

Abstract
Chronic lymphocytic leukemia is a malignancy of mature auto-reactive B cells. Genetic and functional studies implicate B-cell receptor signaling as a pivotal pathway in its pathogenesis. Full B-cell receptor activation requires tumor-microenvironment interactions in lymphoid tissues. Spleen tyrosine kinase, Bruton’s tyrosine kinase, and the phosphatidylinositol 3-kinase (PI3K) δ isoform are essential for B-cell receptor signal transduction but also mediate the effect of other pathways engaged in chronic lymphocytic leukemia cells in the tissue-microenvironment. Orally bioavailable inhibitors of spleen tyrosine kinase, Bruton’s tyrosine kinase, or PI3Kδ, induce high rates of durable responses. Ibrutinib, a covalent inhibitor of Bruton’s tyrosine kinase, and idelalisib, a selective inhibitor of PI3Kδ, have obtained regulatory approval in chronic lymphocytic leukemia. Ibrutinib and idelalisib are active in patients with high-risk features, achieving superior disease control in difficult-to-treat patients than prior best therapy, making them the preferred agents for chronic lymphocytic leukemia with TP53 aberrations and for patients resistant to chemoimmunotherapy. In randomized trials, both ibrutinib, versus ofatumumab, and idelalisib in combination with rituximab, versus placebo with rituximab improved survival in relapsed/refractory chronic lymphocytic leukemia. Responses to B-cell receptor inhibitors are mostly partial, and within clinical trials treatment is continued until progression or occurrence of intolerable side effects. Ibrutinib and idelalisib are, overall, well tolerated; notable adverse events include increased bruising and incidence of atrial fibrillation on ibrutinib and colitis, pneumonitis and transaminase elevations on idelalisib. Randomized trials investigate the role of B-cell receptor inhibitors in first-line therapy and the benefit of combinations. This review discusses the biological basis for targeted therapy of chronic lymphocytic leukemia with B-cell receptor inhibitors, and summarizes the clinical experience with these agents.
Introduction
Chronic lymphocytic leukemia (CLL) is a tumor of auto-reactive mature B cells. B-cell receptor (BCR) signaling in the lymph node microenvironment plays a central role in its pathogenesis and in disease progression. The diagnosis of CLL requires the presence of 5000 or more tumor cells/uL of blood with a characteristic immunophenotype (CD19+, CD5+, CD23+, weak CD20 expression). Small lymphocytic lymphoma (SLL) shares the biological characteristics of CLL, albeit with less than 5000 tumor cells/uL of blood in the presence of pathological lymphadenopathy, splenomegaly, or bone marrow disease. The standard of care for CLL is ‘watchful waiting’ of asymptomatic patients. Treatment is reserved for patients presenting symptomatic disease or compromised bone marrow function.1 This approach is based on clinical trials that did not find any benefit for early treatment in asymptomatic patients, and the relatively long and heterogeneous natural history of the disease. While the median survival of all patients in a large referral center was 11 years,2 survival is shorter for patients with high-risk disease. In contrast, patients with indolent CLL may have a life-expectancy comparable to age-matched controls.3,4
Chemoimmunotherapy, the combination of chemotherapy with an anti-CD20 monoclonal antibody (mAb), is the standard first-line treatment of CLL.5–7 However, most patients relapse within years of first-line chemoimmunotherapy. The median progression-free survival (PFS) after first-line chemoimmunotherapy can be less than two years in patients with adverse cytogenetic markers, in particular in those with deletion of chromosome 17p (del17p), or in those carrying somatic mutations in TP53, NOTCH1, or SF3B1.8 During treatment, cells with genetic lesions that confer relative resistance have a survival advantage and can become the dominant population at relapse. For example, del17p or TP53 mutations are present in less than 10% of patients at the time of first-line therapy but in up to one-third of patients with relapsed disease.9,10 There is a major need to identify treatment options for patients with relapsed/refractory disease and for those with TP53 aberrations.11,12
The BCR is a master regulator of B-cell development, survival, proliferation, functional differentiation, and migration, and plays an important role in the pathogenesis of several B-cell malignancies.13,14 Here I will review the role of BCR signaling in CLL, summarize the clinical experience with BCR inhibitors, and provide an outlook on the possible future role of these targeted agents in the treatment of CLL.
The role of BCR signaling in the pathogenesis of CLL
Genetic evidence for a role of antigenic signaling in the pathogenesis of CLL derives from the analysis of the immunoglobulin genes that encode the antigen-binding domains of the BCR.15,16 In contrast, somatic mutations in genes encoding components of the BCR signaling pathway are uncommon in CLL.17 During development, each B cell recombines immunoglobulin variable (V), diversity (D), and junction genes (J) in order to form a novel, unique sequence that encodes the antigen binding domain of the BCR (Figure 1). In theory, billions of different combinations are possible, generating a vastly diverse repertoire of possible antigen binding sites. However, CLL cells display a highly restricted, non-random repertoire of different immunoglobulin heavy chain variable (IGHV) region genes, suggesting that CLL cells have distinct antigen specificities.15,18,19 Furthermore, the presence or absence of somatic mutations in the clonal IGHV gene, a mark of antigenic selection, distinguishes two major CLL subtypes; IGHV mutated (M-CLL) and IGHV unmutated (U-CLL); the latter having more than 98% sequence homology of the clonal IGHV gene to germline. M-CLL cells appear to be “anergic”, that is in a state of hypo-responsiveness to BCR activation, which may be due to frequent BCR stimulation.20 In contrast, U-CLL express BCR structures found in polyreactive, natural antibody producing B cells that weakly bind many antigens, possibly resulting in low level chronic stimulation.21,22 Some antigens bound by BCRs expressed on CLL cells include microbial structures, molecules expressed on dying cells, and autoantigens.15 In addition, the BCR of many CLL cells recognizes an epitope that is part of the CLL BCR itself, possibly contributing to auto-stimulation on a single cell level.23 The observation that U-CLL is a more rapidly progressive disease with inferior survival compared to M-CLL suggests that the degree of BCR activation and/or the type of antigen may be important.
Figure 1.
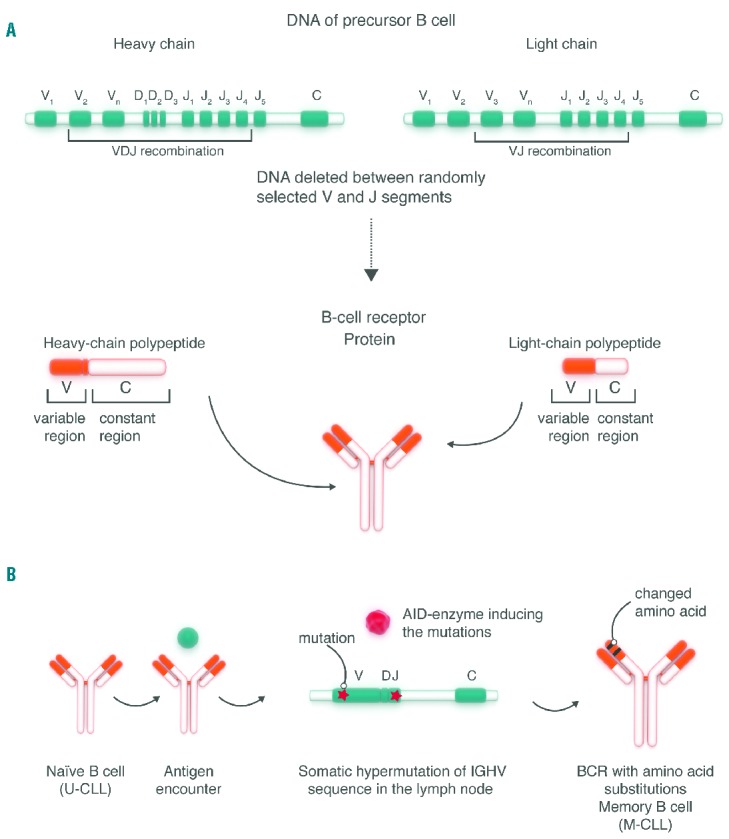
Generation of the BCR repertoire and chronic lymphocytic leukemia (CLL) subtypes. (A) B-cell precursors rearrange genetic sequences (V; variable; D: diversity; J: joining; C: constant) to form heavy chain (VDJ recombination) and light chain (VJ recombination) sequences that encode the antigen binding structures of the BCR. (B) Upon antigen encounter naïve B cells undergo further maturation in lymphoid tissues. BCR activation induces expression of the enzyme adenosine deaminase (AID) which introduces somatic mutations into the gene segments encoding the variable domain of the BCR. BCRs carrying amino acid substitutions that confer stronger antigen binding preferentially expand. The presence or absence of somatic mutations in the immunoglobulin heavy chain variable region (IGHV) has been used to differentiate between IGHV unmutated (U-CLL) and IGHV mutated (M-CLL) subtypes, the former apparently derived from a precursor having undergone antigenic selection, the latter carrying IGHV sequences in germline configuration as found in naïve B cells. However, the cellular derivation of CLL cells is more complex, and there is good evidence that antigen selection plays a role in both CLL subtypes.15,16,116
Gene expression profiles of CLL cells isolated from blood and lymph node provided direct evidence for ongoing antigen-dependent signaling through the BCR in vivo and suggested the lymph node as the primary site of BCR activation.24 Further evidence for ongoing BCR activation in CLL are the reversible downmodulation of surface IgM expression on CLL cells and the anergic state of some CLL cells, both a reflection of chronic antigenic stimulation.20,25 Consistent with the role of BCR signaling as a driver of CLL progression, strong cellular response to BCR activation correlates with a more aggressive disease course.24,26
The transduction of signals from the BCR involves a network of kinases and adaptor molecules that connect antigen stimulation to intracellular responses (Figure 2). Spleen tyrosine kinase (SYK), phosphatidylinositol 3-kinase (PI3K) δ isoform, and Bruton’s tyrosine kinase (BTK) are essential for BCR signal transduction, and the inactivation of the respective genes in mice leads to impaired antigen-dependent maturation and expansion of B cells.27 Soluble, structural, and cellular components of the tissue microenvironment co-operate with the BCR to influence the cellular response (Figure 3).28 Notably, T cells aggregate with proliferating CLL cells in so-called proliferation centers in the bone marrow and lymphoid tissues, and are required for CLL cell proliferation in mice xenografted with PBMCs from CLL patients.29 Extensive in vitro studies identified many pathways and factors that enhance CLL cell survival and promote limited proliferation, including the BCR, Toll-like receptors (TLRs), CD40, CD49d, cytokines, chemokines, and components of the extracellular matrix (Figure 3).30–38 Many of these signals are transmitted through SYK, and/or PI3Kδ, and/or BTK, and activate similar intracellular pathways, most prominently the PI3K/AKT/mTOR, NF-κB, and MAPK pathways. It is, therefore, difficult to estimate to what degree any single factor or pathway may be necessary or sufficient for CLL pathogenesis.27 Notably, many adverse prognostic markers in CLL, in particular use of unmutated IGHV genes, expression of ZAP70, and CD49d, relate to BCR signaling and tumor-microenvironment interactions, supporting the importance of these pathways in tumor biology.39–41
Figure 2.

BCR signaling and downstream pathways. The BCR consists of a surface transmembrane immunoglobulin (Ig) receptor associated with the Ig alpha (Igα, CD79A) and Ig beta (Igβ, CD79B) chains. BCR signaling in response to antigen binding induces LYN and SYK-dependent phosphorylation of tyrosine motifs (phosphorylation denoted by “P” in yellow circle) on CD79A and CD79B. A number of protein tyrosine kinases and the lipid kinase PI3Kδ transmit survival and proliferation signals and regulate cell maturation and migration. Small molecule inhibitors of select kinases in the BCR pathway that have demonstrated clinical activity are indicated (See text for details and references).
Figure 3.

Chronic lymphocytic leukemia (CLL) cells interact with soluble, structural, and cellular elements in the tissue microenvironment. Shown is a selection of possible interactions between CLL cells and components of the microenvironment investigated primarily in in vitro models and supportive evidence from in vivo models or observations in patients. (See text for details and references).
Inhibitors of BCR signaling
Functional studies with CLL cells in vitro, ongoing activation of CLL cells through the BCR in vivo, and the genetic evidence for antigenic selection gleaned from the clonal IGHV gene are strong indicators that BCR signaling is a pivotal pathway in CLL pathogenesis and disease progression (reviewed in more detail elsewhere13–15,27,42). Several kinases in the BCR pathway can be targeted with small molecules to effectively interrupt BCR signaling in vivo, resulting in the inhibition of activation, proliferation, and survival of the tumor cells.43,44 Most advanced are inhibitors of BTK, PI3K, and SYK (Figure 1). Because the same kinases also play important roles in pathways activated by tumor-microenvironment interactions, the inhibitory effects of the small molecules extend beyond BCR signaling interfering with signals from chemokines, CD40 ligand, BAFF, fibronectin, and TLR ligands.45–47 In particular, kinase inhibitors can reduce chemokine-mediated migration and integrin-dependent adhesion of CLL cells, which could reduce the ability of tumor cells to “home” to the microenvironment.47–49 In addition, TLR signaling, which can enhance the cellular response to BCR activation in B cells,50 may be a critical target of ibrutinib in Waldenstrom’s macroglobulinemia where mutations in the adaptor protein MyD88 downstream of TLR engagement promote BTK signaling and tumor progression.51,52 It is possible that inhibitors of SYK, BTK, and PI3Kδ are so efficacious because these kinases constitute “signaling hubs” of many different pathways important to B-cell biology. In addition, the effect of kinase inhibitors may be amplified because they interfere with the ability of CLL cells to modulate the composition of the microenvironment. For example, CCL3/CCL4 are chemokines produced by CLL cells in response to BCR activation,24,53 and both ibrutinib and idelalisib have been shown to greatly reduce their secretion,45,54 which may contribute to changes in the composition of the microenvironment and reduce its pro-tumor effects.55 Furthermore, most inhibitors are not entirely specific to only one kinase but may inhibit additional (related) kinases to some degree and kinases targeted by these small molecules have roles in cells other than B cells.27,42 To what degree on-target but tumor extrinsic effects contribute to efficacy and adverse events are becoming an important area of investigation.
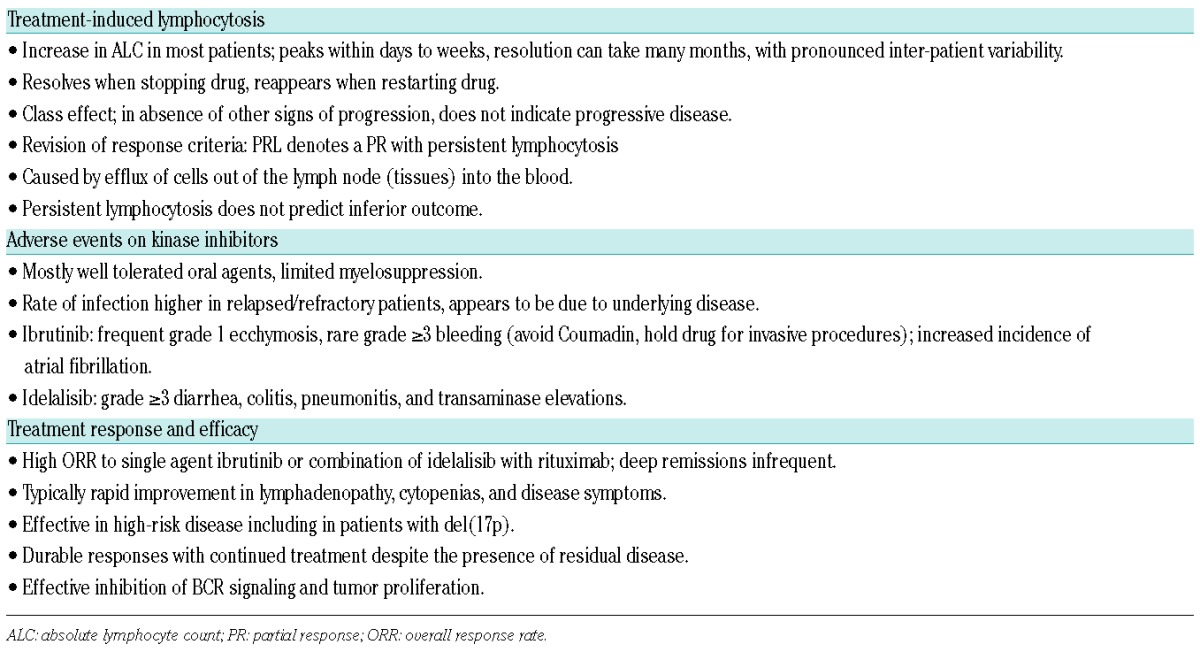
Patients treated with BCR inhibitors typically experience symptomatic improvement and rapid reduction in lymphadenopathy in parallel with a transient increase in absolute lymphocyte count (ALC) (Table 1).56–58 Thus, this treatment-induced lymphocytosis is a class effect of kinase inhibitors that target BCR signaling, with the caveat that it may be missed as the peak ALC can occur within hours of starting drug.58 Treatment-induced lymphocytosis, in the absence of signs of progression in other disease sites, does not indicate disease progression, and patients can safely continue therapy.59 Furthermore, there is no evidence that treatment-induced lymphocytosis leads to morbidity in CLL patients.56–58,60–62 In most patients, the treatment induced lymphocytosis peaks within the first two months before gradually resolving, albeit with considerable inter-patient variability.58 However, a subset of patients may show persistent lymphocytosis for more than a year on single agent ibrutinib. Importantly, this persistent lymphocytosis is not associated with treatment failure,63 and is composed of quiescent cells with inhibited signaling, activation, and proliferation.44 Persistent lymphocytosis, therefore, is not an indication of treatment resistance or imminent relapse. Treatment-induced lymphocytosis is not limited to CLL; an increase in ALC, albeit less common and more moderate, is also seen in patients with indolent non-Hodgkin’s lymphoma (NHL) and mantle cell lymphoma.64,65
Table 1.
Characteristic clinical observations in patients with chronic lymphocytic leukemia (CLL) treated with BCR inhibitors.
Decreased cell adhesion and migration of CLL cells contribute to the lymphocytosis on BCR inhibitors.48,54,66 The rise in ALC within hours of starting drug treatment and changes in the expression of immunophenotypic markers indicate that the increase in circulating CLL cells, at least initially, is fueled by the egress of cells from lymphoid tissues.58 The lymphocytosis is clearly not an indicator of treatment failure as, concurrent with the increase in ALC, there is often a dramatic and rapid reduction of disease in lymph nodes, spleen, and to a lesser extent also in the bone marrow, resulting, overall, in a substantial reduction in total tumor burden.58,67 Furthermore, tumor proliferation is virtually abolished within days of starting treatment.43,44 The degree of disease reduction suggests that a substantial number of cells are dying, but frank apoptosis has been difficult to detect in vivo. In patients starting treatment with ibrutinib, we found that the frequency of dead or dying cells in circulation more than doubled from a median 2.4% at baseline to 5.4% on treatment.58 This increase in the rate of cell death fits well with the initial kinetics of tumor response seen in many patients. A comparable rate of cell loss has been estimated using mathematical modeling.68 Decreased proliferation and moderately increased cell death provide a good explanation for the observed gradual attrition of the tumor over time and the absence of tumor lysis syndrome when single agent therapy is initiated.58,63
Most early responses to monotherapy with kinase inhibitors are partial (PR) and, despite clear and substantial clinical benefit, many patients do not formally achieve PR according to the 2008 IWCLL criteria because of the treatment-induced lymphocytosis.56,57,61 Recently, the authors of the IWCLL guidelines have clarified that patients meeting standard criteria for PR, except for persistent lymphocytosis, can be considered responders,69 and are now often classified as PR with lymphocytosis (PR-L). With continued therapy, most patients with PR-L convert to PR or CR.57,63,67
In the next sections, I will focus on ibrutinib and idelalisib, both approved in the USA and Europe for the treatment of CLL.
Ibrutinib
Ibrutinib (formerly PCI-32765), an orally bioavailable small molecule, irreversibly inactivates BTK through covalent binding of a cysteine residue (Cys481) near the active site (Figure 4A).70 BTK is a cytoplasmic tyrosine kinase of the TEC family that is essential for BCR signaling, NF-kB activation, and cellular proliferation.71 Loss-of-function mutations in BTK cause X-linked agammaglobulinemia (XLA; Bruton’s agammaglobulinemia) characterized by the virtual absence of mature B cells and immunoglobulins resulting in recurrent bacterial infections. While expressed in other cells, the essential functions of BTK appear to be limited to B cells.71
Figure 4.

Mechanism of BTK inhibition by ibrutinib and effect of resistance mutations. (A) Ibrutinib covalently binds to a cysteine at position 481 (Cys481) in the ATP binding pocket of BTK which leads to irreversible inhibition of the kinase molecule. Reversal of inhibition requires synthesis of new BTK molecules. (B) In patients with ibrutinib resistance, mutations that lead to replacement of Cys481 by another amino acid have been identified. In the absence of Cys481, ibrutinib cannot form a covalent bond and inhibition of the kinase is reversible and short-lived consistent with the rapid clearance of ibrutinib.
Ibrutinib inhibits BCR signaling with an IC50 of less than 10 nM. In vitro, ibrutinib can inhibit a limited number of other kinases. Few kinases have a cysteine residue aligning with Cys481 in BTK and can be expected to be covalently bound by ibrutinib, including the TEC family kinases BLK, BMX, ITK, and TEC, as well as EGFR, ERBB2, and JAK3.70 ITK, which is expressed in T cells and NK cells, has been shown to be inhibited at concentrations achievable in vivo.72 For other kinases, in vivo inhibition has not been reported. In spite of this, inhibition of BTK is likely responsible for the direct anti-tumor effects, as indicated by genetic mouse models,73,74 and the Cys481 mutations in BTK identified in patients with ibrutinib resistance (discussed below).
Owing to its covalent binding to BTK, ibrutinib can be dosed once daily, despite a short half-life. Up to 12.5 mg/kg, no dose limiting toxicities were recorded.75 Full occupancy of the target-binding site in BTK, a surrogate of complete kinase inhibition, was achieved at doses of 2.5 mg/kg or more. Subsequent studies in CLL used fixed doses of 420 and 840 mg once daily.57 At steady state, pre-dose occupancy of BTK ranged from approximately 60% to 99%, with no correlation between the degree of inhibition and best objective response.
Safety: ibrutinib is generally well tolerated. The most commonly reported side effects (≥20% of patients on single agent57,61,63,67,76), are mostly grade 1–2 and transient, and include diarrhea, nausea, vomiting, dyspepsia, hypertension, upper respiratory tract infections, urinary tract infections, fatigue, cough, arthralgia, muscle cramps, nail ridging, bruising, rash, pyrexia, and peripheral edema. The most commonly reported (≥5% of patients) grade 3 or greater events include anemia, atrial fibrillation, bleeding, diarrhea, hypertension, neutropenia, pneumonia, and thrombocytopenia. In 132 patients, mostly from the phase Ib/II study followed for a median of three years, bleeding AEs were recorded in 61%, with 7% of AEs grade 3, and one grade 5 event after drug discontinuation for PD.63 In the same group of patients, grade 3 or greater infections were seen in 51% of patients with relapsed/refractory disease and in 13% of previously untreated patients, and were more frequent during the first year on treatment than in subsequent years. Treatment was discontinued because of AEs in 13% of patients.63 In the RESONATE study, grade 3 or greater AEs that were more common in ibrutinib compared to ofatumumab treated patients included diarrhea (4% vs. 2%) and atrial fibrillation (3% vs. 0%).76 The frequency of grade 3 or greater infections was similar in the two study groups (24% vs. 22%).76 Bleeding-related AEs of any grade were more common in the ibrutinib group than in the ofatumumab group (44% vs. 12%). However, there was no difference in grade 3 or greater hemorrhagic events between the two groups (1% vs. 2%). In our own experience in 85 patients followed for a median of 24 months, grade 1 or 2 bleeding-related AEs occurred in 55% with no grade 3 or greater events. Interestingly, the cumulative incidence of an event plateaued by six months, suggesting that the risk of bleeding decreases with continued therapy.77
Studies investigating the mechanism of bleeding diathesis in CLL patients treated with ibrutinib reported a decrease in collagen-induced platelet aggregation by the drug.78–80 However, collagen-induced platelet aggregation is decreased in all patients with CLL, whether on ibrutinib or not, with a mild further decrement in collagen response on ibrutinib.77 Notably, patients with XLA carrying loss-of-function mutations in BTK also have impaired collagen-induced platelet aggregation but do not have a bleeding diathesis. The contribution of ibrutinib to serious bleeding events remains unclear, and the overall risk may depend on both disease and treatment-related factors.77
Single agent efficacy: a recent update on patients participating in the phase Ib/II study reported 90% overall response rate (ORR) in relapsed or refractory CLL [7% complete remission (CR), 80% PR, 3% PR-L] and 84% ORR in previously-untreated patients (23% CR, 55% PR, 6% PR-L).63 Median time to initial response was 1.9 months and 7.4 months to best response. Notably, responses were seen across different risk groups, including in patients with high-risk disease. The estimated PFS rate at 30 months was 96% for previously-untreated patients, and 69% for relapsed/refractory CLL. The estimated PFS rate for patients with relapsed/refractory disease with del(17p) was 48%, with del(11q) 74%, and without either of these aberrations 87%. Disease progression was seen in 24 patients, appearing as Richter’s transformation in 8.63 Farooqui and colleagues enrolled 51 patients with del(17p) or TP53 mutations; 35 were previously untreated and 16 had relapsed/refractory CLL.67 The ORR at 24 weeks of treatment was 92% (50% PR, 42% PR-L). While patients with M-CLL CLL had slower resolution of the treatment-induced lymphocytosis and consequently were slower to convert from PR-L to PR, there was no difference between the two CLL subtypes in the degree of tumor reduction in bone marrow, lymph nodes, and spleen. The estimated PFS for all patients at 24 months was 82%. Five patients (10%) progressed, 3 with Richter’s transformation and 2 with prolymphocytic transformation. The cumulative incidence of progression was 20% for relapsed/refractory CLL, 9% in previously untreated patients, and 20% for patients with U-CLL. None of the patients with M-CLL had progressed.67
In an open label phase III study (RESONATE), 391 patients with relapsed or refractory CLL or SLL were randomly assigned to receive daily ibrutinib or the anti-CD20 mAb ofatumumab. The ORR was 42.6% in the ibrutinib arm and 4.1% in the ofatumumab arm. An additional 20% of ibrutinib-treated patients had PR-L. At six months, the PFS with ibrutinib was 88% compared to 65% with ofatumumab. Ibrutinib also significantly improved OS at 12 months (90% vs. 81%).76
Combination therapy: the combination of ibrutinib with rituximab for 6 cycles followed by ibrutinib until disease progression was well tolerated and resulted in an ORR of 95% with 8% CRs in patient with high-risk disease features.81 The estimated PFS at 18 months in all patients was 78% and was 72% in patients with del(17p) or TP53 mutation. The combination of ibrutinib 420 mg with ofatumumab was explored using 3 different administration sequences; ibrutinib lead-in, concurrent start, or ofatumumab lead-in. Both ORR at 100% and 12-month PFS at 89% was best in the ibrutinib lead-in cohort.82
In a phase Ib study, 30 patients with relapsed/refractory CLL received ibrutinib with bendamustine and rituximab (BR).83 BR was given for up to 6 cycles and ibrutinib at 420 mg was given continuously from day 1 onwards. No added toxicities were observed beyond what would be expected with BR alone. At a median follow up of 15.8 months, the ORR and CR rates were 93% and 17%. Responding patients continued ibrutinib on an extension study, increasing the median follow up to 37.3 months and the rate of CRs to 40%. The median time to CR was 18.2 months. The estimated PFS was 86.3% at 12 months, 78.6% at 24 months, and 70.3% at 36 months. More recently, first results from a randomized phase III study evaluating 578 patients with previously treated CLL randomized to ibrutinib in combination with BR (BR+ibr) versus BR plus placebo (BR+plb) have been presented.84 The median age was 64 years; 38% had advanced Rai stage; notably, patients with del17p were excluded. At a median follow up of 17.2 months, the PFS, the pre-defined primary end point of the study, was significantly longer with BR+ibr versus BR+plb (median not reached vs. 13.3 months; HR: 0.203, 95%CI: 0.150–0.276, P<0.0001). The ORR was 82.7% versus 67.8% (P<0.0001). Incidence of most AEs was similar between the two arms. The most common AEs of any grade with BR+ibr and BR+plb were neutropenia (58.2% vs. 54.7%) and nausea (36.9% vs. 35.2%). Rates of grade 3/4 atrial fibrillation were 2.8% and 0.7%, and of major hemorrhage were 2.1% and 1.7%.84
Idelalisib
Idelalisib (formerly GS-1101 or CAL-101) is an orally bioavailable, selective, and reversible inhibitor of the PI3Kδ isoform. The PI3K pathway regulates cellular growth, proliferation, and survival in response to different stimuli.85,86 PI3K isoforms α and β are ubiquitously expressed, whereas the PI3Kδ isoform is primarily expressed in leukocytes. PI3Kδ is essential for antigen-induced BCR signaling. Both PI3Kδ and PI3Kα participate in the so-called tonic BCR signaling required for B cell survival (discussed in 27). In a dose escalation phase I study of oral idelalisib in NHL87 and CLL,56 patients were treated at 6 dose levels ranging from 50–350 mg once or twice daily, and remained on continuous therapy while deriving clinical benefit. No maximum tolerated dose (MTD) was established. Based on consistent reductions in lymphadenopathy, long PFS, and pharmacokinetic considerations a dose of 150 mg twice daily was chosen for subsequent studies.87
Safety: idelalisib US prescribing information contains a black box warning for fatal and/or severe diarrhea or colitis, hepatotoxicity, pneumonitis, and intestinal perforation.88,89 Additional warnings and precautions from the US prescribing information include severe cutaneous reactions, anaphylaxis, neutropenia and embryo-fetal toxicity. Severe diarrhea or colitis has been reported in a combined 14% of patients being treated on phase I and II studies and the phase III CLL study.88,90 Diarrhea was one of the most common AEs that led to idelalisib dose reduction and treatment discontinuation. Two types of diarrhea are observed with idelalisib. The first type tends to be mild, self-limiting and generally occurs within the first eight weeks; the second type tends to occur relatively late, responds poorly to antidiarrheal or antimicrobial therapy, and is considered to be idelalisib-related. Intestinal perforation has been reported in 0.5% of patients with hematologic malignancies enrolled in phase I, II and III clinical trials.88 Endoscopies in patients with treatment-emergent, late-onset diarrhea revealed increased intraepithelial lymphocytes and villous blunting in the small intestine, and a spectrum of changes including prominent apoptosis with acute cryptitis, crypt abscesses, and increased intraepithelial CD8 T cells in the colon.91 The histological features of idelalisib-associated diarrhea overlapped with those found in autoimmune enterocolitis, graft-versus-host disease (GvHD), and cytomegalovirus (CMV) colitis. Similar histological findings were noted in PI3Kδ knock-out mice that develop spontaneous colitis due to altered macrophage function resulting in a disturbed intestinal immune microenvironment.92,93 These data suggest that colitis might be a class effect of PI3Kδ inhibitors. In fact, grade 3 or greater diarrhea is reported as an important side effect with other PI3Kδ inhibitors.94
Serious, including fatal, hepatotoxicity and pneumonitis have occurred in patients treated with idelalisib. Across idelalisib clinical trials, serious hepatotoxicity occurred in 14% of patients and was one of the most common reasons for idelalisib dose reduction and treatment discontinuation.88 Across clinical trials, pneumonitis occurred in 3% of idelalisib-treated patients with fatal events in 0.5%. The combination of idelalisib with the SYK inhibitor GS-9973 resulted in an unexpectedly high rate of pneumonitis and resulted in stopping the combination study.95 The precise mechanism of idelalisib-related pneumonitis remains to be defined. However, drug-induced pneumonitis has also been observed with mTOR inhibitors, where immune- and host-mediated factors have been implicated, possibly through pro-inflammatory activation of the innate immune system, mediated by effects on monocytes or macrophages.96 Thus, pneumonitis may be a toxicity resulting from the inhibition of PI3K/AKT/mTOR signaling.
Recommendations on the monitoring for and management of treatment-emergent AEs with idelalisib use are found in the idelalisib US prescribing information and in the report of an expert panel.88,89
Single agent efficacy: in CLL, the ORR according to the 2008 IWCLL criteria was 39%.56 An additional 33% of patients had PR-L. The median PFS for all CLL patients enrolled was 15.8 months and 32 months for those receiving continuous dosing with idelalisib 150 or more mg twice daily. While patients with del(17p) or a TP53 mutation responded to treatment, the median PFS of five months was shorter than the 41 months in patients without this abnormality.
Combination therapy: idelalisib combined with rituximab was compared to rituximab with placebo in 220 frail patients with relapsed CLL.90 The study was stopped early due to excess events in the placebo group. The ORR (all PRs) was 81% in the idelalisib group, as compared with 13% in the placebo group. At 24 weeks, the PFS was 93% in the idelalisib group and 46% in the placebo group. The benefit of idelalisib and rituximab was similar in groups stratified by status of del(17p), presence of TP53 mutation, and IGHV mutation status. Consistent with the single agent experience, those grade 3 or greater AEs more often encountered in the idelalisib and rituximab group were pneumonitis (4% vs. 1%), diarrhea (4% vs. 0%), and transaminase elevations (5% vs. 1%). In contrast, infusion reactions during rituximab administration appeared to be milder in patients receiving idelalisib.90
Select additional kinase inhibitors in clinical trials
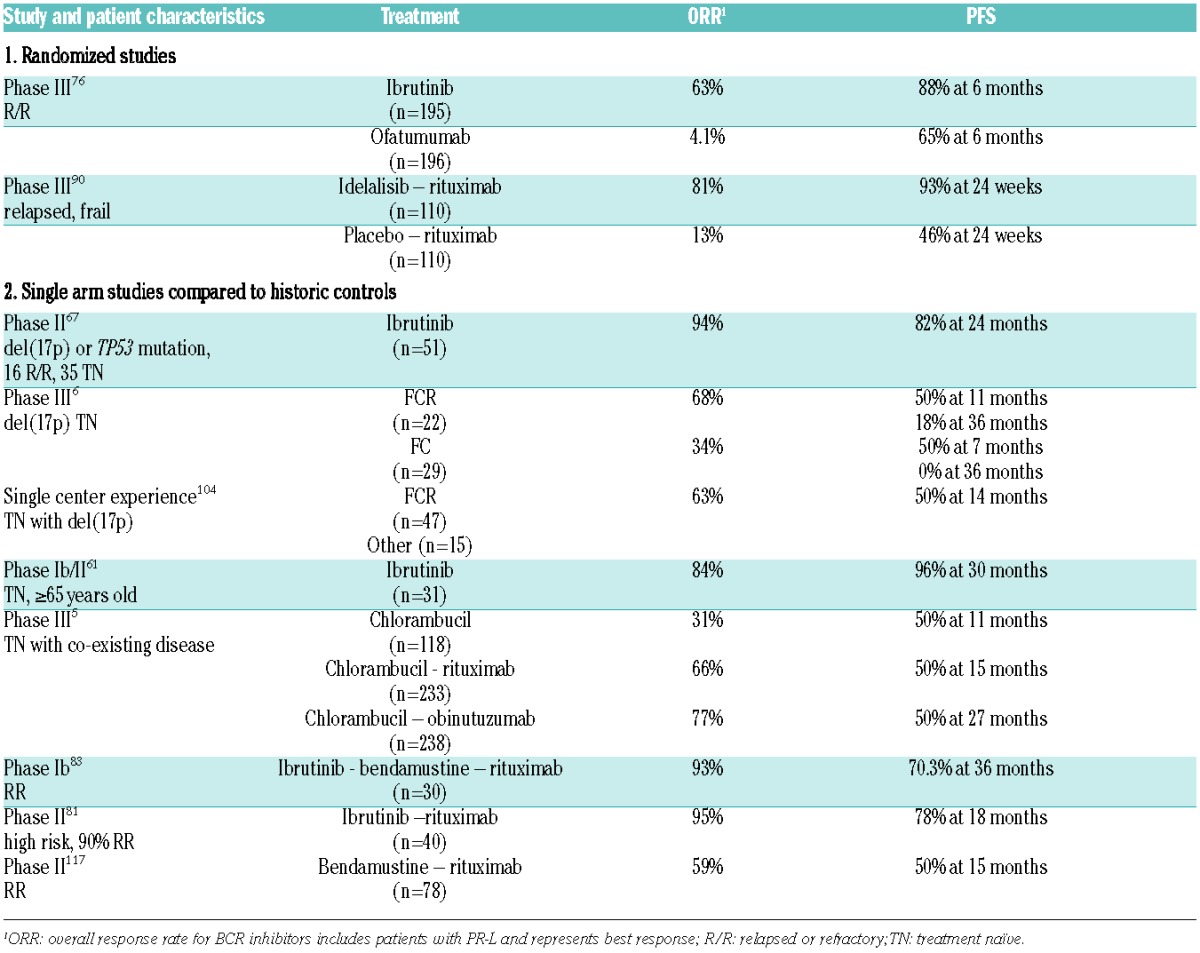
In the eight years since the first patient received the SYK inhibitor fostamatinib,60 there has been a rapid clinical development of multiple BCR inhibitors. In addition to ibrutinib, at least three other covalent BTK inhibitors have entered clinical trials (CC-292, ONO-4059, and ACP-196). In addition, several non-covalent BTK inhibitors are in preclinical or very early clinical development (reviewed in 97). Additional PI3Kδ inhibitors in clinical testing include duvelisib (formerly IPI-145, also inhibits PI3Kγ), TGR-1202, and AMG-319. Isoform selective targeting of the PI3K pathway is attractive because it avoids toxicities of pan PI3K inhibitors. However, in transformed cells, the dominant role of a specific isoform may be lost and different PI3K isoforms can assume redundant functions.98 Whether this will give rise to treatment failure remains to be defined. Table 2 lists select inhibitors in clinical trials and some of these are briefly discussed below.
Table 2.
Outcomes of treatment with BCR inhibitors and comparable treatments.
SYK inhibitors: upon antigen binding to the BCR, LYN phosphorylates SYK, which in turn amplifies the initial BCR signal and activates the downstream signaling cascade. In addition, SYK is involved in chemokine, integrin, and Fc-receptor signaling.99 Fostamatinib, which inhibits SYK and dozens of other kinases, was the first BCR inhibitor to be studied in patients with relapsed/refractory NHL and CLL.60 The dose limiting toxicity was a combination of diarrhea, neutropenia, and thrombocytopenia. The ORR in 11 patients with CLL was 55%. In tumor cells sampled on treatment fostamatinib effectively inhibited BCR signaling, and reduced tumor proliferation.100 The subsequent development of fostamatinib focused on rheumatoid arthritis, with mixed results.
Entospletinib (GS-9973) is a highly selective, orally bioavailable SYK inhibitor. In a phase II study, patients with CLL (n=41) or indolent NHL (n=145) were treated at 800 mg bid. Serious adverse events were reported in 29% of patients, most commonly dyspnea, pneumonia, febrile neutropenia, dehydration and pyrexia. Grade 3 or greater transaminae eleveations were seen in 13.4% of patients. The ORR in CLL was 61% and the median PFS was 13.8 months.62
Resistance to BCR inhibitors and disease transformation
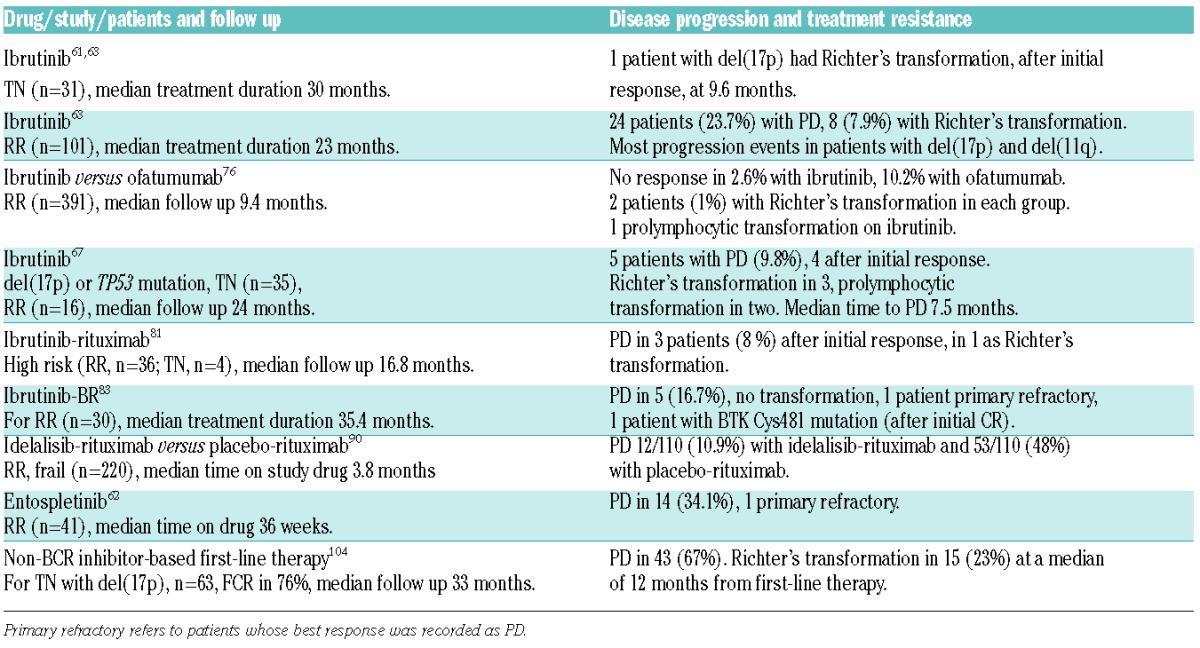
A consistent observation in studies with BCR inhibitors has been that patients with del(17p) or del(11q), bulky disease, or unmutated IGHV respond as well as patients without these adverse disease features.57,90 With longer follow up, however, disease progression occurred more often in patients with high-risk genetic features, and in patients with U-CLL.63,67,101 Patients progressing on ibrutinib are more likely to have U-CLL, del(17p), complex karyotype, or relapsed/refractory treatment status.63,67,101–103 In some patients, disease progression manifests as large B-cell lymphoma or Hodgkin’s lymphoma (Richter’s transformation). Progression with Richter’s transformation tends to occur within the first 12–18 months on treatment, while progression with CLL tends to occur later.101 Overall, the incidence of transformation in patients treated with ibrutinib appears to be less than 10% (Table 3).67,101 In our cohort of 35 patients with CLL and del(17p) receiving ibrutinib in first-line therapy, 2 (5.7%) progressed with Richter’s transformation.67 In contrast, 23% of 63 patients who received first-line treatment at the MD Anderson Cancer Center, most often with FCR, had Richter’s transformation.104 In fact, ibrutinib may have some activity against Richter’s transformation, possibly contributing to fewer events of transformation than that seen in patients after chemoimmunotherapy.105
Table 3.
Disease progression and transformation on BCR inhibitors.
Mechanisms of resistance differ depending on which kinase is targeted. Recently, acquired mutations in BTK and in phospholipase C, gamma 2 (PLCγ2), a direct downstream target of BTK, were identified in CLL patients who developed secondary resistance to ibrutinib.106,107 Five patients showed a cysteine-to-serine mutation in BTK (C481S) at the binding site of ibrutinib that prevents the covalent binding of the drug and thereby dramatically decreases the potency of ibrutinib (Figure 4). Two patients had gain-of-function mutations in PLCγ2 that lead to autonomous BCR signaling.106,108 In a recent update, the Ohio State University group identified BTK and/or PLCγ2 mutations in all 11 patients progressing with CLL. In contrast, BTK mutations were less common in patients with Richter’s transformation,101 suggesting that some of the biological changes occurring during transformation lead to BTK independent tumors. However, at least for the first few years, the incidence of resistance is quite low. Furthermore, the C481S mutation indicates that functional BTK is essential for CLL cells and validates BTK as a therapeutic target. Less is known about resistance mechanisms to PI3Kδ inhibitors. In MCL, high expression of PI3Kα has been associated with resistance to idelalisib.109 Whether overexpression of PI3Kα could play a similar role in CLL is not clear. Finally, patients who failed to respond to the SYK inhibitor fostamatinib had much higher CD38 expression than responding patients.100 Consistent with the appearance that the mechanism of resistance differs between different BCR inhibitors, switching from one inhibitor to another can be effective. For example, 5 of 6 patients with prior treatment with idelalisib achieved a PR with ibrutinib.63
Patients progressing with disease transformation on ibrutinib have been reported to have a median survival of less than four months.101,102 The poor outcome for these patients may reflect the adverse biological characteristics of their particular disease and, at least for patients in the early studies, the absence of effective alternative agents. In contrast, the survival of patients progressing with CLL has recently been estimated as 17.6 months.101 With the availability of different kinase inhibitors, novel agents such as the BCL-2 antagonist ABT-199, or more effective immunotherapies, the outlook for these patients is expected to improve.
Outlook on the role of BCR inhibitors in the treatment of CLL
The clinical benefit of kinase inhibitors is immediately evident in patients who exhausted all other treatment options and now have access to new effective and tolerable agents. BCR inhibitors are clearly among the most effective agents developed for the treatment of CLL and could become the backbone of hopefully curative combination therapy. However, for many patients, especially the elderly and frail, single agent therapy may offer a well-tolerated chronic therapy for a chronic condition. Important variables impacting the choice of treatment strategy have still not been completely defined. First, assessing the safety and tolerability of chronic administration of BCR inhibitors still requires longer follow up. Second, the rate of resistance to single agent therapy will be an important consideration. While, at least for a subset of high-risk patients, resistance may develop at an appreciable rate, resistance appears to be less common in patients being treated with ibrutinib in first-line therapy. Given the excellent results with single agent BCR inhibitor therapy in CLL, clear measures of success of combination therapy should be established. For example, pending evidence of cure, one goal could be the induction of responses deep enough to permit treatment discontinuation for extended periods of time, a paradigm set by minimal-residual disease negative remissions after chemoimmunotherapy. Another important goal of combination therapy could be to reduce the incidence of drug resistance.
The rapidly expanding treatment options for CLL provide the tools for “individualized”, or maybe better, goal-and risk-adapted treatment strategies. For example, in our experience, patients treated with single agent ibrutinib in first-line therapy for CLL with TP53 aberrations had superior PFS than patients treated with FCR.67 With an estimated rate of progression of 9% at two years in previously untreated patients, chemoimmunotherapy for patients with CLL and TP53 aberrations should be avoided.67
Unfortunately, patients with relapsed/refractory disease and del(17p) appear to be at higher risk of progression; approximately 41% of these patients treated with ibrutinib progressed after a median treatment duration of 23 months.63 Thus, patients at high risk of progression on BCR inhibitors may benefit from combination therapy, and, at least for them, allogeneic stem cell transplantation may still be an option for long-term disease control.110 For many patients without high-risk disease features, single agent ibrutinib appears to offer a low intensity, tolerable and durable treatment option. Conceivably, these patients, once they achieve a stable remission, could safely suspend treatment and resume again once progression of the disease justifies re-institution of therapy. However, there are very little data on such voluntary interruptions, and the impact on drug resistance and long-term disease control is not known. On the other hand, maturing data with FCR suggest that a subset of patients can be relapse-free at ten years, raising the possibility that a proportion of patients are “cured” with FCR.8,111 For these patients, typically having lower-risk disease (M-CLL, Rai stage <3, serum β2-microglobulin <4), a relatively short treatment course could achieve long-lasting remissions and obviate the need of chronic drug administration.111
Current data on combinations of BCR inhibitors in CLL are limited. The initial response rates in combination with anti-CD20 antibodies are not that different from single agent data and more relevant endpoints such as PFS await longer follow up. Notably, mechanistic studies on the interaction between BCR inhibitors and anti-CD20 antibodies revealed both potentially negative as well as potentially favorable interactions between the two classes in in vitro models.112–115 These data, and the observation that the sequencing of antibody and kinase inhibitor may matter,82 suggest that optimizing the benefit of combination therapy could be more complex than initially thought. Results from ongoing randomized trials should answer the question as to how much the addition of an anti-CD20 antibody can increase the benefit of BCR inhibitors. Preliminary data suggest that the combination of BCR inhibitors with chemotherapy is safe and more effective than chemotherapy alone. However, it remains unclear what the addition of chemotherapy to the BCR inhibitor contributes; with the caveat that these studies have been carried out in patients with relapsed disease. Conceivably, combining kinase inhibitors with chemoimmunotherapy in first-line therapy could be more effective and possibly become a curative strategy for younger patients. Whether combinations of BCR inhibitors with other agents in development, especially the BCL-2 antagonist ABT-199, or emerging immunotherapies such as checkpoint inhibitors and chimeric antigen receptor modified T cells, could become the combination therapy of choice will require a great deal of additional investigation. The first reported attempt at combining two BCR inhibitors, idelalisib and the SYK inhibitor GS-9973, was discontinued due to an unexpected pneumonitis syndrome,62 serving as a reminder that combination therapy may result in additional toxicities.
In summary, there is an unprecedented abundance of options for the treatment of patients with CLL and there are many good reasons to explore very different strategies. Clearly, advantages and disadvantages of different approaches still have to be defined in clinical trials. At the same time, we are entering an area where patients may be able to choose between different treatment options. Thus, goal- and risk-adapted strategies need to be combined with patient preferences to formulate truly personalized therapy.
Acknowledgments
The author is supported by the intramural research program of the Heart, Lung, and Blood Institute, National Institutes of Health.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the author and is available with the online version of this article at www.haematologica.org.
References
- 1.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12): 5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wierda WG, O’Brien S, Wang X, et al. Prognostic nomogram and index for overall survival in previously untreated patients with chronic lymphocytic leukemia. Blood. 2007;109(11):4679–4685. [DOI] [PubMed] [Google Scholar]
- 3.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. [DOI] [PubMed] [Google Scholar]
- 4.Orchard JA, Ibbotson RE, Davis Z, et al. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet. 2004;363(9403):105–111. [DOI] [PubMed] [Google Scholar]
- 5.Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101–1110. [DOI] [PubMed] [Google Scholar]
- 6.Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. [DOI] [PubMed] [Google Scholar]
- 7.Tam CS, Keating MJ. Chemoimmunotherapy of chronic lymphocytic leukemia. Nat Rev Clin Oncol. 2010;7(9):521–532. [DOI] [PubMed] [Google Scholar]
- 8.Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247–3254. [DOI] [PubMed] [Google Scholar]
- 9.Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schnaiter A, Stilgenbauer S. 17p deletion in chronic lymphocytic leukemia: risk stratification and therapeutic approach. Hematol Oncol Clin North Am. 2013;27(2):289–301. [DOI] [PubMed] [Google Scholar]
- 11.Badoux XC, Keating MJ, Wierda WG. What is the best frontline therapy for patients with CLL and 17p deletion¿ Curr Hematol Malig Rep. 2011;6(1):36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fegan C, Pepper C. Understanding cancer cell survival is key to patient survival. Lancet Oncol. 2015;16(2):122–124. [DOI] [PubMed] [Google Scholar]
- 13.Niemann CU, Wiestner A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin Cancer Biol. 2013;23(6):410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong Y, Byrd JC, Dubovsky JA. The B-cell receptor pathway: a critical component of healthy and malignant immune biology. Semin Hematol. 2014;51(3):206–218. [DOI] [PubMed] [Google Scholar]
- 15.Stevenson FK, Forconi F, Packham G. The meaning and relevance of B-cell receptor structure and function in chronic lymphocytic leukemia. Semin Hematol. 2014;51(3):158–167. [DOI] [PubMed] [Google Scholar]
- 16.Vardi A, Agathangelidis A, Sutton LA, Ghia P, Rosenquist R, Stamatopoulos K. Immunogenetic studies of chronic lymphocytic leukemia: revelations and speculations about ontogeny and clinical evolution. Cancer Res. 2014;74(16):4211–4216. [DOI] [PubMed] [Google Scholar]
- 17.Gruber M, Wu CJ. Evolving understanding of the CLL genome. Semin Hematol. 2014;51(3):177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fais F, Ghiotto F, Hashimoto S, et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J Clin Invest. 1998;102(8):1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tobin G, Thunberg U, Karlsson K, et al. Subsets with restricted immunoglobulin gene rearrangement features indicate a role for antigen selection in the development of chronic lymphocytic leukemia. Blood. 2004;104(9):2879–2885. [DOI] [PubMed] [Google Scholar]
- 20.Muzio M, Apollonio B, Scielzo C, et al. Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood. 2008;112(1):188–195. [DOI] [PubMed] [Google Scholar]
- 21.Agathangelidis A, Darzentas N, Hadzidimitriou A, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012;119(19):4467–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Messmer BT, Albesiano E, Efremov DG, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200(4):519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duhren-von Minden M, Ubelhart R, Schneider D, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–312. [DOI] [PubMed] [Google Scholar]
- 24.Herishanu Y, Perez-Galan P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. 2007;109(10):4424–4431. [DOI] [PubMed] [Google Scholar]
- 26.Le Roy C, Deglesne PA, Chevallier N, et al. The degree of BCR and NFAT activation predicts clinical outcomes in chronic lymphocytic leukemia. Blood. 2012;120(2):356–365. [DOI] [PubMed] [Google Scholar]
- 27.Wiestner A. Emerging role of kinase-targeted strategies in chronic lymphocytic leukemia. Blood. 2012;120(24):4684–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herishanu Y, Katz BZ, Lipsky A, Wiestner A. Biology of chronic lymphocytic leukemia in different microenvironments: clinical and therapeutic implications. Hematol Oncol Clin North Am. 2013;27(2):173–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bagnara D, Kaufman MS, Calissano C, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood. 2011;117(20):5463–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernal A, Pastore RD, Asgary Z, et al. Survival of leukemic B cells promoted by engagement of the antigen receptor. Blood. 2001;98(10):3050–3057. [DOI] [PubMed] [Google Scholar]
- 31.Buchner M, Baer C, Prinz G, et al. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood. 2010;115(22):4497–4506. [DOI] [PubMed] [Google Scholar]
- 32.Endo T, Nishio M, Enzler T, et al. BAFF and APRIL support chronic lymphocytic leukemia B-cell survival through activation of the canonical NF-kappaB pathway. Blood. 2007;109(2):703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herishanu Y, Gibellini F, Njuguna N, et al. Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL-1. Leuk Lymphoma. 2011;52(9):1758–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating anti-apoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111(2):846–855. [DOI] [PubMed] [Google Scholar]
- 35.Muzio M, Scielzo C, Bertilaccio MT, Frenquelli M, Ghia P, Caligaris-Cappio F. Expression and function of toll like receptors in chronic lymphocytic leukaemia cells. Br J Haematol. 2009;144(4):507–516. [DOI] [PubMed] [Google Scholar]
- 36.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. [PubMed] [Google Scholar]
- 37.Ferrer G, Bosch R, Hodgson K, et al. B cell activation through CD40 and IL4R ligation modulates the response of chronic lymphocytic leukaemia cells to BAFF and APRIL. Br J Haematol. 2014;164(4):570–578. [DOI] [PubMed] [Google Scholar]
- 38.Zucchetto A, Benedetti D, Tripodo C, et al. CD38/CD31, the CCL3 and CCL4 chemokines, and CD49d/vascular cell adhesion molecule-1 are interchained by sequential events sustaining chronic lymphocytic leukemia cell survival. Cancer Res. 2009;69(9):4001–4009. [DOI] [PubMed] [Google Scholar]
- 39.Bulian P, Shanafelt TD, Fegan C, et al. CD49d Is the Strongest Flow Cytometry–Based Predictor of Overall Survival in Chronic Lymphocytic Leukemia. J Clin Oncol. 2014;32(9):897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sivina M, Hartmann E, Kipps TJ, et al. CCL3 (MIP-1alpha) plasma levels and the risk for disease progression in chronic lymphocytic leukemia. Blood. 2011;117(5):1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun C, Wiestner A. Prognosis and therapy of chronic lymphocytic leukemia and small lymphocytic lymphoma. Cancer Treat Res. 2015;165:147–175. [DOI] [PubMed] [Google Scholar]
- 42.Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013;34(12):592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649–657. [DOI] [PubMed] [Google Scholar]
- 44.Herman SE, Mustafa RZ, Gyamfi JA, et al. Ibrutinib inhibits BCR and NF-kappaB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123(21):3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herman SE, Gordon AL, Wagner AJ, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119(11): 2590–2594. [DOI] [PubMed] [Google Scholar]
- 49.Herman SE, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with Ibrutinib Inhibits BTK- and VLA-4-Dependent Adhesion of Chronic Lymphocytic Leukemia Cells In Vivo. Clin Cancer Res. 2015;21(20):4642–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416(6881):603–607. [DOI] [PubMed] [Google Scholar]
- 51.Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. [DOI] [PubMed] [Google Scholar]
- 52.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. [DOI] [PubMed] [Google Scholar]
- 53.Burger JA, Quiroga MP, Hartmann E, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113(13):3050–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niemann CU, Biancotto A, Chang BY, et al. Cytokine and T-Cell Phenotypic Changes Upon In Vivo Ibrutinib Therapy For CLL – Targeting Both CLL Cells and The Tumor-Microenvironment. Blood. 2013;122(21): 2856–2856.24004665 [Google Scholar]
- 56.Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herman SE, Niemann CU, Farooqui M, et al. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia. 2014;28(11):2188–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheson BD, Byrd JC, Rai KR, et al. Novel targeted agents and the need to refine clinical end points in chronic lymphocytic leukemia. J Clin Oncol. 2012;30(23):2820–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharman J, Hawkins M, Kolibaba K, et al. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2015;125(15):2336–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125(16):2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang BY, Francesco M, De Rooij MF, et al. Egress of CD19+CD5+ cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood. 2013;122(14):2412–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiestner A. BCR pathway inhibition as therapy for chronic lymphocytic leukemia and lymphoplasmacytic lymphoma. Hematology Am Soc Hematol Educ Program. 2014;2014(1):125–134. [DOI] [PubMed] [Google Scholar]
- 66.Mustafa R, Herman SEM, Jones J, Gyamfi J, Farooqui M, Wiestner A. Ibrutinib Inhibits B-Cell Adhesion and Causes An Efflux Of Chronic Lymphocytic Leukemia Cells From The Tissue Microenvironment Into The Blood Leading To a Transient Treatment-Induced Lymphocytosis. Blood. 2013;122(21):(Abstract) 674.23775714 [Google Scholar]
- 67.Farooqui MZ, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16(2):169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wodarz D, Garg N, Komarova NL, et al. Kinetics of chronic lymphocytic leukemia (CLL) cells in tissues and blood during therapy with the BTK inhibitor ibrutinib. Blood. 2014;123(26):4132–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hallek M, Cheson BD, Catovsky D, et al. Clarification Of iwCLL Criteria For A Partial Response To Therapy. Blood. 2013; Available from: (http://www.bloodjournal.org/content/111/12/5446.e-letters)
- 70.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107(29):13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buggy JJ, Elias L. Bruton Tyrosine Kinase (BTK) and Its Role in B-cell Malignancy. Int Rev Immunol. 2012;31(2):119–132. [DOI] [PubMed] [Google Scholar]
- 72.Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kil LP, de Bruijn MJ, van Hulst JA, Langerak AW, Yuvaraj S, Hendriks RW. Bruton’s tyrosine kinase mediated signaling enhances leukemogenesis in a mouse model for chronic lymphocytic leukemia. Am J Blood Res. 2013;3(1):71–83. [PMC free article] [PubMed] [Google Scholar]
- 74.Woyach JA, Bojnik E, Ruppert AS, et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood. 2014;123(8):1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med. 2014;371(3):213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lipsky AH, Farooqui MZ, Tian X, et al. Incidence and risk factors of bleeding-related adverse events in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. 2015. October 1 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783–787. [DOI] [PubMed] [Google Scholar]
- 79.Levade M, David E, Garcia C, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014;124(26):3991–3995. [DOI] [PubMed] [Google Scholar]
- 80.Rushworth SA, MacEwan DJ, Bowles KM. Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(13):1277–1278. [DOI] [PubMed] [Google Scholar]
- 81.Burger JA, Keating MJ, Wierda WG, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15(10):1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jaglowski SM, Jones JA, Nagar V, et al. Safety and activity of BTK inhibitor ibrutinib combined with ofatumumab in chronic lymphocytic leukemia: a phase 1b/2 study. Blood. 2015;126(7):842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown JR, Barrientos JC, Barr PM, et al. The Bruton tyrosine kinase inhibitor ibrutinib with chemoimmunotherapy in patients with chronic lymphocytic leukemia. Blood. 2015;125(19):2915–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chanan-Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab (BR) in previously treated chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): First results from a randomized, double-blind, placebo-controlled, phase III study. J Clin Oncol. 2015;33(18 suppl):(Abstract)7005. [Google Scholar]
- 85.Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol. 2011;23(2):178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442(3):465–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flinn IW, Kahl BS, Leonard JP, et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-delta, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood. 2014;123(22):3406–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coutre SE, Barrientos JC, Brown JR, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. 2015;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.ZYDELIG. (Full prescription details for idelalisib tablets.). Full prescribing information, Gilead Sciences, Inc., Foster City, CA: 2014. [Google Scholar]
- 90.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014; 370(11):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Louie CY, DiMaio MA, Matsukuma KE, Coutre SE, Berry GJ, Longacre TA. Idelalisib-associated Enterocolitis: Clinicopathologic Features and Distinction From Other Enterocolitides. Am J Surg Pathol. 2015. September 29 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 92.Steinbach EC, Kobayashi T, Russo SM, et al. Innate PI3K p110delta regulates Th1/Th17 development and microbiota-dependent colitis. J Immunol. 2014;192(8):3958–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Uno JK, Rao KN, Matsuoka K, et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110delta. Gastroenterology. 2010;139(5):1642–1653, 1653.e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flinn I, Oki Y, Patel M, et al. A Phase 1 Evaluation of Duvelisib (IPI-145), a PI3K-δ,γ Inhibitor, in Patients with Relapsed/ Refractory iNHL. Blood. 2014;124(21):802–802. [Google Scholar]
- 95.Barr PM, Saylors G, Spurgeon S, et al. Phase 2 trial of GS-9973, a selective syk inhibitor, and idelalisib (idela) in chronic lymphocytic leukemia (CLL) and non-Hodgkin lymphoma (NHL). J Clin Oncol. 2014;32(5s): abstr 7059. [Google Scholar]
- 96.Duran I, Goebell PJ, Papazisis K, et al. Drug-induced pneumonitis in cancer patients treated with mTOR inhibitors: management and insights into possible mechanisms. Expert Opin Drug Saf. 2014;13(3):361–372. [DOI] [PubMed] [Google Scholar]
- 97.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–232. [DOI] [PubMed] [Google Scholar]
- 98.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. [DOI] [PubMed] [Google Scholar]
- 99.Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10(6):387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Herman SE, Barr PM, McAuley EM, Liu D, Wiestner A, Friedberg JW. Fostamatinib inhibits B-cell receptor signaling, cellular activation and tumor proliferation in patients with relapsed and refractory chronic lymphocytic leukemia. Leukemia. 2013;27(8):1769–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol. 2015;1(1):80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jain P, Keating M, Wierda W, et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood. 2015;125(13):2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thompson PA, O’Brien SM, Wierda WG, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer. 2015;121(20):3612–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Strati P, Keating MJ, O’Brien SM, et al. Outcomes of first-line treatment for chronic lymphocytic leukemia with 17p deletion. Haematologica. 2014;99(8):1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsang M, Shanafelt TD, Call TG, et al. The efficacy of ibrutinib in the treatment of Richter syndrome. Blood. 2015;125(10): 1676–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Furman RR, Cheng S, Lu P, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370(24):2352–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu TM, Woyach JA, Zhong Y, et al. Hypermorphic mutation of phospholipase C, gamma2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood. 2015;126(1):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Iyengar S, Clear A, Bodor C, et al. P110alpha-mediated constitutive PI3K signaling limits the efficacy of p110delta-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood. 2013;121(12):2274–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dreger P, Schetelig J, Andersen N, et al. Managing high-risk CLL during transition to a new treatment era: stem cell transplantation or novel agents¿ Blood. 2014;124(26): 3841–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Keating MJ, Tam CS, Wierda WG, et al. Is Chronic Lymphocytic Leukemia Still Incurable¿ ASH Annual Meeting Abstracts. 2012;120(21):3929. [Google Scholar]
- 112.Bojarczuk K, Siernicka M, Dwojak M, et al. B-cell receptor pathway inhibitors affect CD20 levels and impair antitumor activity of anti-CD20 monoclonal antibodies. Leukemia. 2014;28(5):1163–1167. [DOI] [PubMed] [Google Scholar]
- 113.Da Roit F, Engelberts PJ, Taylor RP, et al. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica. 2015;100(1):77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kohrt HE, Sagiv-Barfi I, Rafiq S, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123(12):1957–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Skarzynski M, Niemann CU, Lee YS, et al. Interactions between ibrutinib and anti-CD20 antibodies; competing effects on the outcome of combination therapy. Clin Cancer Res. 2015. August 17 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117(6):1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fischer K, Cramer P, Busch R, et al. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2011;29(26):3559–3566. [DOI] [PubMed] [Google Scholar]