

Abstract
Intravascular hemolysis increases the risk of hypercoagulation and thrombosis in hemolytic disorders. Our study shows a novel mechanism by which extracellular hemoglobin directly affects platelet activation. The binding of Hb to glycoprotein1bα activates platelets. Lower concentrations of Hb (0.37–3 μM) significantly increase the phosphorylation of signaling adapter proteins, such as Lyn, PI3K, AKT, and ERK, and promote platelet aggregation in vitro. Higher concentrations of Hb (3–6 μM) activate the pro-apoptotic proteins Bak, Bax, cytochrome c, caspase-9 and caspase-3, and increase platelet clot formation. Increased plasma Hb activates platelets and promotes their apoptosis, and plays a crucial role in the pathogenesis of aggregation and development of the procoagulant state in hemolytic disorders. Furthermore, we show that in patients with paroxysmal nocturnal hemoglobinuria, a chronic hemolytic disease characterized by recurrent events of intravascular thrombosis and thromboembolism, it is the elevated plasma Hb or platelet surface bound Hb that positively correlates with platelet activation.
Introduction
Thrombotic complications are the hallmark of many hemolytic disorders associated with elevated plasma hemoglobin (Hb). In intravascular hemolysis, lysed red blood cells (RBCs) release Hb directly into circulation.1 After release into plasma, Hb forms a complex with haptoglobin and is subsequently cleared by macrophages.2 Once the Hb-scavenging capacity of haptoglobin and other Hb-binding plasma proteins is saturated, an elevated plasma Hb is associated with vascular dysfunction and thrombotic complications3–8 that is particularly well described in paroxysmal nocturnal hemoglobinuria (PNH), a prototype of intravascular hemolysis. Direct correlation of a broad range of plasma Hb concentrations with the occurrence and severity of thromboembolisms and intravascular thrombosis in PNH patients was reported,9,10 where venous11,12 and arterial13,14 thrombosis are principal causes of PNH morbidity and mortality.
The pathophysiology of thrombosis in PNH is complex. It could be due to increased platelet activation and adhesiveness. Reports suggest that intravascular hemolysis and activation of platelets are potential causes of thrombosis in PNH.15–17 The major untoward effect of free Hb on platelet functions is most likely mediated via the scavenging of nitric oxide (NO) by Hb18–20 or the redox effects of Hb.21 It has been shown that NO inhibits platelet aggregation, induces disaggregation of aggregated platelets, and inhibits platelet adhesion via the cyclic guanosine monophosphate (GMP) pathway.22 On the other hand, it has also been suggested that the reduction in NO level may not play a causative role in thrombosis in hemolytic disease such as β-thalassemia.23
In order to understand further the complex pathophysiology of the thrombotic complications in intravascular hemolysis, we analyzed the direct interactions between Hb and platelets. Our study describes for the first time that the binding of Hb to glycoprotein (GP)1bα induces platelet activation and apoptosis in a concentration-dependent manner and modulates platelet functions. The above observation is further supported by data from paroxysmal nocturnal hemoglobinuria (PNH) patients showing the parallel correlation between plasma Hb and platelet activity.
Methods
Materials
Antibodies: phospho or non-phospho Lyn/PI3K/AKT/MAPK, caspase-9, caspase-3, Src, Bak, Bax, cytochrome C, β-Actin, goat anti-rabbit and goat anti-mouse HRP conjugated antibody, anti-human CD62P-FITC, PE anti-human CD41a, annexin V-FITC, annexin V-PE-Cy5, anti-Hb β-PE, and GP1bα monoclonal antibodies such as AN51, AK2, HIP1 and SZ2 were purchased from respective vendors; details in the Online Supplementary Appendix. The GP1bα antibody, 6D1 was provided by Dr. Barry Coller, Rockefeller University, New York, NY, USA. The anti-GP1bα WM23 was a gift from Dr. Michael Berndt, Curtin University, Perth, Australia. The GPIIbIIIa antibody ReoPro (Janssen Biotech, Inc., Leiden, the Netherland), peptide inhibitor integrilin (Millennium Pharma, Cambridge, USA) were purchased. The synthetic peptides (designed from N-terminal region of GP1bα) including AA1-50 (mpllllllllpsplhphpicwvskvashlevncdkrnltalppdlpkdtt) and scrambled control peptide (msplekcplstvcldtplnhkvlpelpvlilplthnalradkplsdlplh) were purchased from (GL BioChem, Shanghai, People’s Republic of China). The majority of other laboratory chemicals including normal adult Hb (HbA with a purity of 98.5% isolated through Sephadex G-25 column) were purchased from Sigma-Aldrich, St. Louis, USA. The HbA was used for all experiments mentioned in this manuscript.
Patients’ samples
To obtain blood samples, approval was obtained from the Institutional Ethics Committee for Human Research of the Regional Centre for Biotechnology as well as from the All India Institute of Medical Sciences, Delhi, India. Informed consent was provided according to the recommendations of the Declaration of Helsinki. A total of 56 patients with type III paroxysmal nocturnal hemoglobinuria (PNH) and 23 normal healthy controls were recruited; 5–10 mL of blood sample were collected in 0.32% sodium citrate anticoagulant. Type III PNH patients were recruited following confirmation of complete absence of GPI-anchoring proteins on erythrocytes using CD55 and CD59 antibodies. The platelet surface P-selectin, PS and Hb, and plasma levels of Hb and CD41+ microparticles were measured from 56 PNH patients and 23 healthy individuals. Patients’ details are available in the Online Supplementary Appendix.
Washed platelet preparation
Platelet-rich plasma (PRP) was isolated from the acid-citrate dextrose (ACD) anti-coagulated blood of normal individuals. PRP was centrifuged, and the platelets were resuspended in calcium-free Tyrode buffer [126 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.38 mM NaH2PO4, 5.6 mM dextrose, 6.2 mM sodium Hepes, 8.8 mM Hepes-free acid, 0.1% bovine serum albumin, pH 6.5] and, gel-filtered on a column of Sepharose 2B (Sigma-Aldrich, St. Louis, USA) equilibrated in calcium-free Tyrode buffer, pH 7.2.
Binding assay
(A) ELISA: various concentrations of Hb were incubated in ELISA plate immobilized with either pre-fixed platelets (Bio/Data Corporation, Horsham, USA) or glycocalicin (extracellular part of platelet GP1bα, purified from plasma; details are available in the Online Supplementary Appendix). HRP conjugated anti-Hb antibody was used to measure Hb binding at optical density of approximately 450 nm.
(B) Surface plasmon resonance: the Hb binding to glycocalicin was measured using a BIAcore T200 system (GE Healthcare Life Sciences, USA). The glycocalicin was covalently coupled to sensor chip CM5 via aldehyde coupling to an 8000 resonance units. The binding assays were performed in 10 mM Hepes, 150 mM NaCl, 0.05% Tween 20, pH 7.4 at 25°C at a flow rate of 30 μL/min. The binding affinity (KD) was calculated using steady state affinity binding model, BIAcore T200 evaluation software (v. 1.0).
Immunoblotting
Washed platelets were incubated with various concentrations of Hb. Platelet pellets were lysed with RIPA Buffer in presence of Halt™ protease-phosphatase inhibitor (Thermo Scientific Life Technologies, Omaha, USA). The lysis products were processed for SDS PAGE and immunoblotting for all above signaling molecules associated with platelet activation (such as phospho-Lyn or ERK) and apoptosis (such as Bak, Bax or caspase 9).
Flow cytometry
Flow cytometry assay was performed to detect P-selectin and PS on platelet surface. Washed platelets, incubated with various concentrations of Hb (incubated for 30 min at 37°C); or platelets isolated directly from patients’ blood were labeled with FITC anti-P-selectin, annexin V-FITC or annexin V-PE-Cy5, or anti-Hb β (37-8)-PE by incubating for 15 min at RT; fixed with 1% paraformaldehyde analyzed by flow cytometer (Becton Dickinson, San Jose, USA).
Confocal fluorescent microscopy
The FITC anti-P-selectin was added to washed platelets (0.5×106/mL) and smeared on glass cover slides immobilized with bovine serum albumin (BSA) or various concentrations of Hb, and incubated for 30 min at 37°C. After gentle wash with Tyrode’s buffer, slides were mounted with fluoromount- G (Southern Biotech Assoc., Birmingham, USA) and visualized under a confocal microscope (TCS-SP8, Leica Microsystems, Wetzlar, Germany).
Platelet aggregation using flow cytometry
Washed platelets (350×109/L) were incubated with various concentrations of Hb for 30 min at 37°C and allowed for aggregation in the presence of purified fibrinogen (500 μg/mL) under constant stirring (1000 rpm). The aggregates (25 μL) were transferred immediately to a buffer containing 1% paraformaldehyde at every 30 seconds (s). The number and size of aggregates was measured using a flow cytometer. We have adopted the above method to measure aggregation, as the light transmission aggregometer was not useful in the presence of Hb in solution.
Thrombin generation assay
Washed platelets (350×109/L) were incubated with Hb (0–8 μM) at 37°C for 30 min. Thrombin generation was evaluated using a commercially available TGA kit (Technoclone, Vienna, Austria). The assay is based on the assessment of fluorescence generated over time by the cleavage of a fluorogenic substrate by thrombin upon activation of the coagulation cascade by a low concentration of recombinant human tissue factor (71.6 pM) and CaCl2. The fluorescence generated was measured at 1-min intervals up to 60 min.
Coagulation assay
The washed platelets (350×109/L) were incubated with Hb (0–8 μM) at 37°C for 1 h and fixed with 1% paraformaldehyde, and washed. The platelets were used as the PS surface, and was incubated with 50 μL of platelet poor plasma and coagulation reagents Kaolin (Diagnostica Stago Inc., Parsippany, USA) including 100 μL of 0.025 M CaCl2 at 37°C for 3 min. The coagulation time was measured using a coagulometer.
Measurement of extracellular Hb level in plasma
A sandwich ELISA assay was used to measure plasma Hb concentration. Briefly, plasma (100 μL of 1:5000 dilutions) from patients or normal individuals was incubated in microtiter plates coated with rabbit anti-Hb antibody (Sigma-Aldrich, St. Louis, USA) for 1 h at RT. The bound Hb was detected by an affinity purified goat anti-Hb conjugated with HRP (Abcam, Cambridge, USA) using TMB as the substrate (450 nm). Plasma Hb concentration was calculated as μM compared to human erythrocyte purified Hb, as mentioned in our earlier work.24
Microparticle measurement from plasma
Plasma microparticles (MP) were isolated from 5 mL of citrate anti-coagulated plasma. Platelet-free plasma (PFP) was obtained by 3 sequential centrifugation of blood at 700 g for 6 min; platelet-rich plasma at 1500 g for 7 min; and platelet-poor plasma at 1500 g for 15 min. Platelet-derived MP were measured from PFP in filtered-PBS using anti-CD41 PE antibody and quantified using flow cytometry.
Statistical analysis
Experimental values were presented as mean±standard error of mean (SEM) or standard deviation (SD). The Student’s t-test and one-way ANOVA was used for data analysis; P<0.05 was considered to be statistically significant. Pearson’s correlation and linear regression analysis was performed with confidence interval 95% using Sigma plot 12.0.
Results
Extracellular Hb activates platelets
To study the platelet activity in hemolytic conditions, we have examined the effects of extracellular Hb on platelets in vitro. Our data show that Hb increased the expression of P-selectin (secreted by α-granules upon platelet activation) in a concentration-dependent manner irrespective of the platelets that were incubated in buffer or plasma (Figure 1A and Online Supplementary Figure S1). Hb also increased the binding of a conformational-dependent antibody PAC-1 to integrin GPIIb-IIIa (Figure 1B), demonstrating the dose-dependent activation of platelets. Furthermore, Hb stimulated platelets to release microparticles in a concentration-dependent manner (Figure 1C).
Figure 1.
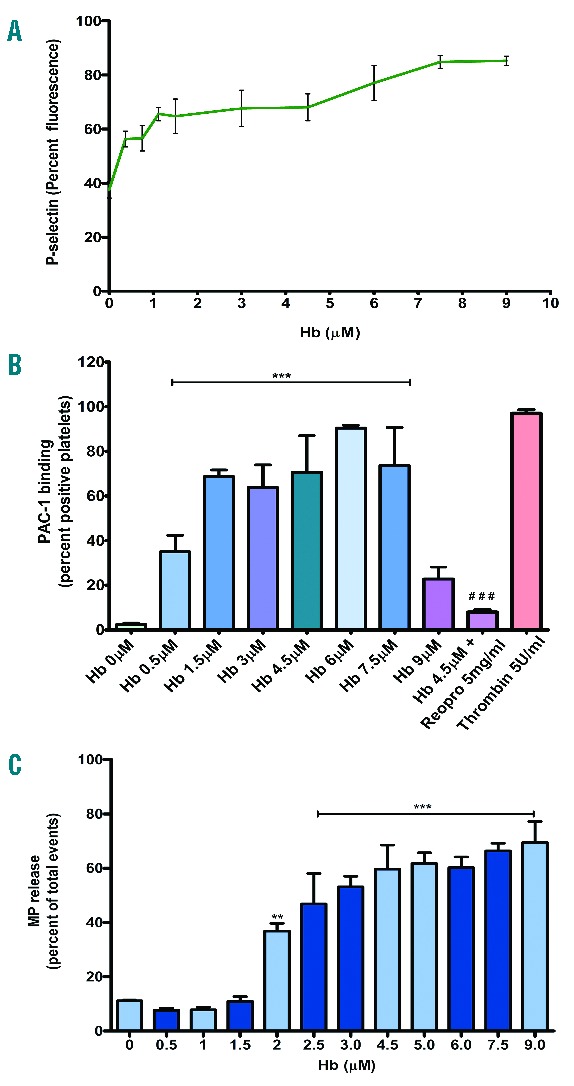
(A) Platelet P-selectin expression in the presence of Hb. Washed platelets were incubated with various concentrations of Hb in buffer; labeled with FITC P-selectin antibody and analyzed by flow cytometry. Data presented as mean ± SEM from 3 independent experiments. Hb increased the expression of P-selectin in a concentration-dependent manner (P<0.001). (B) PAC-1 binding to platelet in the presence of Hb. Washed platelets were incubated with various concentrations of Hb. The PAC-1 binding was measured using flow cytometry. The Hb increased the PAC-1 binding in a concentration-dependent manner, ***P<0.0001 (compared to Hb 0 μM) and ###P<0.0001 (Hb 4.5 μM alone). (C) Microparticle generation by platelets treated with Hb. Washed platelets were incubated with various concentrations of Hb. The microparticles (MPs) were analyzed in a flow cytometer. Hb increased the MP generation in a concentration-dependent manner, **P<0.001 and ***P<0.0001 (compared to Hb 0 μM).
Hb binds to GP1bα and activates platelets
In order to understand the Hb-platelet interaction, we have examined the direct binding of Hb to platelet surface. We show that Hb bound to immobilized platelets in a concentration-dependent manner (Figure 2A). The antibodies (AN51, AK2, and HIP1) that bind to the N-terminal region of GP1bα (between amino acid M1-C80) inhibited significantly the Hb binding to platelets. Other GP1bα antibodies, such as 6D1 that binds between Q104-L128 and SZ2 that binds between D268-T282, did not affect Hb binding to platelets. Neither abciximab (antibody against GPIIb-IIIa) nor integrilin (peptide inhibitor to GPIIb-IIIa) had any effect on Hb binding to platelets (Figure 2A). The effect of the last 2 inhibitors was expected because Hb did not block the binding of PAC-1 to activated GPIIb-IIIa. Next, we used synthetic peptides encompassing different amino acid sequences of N-terminal region of GP1bα to determine Hb binding region. The peptide AA1-50 maximally inhibited the binding of Hb to platelets or glycocalicin (extracellular part of GP1bα) (Figure 2B). An additional experiment revealed that Hb bound to immobilized glycocalicin in a concentration-dependent manner under flow with a KD = 44 μM (Figure 2C) and this binding was inhibited by peptide AA1-50 (Figure 2D). We further examined the consequences of Hb-GP1bα interaction on platelets. We show that the Hb concentrations 0.37 to 3.0 μM significantly increased the phosphorylation of signaling proteins such as Lyn, PI3K, AKT, and ERK (Figure 2E). The effect of Hb was further reduced by the peptide AA1-50 (Figure 2F), which also inhibited the platelet spreading on immobilized Hb (Figure 2G). Furthermore, the blocking of ERK phosphorylation by the Src inhibitor PP2 confirmed the involvement of Lyn-ERK transduction pathway in Hb-mediated platelet activation (Online Supplementary Figure S2).
Figure 2.
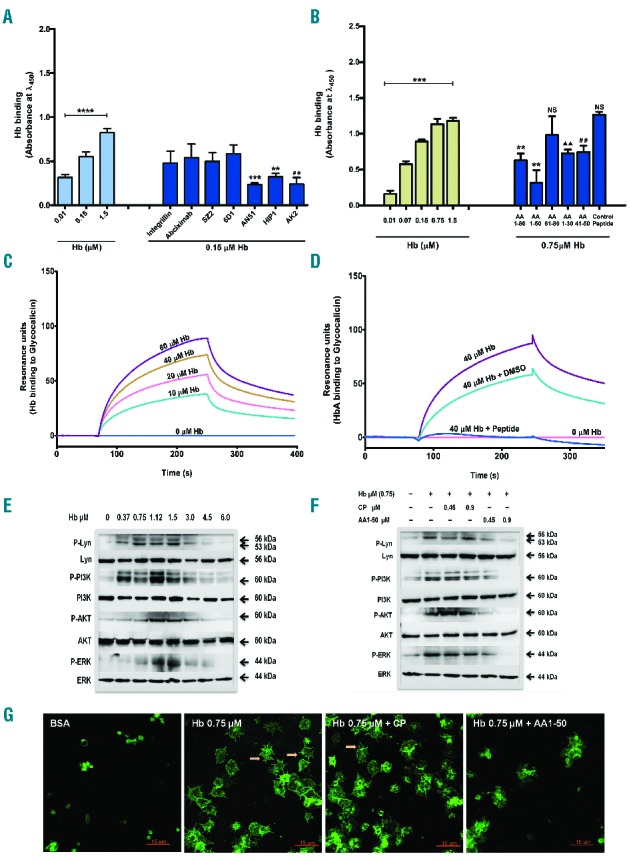
(A) Hb binding to platelet GP1bα: Effect of antibodies. In ELISA, Hb was incubated with immobilized platelets in the presence of 10 μg/mL of GP1bα or GPIIb-IIIa antibodies. Hb binding was detected using anti-Hb HRP antibody. Data are the mean absorbance ± SD from 3 independent experiments. Hb bound in a concentration-dependent manner, ****P<0.0001. Specifically, AN51, HIP1, and AK2 inhibited the binding with ***P<0.0007, **P<0.004, or ##P<0.0045 compared to Hb 0.15 μM alone. (B) Hb binding to glycocalicin: effect of peptides. In glycocalicin-coated ELISA plate, Hb was incubated. Hb bound to glycocalicin in a concentration dependent manner, ***P<0.0001. When Hb (0.75 μM) was incubated in the presence of equimolar peptides (designed from GP1bα N-terminal), the peptides inhibited Hb binding with **P<0.002, P<0.0017, ##P<0.0045, or NS (non-significant). (C) Hb binding to glycocalicin under flow. Hb was perfused under flow over glycocalicin-coated CM5 chip and binding was measured using BIAcore. Hb bound to glycocalicin with KD = 44 μM. (D) Hb binding to glycocalicin under flow inhibited by AA1-50. Hb (40 μM) was perfused under flow with and without AA1-50 (9 μM). The AA1-50 blocked completely Hb binding to glycocalicin. (E) Hb binding activates platelets. Washed platelets were incubated with various concentrations of Hb. The platelet lysate was immunoblotted for p/non-p-Lyn, PI3K, AKT, or ERK. The Hb (0.37–3.0 μM) increased phosphorylation of the above proteins. Densitometry data are available in Online Supplementary Figure S3. (F) AA1-50 inhibits platelet activation. Washed platelets were incubated with Hb in the presence of control peptide (CP) or AA1-50. The expression of above signaling proteins was measured by immunoblotting. The peptide AA1-50 inhibited the phosphorylation of above proteins dose dependently. Densitometry data are available in Online Supplementary Figure S4. (G) AA1-50 inhibits platelet spreading on immobilized Hb. Washed platelets were incubated over surface coated with Hb (0.75 μM) in the presence of CP or AA1-50. Stained with anti-P selectin FITC. The peptide AA1-50 inhibited the platelet spreading. Arrows indicate the filopodia of spread platelets.
Hb potentiates platelet aggregation
We next examined the effects of Hb on platelet functions. Extracellular Hb at 0.37–1.5 μM showed a concentration-dependent increase in the fibrinogen-mediated platelet aggregation (Figure 3A and B), which was inhibited by the blocking peptide AA1-50 (Figure 3C and D). Furthermore, the Hb at the concentration ranging from more than 1.5–6 μM significantly increased the size of the platelet-fibrinogen aggregates in a concentration-dependent manner (Online Supplementary Figure S6).
Figure 3.
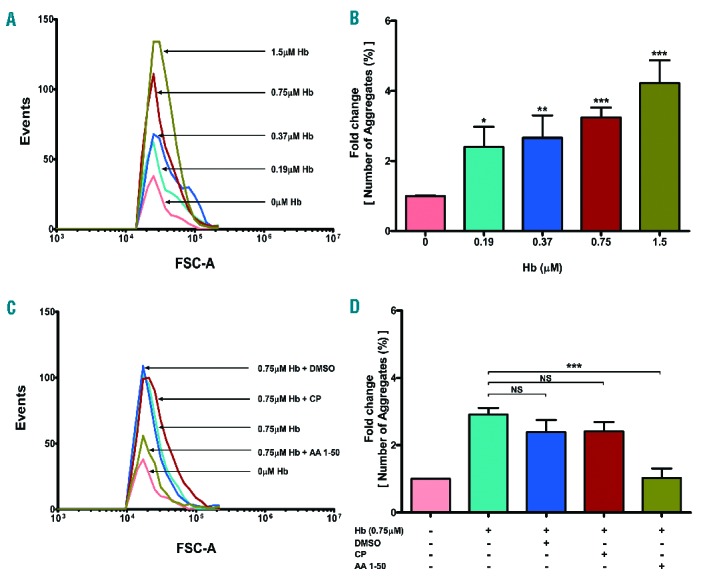
Hb mediated platelet aggregation. Washed platelets were incubated with various concentrations of Hb (0–1.5 μM) and stirred in the presence of fibrinogen (500 μg/mL). The aggregates were measured using flow cytometry (method described in Online Supplementary Figure S5), represented as (A) events and (B) percent aggregation (mean ± SEM) from 3 different experiments, *P<0.01, **P<0.001, ***P< 0.002. AA1-50 inhibits aggregation. The above aggregation was performed in the presence of equimolar Hb and peptides (0.75 μM). Data measured as mentioned above, represented as (C) events and (D) percent aggregation. The peptide AA1-50 significantly decreased the Hb-mediated platelet aggregation, ***P< 0.001, NS: non-significant.
Hb induces apoptosis
Further studies showed that Hb has significantly increased the expression of platelet surface phosphatidylserine (PS) in a concentration-dependent manner (Figure 4A and Online Supplementary Figure S7). Furthermore, the higher concentrations of Hb (3–6 μM) significantly increased the expression of proapoptotic proteins Bak, Bax, cytochrome C and activated caspase-9 and caspase-3 in platelets (Figure 4B). The increase in the cytochrome C oxidase activity by Hb corresponded to the mitochondrial membrane potential in apoptotic platelets (Online Supplementary Figure S10). We show that the peptide AA1-50, which blocks Hb-GP1bα binding, significantly decreased the Hb mediated expression of Bak and Bax, and activation of caspase-9 in platelets (Figure 4C).
Figure 4.
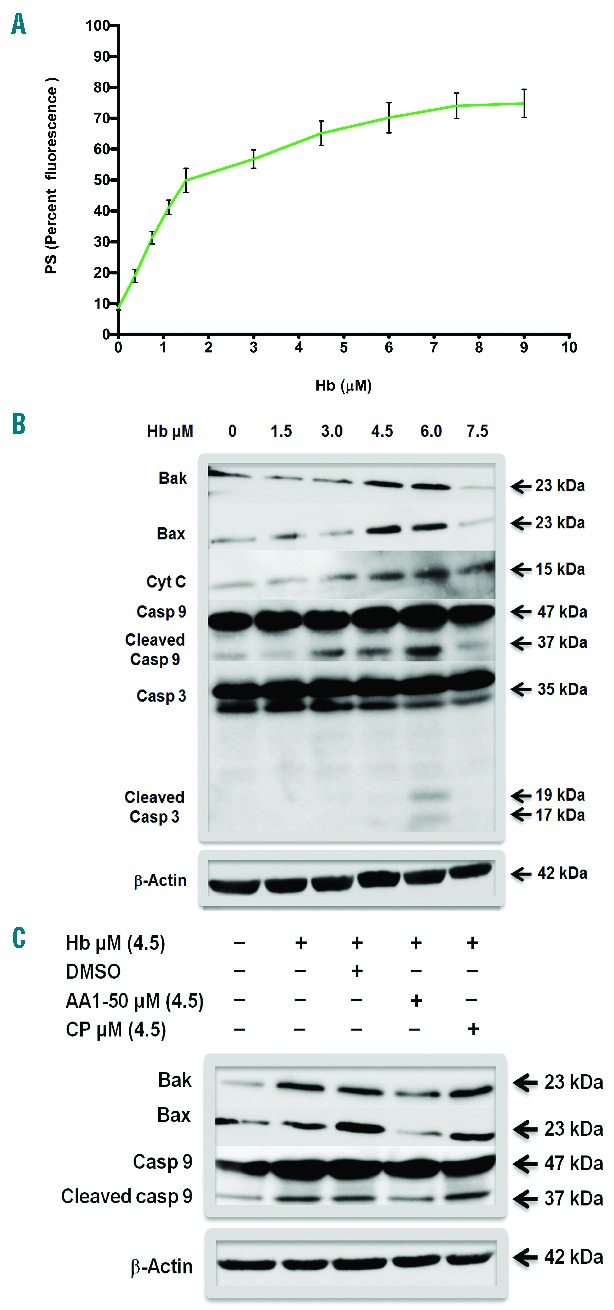
(A) PS expression in platelets treated with Hb. Washed platelets were incubated with various concentrations of Hb in buffer. Labeled with PE-Cy5 annexin V and analyzed by flow cytometry. The Hb increased PS expression in a concentration-dependent manner, P<0.001. (B) Expression of proapoptotic proteins in platelets in the presence of Hb. Washed platelets were incubated with various concentrations of Hb. The platelet lysate was immunoblotted for Bak, Bax, cytochrome C, caspase-9, and caspase-3. Hb concentrations, 4.5 and 6 Μm, showed increased expression of above proteins including the cleavage upon activation of caspase-9 and caspase-3. The β-Actin is the loading control. Densitometry data are available in Online Supplementary Figure S8. (C) AA1-50 blocks platelet apoptosis. Washed platelets were incubated with Hb in the presence of AA1-50 or CP. The expression of Bak, Bax, and caspase-9 were measured by immunoblotting. The peptide AA1-50 inhibited the expression of above proteins. Densitometry data are available in Online Supplementary Figure S9.
Hb promotes thrombin generation
Because the increasing concentrations of Hb induce PS exposure on platelets (Figure 4A), we further examined whether Hb-treated platelets promote thrombin generation. The Hb in the range of 0.5–8 μM increased the thrombin generation in a concentration-dependent manner (Figure 5A). Furthermore, data show that the platelets incubated with increasing concentrations of Hb (0–8 μM) mediated faster clot formation (Figure 5B).
Figure 5.
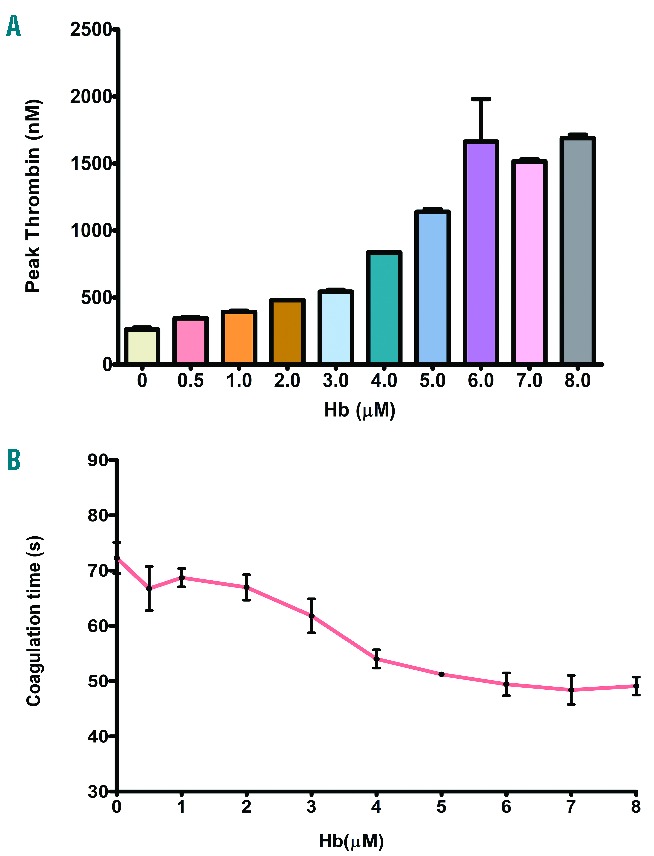
(A) Thrombin generation by platelets treated with Hb. Washed platelets treated with Hb have increased thrombin generation dose dependently as measured by the cleavage of fluorogenic substrate by thrombin. The thrombin levels are the mean ± SEM from 3 independent experiments (P<0.001). (B) Coagulation reaction by platelets treated with Hb. Washed platelets treated with Hb were incubated with platelet-poor plasma and coagulation reagents. Data show the mean ± SEM of clot formation time from 3 independent experiments. Hb increased faster coagulation in a concentration-dependent manner (P<0.001).
Plasma Hb concentrations or platelet surface bound Hb correlate with platelet activation in PNH patients
To assess the in vivo correlations between extracellular Hb and platelet activation, we measured the plasma Hb concentration/platelet surface bound Hb as well as platelet surface P-selectin/PS expressions in PNH patients and healthy controls. We show a positive correlation between the plasma Hb and the platelet surface P-selectin (correlation coefficient, r = 0.813) (Figure 6A) as well as PS (r = 0.806) (Figure 6B). We also show positive correlation between bound-Hb and P-selectin on platelet surface (r = 0.75) (Figure 6D). We then assessed platelet activation in these hemolytic patients showing a positive correlation between Hb levels and platelet-derived microparticles (r = 0.641) in plasma (Figure 6C). The above in vivo observation was further supported by the in vitro data showing the concentration-dependent increase in Hb-binding on platelet surface as well as expression of P-selectin by Hb. The Hb-binding to platelet and P-selectin expression was inhibited by the inhibitory peptide that blocks Hb-binding to GP1bα (Online Supplementary Figure S11).
Figure 6.
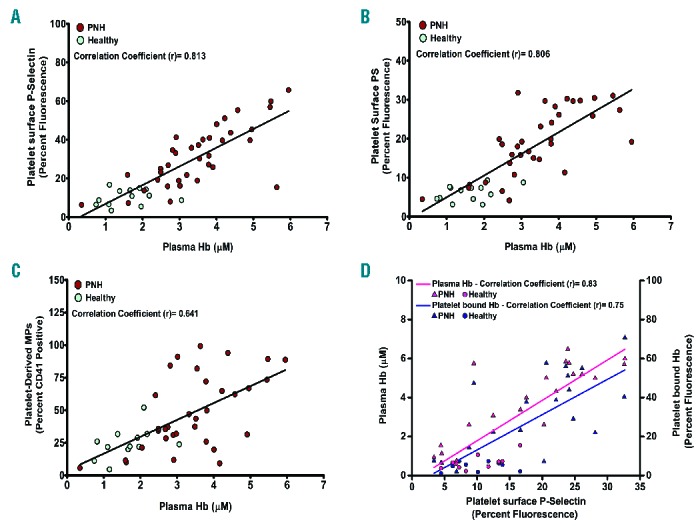
Correlation between plasma Hb/platelet-surface Hb and platelet surface P-selectin, or PS, or platelet-derived MPs. Extracellular Hb concentration was measured from plasma of PNH patients and healthy individuals using sandwich ELISA. Plasma Hb shows positive correlations with (A) P-selectin and (B) PS expression on platelets (freshly isolated from individuals) measured by flow cytometry using FITC-CD62P or PE-Cy5 annexin V antibody. (C) Plasma Hb also correlates positively with platelet-derived microparticles (MPs) in plasma, measured by flow cytometry using CD41-PE antibody. (D) Both platelet surface-bound Hb and plasma extracellular Hb correlates positively with platelet surface P-selectin. Each dot represents individual data. (A–C) Data from 36 PNH patients and 13 healthy controls. (D) Data from another pool of 20 PNH and 10 healthy controls.
Discussion
Our study shows that binding of Hb to GP1bα modulates platelet activity. Extracellular Hb (0.37–3 μM) stimulated platelet activation in vitro. The Hb-GP1bα interaction activated platelets via the Lyn, PI3K, AKT, and ERK path way (Figure 2E and Online Supplementary Figure S2) generally seen with other platelet receptor-ligand interactions such as GP1bαIX-VWF and GPVI-collagen.25–27 As described by others, the VWF-GP1bIX interaction induces platelet activation through this signaling pathway, leading to the integrin activation and stabilization of platelet adhesion and aggregation.27–29 Our observation also describes a similar signaling for the Hb in inducing platelet activation upon binding to GP1bα. This Hb-GP1bα interaction initiated a change in platelet shape (Figure 2G), granule secretion (Figure 1A) and the inside out signaling process (Figure 2E), and activated integrin GPIIb-IIIa (Figure 1B). The binding of ligand such as fibrinogen/VWF to GPIIb-IIIa further induces stable platelet adhesion and aggregation.30–32 Our data show that the Hb stimulates platelet-fibrinogen aggregates formation in a concentration-dependent manner in vitro (Figure 3A and B, and Online Supplementary Figure S6).
Our data also show that the Hb concentrations (3–6 μM) induced apoptotic signals in platelets. The Hb-GP1bα interaction stimulated the intrinsic (mitochondria-dependent) pathway of apoptotic signals (Figure 4), as other studies have demonstrated for VWF-GP1bα interaction-mediated apoptosis in platelets.33,34 In fact, the Hb-GP1bα interactions stimulated the activation of proapoptotic proteins Bak and Bax, release of apoptogenic cytochrome c from mitochondrial intermembrane space to the cytosol, and the activation of caspase-9 and caspase-3 (Figure 4B and Online Supplementary Figure S10). The Hb-mediated apoptotic responses in turn induced the cytoskeleton expression of PS in platelets (Figure 4A) and generation of platelet microparticles (Figure 1C) as described in other agonists (such as thrombin, collagen, or ristocetin) that induced signal transduction.35,36 Furthermore, our data show that the Hb-induced apoptotic platelets enhanced thrombin generation and clot formation (Figure 5), which are the hallmarks of a procoagulant state and thrombosis.
The plasma Hb concentration in circulation appears to be the crucial determinant of activation and apoptosis of platelets contributing to the pathogenesis of platelet aggregation and procoagulant state in hemolytic disorders. The effects of plasma extracellular Hb on platelet functions are further supported by our data from patients with PNH, a prototype of intravascular hemolytic disease associated with recurrent events of thrombosis and thromboembolic complications.9,10 Several studies demonstrated that activation of blood cells including platelets and generation of microparticles by these cells are associated with the pathogenesis of thrombosis in PNH patients.37,38 Our data show a unique parallel correlation between plasma Hb or platelet surface-bound Hb and platelet activation markers (such as surface expression of P-selectin and PS, and the platelet-derived microparticles) in PNH patients (Figure 6).
In conclusion, Hb binding to GP1bα induces platelet activation and apoptosis in a concentration-dependent manner. The lower Hb concentrations activate platelets and higher concentrations induce apoptosis, and in turn promote platelet aggregation and clot formation (see schematic representation in Figure 7). The plasma extracellular Hb or platelet surface-bound Hb correlates with platelet activation in PNH patients.
Figure 7.
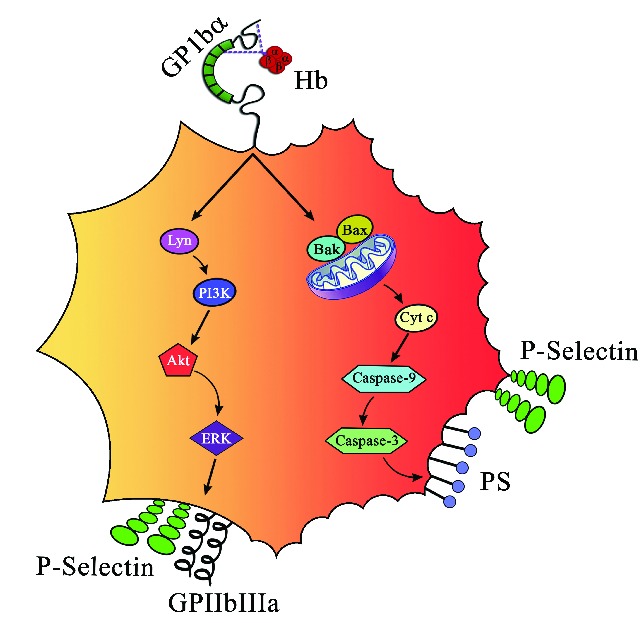
Schematic representation of platelet activation and apoptosis by Hb. Hb at lower concentrations (0.37–3 μM) induces inside out signaling via Lyn, PI3K, AKT, and ERK by binding to GP1bα and increases surface expression of P-selectin and GPIIb-IIIa. On the other hand, concentrations of Hb (3-6 μM) induce apoptosis and increase expression of Bak and Bax, release cytochrome C, activate caspase-9 and caspase-3, and expose PS on platelets.
Acknowledgments
The authors wish to thank the patients and healthy individuals who participated in this study. Authors thank Dr. Satyajit Rath of National Institute of Immunology, New Delhi, India, for important suggestions.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This study was financially supported by a grant from the Department of Biotechnology, Govt. of India to Prasenjit Guchhait.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293(13):1653–1662. [DOI] [PubMed] [Google Scholar]
- 2.Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817): 198–201. [DOI] [PubMed] [Google Scholar]
- 3.Cappellini MD. Coagulation in the pathophysiology of hemolytic anemias. Hematology Am Soc Hematol Educ Program. 2007:74–78. [DOI] [PubMed] [Google Scholar]
- 4.Barker JE, Wandersee NJ. Thrombosis in heritable hemolytic disorders. Curr Opin Hematol. 1999;6(2):71–75. [DOI] [PubMed] [Google Scholar]
- 5.Regoeczi E, Rubenberg ML, Brain MC. Intravascular haemolysis and disseminated intravascular coagulation. Lancet. 1967;1(7490):601–602. [DOI] [PubMed] [Google Scholar]
- 6.Brain MC, Esterly JR, Beck EA. Intravascular haemolysis with experimentally produced vascular thrombi. Br J Haematol. 1967;13(6):868–891. [DOI] [PubMed] [Google Scholar]
- 7.Amris CJ, Hansen NE. Coagulation and fibrinolytic studies in paroxysmal nocturnal haemoglobinuria. Acta Med Scand. 1968;184(6):551–559. [PubMed] [Google Scholar]
- 8.Van Vleymen B, Dehaene I, Van Hoof A, Pattyn G. Cerebral venous thrombosis in paroxysmal nocturnal haemoglobinuria. Acta Neurol Belg. 1987;87(2):80–87. [PubMed] [Google Scholar]
- 9.Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(25):4985–4996. [DOI] [PubMed] [Google Scholar]
- 10.Hartmann RC, Jenkins DE, Jr, McKee LC, Heyssel RM. Paroxysmal nocturnal hemoglobinuria: clinical and laboratory studies relating to iron metabolism and therapy with androgen and iron. Medicine (Baltimore). 1966;45(5):331–363. [DOI] [PubMed] [Google Scholar]
- 11.Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005; 106(12):3699–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poulou LS, Xila V, Rokas GI, et al. Temporal trends in mortality rates from visceral vein thrombosis in paroxysmal nocturnal haemoglobinuria: an optimistic view. Thromb Haemost. 2008;99(3):642–645. [DOI] [PubMed] [Google Scholar]
- 13.Ziakas PD, Poulou LS, Rokas GI, et al. Thrombosis in paroxysmal nocturnal hemoglobinuria: sites, risks, outcome. An overview. J Thromb Haemost. 2007; 5(3):642–645. [DOI] [PubMed] [Google Scholar]
- 14.Audebert HJ, Planck J, Eisenburg M, et al. Cerebral ischemic infarction in paroxysmal nocturnal hemoglobinuria report of 2 cases and updated review of 7 previously published patients. J Neurol. 2005;252(11):1379–1386. [DOI] [PubMed] [Google Scholar]
- 15.Stormorken H. Platelets, thrombosis and hemolysis. Fed Proc. 1971;30(5):1551–1556. [PubMed] [Google Scholar]
- 16.Gralnick HR, Vail M, McKeown LP, et al. Activated platelets in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1995;91(3):697–702. [DOI] [PubMed] [Google Scholar]
- 17.Dixon RH, Rosse WF. Mechanism of complement-mediated activation of human blood platelets in vitro: comparison of normal and paroxysmal nocturnal hemoglobinuria platelets. J Clin Invest. 1977;59(2): 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villagra J, Shiva S, Hunter LA, et al. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110(6):2166–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2(8567):1057–1058. [DOI] [PubMed] [Google Scholar]
- 20.Helms CC, Marvel M, Zhao W, et al. Mechanisms of hemolysis associated platelet activation. J Thromb Haemost. 2013;11(12):2148–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iuliano L, Violi F, Pedersen JZ, et al. Free radical-mediated platelet activation by hemoglobin released from red blood cells. Arch Biochem Biophys. 1992;299(2):220–224. [DOI] [PubMed] [Google Scholar]
- 22.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA. 1977;74(8):3203–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002; 99(1):36–43. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Z, Han H, Cruz MA, et al. Haemoglobin blocks von Willebrand factor proteolysis by ADAMTS-13: a mechanism associated with sickle cell disease. Thromb Haemost. 2009;101(6):1070–1077. [PubMed] [Google Scholar]
- 25.Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor gamma chain complex on human platelets. J Exp Med. 1988;188(2):267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84(2):289–297. [DOI] [PubMed] [Google Scholar]
- 27.Du X. Signaling and regulation of the platelet glycoprotein Ib-IX-V complex. Curr Opin Hematol. 2007;14(3):262–269. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Zhang CZ, Zhang X, Springer TA. A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature. 2010;466(7309):992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Fitzgerald ME, Berndt MC, et al. Bruton tyrosine kinase is essential for botrocetin/VWF-induced signaling and GPIb-dependent thrombus formation in vivo. Blood. 2006;108(8);2596–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104(6):1606–1615. [DOI] [PubMed] [Google Scholar]
- 31.Leisner TM, Wencel-Drake JD, Wang W, Lam SC. Bidirectional transmembrane modulation of integrin alphaIIbbeta3 conformations. J Biol Chem. 1999;274(18):12945–12949. [DOI] [PubMed] [Google Scholar]
- 32.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11(4):288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leytin V, Allen DJ, Mutlu A, et al. Mitochondrial control of platelet apoptosis: effect of cyclosporin A, an inhibitor of the mitochondrial permeability transition pore. Lab Invest. 2009;89(4):374–384. [DOI] [PubMed] [Google Scholar]
- 34.Li S, Wang Z, Liao Y, et al. The glycoprotein Ibalpha-von Willebrand factor interaction induces platelet apoptosis. J Thromb Haemost. 2010;8(2):341–350. [DOI] [PubMed] [Google Scholar]
- 35.Shcherbina A, Remold-O’Donnell E. Role of caspase in a subset of human platelet activation responses. Blood. 1999;93(12): 4222–4231. [PubMed] [Google Scholar]
- 36.Keuren JF, Wielders SJ, Ulrichts H. Synergistic effect of thrombin on collagen-induced platelet procoagulant activity is mediated through protease-activated receptor-1. Arterioscler Thromb Vasc Biol. 2005;25(7):1499–1505. [DOI] [PubMed] [Google Scholar]
- 37.Preis M, Ornstein DL. The different faces of myelodysplasia in peripheral blood granulocytes. Am J Hematol. 2013;89(3):342. [DOI] [PubMed] [Google Scholar]
- 38.Aster RH, Enright SE. A platelet and granulocyte membrane defect in paroxysmal nocturnal hemoglobinuria: usefulness for the detection of platelet antibodies. J Clin Invest. 1969;48(7):1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]