exchange GDP for GTP, thereby activating the RAS protein. GAPs stimulate the RAS protein’s intrinsic GTPase activity, causing the hydrolysis of GTP to GDP, thereby inactivating the protein.
KRAS has many downstream effector proteins that it can interact with to alter cell survival and proliferation (Figure 3).3,37,38 Active GTP-bound KRAS predominantly signals through RAF protein kinases, phosphoinositide 3-kinases (PI3K), guanine nucleotide exchange factors for the RAS-related protein Ral (RalGDS), and phospholipase Cε (PLCε). RAF initiates the mitogen-activated protein (MAP) kinase cascade, which activates extracellular signal-regulated kinase (ERK). This active kinase has numerous targets, including the transcription factor ELK1, which regulates the expression of cell-cycle progression genes.39–43 The PI3K pathway activates AKT and leads to the transcription of prosurvival genes, cytoskeletal remodeling, and the activation of numerous transcription-factor pathways, most notably NF-κB.44,45 RalGDS activation leads to the activation of the RAL1 binding protein: this in turn leads the inhibition of FOX transcription factors46 which are involved in cell growth, proliferation, and differentiation. RalGDS can also signal through the JNK pathway to stimulate the transcription of prosurvival and cell-cycle progression genes.47,48 The activation of PLCε results in protein kinase C (PKC) activation and the mobilization of calcium from intracellular stores.6 It is clear from this brief overview that there are numerous downstream targets that can be inhibited in order to try and overcome aberrant KRAS signaling.
Figure 3.

Downstream signaling of RAS. Ras interacts with a number of downstream effectors. The main ones include RAF protein kinases, phosphoinositide 3-kinases (PI3Ks), guanine nucleotide exchange factors for the RAS-related protein Ral (RalGDS), and phospholipase Cε (PLCε).
HIGH-THROUGHPUT SCREENING
The screening of chemical compounds for pharmacological activity has been ongoing in various forms for the past 40 years. The recent advances in molecular biology, computer science, robotics, instrumentation, and overall process engineering have established high-throughput screening as a predominant tool in the field of drug discovery to identify compounds that are active against various biological targets or phenotypes. High-throughput screening (HTS) has been used to identify structurally diverse chemical compounds that are capable of blocking different stages of KRAS signaling in order to inhibit the proliferation of mutant KRAS-driven cancer cells both in vitro and in vivo.
Synthetic Lethality Screening
Synthetic lethality screening aims to identify small molecules that selectively kill oncogene-expressing engineered cell lines.49 Different groups have performed HTS screening, using different cell line models, in an attempt to find compounds that are lethal to mutant KRAS-driven cell lines. A few such examples and their successes are discussed below.
Torrance et al. utilized DLD-1 colon cancer cells that harbor mutant KRAS and an isogenic derivative in which the mutant KRAS allele was deleted (KO cells).50 A yellow fluorescent protein expression vector was introduced into the DLD-1 cells, while a blue fluorescent protein was introduced into the isogenic derivative. Equal volumes of the two cell lines were cocultured and tested against 29 440 compounds from the ChemBridge and NCI libraries, and results were assessed by analyzing the differential fluorescence intensity for blue and yellow fluorescent protein. Positive hits were then screened as above but over a range of drug concentrations. The main hits from this screen were a sulfinyl cytidine (1) derivative (2 or 3) and triphenyltetrazolium (4) (Figure 4).50 A mixture of the isomers 2 and 3 on DLD-1 and KO cells demonstrated IC50 values of 125 and ~750 ng/mL, respectively, while the IC50 values for 4 on DLD-1 and KO cells were ~2 and ~12 μg/mL. Both compounds demonstrated a 6-fold selectivity for the mutant KRAS-cell-lines-containing DLD-1 cells over the KO cells. Further research demonstrated that the mixture of isomers 2 and 3 selectively inhibited the growth of the KRASG12 V-transformed rat kidney epithelial cells compared to the parental RK3E cell line, while compound 4 had no effect in these two assays. The antitumor activity of the mixture of isomers 2 and 3 was also assessed in vivo using HCT116 and DLD-1 xenograft models, both of which harbor a single G13D point mutation in the KRAS gene; at 150 mg/kg, tumor volumes were reduced by approximately 65% and 45%, respectively.
Figure 4.

Structures of 1; 2; 3; and 4.
Guo et al. utilized human ovarian surface epithelial cells that were immortalized with human telomerase reverse transcriptase (hTERT) and SV40 (T29) transformed with either mutant KRAS (T29Kt1) or mutant HRAS (T29Ht1).51 Ten-thousand compounds from the ChemBridge library were screened against the three cell lines, and a sulforhodamine B (SRB) colorimetric assay was used to determine cytotoxicity. Initial hits found to inhibit >50% of cell growth were confirmed by two more SRB dose–response tests. The hit identified was oncrasin 1 (5; Figure 5). Compound 5 induced cytotoxicity in T29Ktl cells in a dose-dependent manner, with an IC50 of 4.81 μM. However, no cytotoxicity was detected in T29 or T29Ht1 cells. Further research demonstrated that 5 effectively killed KRAS mutant lung cancer cells (H460, H2122, H2887, and A549) with an IC50 of ≤3 μM but had little effect on the cell viability of lung cancer cells with wild-type RAS (H322 and H1395) or lung cancer cells that harbor mutant NRAS (H1299 and H2087). These results illustrate that 5 is highly selective for cells transformed with mutant KRAS. The in vivo antitumor activity of 5 was assessed using an H460 lung cancer xenograft model with daily intraperitoneal (ip) injections of 100 mg/kg. Compound 5 inhibited tumor growth by 75.4% compared to a solvent control, prolonged survival from 24 to 32 days and was well tolerated as evidenced by no weight loss. Additionally, the researchers noted that 5 was also synthetically lethal to protein kinase Cι; but the biological mechanisms behind this dual synthetic lethality remain to be determined.51
Figure 5.
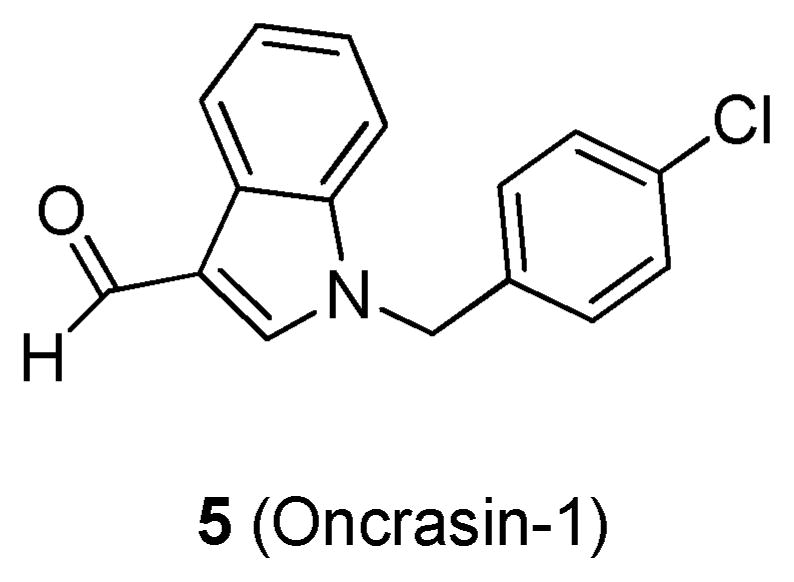
Structure of 5.
Shaw et al. utilized mouse embryonic fibroblasts (MEFs) that expressed oncogenic KRASG12D to identify effective therapeutic agents for patients with KRAS mutant tumors.52 Over 50 000 compounds were screened against KRAS wild-type and mutant expressing MEF cell lines using a high-throughput ATP-based cell viability assay (CellTiter-Glo (Promega)). This assay identified tolperisone (6; Figure 6) as a lead, which showed significant differential activity in the KRAS WT cell line compared to the KRASG12D cell line in the viability assays, as well as in a bromodeoxyuridine (BrdU) incorporation assay. Subsequently, 10 derivatives of 6 were examined to explore structure–activity relationships. The derivative lanperisone (7; Figure 6)52 showed the most potent and selective activity against KRASG12D MEFs, with an IC50 of 4 μM in an ATP-based cell viability assay. Compound 7; a single isomer with a minor structural modification from 6; was shown to induce reactive oxygen species that are inefficiently scavenged in KRAS mutant cells leading to apoptosis. This increase in oxidative stress is the putative mechanism of action for compound 7. In vivo pharmacological studies were carried out using compound mutant MEFs carrying KRASG12D in a p53-null background. Results showed that tumors from mice treated with 7 were 24% smaller (by volume) and 37% smaller (by weight) compared to tumors from control animals. With human safety and pharmacokinetic data already completed, 7 is poised for rapid clinical development.
Figure 6.

Structures of 6 and 7.
In summary, researchers have identified several lead compounds to treat KRAS driven cancer using synthetic lethality screening. However, it should be noted that the synthetic lethality screening approach is based on a phenotype, not a molecule target. Thus, while there is selectivity based on the expression of mutant KRAS, the exact target at a molecular level is unknown. This fact can limit the full understanding of the biology involved in the selectivity of the compounds and accordingly the design of more druglike analogues.
Blocking KRAS/GEF Interactions
While PPIs are challenging to target because they often lack a defined binding pocket for small molecules, fragment-based screening is well-suited for this endeavor.11 Outside the guanine nucleotide-binding domain, the KRAS protein has no known allosteric sites for potential modulation.53 Several RAS-GEFs have been identified, and most research efforts have been focused on son of sevenless (SOS). The pockets adjacent to the KRAS/SOS contact surface are amenable to small-molecule binding.53 In 2012, two groups at Genentech and Vanderbilt independently utilized NMR-based fragment screening to identify small molecules that bind to the pocket adjacent to the KRAS/SOS interaction surface and subsequently interfere with the KRAS/SOS interaction.10,53
Fragments are defined as low molecular weight (120–250 Da), moderately lipophilic (clogP < 3), highly soluble organic molecules that typically bind to their target protein with low affinity (100 μM to millimolar).54,55 In order to identify fragments that bind to KRASG12D, both groups utilized a target-based NMR method, measuring the changes in the chemical shift of a labeled target protein upon ligand binding, with 15N-labeled GDP-bound KRAS12D. Fragment binding was assessed by changes in the resonance position of cross-peaks in the 1H/15N two-dimensional heteronuclear single-quantum coherence spectra.56,57 The Genentech group also used the ligand-based approach of saturation transfer difference.53 In this approach, the protein is selectively irradiated and magnetization is transferred from the protein to the bound ligand. The magnetized ligand is then detected in the free solution, and difference spectroscopy (with an unirradiated protein reference spectrum) is utilized to identify the bound ligand. Although these methods are not considered to be a high-throughput methodology, NMR automation and the pooling of test fragments for screening allowed for large fragment libraries to be screened relatively quickly.
DCAI (8; Figure 7a), identified by Genentech, was found to bind in the hydrophobic pocket located between the α2 helix (of the switch II region) and the core β-sheet of the KRAS protein and thereby interrupt KRAS/SOS association.53 Titration studies with 8 indicated that it blocked both nucleotide exchange and release reactions, with IC50 values of 342 ± 22 and 155 ± 36 μM, respectively. It was also noted that a secondary electronegative binding cleft is formed in the protein upon fragment binding. It is proposed that 8 blocks the first phase of the exchange reaction by inhibiting nucleotide release from KRAS.
Figure 7.

(a) Structures of compounds shown to interfere with the KRAS/SOS interaction. (b) Overlay of 8 (pink) and a compound similar to 11 (teal) in the hydrophobic binding pocket of KRAS. Reproduced with permission of D. Erlanson.58
The Vanderbilt group10 screened 11 000 fragments of which 140 were shown to bind to GDP-bound KRASG12D. Indole derivative 9 (Figure 7a) is a representative example of the hits that exhibit binding affinity to KRASG12D at 1.3–2 mM. Furthermore, compound 9 was shown to inhibit SOS-mediated nucleotide exchange, thereby preventing the activation of KRAS. Using structure-based design, the Vanderbilt group added functionality to their original fragment hit in order to target the secondary electronegative binding cleft formed upon fragment binding. The resulting S-isoleucin derivative 10 and D-proline analogue 11 (Figure 7a)10 displayed improved binding affinity (190 and 340 μM, respectively) and were capable of inhibiting a greater percent of SOS-catalyzed nucleotide exchange. Additionally, a molecular modeling overlay indicates that compound 11 binds to the same hydrophobic pocket as 8; however, they appear to make very different contacts (Figure 7b).58
Inhibiting KRAS Effectors
Because of the difficulty of targeting KRAS directly, efforts have been put into targeting the downstream effector pathways, particularly the Raf-MEK-ERK59–61 and PI3K pathways.44,45 Raf kinase inhibitors, such as sorafenib (12)62,63 and vemurafenib (13)64 (Figure 8), have shown impressive antitumor activity. However, the use of Raf inhibitors in RAS mutant cancer cells has been shown, paradoxically, to promote Raf-MEK-ERK signaling, thus precluding their use.9 Furthermore, data suggest that RAS mutant tumors are insensitive to single-agent PI3K inhibitors.9 On the other hand, certain inhibitors of MEK, such as selumetinib (14)65 and GDC-0973 (15)66 (Figure 8), have shown antiproliferative efficacy in RAS mutant cancers.
Figure 8.

Structures of 12; 13; 14; and 15.
Daouti et al. recently screened compounds in an IMAP kinase assay (Molecular Devices) with a cRaf/MEK/ERK cascade.67 This unique assay detects phosphorylation by the binding of an FITC-labeled ERK substrate peptide to a proprietary binding reagent bead.68 To determine MEK specificity, secondary assays with Caliper LabChip mobility shift kinase kits were utilized with cRaf and MEK/ERK cascades independently.67 These assays measure the conversion of a fluorescent peptide substrate to a phosphorylated product on a microfluidic chip by separating the nonphosphorylated substrate from the phosphorylated product via electrophoresis and detection via laser-induced fluorescence. 69 Of the compounds screened, a new class of potent and selective MEK1/2 inhibitors, represented by RO4927350 (16; Figure 9) was identified.67 In a cRaf/MEK/ERK cascade assay, compound 16 potently inhibited MEK1 kinase activity with an IC50 of 23 nM. Interestingly, it also showed consistent MEK inhibition with ATP concentrations ranging from 2 to 250 μM, which suggests that 16 is a non-ATP-competitive inhibitor and induces or stabilizes a novel fold in the MEK kinase. Compound 16 showed activity in the cRaf/MEK/ERK and MEK/ERK assays (with Ki values of 53 and 184 nM, respectively) but was not active in the cRaf assay (Ki > 50 μM) when the compound was tested in the Caliper cRaf assay. Ambit’s proprietary kinase profiling platform was used to assess the effects of 16 (10 μM) on 227 different kinases, and it was found to be inactive against all other kinases, including cRaf and bRaf. The antitumor activity of 16 was investigated in vivo in a LOX human melanoma xenograft model in nude mice, containing the most common activating bRaf mutation (V600E). When 16 was administered orally to LOX tumor-bearing mice twice daily with doses ranging from 12.5 to 200 mg/kg for 11 days, dose-dependent antitumor activity was observed and partial or complete tumor regression was elicited at doses above 50 mg/kg. This compound was determined to have a unique mechanism of action: inhibiting both MEK and ERK phosphorylation simultaneously.67
Figure 9.

Structures of 16; 17; and 18.
Lead optimization to improve the potency and PK–PD profile of 16 resulted in RO5068760 (17; Figure 9),70 which contains a phenyl group in place of the thiazole in 16. In vitro, 17 demonstrates MEK1 kinase inhibitory activity with an IC50 of 0.025 ± 0.012 μM in a cRaf/MEK/ERK cascade assay, a 2-fold improvement in potency compared to 16. In vivo characterization of MEK 1/2 inhibition (as measured by ERK phosphorylation) in LOX melanoma and HT-29 colorectal cancer models gave estimated EC50 values in plasma of 1.36 and 3.35 μM, respectively. Furthermore, to produce ≥90% of tumor inhibition in vivo, plasma concentrations of 17 needed to be 0.65 or 5.23 μM for bRafV600E or mutant KRAS tumor models, respectively. These data were similar to the IC95 values (0.64 or 4.1 μM) determined for cellular growth inhibition in vitro. Compound 17 is currently in preclinical development.
Tumor cell lines that harbor oncogenic mutation in both RAS and PIK3CA have been shown to be resistant to MEK inhibition.9 Thus, dual combinations of cRaf-MEK-ERK and PI3K inhibitors may be needed to effectively treat RAS mutant cancers. Indeed, combined inhibition of MEK and PI3K signaling showed good antitumor activity in mice with mutant KRAS-driven lung tumors.71 For example, when NVP-BEZ235 (18; Figure 9),72 a potent dual inhibitor of PI3K and mTOR, was combined with the MEK inhibitor 14; there was marked synergy in shrinking mutant KRAS lung cancers.71 Additionally, after 2 days of the combination treatment, there was notable down-regulation of PI3K and ERK.
Inhibiting the Post-Translational Processing of KRAS
As shown in Figure 1, RAS proteins undergo a four-stage post-translational lipid modification: (1) farnesylation by FTase, (2) proteolytic cleavage of the terminal AAX motif by the CAAX protease RCE1, (3) carboxymethylation of the terminal cysteine by ICMT1, and (4) palmitoylation of cysteine residues close to the C-terminus of the protein.4,6 As membrane localization is required for the signaling and transforming capabilities of KRAS,73 any of these points represent viable targets for therapeutic intervention.
FTase is an essential enzyme that attaches a 15-carbon isoprenoid farnesyl group to KRAS and is required for its proper localization and activity. Farnesyltransferase inhibitors (FTIs)74,75 fall into two categories: the CAAX peptidomimetics, including FTI-276 (19),76 FTI-277 (20),77 L-744832 (21),78 B956 (22),79 and FTI-2153 (23)80 (Figure 10), and the non-peptidomimetics, which include tipifarnib (24),81 lonafarnib (25),82 and BMS-214662 (26)83 (Figure 10). A number of recent publications have described their preclinical efficacy. 4,75,84,85 In the case of compound 26; the mean IC50 values for HRAS farnesylation and KRAS farnesylation were determined to be 1.3 and 8.4 nM, respectively. Intravenous administration at 75 mg/kg compound 26 for 24 h led to 90% tumor cell kill in HCT-116 xenograft models.83 While compound 26 was able to reverse the HRAS-transformed phenotype in vivo, it was unable to do the same for the KRAS-transformed phenotype, likely because of the potential geranylation of KRAS as an alternative to farnesylation. In fact, in the presence of FTIs the geranylation of KRAS is able to rescue its processing.86
Figure 10.

Structures of selected farnesyltransferse inhibitors.
Compound 24 has displayed clinical efficacy as a single agent in elderly patients with acute myeloid leukemia (AML),87–89 myeloproliferative disorders,90,91 high-risk myelodysplasia,92–94 and multiple myeloma.95 It has also been investigated in combination with etoposide for the treatment of AML: in a phase I trial 21 of 84 patients (25%) achieved complete remission, with a median complete remission duration of 9.8 months.96 Additionally, compound 24 was investigated in combination with gemcitabine for the treatment of advanced pancreatic cancer: while the combination gave a favorable toxicity profile, overall survival was not prolonged compared to gemcitabine as a single agent.97 Compound 25 has not shown promising clinical acitivty as a monotherapy for solid tumors, as phase II monotherpy trials for advanced pancreatic,98 advanced colorectal,99 and head and neck cancers100 indicated single-agent activity to be <10%.101 However, 25 has shown some single agent activity in advanced hematologic malignancies.102 Combination of 25 and gemcitabine in a phase II trial as a second line treatment for advanced urothetial cancer gave a good response rate,103 but a randomized phase III trial will be needed to assess the contribution of 25.101
The loss of RCE1 has been shown to cause a mislocalization of RAS proteins and to reduce RAS-induced transformation in cells.9,104 In an effort to find small-molecule inhibitors of RCE1, Manandhar et al. screened the National Cancer Institute Diversity Set compound library in an in vitro assay utilizing yeast Rce1p.105 In this high-throughput assay, diluted yeast (SM3164) membranes overexpressing the CAAX protease Rce1p were incubated with the compounds prior to the addition of a quenched fluorogenic substrate based on the KRAS C-terminal sequence. Results were determined by measuring the fluorescence every 30–60 s over a 60 min time-course. Of the almost 2000 compounds screened, 46 inhibitors were initially identified in a RAS-based assay and 9 compounds (selected examples 27–30; Figure 11)105 were effective in a secondary assay with a yeast a-factor mating pheromone substrate peptide. The IC50 values of these nine compounds were in the low micromolar range for both yeast (6–35 μM) and human Rce1p (0.4–46 μM). Additionally, copper complex 30 (Figure 11) showed good selectivity for Rce1p compared to the competitive CAAX protease Ste24p.
Figure 11.

Structures of RCE1 inhibitors.
Disruption of ICMT1 results in a significant mislocalization of RAS proteins and has been shown to inhibit cell growth and KRAS-induced oncogenic transformation in nude mice.9,27 Using a radioactive HTS, Winter-Vann et al. discovered a small-molecule inhibitor of ICMT1.27 The assay used measured ICMT1 activity by the incorporation of an [3H]methyl group into a farnesylated RCE1-proteolyzed KRAS substrate. In this assay, Sf9 insect cell membranes containing either recombinant RCE1 or ICMT1 were prepared and incubated with the library compounds. Results were assessed by the incorporation of a [3H]methyl group into a farnesylated, Rce1-proteolyzed KRAS substrate. Compounds displaying >50% inhibition of ICMT were screened in a secondary assay with an ICMT substrate biotin S-farnesyl-L-cysteine (BFC). The most potent compound was determined to be cysmethynil (31; Figure 12),27 which gave an IC50 of 2.4 μM in the BFC assay. This compound was also shown to inhibit cell growth in an ICMT1-dependent fashion, cause mislocalization of RAS, and impair epidermal growth factor-induced MAPK signaling. Additionally, compound 31 displayed high specificity for ICMT, as it did not inhibit other enzymes in the prenylation pathway at 50 μM including FTase, geranylgeranyltransferase type I, RCE1, an AdoMet-dependent DNA methyltransferase, and an unrelated protein methyltransferase.
Figure 12.

Structure of 31.
As membrane localization is essential for KRAS activity, van der Hoeven et al.106 developed a high-content assay to identify compounds capable of inhibiting KRAS membrane localization. The Prestwick chemical library was screened against wild-type Madin–Darby canine kidney epithelial (MDCK) cells or MDCK cells expressing green fluorescent protein (GFP) attached to the C-terminal 24 or 25 amino acids of KRAS (GFP-CTK) or HRAS (GFP-CTH). The cell lines were seeded in 96-well plates and exposed to the chemical library compounds for 48 h at 4 μg/mL. Plates were imaged to assess the compounds’ ability to displace the GFP-CTK or GFP-CTH from the plasma membrane. The hit identified was fendiline (32; Figure 13), which selectively and significantly mislocalized GFP-CTK compared to GFP-CTH. GFP-CTK was shown to be redistributed to endomembranes including the endoplasmic reticulum, Golgi apparatus, and cytosol. Furthermore, compound 32 inhibited downstream KRAS signaling and was able to block the proliferation of pancreatic, colon, lung, and endometrial cells. Compound 32 is a known L-type calcium channel blocker; however, as other classes of L-type calcium channel blockers were not shown to mislocalize KRAS, it is likely that the mechanism of disrupting KRAS membrane localization is not related to calcium channel blockade.
Figure 13.

Structures of 32; 33; and 34.
The farnesyl isoprenoid-containing small molecules salirasib (33) and TLN-4601 (34) (Figure 13) have also been shown to disrupt RAS membrane anchorage, leading to an inhibition of tumor cell growth.107–109 More specifically, these compounds are proposed to act as antagonist of RAS function by competing for membrane-bound farnesyl-binding proteins located in the cell membrane.110 These compounds have been evaluated in a number of clinical trials with varying results.111–113
EMERGING TARGETS AND NOVEL MECHANISMS
Many attempts to target mutant KRAS have been made; however, KRAS-related treatments have proved challenging in the clinic. Inhibitors of the upstream target epidermal growth factor receptor (EGFR), gefitinib (35) and erlotinib (36) (Figure 14), can significantly block KRAS signaling,114 but their effect is brief because of the rapid development of drug resistance. This often leads patients to develop a tumor recurrence that is generally refractory to chemotherapy and/or targeted therapy.
Figure 14.

Structures that are active in the KRAS mutant synthetic lethal assays.
To elucidate the biological mechanisms that play a role in regulating aberrant KRAS signaling but that are independent from the KRAS signaling cascade, RNAi screening has been the technique of choice.115 RNAi research can be completed in a high-throughput manner and aims to identify a novel synthetically lethal interaction. Using this technique, Luo et al.115 found KRAS mutant cells to be hypersensitive to the inhibition of the mitotic kinase polo-like kinase 1 (PLK1).116 PLK1 plays an important role in the G2/M and M phases of the cell cycle and has been reported to have deregulated activity in various cancer cells.117 The PLK1 inhibitor BI-2536 (37; Figure 14)115 was shown to inhibit the proliferation of KRAS mutant DLD-1 cells at 25 nM by >50%. In contrast, wild-type KRAS DLD-1 cells were unaffected by the treatment. Compound 37 also showed a statistically significant reduction in tumor volume in a KRAS mutants DLD-1 xenograft model.
Luo et al. also showed that KRAS mutant cells are sensitive to proteasome inhibition.115 By use of the proteasome inhibitors MG132 (38) and bortezomib (39) (Figure 14), proteasome inhibition was shown to exhibit synthetic lethality in cells that harbor mutant KRAS.115 Additionally, researchers showed that paclitaxel (40; Figure 14)115 produced a specific and marked reduction in cell fitness for mutant KRAS cells. This indicates that cells harboring mutant KRAS are hypersensitive to mitotic disruption, as the mitotic cycle of wild-type KRAS cells was not statistically affected.
Kumar et al.118 identified the transcription factor GATA binding protein 2 (GATA2) as being essential for the survival of non-small-cell lung carcinomas (NSCLC) that harbor mutant KRAS. Indeed, deletion of GATA2 in KRAS mutant NSCLC xenografts models (via tamoxifen induction of GATA2Flox/Flox mice) led to near-complete tumor regression. In another study investigating NSCLC, Puyol et al.119 identified cyclin-dependent kinase 4 (CDK4) as essential for propagating the survival of lung cells harboring mutant KRAS, as deletion of CDK4 led to immediate senescence and halted tumor progression in mouse models.
FUTURE PROSPECTS
HTS has been used to identify numerous compounds capable of disrupting the KRAS signaling pathway. However, no therapy capable of specifically and effectively targeting mutant KRAS is currently available. The problem is not due to a lack of therapeutic targets but rather due to a lack of a comprehensive understanding of the biology underlying the disease. Cell signaling is often depicted as a linear cascade, and this model is used to identify potential therapeutic targets. However, the linear depiction of cell signaling is an oversimplification. In reality, cell signaling is complex and dynamic, in which one protein interacts with many others in a nonlinear, circuit-like fashion. Cancer is not caused by just one genetic mutation or aberrant protein that drives the disease. Instead, multiple mutations existing together form and drive the progression of cancer. Thus, effective treatment will require the regulation of many targets to attain a sustained response. Therefore, a therapy capable of inhibiting multiple pathways/targets is likely the best option for treating mutant KRAS-driven cancers.
KRAS can activate both PI3K and RAF, which lie in different signaling cascades. If only one pathway is inhibited, the cell can circumvent this event by overexpressing the other pathway and thereby create a resistant phenotype. For example, treatment with just the multikinase inhibitor PP-121 (41; Figure 15),120 which is active against PI3K without blocking the RAS/RAF pathway, will likely be ineffective in patients. A better treatment option would be blocking both the PI3K and the RAS/RAF pathways, which could be achieved by the combination of a MEK inhibitor, such as 14; with an AKT inhibitor, such as MK-2206 (42; Figure 15).120 Inhibition of both pathways simultaneously may be able to produce better efficacy in mutant KRAS-driven cancers, as opposed to the inhibition of only one pathway.
Figure 15.

Structures of 41 and 42.
The design of a successful cancer therapeutic relies on an understanding of the biology underlying the disease. The idea that cells signal in a linear fashion is quickly becoming antiquated and is being replaced with circuit-type models. Likewise, cancer treatments need to be designed under the notion of circuit signaling. We expect that employing this rationale will lead to better treatments, more effective drug combinations, and better outcomes for patients.
Acknowledgments
We are grateful to Professor Laurence Hurley of the University of Arizona College of Pharmacy for his critical reading and suggestions.
ABBREVIATIONS USED
- RAS
rat sarcoma
- GTP
guanosine triphosphate
- GDP
guanosine diphosphate
- PPI
protein–protein interaction
- GEF
guanine nucleotide exchange factor
- GAP
guanosine triphosphatase activating protein
- FTase
farnesyltransferase
- RCE1
RAS-converting enzyme 1
- ICMT1
isoprenylcysteine carboxylmethyltransferase 1
- PTase
palmitoyl transferase
- SH2
sarcoma homology 2
- GRB2
growth factor receptor bound 2
- RAF
rapidly accelerated fibrosarcoma
- MAP
mitogen-activated protein
- MEK
also known as MAPK, mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- ELK1
E twenty-six (ETS)-like transcription factor 1
- PI3K
phosphoinositide 3-kinase
- AKT
protein kinase B
- NF-κB
nuclear factor κ light-chain enhancer of activated B cells
- RalGDS
Ral guanine nucleotide dissociation stimulator
- FOX
Forkhead box
- PLC
phospholipase C
- HTS
high-throughput screening
- SC
sulfinyl cytidine
- TPT
triphenyl
- hTERT
human telomerase reverse transcriptase
- ip
intraperitoneal
- LT
large T
- ST
small T
- HPV
human papillomavirus
- BJ
Bence Jones protein
- RB
retinoblastoma
- ROS
reactive oxygen species
- MEF
mouse embryonic fibroblast
- CTG
chemotaxigenesis
- BrdU
bromodeoxyuridine
- SOS
son of sevenless
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- IMAP
immobilized metal ion affinity based fluorescence polarization
- FITC
fluorescein isothiocyanate
- LOX
lipoxygenase
- PK
pharmacokinetics
- PD
pharmacodynamics
- PIK3CA
phosphoinositide 3-kinase, catalytic α polypeptide
- mTOR
mammalian target of rapamycin
- FTI
farnesyltransferse inhibitor
- BFC
biotin S-farnesyl-L-cysteine
- AdoMet
S-adenosylmethionine
- RNAi
RNA interference
- PLK1
polo-like kinase 1
- MDCK
Madin–Darby canine kidney epithelial
- EGFR
epidermal growth factor receptor
- NSCLC
non-small-cell lung cancer
- CDK4
cyclin-dependent kinase 4
- RTK
receptor tyrosine kinase
Biographies
Yuanxiang Wang obtained his Ph.D. in Medicinal Chemistry from Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China, under the supervision of Professor Ao Zhang in 2012. He is currently in a postdoctoral position under the direction of Professor Hong-yu Li at the University of Arizona. His main areas of expertise are development of novel synthetic methods to produce druglike heterocyclic chemical scaffolds, kinase inhibitor design, and total synthesis of bioactive natural products.
Christine E. Kaiser is currently persuing a Ph.D. degree in the Drug Discovery and Development program at the University of Arizona under the advisement of Professor Laurence Hurley. She has an interest in targeting novel DNA secondary structures to regulate the transcription of various oncogenes. Her expertise is in the characterization of DNA secondary structures, computational modeling, and optimization of in vitro and in vivo biological assays for use in high-throughput screening.
Brendan Frett is currently perusing a Ph.D. degree in the Drug Discovery and Development program at the University of Arizona under the advisement of Professor Hong-yu Li. He has a strong interest in small molecule inhibitors for novel oncology-related targets. His expertise is in the development and synthesis of druglike compounds, developing biological assays for high-throughput compound screening, computational modeling, kinase inhibitor design, and the discovery of unique synthetic methodologies.
Hong-yu Li is currently an Associate Professor of Medicinal Chemistry at the College of Pharmacy at the University of Arizona. He received his Ph.D. degree from the University of Tokyo, Japan, and did his postdoctoral trainings at Columbia University, NY (with Professor Koji Nakanishi), and Harvard University, MA(with Professor Yoshito Kishi). He previously worked at Eli Lilly where he focused on oncology drug discovery. His current research interests are in chemical biology and drug discovery, especially for oncology related targets and phenotypes. In his lab at the University of Arizona, a highly potent (picomolar activity in an oncogene-driven cancer cell line), selective, and orally bioavailable small molecule drug inhibitor was discovered and is currently ready for preclinical development and further IND filing.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- 1.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 2.Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 3.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friday BB, Adjei AA. K-ras as a target for cancer therapy. Biochim Biophys Acta. 2005;1756:127–144. doi: 10.1016/j.bbcan.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev, Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 7.Podsypanina K, Politi K, Beverly LJ, Varmus HE. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci US A. 2008;105:5242–5247. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, Horner JW, 2nd, Cordon-Cardo C, Yancopoulos GD, DePinho RA. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 9.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW, Fesik SW. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem, Int Ed. 2012;51:6140–6143. doi: 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletcher S, Hamilton AD. Targeting protein–protein interactions by rational design: mimicry of protein surfaces. J R Soc Interface. 2006;3:215–233. doi: 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghobrial IM, Adjei AA. Inhibitors of the ras oncogene as therapeutic targets. Hematol Oncol Clin North Am. 2002;16:1065–1088. doi: 10.1016/s0889-8588(02)00050-3. [DOI] [PubMed] [Google Scholar]
- 13.Adjei AA. Farnesyltransferase inhibitors. Cancer Chemother Biol Response Modif. 2001;19:149–164. [PubMed] [Google Scholar]
- 14.Cho KN, Lee KI. Chemistry and biology of Ras farnesyltransferase. Arch Pharm Res. 2002;25:759–769. doi: 10.1007/BF02976989. [DOI] [PubMed] [Google Scholar]
- 15.Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci US A. 1989;86:8323–8327. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox AD, Der CJ. Farnesyltransferase inhibitors and cancer treatment: targeting simply Ras? Biochim Biophys Acta. 1997;1333:F51–F71. doi: 10.1016/s0304-419x(97)00011-5. [DOI] [PubMed] [Google Scholar]
- 17.Hancock JF, Magee AI, Childs JE, Marshall CJ. All RAS proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 18.Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133–139. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- 19.Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science. 1997;275:1796–1800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 20.Otto JC, Kim E, Young SG, Casey PJ. Cloning and characterization of a mammalian prenyl protein-specific protease. J Biol Chem. 1999;274:8379–8382. doi: 10.1074/jbc.274.13.8379. [DOI] [PubMed] [Google Scholar]
- 21.Clarke S, Vogel JP, Deschenes RJ, Stock J. Posttranslational modification of the Ha-ras oncogene protein: evidence for a third class of protein carboxyl methyltransferases. Proc Natl Acad Sci US A. 1988;85:4643–4647. doi: 10.1073/pnas.85.13.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hrycyna CA, Sapperstein SK, Clarke S, Michaelis S. The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. EMBO J. 1991;10:1699–1709. doi: 10.1002/j.1460-2075.1991.tb07694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, Michaelis S, Philips MR. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem. 1998;273:15030–15034. doi: 10.1074/jbc.273.24.15030. [DOI] [PubMed] [Google Scholar]
- 24.Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: a review. Oncologist. 2005;10:565–578. doi: 10.1634/theoncologist.10-8-565. [DOI] [PubMed] [Google Scholar]
- 25.Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47:883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Manandhar SP, Hildebrandt ER, Jacobsen WH, Santangelo GM, Schmidt WK. Chemical inhibition of CaaX protease activity disrupts yeast Ras localization. Yeast. 2010;27:327–343. doi: 10.1002/yea.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winter-Vann AM, Baron RA, Wong W, dela Cruz J, York JD, Gooden DM, Bergo MO, Young SG, Toone EJ, Casey PJ. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc Natl Acad Sci US A. 2005;102:4336–4341. doi: 10.1073/pnas.0408107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuther GW, Der CJ. The Ras branch of small GTPases: RAS family members don’t fall far from the tree. Curr Opin Cell Biol. 2000;12:157–165. doi: 10.1016/s0955-0674(99)00071-x. [DOI] [PubMed] [Google Scholar]
- 29.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 30.Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 31.Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 32.Lemmon MA, Schlessinger J. Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem Sci. 1994;19:459–463. doi: 10.1016/0968-0004(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 33.Weiss FU, Daub H, Ullrich A. Novel mechanisms of RTK signal generation. Curr Opin Genet Dev. 1997;7:80–86. doi: 10.1016/s0959-437x(97)80113-x. [DOI] [PubMed] [Google Scholar]
- 34.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 35.Sawyers CL. Rational therapeutic intervention in cancer: kinases as drug targets. Curr Opin Genet Dev. 2002;12:111–115. doi: 10.1016/s0959-437x(01)00273-8. [DOI] [PubMed] [Google Scholar]
- 36.Quilliam LA, Huff SY, Rabun KM, Wei W, Park W, Broek D, Der CJ. Membrane-targeting potentiates guanine nucleotide exchange factor CDC25 and SOS1 activation of Ras transforming activity. Proc Natl Acad Sci US A. 1994;91:8512–8516. doi: 10.1073/pnas.91.18.8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding RAS: “it ain’t over ‘til it’s over”. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 38.Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639–647. doi: 10.1016/j.tcb.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 40.Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finney R, Herrera D. Ras-Raf complexes: analyses of complexes formed in vivo. Methods Enzymol. 1995;255:310–323. doi: 10.1016/s0076-6879(95)55034-8. [DOI] [PubMed] [Google Scholar]
- 42.Johnson LM, James KM, Chamberlain MD, Anderson DH. Identification of key residues in the A-Raf kinase important for phosphoinositide lipid binding specificity. Biochemistry. 2005;44:3432–3440. doi: 10.1021/bi0487692. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh S, Moore S, Bell RM, Dush M. Functional analysis of a phosphatidic acid binding domain in human Raf-1 kinase: mutations in the phosphatidate binding domain lead to tail and trunk abnormalities in developing zebrafish embryos. J Biol Chem. 2003;278:45690–45696. doi: 10.1074/jbc.M302933200. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 45.Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase γ. Cell. 2000;103:931–943. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 46.De Ruiter ND, Burgering BM, Bos JL. Regulation of the Forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol Cell Biol. 2001;21:8225–8235. doi: 10.1128/MCB.21.23.8225-8235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.González-García A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219–226. doi: 10.1016/j.ccr.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 48.Hofer F, Fields S, Schneider C, Martin GS. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc Natl Acad Sci US A. 1994;91:11089–11093. doi: 10.1073/pnas.91.23.11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bittker JA, Weiwer M, Shimada K, Yang WS, MacPherson L, Dandapani S, Munoz B, Palmer M, Stockwell BR, Schreiber SL. Probe Reports from the NIH Molecular Libraries Program [Internet] National Center for Biotechnology Information, U.S. National Library of Medicine; Bethesda, MD: 2011. Screen for RAS-Selective Lethal Compounds and VDAC Ligands—Probe 1. http://www.ncbi.nlm.nih.gov/books/NBK55069/ [Google Scholar]
- 50.Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol. 2001;19:940–945. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 51.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-RAS and protein kinase C iota. Cancer Res. 2008;68:7403–7408. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N, Jacks T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci US A. 2011;108:8773–8778. doi: 10.1073/pnas.1105941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJ, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W, Fang G. Small-molecule ligands bind to a distinct pocket in RAS and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci US A. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rees DC, Congreve M, Murray CW, Carr R. Fragment based lead discovery. Nat Rev Drug Discovery. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 55.Chessari G, Woodhead AJ. From fragment to clinical candidate—a historical perspective. Drug Discovery Today. 2009;14:668–675. doi: 10.1016/j.drudis.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 56.Zartler ER, Mo H. Practical aspects of NMR-based fragment discovery. Curr Top Med Chem. 2007;7:1592–1599. doi: 10.2174/156802607782341055. [DOI] [PubMed] [Google Scholar]
- 57.Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem, Int Ed. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 58.Erlanson D. Fragments versus Ras—Part 2. Zartler T, Erlanson D, editors. Practical Fragments. 2012 Jun 6; BlogSpot, June 10, 2012, http://practicalfragments.blogspot.com/2012/06/fragments-versus-ras-part-2.html.
- 59.Sridhar SS, Hedley D, Siu LL. Raf kinase as a target for anticancer therapeutics. Mol Cancer Ther. 2005;4:677–685. doi: 10.1158/1535-7163.MCT-04-0297. [DOI] [PubMed] [Google Scholar]
- 60.Khosravi-Far R, White MA, Westwick JK, Solski PA, Chrzanowska-Wodnicka M, Van Aelst L, Wigler MH, Der CJ. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923–3933. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for RAS transformation. Mol Cell Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heim M, Sharifi M, Hilger RA, Scheulen ME, Seeber S, Strumberg D. Antitumor effect and potentiation or reduction in cytotoxic drug activity in human colon carcinoma cells by the Raf kinase inhibitor (RKI) BAY 43-9006. Int J Clin Pharmacol Ther. 2003;41:616–617. doi: 10.5414/cpp41616. [DOI] [PubMed] [Google Scholar]
- 63.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 64.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, Minasyan A, Graham NA, Graeber TG, Chodon T, Ribas A. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:3928–3937. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holt SV, Logié A, Odedra R, Heier A, Heaton SP, Alferez D, Davies BR, Wilkinson RW, Smith PD. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106:858–866. doi: 10.1038/bjc.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, Kasman I, Koeppen H, Rice K, Yang NY, Engst S, Johnston S, Friedman LS, Belvin M. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012;72:210–219. doi: 10.1158/0008-5472.CAN-11-1515. [DOI] [PubMed] [Google Scholar]
- 67.Daouti S, Wang H, Li WH, Higgins B, Kolinsky K, Packman K, Specian A, Jr, Kong N, Huby N, Wen Y, Xiang Q, Podlaski FJ, He Y, Fotouhi N, Heimbrook D, Niu H. Characterization of a novel mitogen-activated protein kinase kinase 1/2 inhibitor with a unique mechanism of action for cancer therapy. Cancer Res. 2009;69:1924–1932. doi: 10.1158/0008-5472.CAN-08-2627. [DOI] [PubMed] [Google Scholar]
- 68.Sportsman JR, Gaudet EA, Boge A. Immobilized metal ion affinity-based fluorescence polarization (IMAP): advances in kinase screening. Assay Drug Dev Technol. 2004;2:205–214. doi: 10.1089/154065804323056549. [DOI] [PubMed] [Google Scholar]
- 69.CDAS: KinaseScreen Programs. http://www.caliperls.com/assets/011/6679.pdf.
- 70.Daouti S, Higgins B, Kolinsky K, Packman K, Wang H, Rizzo C, Moliterni J, Huby N, Fotouhi N, Liu M, Goelzer P, Sandhu HK, Li JK, Railkar A, Heimbrook D, Niu H. Preclinical in vivo evaluation of efficacy, pharmacokinetics, and pharmacodynamics of a novel MEK1/2 kinase inhibitor RO5068760 in multiple tumor models. Mol Cancer Ther. 2010;9:134–144. doi: 10.1158/1535-7163.MCT-09-0601. [DOI] [PubMed] [Google Scholar]
- 71.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, García-Echeverría C, Weissleder R, Mahmood U, Cantley LC, Wong KK. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 73.Seabra MC. Membrane association and targeting of prenylated Ras-like GTPases. Cell Signalling. 1998;10:167–172. doi: 10.1016/s0898-6568(97)00120-4. [DOI] [PubMed] [Google Scholar]
- 74.Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene. 2000;19:6584–6593. doi: 10.1038/sj.onc.1204146. [DOI] [PubMed] [Google Scholar]
- 75.Cox AD, Der CJ. Farnesyltransferase inhibitors: promises and realities. Curr Opin Pharmacol. 2002;2:388–393. doi: 10.1016/s1471-4892(02)00181-9. [DOI] [PubMed] [Google Scholar]
- 76.Sun J, Qian Y, Hamilton AD, Sebti SM. Both farnesyltransferase and geranylgeranyltransferase I inhibitors are required for inhibition of oncogenic K-Ras prenylation but each alone is sufficient to suppress human tumor growth in nude mouse xenografts. Oncogene. 1998;16:1467–1473. doi: 10.1038/sj.onc.1201656. [DOI] [PubMed] [Google Scholar]
- 77.Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton AD, Sebti SM. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem. 1995;270:26802–26806. doi: 10.1074/jbc.270.45.26802. [DOI] [PubMed] [Google Scholar]
- 78.Song SY, Meszoely IM, Coffey RJ, Pietenpol JA, Leach SD. K-Ras-independent effects of the farnesyl transferase inhibitor L-744,832 on cyclin B1/Cdc2 kinase activity, G2/M cell cycle progression and apoptosis in human pancreatic ductal adenocarcinoma cells. Neoplasia. 2000;2:261–272. doi: 10.1038/sj.neo.7900088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagasu T, Yoshimatsu K, Rowell C, Lewis MD, Garcia AM. Inhibition of human tumor xenograft growth by treatment with the farnesyl transferase inhibitor B956. Cancer Res. 1995;55:5310–5314. [PubMed] [Google Scholar]
- 80.Crespo NC, Ohkanda J, Yen TJ, Hamilton AD, Sebti SM. The farnesyltransferase inhibitor, FTI-2153, blocks bipolar spindle formation and chromosome alignment and causes prometaphase accumulation during mitosis of human lung cancer cells. J Biol Chem. 2001;276:16161–16167. doi: 10.1074/jbc.M006213200. [DOI] [PubMed] [Google Scholar]
- 81.Beaupre DM, Cepero E, Obeng EA, Boise LH, Lichtenheld MG. R115777 induces Ras-independent apoptosis of myeloma cells via multiple intrinsic pathways. Mol Cancer Ther. 2004;3:179–186. [PubMed] [Google Scholar]
- 82.Wang EJ, Johnson WW. The farnesyl protein transferase inhibitor lonafarnib (SCH66336) is an inhibitor of multidrug resistance proteins 1 and 2. Chemotherapy. 2003;49:303–308. doi: 10.1159/000074531. [DOI] [PubMed] [Google Scholar]
- 83.Rose WC, Lee FY, Fairchild CR, Lynch M, Monticello T, Kramer RA, Manne V. Preclinical antitumor activity of BMS-214662.; a highly apoptotic and novel farnesyltransferase inhibitor. Cancer Res. 2001;61:7507–7517. [PubMed] [Google Scholar]
- 84.Haluska P, Dy GK, Adjei AA. Farnesyl transferase inhibitors as anticancer agents. Eur J Cancer. 2002;38:1685–1700. doi: 10.1016/s0959-8049(02)00166-1. [DOI] [PubMed] [Google Scholar]
- 85.Sebti SM. Blocked pathways: FTIs shut down oncogene signals. Oncologist. 2003;8(Suppl 3):30–38. doi: 10.1634/theoncologist.8-suppl_3-30. [DOI] [PubMed] [Google Scholar]
- 86.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 87.Lancet JE, Gojo I, Gotlib J, Feldman EJ, Greer J, Liesveld JL, Bruzek LM, Morris L, Park Y, Adjei AA, Kaufmann SH, Garrett-Mayer E, Greenberg PL, Wright JJ, Karp JE. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemia. Blood. 2007;109:1387–1394. doi: 10.1182/blood-2006-04-014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Karp JE, Lancet JE, Kaufmann SH, End DW, Wright JJ, Bol K, Horak I, Tidwell ML, Liesveld J, Kottke TJ, Ange D, Buddharaju L, Gojo I, Highsmith WE, Belly RT, Hohl RJ, Rybak ME, Thibault A, Rosenblatt J. Clinical and biologic activity of the farnesyltransferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: a phase 1 clinical-laboratory correlative trial. Blood. 2001;97:3361–3369. doi: 10.1182/blood.v97.11.3361. [DOI] [PubMed] [Google Scholar]
- 89.Karp JE, Smith BD, Gojo I, Lancet JE, Greer J, Klein M, Morris L, Levis MJ, Gore SD, Wright JJ, Garrett-Mayer E. Phase II trial of tipifarnib as maintenance therapy in first complete remission in adults with acute myelogenous leukemia and poor-risk features. Clin Cancer Res. 2008;14:3077–3082. doi: 10.1158/1078-0432.CCR-07-4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cortes J, Albitar M, Thomas D, Giles F, Kurzrock R, Thibault A, Rackoff W, Koller C, O’Brien S, Garcia-Manero G, Talpaz M, Kantarjian H. Efficacy of the farnesyl transferase inhibitor R115777 in chronic myeloid leukemia and other hematologic malignancies. Blood. 2003;101:1692–1697. doi: 10.1182/blood-2002-07-1973. [DOI] [PubMed] [Google Scholar]
- 91.Mesa RA, Camoriano JK, Geyer SM, Wu W, Kaufmann SH, Rivera CE, Erlichman C, Wright J, Pardanani A, Lasho T, Finke C, Li CY, Tefferi A. A phase II trial of tipifarnib in myelofibrosis: primary, post-polycythemia vera and post-essential thrombocythemia. Leukemia. 2007;21:1964–1970. doi: 10.1038/sj.leu.2404816. [DOI] [PubMed] [Google Scholar]
- 92.Kurzrock R, Albitar M, Cortes JE, Estey EH, Faderl SH, Garcia-Manero G, Thomas DA, Giles FJ, Ryback ME, Thibault A, De Porre P, Kantarjian HM. Phase II study of R115777, a farnesyl transferase inhibitor, in myelodysplastic syndrome. J Clin Oncol. 2004;22:1287–1292. doi: 10.1200/JCO.2004.08.082. [DOI] [PubMed] [Google Scholar]
- 93.Fenaux P, Raza A, Mufti GJ, Aul C, Germing U, Kantarjian H, Cripe L, Kerstens R, De Porre P, Kurzrock R. A multicenter phase 2 study of the farnesyltransferase inhibitor tipifarnib in intermediate-to high-risk myelodysplastic syndrome. Blood. 2007;109:4158–4163. doi: 10.1182/blood-2006-07-035725. [DOI] [PubMed] [Google Scholar]
- 94.Kurzrock R, Kantarjian HM, Cortes JE, Singhania N, Thomas DA, Wilson EF, Wright JJ, Freireich EJ, Talpaz M, Sebti SM. Farnesyltransferase inhibitor R115777 in myelodysplastic syndrome: clinical and biologic activities in the phase 1 setting. Blood. 2003;102:4527–4534. doi: 10.1182/blood-2002-11-3359. [DOI] [PubMed] [Google Scholar]
- 95.Alsina M, Fonseca R, Wilson EF, Belle AN, Gerbino E, Price-Troska T, Overton RM, Ahmann G, Bruzek LM, Adjei AA, Kaufmann SH, Wright JJ, Sullivan D, Djulbegovic B, Cantor AB, Greipp PR, Dalton WS, Sebti SM. Farnesyltransferase inhibitor tipifarnib is well tolerated, induces stabilization of disease, and inhibits farnesylation and oncogenic/tumor survival pathways in patients with advanced multiple myeloma. Blood. 2004;103:3271–3277. doi: 10.1182/blood-2003-08-2764. [DOI] [PubMed] [Google Scholar]
- 96.Karp JE, Flatten K, Feldman EJ, Greer JM, Loegering DA, Ricklis RM, Morris LE, Ritchie E, Smith BD, Ironside V, Talbott T, Roboz G, Le SB, Meng XW, Schneider PA, Dai NT, Adjei AA, Gore SD, Levis MJ, Wright JJ, Garrett-Mayer E, Kaufmann SH. Active oral regimen for elderly adults with newly diagnosed acute myelogenous leukemia: a preclinical and phase 1 trial of the farnesyltransferase inhibitor tipifarnib (R115777, Zarnestra) combined with etoposide. Blood. 2009;113:4841–4852. doi: 10.1182/blood-2008-08-172726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H, Safran H, Humblet Y, Perez Ruixo J, Ma Y, Von Hoff D. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 98.Lersch C, Van Cutsem E, Amado R, Ehninger G, Heike M, Kerr D, Rothenberg ML, Baum CM, Zaknoen SL. Randomized phase II study of SCH 66336 and gemcitabine in the treatment of metastatic adenocarcinoma of the pancreas. Proc Am Soc Clin Oncol. 2001;20:608. [Google Scholar]
- 99.Sharma S, Kemeny N, Kelsen DP, Ilson D, O’Reilly E, Zaknoen S, Baum C, Statkevich P, Hollywood E, Zhu Y, Saltz LB. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann Oncol. 2002;13:1067–1071. doi: 10.1093/annonc/mdf173. [DOI] [PubMed] [Google Scholar]
- 100.Hanrahan EO, Kies MS, Glisson BS, Khuri FR, Feng L, Tran HT, Ginsberg LE, Truong MT, Hong WK, Kim ES. A phase II study of lonafarnib (SCH66336) in patients with chemo-refractory, advanced squamous cell carcinoma of the head and neck. Am J Clin Oncol. 2009;32:274–279. doi: 10.1097/COC.0b013e318187dd57. [DOI] [PubMed] [Google Scholar]
- 101.Wong NS, Morse MA. Lonafarnib for cancer and progeria. Expert Opin Invest Drugs. 2012;21:1043–1055. doi: 10.1517/13543784.2012.688950. [DOI] [PubMed] [Google Scholar]
- 102.Cortes J, Jabbour E, Daley GQ, O’Brien S, Verstovsek S, Ferrajoli A, Koller C, Zhu Y, Statkevich P, Kantarjian H. Phase 1 study of lonafarnib (SCH 66336) and imatinib mesylate in patients with chronic myeloid leukemia who have failed prior single-agent therapy with imatinib. Cancer. 2007;110:1295–1302. doi: 10.1002/cncr.22901. [DOI] [PubMed] [Google Scholar]
- 103.Theodore C, Geoffrois L, Vermorken JB, Caponigro F, Fiedler W, Chollet P, Ravaud A, Peters GJ, de Balincourt C, Lacombe D, Fumoleau P. Multicentre EORTC study 16997: feasibility and phase II trial of farnesyl transferase inhibitor & gemcitabine combination in salvage treatment of advanced urothelial tract cancers. Eur J Cancer. 2005;41:1150–1157. doi: 10.1016/j.ejca.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 104.Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A, Young SG. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 2002;22:171–181. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Manandhar SP, Hildebrandt ER, Schmidt WK. Small-molecule inhibitors of the Rce1p CaaX protease. J Biomol Screening. 2007;12:983–993. doi: 10.1177/1087057107307226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van der Hoeven D, Cho KJ, Ma X, Chigurupati S, Parton RG, Hancock JF. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol. 2013;33:237–251. doi: 10.1128/MCB.00884-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marciano D, Ben-Baruch G, Marom M, Egozi Y, Haklai R, Kloog Y. Farnesyl derivatives of rigid carboxylic acids—inhibitors of ras-dependent cell growth. J Med Chem. 1995;38:1267–1272. doi: 10.1021/jm00008a004. [DOI] [PubMed] [Google Scholar]
- 108.Marom M, Haklai R, Ben-Baruch G, Marciano D, Egozi Y, Kloog Y. Selective inhibition of Ras-dependent cell growth by farnesylthiosalisylic acid. J Biol Chem. 1995;270:22263–22270. doi: 10.1074/jbc.270.38.22263. [DOI] [PubMed] [Google Scholar]
- 109.Boufaied N, Wioland MA, Falardeau P, Gourdeau H. TLN-4601, a novel anticancer agent, inhibits Ras signaling post Ras prenylation and before MEK activation. Anti-Cancer Drugs. 2010;21:543–552. doi: 10.1097/CAD.0b013e328337f373. [DOI] [PubMed] [Google Scholar]
- 110.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Laheru D, Shah P, Rajeshkumar NV, McAllister F, Taylor G, Goldsweig H, Le DT, Donehower R, Jimeno A, Linden S, Zhao M, Song D, Rudek MA, Hidalgo M. Integrated preclinical and clinical development of S-trans:trans-farnesylthiosalicylic acid (FTS, salirasib) in pancreatic cancer. Invest New Drugs. 2012;30:2391–2399. doi: 10.1007/s10637-012-9818-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG, Pietanza MC, Azzoli CG, Krug LM, Pao W, Ginsberg MS. A phase II trial of salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011;6:1435–1437. doi: 10.1097/JTO.0b013e318223c099. [DOI] [PubMed] [Google Scholar]
- 113.Mason WP, Belanger K, Nicholas G, Vallières I, Mathieu D, Kavan P, Desjardins A, Omuro A, Reymond D. A phase II study of the Ras-MAPK signaling pathway inhibitor TLN-4601 in patients with glioblastoma at first progression. J Neuro-Oncol. 2012;107:343–349. doi: 10.1007/s11060-011-0747-6. [DOI] [PubMed] [Google Scholar]
- 114.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barr FA, Silljé HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 117.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discovery. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 118.Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, Armenteros-Monterroso E, Lassailly F, Matthews N, Nye E, Stamp G, Behrens A, Downward J. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642–655. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 119.Puyol M, Martín A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaría D, Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 120.Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10:130–137. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]