

Abstract
Effects of concurrent inhibition of mTORC1/2 and Bcl-2/Bcl-xL in human acute myeloid leukemia cells were examined. Tetracycline-inducible Bcl-2/Bcl-xL dual knockdown markedly sensitized acute myeloid leukemia cells to the dual TORC1/2 inhibitor INK128 in vitro as well as in vivo. Moreover, INK128 co-administered with the Bcl-2/xL antagonist ABT-737 sharply induced cell death in multiple acute myeloid leukemia cell lines, including TKI-resistant FLT3-ITD mutants and primary acute myeloid leukemia blasts carrying various genetic aberrations e.g., FLT3, IDH2, NPM1, and Kras, while exerting minimal toxicity toward normal hematopoietic CD34+ cells. Combined treatment was particularly active against CD34+/CD38−/CD123+ primitive leukemic progenitor cells. The INK128/ABT-737 regimen was also effective in the presence of a protective stromal microenvironment. Notably, INK128 was more potent than the TORC1 inhibitor rapamycin in down-regulating Mcl-1, diminishing AKT and 4EBP1 phosphorylation, and potentiating ABT-737 activity. Mcl-1 ectopic expression dramatically attenuated INK128/ABT-737 lethality, indicating an important functional role for Mcl-1 down-regulation in INK128/ABT-737 actions. Immunoprecipitation analysis revealed that combined treatment markedly diminished Bax, Bak, and Bim binding to all major anti-apoptotic Bcl-2 members (Bcl-2/Bcl-xL/Mcl-1), while Bax/Bak knockdown reduced cell death. Finally, INK128/ABT-737 co-administration sharply attenuated leukemia growth and significantly prolonged survival in a systemic acute myeloid leukemia xenograft model. Analysis of subcutaneous acute myeloid leukemia-derived tumors revealed significant decrease in 4EBP1 phosphorylation and Mcl-1 protein level, consistent with results obtained in vitro. These findings demonstrate that co-administration of dual mTORC1/mTORC2 inhibitors and BH3-mimetics exhibits potent anti-leukemic activity in vitro and in vivo, arguing that this strategy warrants attention in acute myeloid leukemia.
Introduction
Acute myelogenous leukemia (AML) is characterized by frequent aberration of the PI3K/AKT/mTOR axis reflecting various mechanisms including FLT3, Ras, and c-KIT mutations,1 PI3K delta isoform amplification,2 or autocrine IGF-1/IGF-1R signaling.3 The mammalian target of rapamycin (mTOR) is a key component of this pathway4 which integrates growth factor signals through AKT and multiple other cellular processes.5 mTOR is a serine/threonine kinase involved in two distinct multi-protein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).5 mTORC1 plays a central role in cap-dependent mRNA translation initiation through 4EBP1 phosphorylation, releasing eukaryotic initiation factor eIF4E.6 mTORC1 also promotes translation elongation by phosphorylating S6 kinase 1 (S6K1).6 mTORC2 is less studied and has distinct substrates e.g., AKT and AGC protein kinase family members.5 Importantly, mTORC2 phosphorylates AKT at serine 473, inducing maximal AKT activation.
First-generation agents, including rapamycin and its analogs (rapalogs) e.g., everolimus, temsirolimus and ridaforolimus, inhibited mTORC1 but not mTORC2. While these agents are approved in RCC,7 leukemic activity has been minimal,8 despite evidence they target leukemia stem cells.9 Limited rapalog activity may reflect absent (mTORC2) or incomplete (4EBP1) target inhibition, or feedback activation of PI3K, AKT and MEK/ERK through p70S6K and IRS1.10,11 Second generation inhibitors targeting both mTORC1 and mTORC2, including AZD8055 and INK128, are currently undergoing clinical evaluation. (www.clinicalTrials.gov).
Anti-apoptotic Bcl-2 members e.g., Bcl-2, Bcl-xL, and Mcl-1 are often overexpressed in hematological malignancies, including AML.12 Loss or diminished expression of pro-apoptotic Bcl-2 members e.g., Bim or Bax have also been observed in many malignancies.13 This prompted development of agents that neutralize anti-apoptotic or activate pro-apoptotic Bcl-2 members. ABT-737 and its clinical derivative ABT-263 target Bcl-2 and Bcl-xL, but not Mcl-1, and show significant pre-clinical activity.14,15 ABT-199 is a BH3-mimetic that specifically targets Bcl-2,16 and displays promising early activity in CLL17 and AML.18 However, BH3-mimetics by themselves are unlikely to be curative, thus arguing for rational combination strategies.
Previously, we demonstrated that combining dual PI3K/mTOR inhibitors and Bcl-2/Bcl-xL antagonists sharply induced cell death in diverse leukemia types in vitro and in vivo.19 Herein, we sought to determine whether mTOR inhibition, which targets primitive AML progenitors,9 could similarly potentiate BH3-mimetic anti-leukemia activity, and whether dual mTORC1/TORC2 inhibition might provide an advantage over selective mTORC1 inhibition.
Methods
Cells
Human acute myeloid leukemia U937, KG-1 and MV4-11 cells were as previously reported.19 MOLM-13 and OCI-AML3 cells were purchased from DSMZ (Braunschweig, Germany). U937 cells exhibiting inducible knockdown of Bcl-2 and Bcl-xL have been described previously.19 U937 cells stably overexpressing wild-type Mcl-1, Bcl-2, Bcl-xL, Bax or Bak were previously described.19
Generation of Ba/F3 mutants
Ba/F3 cells carrying FLT3 mutations were generated as described in Online Supplementary Methods.
Stromal cells
As described in Online Supplementary Methods.
Isolation of patient-derived leukemic blast cells
These studies are sanctioned by the Virginia Commonwealth University Investigational Review Board. Bone marrow or peripheral blood were collected with informed consent from 11 patients with acute myeloblastic leukemia (AML) with a preponderance of blasts (e.g., ≥ 80%). Normal hematopoietic cells were isolated from cord blood. Mononuclear cells were isolated by Ficoll-Hypaque gradient separation as described.19
Mutation analysis
Primary AML samples were analyzed for mutations in 50 cancer associated genes using next generation sequencing (NGS) as in Online Supplementary methods.
Reagents
ABT-737 and ABT-199 were provided by Abbott Laboratories (Abbott Park, IL). INK128 was purchased from ChemieTek (Indianapolis, IN). AKT inhibitor VIII was purchased from EMD Millipore.
Assessment of apoptosis and cell growth and viability
Apoptosis was assessed by Annexin V/PI analysis.20 Cell growth and viability were monitored by the CellTiter-Glo Luminescent Assay (Promega Corporation).19
Immunoprecipitation and immunoblotting were performed as before.20,21 Primary antibodies are listed in Online Supplementary Methods.
Subcellular fractionation
Cytosolic and membrane fractions were separated as previously described.21
In vivo studies
These studies were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee and conducted as previously described.19 Briefly, female NOD/SCID-gamma (Jackson laboratories) were injected intravenously via tail vein with 5×106 luciferase-expressing U937 cells in which dual knockdown of Bcl-2 and Bcl-xL is achieved by doxycycline. The mice were monitored using the IVIS 200 imaging system (Xenogen Corporation, Alameda, CA), and separated into 2 groups, one of which was fed with doxycycline-supplemented pellets (200 mg/kg, Bio-Serv, Frenchtown, NJ). Both groups were treated with INK128 administered by gavage every 24 hours, 5 days a week. NOD/SCID-gamma mice were inoculated via tail vein with 5×106 luciferase-expressing MV4-11 cells. 5 days later, the mice were randomly separated into 4 groups; each group was treated with vehicle, ABT-737 (intraperitoneal), INK128 (oral), or ABT-737 + INK128. Tumor growth was monitored by the IVIS 200 imaging system. In some cases, female athymic nude mice (Charles River laboratories) were injected subcutaneously in the flank with 5 × 106 MV4-11 cells. Once tumors reached 1 cm in diameter, the mice were treated as above, and 4 hours later tumors were excised, lysed and subjected to Western blot analysis.
Statistical analysis is described in Online Supplementary Methods.
Results
Dual knockdown of Bcl-2/Bcl-xL markedly potentiates the anti-leukemic activity of the mTORC1/TORC2 inhibitor INK128 in vitro and in vivo
To determine whether Bcl-2/xL inhibition sensitized AML cells to mTOR inhibitors, U937 cells exhibiting tet-inducible Bcl-2 and Bcl-xL dual knockdown were employed. Doxycycline exposure (48 hrs) sharply downregulated both Bcl-2 and Bcl-xL. In dual knockdown cells, the dual mTORC1/TORC2 inhibitor INK128 (50 nM) triggered rapid (e.g., within 4 hrs) and pronounced apoptosis, reflected by caspase-3 and PARP cleavage (Figure 1A) and cytochrome c and AIF cytosolic release (Figure 1B). In sharp contrast, INK128 effects on caspase-3 and PARP in cells with intact Bcl-2 and Bcl-xL were minimal. Dose-response studies confirmed that knockdown of Bcl-2/Bcl-xL rendered cells exquisitely sensitive to INK128-mediated lethality (Figure 1C, and Online Supplementary Figure S1A). In contrast, effects of the selective mTORC1 inhibitor rapamycin were only modestly enhanced by dual knockdown of Bcl-2 and Bcl-xL (Figure 1C, and Online Supplementary Figure S1A). Notably, INK128 markedly reduced both AKT-activating sites (serine 473 and threonine 308) phosphorylation in the presence or absence of doxycycline (Figure 1A).
Figure 1.

Bcl-2/Bcl-xL dual knockdown strikingly enhances anti-tumor activity of INK128, but not rapamycin, in vitro as well as in vivo. A–B. U937 cells displaying tet-inducible Bcl-2 and Bcl-xL dual knockdown were left untreated or pre-treated with 1 μg/mL doxycycline (Dox) for 48 hrs, then exposed to 50 nM INK128 for varying intervals after which Western blot analysis was performed on whole cell lysates (A) or on the cytosolic fractions (B). Ns = Non-specific. C) Alternatively, tet-inducible Bcl-2 and Bcl-xL U937 dual knockdown cells were treated with the designated concentrations of INK128 or rapamycin for 24 hrs, after which cell growth and viability was assessed using the CellTiter-Glo luminescent assay. D) NOD/SCID-gamma (NSG) mice were inoculated via the tail vein with U937 cells exhibiting tet-inducible Bcl-2/Bcl-xL dual knockdown and expressing luciferase. 5 days following cell injection, mice were treated with 1 mg/kg INK128 in the presence (5 mice) or absence (4 mice) of doxycycline and imaged using the IVIS 200 system. E) Quantification of the luminescent signals. Data represent the mean ± SD performed on all mice for each group. *P<0.0005. F) Kaplan-Meier survival plot. Survival curves for treatments with or without doxycycline differed very significantly e.g., P=0.0027, log-rank test. The median survival was prolonged from 14 to 21 days for mice in the presence of doxycycline. G) Animal body weights during the course of treatment.
In vivo studies employing a systemic xenograft mouse model bearing luciferase-labeled U937 cells exhibiting inducible Bcl-2/Bcl-xL dual knockdown revealed that doxycycline significantly enhanced INK128 anti-leukemia effects compared to controls (Figure 1D,E). Knockdown of Bcl-2/Bcl-xL also significantly prolonged median survival of INK128-treated mice i.e., from 14 to 21 days (P = 0.0027 log-rank test; Figure 1F). Doxycycline alone had no effect on tumor growth or survival (Online Supplementary Figure S1B). Finally, INK128 ± doxycycline did not induce weight loss (Figure 1G) or other signs of toxicity. These findings indicate that dual knockdown of Bcl-2/Bcl-xL strikingly enhances INK128-mediated AML cell death in vitro and inhibits AML growth while prolonging survival in vivo.
INK128/ABT-737 co-administration sharply induces cell death in AML but not normal hematopoietic CD34+ cells
Dose-response studies of MV4-11 and U937 cells using a range of clinically relevant INK128 concentrations (e.g., 50–200 nM)22 revealed that minimally toxic concentrations of the dual Bcl-2/Bcl-xL inhibitor ABT-737 (10 nM for MV4-11, and 500 nM for U937 cells) sharply potentiated INK128 inhibition of cell growth and viability (Figure 2A,B). The disparate ABT-737 concentrations employed reflect the known variability of leukemic cell vulnerability to this agent,19,23 and were selected to achieve comparable single-agent responses (e.g., marginal toxicity; <15% cell death). These effects were highly synergistic by median dose-effect analysis (Online Supplementary Figure S1C,D). In contrast, identical concentrations of the selective mTORC1 inhibitor rapamycin recapitulating or exceeding clinically achievable concentrations (20 – 400 nM)24 failed to enhance ABT-737 lethality (Figure 2A,B). Similar results were obtained in multiple other leukemia cell lines i.e., MOLM-13, KG-1, and AML3 (Figure 2C). Annexin V/PI analysis yielded equivalent results (data not shown). INK128/ABT-737 co-exposure induced pronounced cas-pase-3 and PARP cleavage, whereas agents alone had minimal effects (Figure 2D). Notably, similar results were obtained with INK128 and the selective Bcl-2 inhibitor ABT-199 in MV4-11, MOLM-13, and KG-1 cells (Online Supplementary Figure S1E). ABT-199 anti-leukemic activity was not significantly enhanced by rapamycin in these cells (data not shown).
Figure 2.
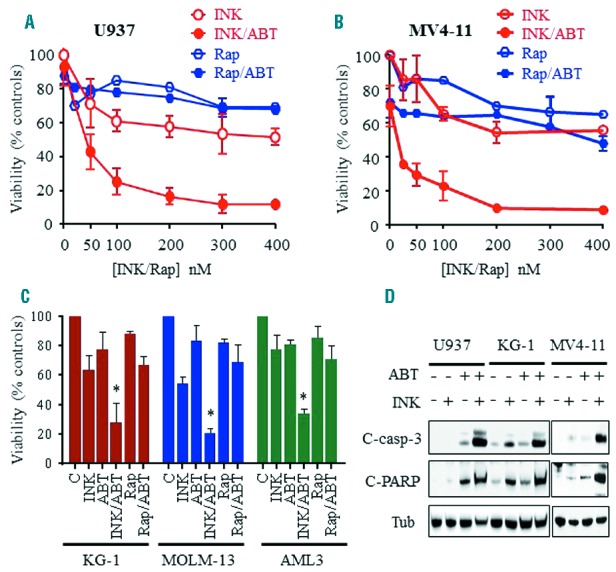
Co-administration of INK128, but not rapamycin, with ABT-737 induces marked inhibition of cell growth and viability in association with pronounced induction of apoptosis in human AML cells. A–B) U937 (A) or MV4-11 (B) cells were exposed to the designated concentrations of INK128 or rapamycin alone or in combination with ABT-737 (500 nM for U937 and 10 nM for MV4-11 cells) for 24 hrs after which cell growth and viability were assessed using the CellTiter-Glo Luminescent Assay. C) KG-1, MOLM-13, or AML3 cells were treated with ABT-737 (200 nM, 10 nM, 2 μM, respectively) ± INK128 or rapamycin (100 nM for MOLM-13, and 200 nM for KG-1 and AML3 cells). For each cell line, agent concentrations were selected based upon minimal toxicity (e.g., ≤ 15% cell death when administered alone) as well as clinical achievability. Cells were exposed to these concentrations for 24 hrs, after which cell growth and viability were assessed using a CellTiter-Glo Luminescent Assay. Error Bars: SD of 3 independent experiments; *P<0.01 for combination vs. either agent alone. Alternatively, cells were lysed and protein lysates were subjected to Western Blot analysis (D).
Parallel studies in primary blasts isolated from AML patients (n=10), revealed that INK128/ABT-737 co-exposure was significantly more effective in diminishing cell viability than single agents; Figure 3A (P < 0.0001). As with cell lines, drug concentrations were selected based upon minimal toxicity when administered alone, and clinical relevance. Furthermore, in the CD34+/CD38−/CD123+ cell population enriched for leukemia progenitor cells,25 combined treatment sharply induced cell death (Figure 3B). Interestingly, this effect appeared more pronounced than in bulk blast populations (Figure 3B). Analysis of three individual primary AML samples (Figure 3C) demonstrated increased sensitivity of CD34+/CD38−/CD123+ cells compared to bulk blasts (P=0.007). In contrast, combined treatment had no major effect on normal hematopoietic CD34+ cells isolated from cord blood (Figure 3B,C). Genetic analysis using next generation sequencing (NGS) revealed that the primary AML blasts assayed carried diverse genetic aberrations including mutations in FLT3, IDH2, NPM1, and Kras, among others (Figure 3D), suggesting activity across a spectrum of leukemia types.
Figure 3.
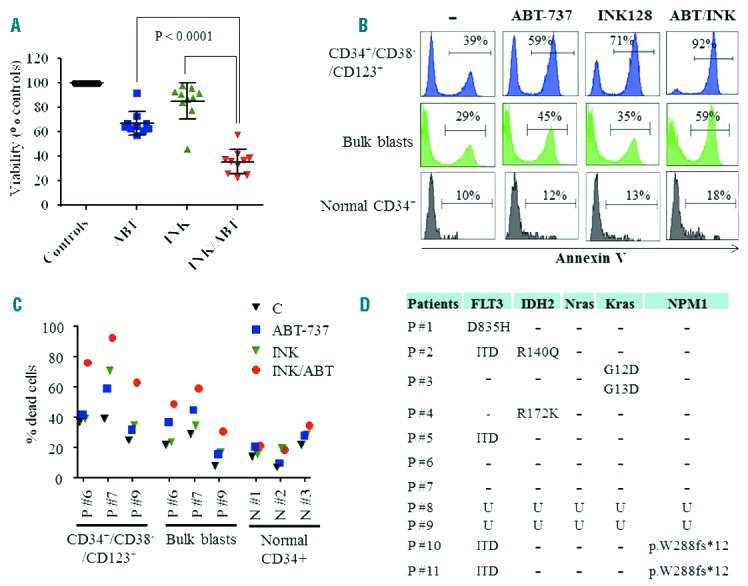
Co-administration of INK128 and ABT-737 kills primary AML blasts and CD34+/CD38−/CD123+ leukemic cells while sparing normal CD34+ progenitor cells. A) Primary AML specimens with a preponderance of blasts (e.g ≥ 80%) were isolated from 10 patients with AML (patients 1 to 8, 10 and 11) and treated with ABT-737 (10 – 500 nM) and/or INK128 (50 – 200 nM) for 24 hrs after which cell viability was assessed using the CellTiter-Glo Luminescent Assay. As in the case of cell lines, ABT-737 and INK128 concentrations were selected based upon marginal toxicity when administered alone as well as clinical relevance. B) Primary AML blasts (patient 7), were treated with INK128 (200 nM) ± ABT-737 (7.5 nM) for 24 hrs, after which cell death was assessed in leukemia CD34+/CD38−/CD123+ progenitors as well as in the bulk blast cell population using an Annexin V/7-AAD staining assay. In parallel studies, normal hematopoietic mononuclear cells isolated from cord blood were treated with INK128 ± ABT-737 (100 nM each) and cell death was assessed in the CD34+ cell population using an Annexin V/7-AAD staining assay. C) Additional studies using the Annexin V/7-AAD staining assay were carried out in the bulk blast populations and in leukemia CD34+/CD38−/CD123+ progenitor cells isolated from 3 patients with AML as well as in normal hematopoietic CD34+ cells isolated from 3 individuals. Agent concentrations were: ABT-737: 50 nM for patients 6 and 9, and 7.5 nM for patient 7; INK128: 100 nM for patients 6 and 9, and 200 nM for patient 7. For normal CD34+ cells, ABT-737 and INK128 were used at 100 nM and 200 nM, respectively. D) Mutation analysis of primary AML specimens using targeted next-generation sequencing (NGS); only AML associated mutations are presented. U = unknown.
The INK128/ABT-737 regimen is active against AML blasts carrying FLT3 mutations and in the presence of a protective microenvironment
Three out of the seven characterized primary AML specimens used carried FLT3-ITD or D835H mutations, each of which exhibited significant sensitivity to the regimen. To test whether ABT-737/INK128 regimen is active in FLT3 mutated AML cells, Ba/F3 cells reliant on FLT3 activation for survival were generated by transfecting wild-type Ba/F3 cells with constructs encoding for FLT3-ITD or FLT3-ITD F691L. As anticipated, FLT3-ITD Ba/F3 cells were sensitive to the FLT3 inhibitors sorafenib (20 nM) and quizartinib (10 nM), whereas FLT3-ITD F691L displayed significant resistance to both (up to 100 nM each; Online Supplementary Figure S2). However, while both leukemia cells exhibited little susceptibility to INK128 or ABT-737, combined treatment sharply increased cell death. (Figure 4A,B). Parallel studies involving bone marrow-derived HS5 cell co-culture revealed that combined INK128/ABT-737 treatment robustly killed AML cells cultured with protective microenvironmental factors (Figure 4C–D). Specifically, GFP-labeled U937 cells co-cultured with HS-5 cells displayed a marked increase in cell death (reflected by Annexin positivity) following combined ABT-737/INK128 exposure (Figure 4C), while luciferase-labeled MV4-11 cells demonstrated a sharp decline in luciferase activity (reflecting both cell viability as well as proliferation) with combined treatment (Figure 4D). As before, drug concentrations were selected based upon minimal toxicity alone and clinical achievability.
Figure 4.
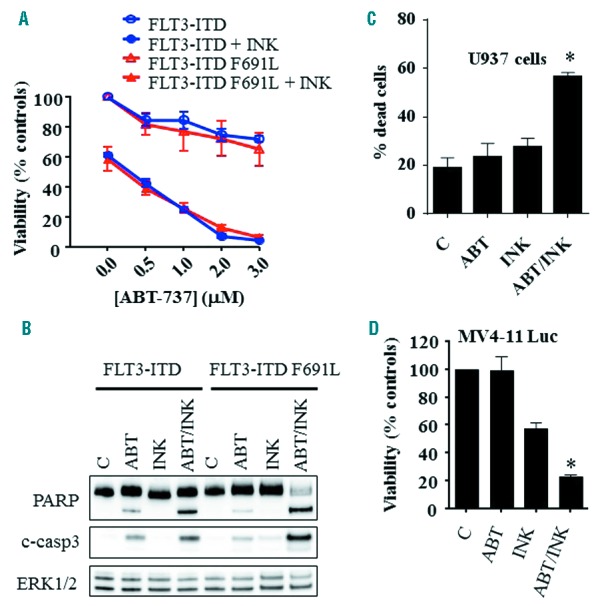
Co-administration of INK128 and ABT-737 effectively kills AML blasts carrying FLT3 mutations or cultured in the presence of a protective microenvironment. A) Ba/F3 cells carrying FLT3-ITD or FLT3-ITD F691L were exposed to the designated concentrations of ABT-737 alone or in combination with 200 nM INK128 for 24 hrs after which cell growth and viability were assessed using the CellTiter-Glo Luminescent Assay. Alternatively, protein lysates were prepared and subjected to Western blot analysis (B). C) U937 cells expressing copGFP were co-cultured with bone marrow-derived stromal HS5 cells for 24 hrs, and exposed to 200 nM INK128 ± 500 nM ABT-737 for an additional 24 hrs. Cell death was then assessed in cop-GFP U937 cells using the Annexin V-APC/7-AAD staining assay. D) MV4-11 cells expressing luciferase were co-cultured with HS5 cells for 24 hrs and treated with INK128 (100 nM) ± ABT-737 (10 nM). After 24 hrs, a luciferase assay was performed using D-luciferin to reflect cell growth and viability. Error Bars: SD of 3 independent experiments; *P<0.0001 in each case.
INK128/ABT-737 co-administration triggers AKT inactivation and Mcl-1 down-regulation in AML cells
U937 cell time course analysis revealed that clinically relevant INK128 concentrations significantly reduced AKT phosphorylation at both activating sites e.g., serine 473 and threonine 308, associated with marked translational factor 4EBP1 dephosphorylation, Mcl-1 down-regulation, and Bak up-regulation (Figure 5A,B). These events were also observed when INK128 was combined with ABT-737, but were not substantially greater (Figure 5A,B). Notably, Bak was also upregulated in MV4-11 cells by combined treatment (Online Supplementary Figure S3A). No major changes were observed in protein levels of other anti-apoptotic Bcl-2 family members i.e., Bcl-2 or Bcl-xL or pro-apoptotic members i.e., Bim or Bax (Figure 5B). Similar results were obtained in MV4-11 cells (Figure 5C), and in primary AML blasts isolated from 2 AML patients (Figure 5D).
Figure 5.
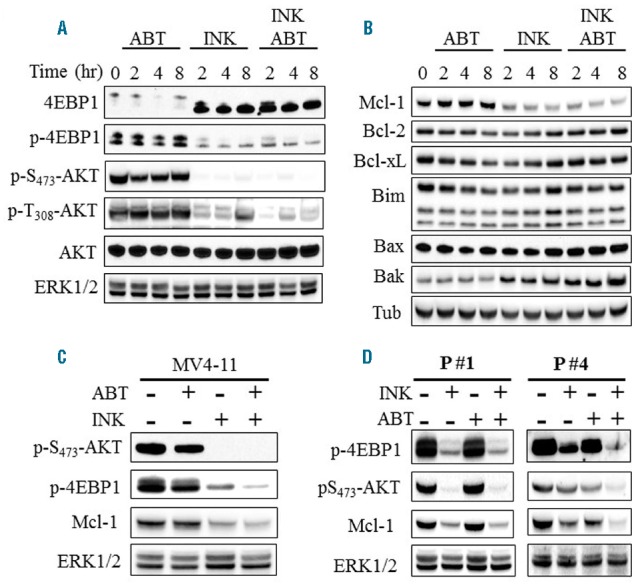
Combined treatment with INK128 and ABT-737 inactivates AKT and down-regulates Mcl-1 in AML cells. Western blot analysis in U937 (A–B) MV4-11 (C), and primary AML blasts isolated from 2 patients (D) following exposure to INK128 ± ABT-737 for the indicated intervals (for U937) or for 8 hrs (MV4-11 and primary blasts). The concentrations used in this study were: INK128: 200 nM for U937, 100 nM for MV4-11 and patient 4, 50 nM for patient 1. ABT-737: 500 nM for U937, 10 nM for MV4-11 and patient 4, and 25 nM for patient 1.
Down-regulation of Mcl-1 plays a critical functional role in INK128/ABT-737-mediated lethality, an event that involves Bax and Bak
To determine whether Mcl-1 down-regulation contributed functionally to INK128/ABT-737 activity, U937 cells ectopically expressing Mcl-1 were employed. These cells were significantly less susceptible to INK128/ABT-737 than control pCEP4 cells, reflected by sharply reduced Annexin V/PI positivity (Figure 6A) and caspase-3 and PARP cleavage (Figure 6B) as well as growth inhibitory effects (Online Supplementary Figure S3B). These findings suggest that Mcl-1 down-regulation contributes functionally to cell death mediated by combined treatment with INK128/ABT-737.
Figure 6.
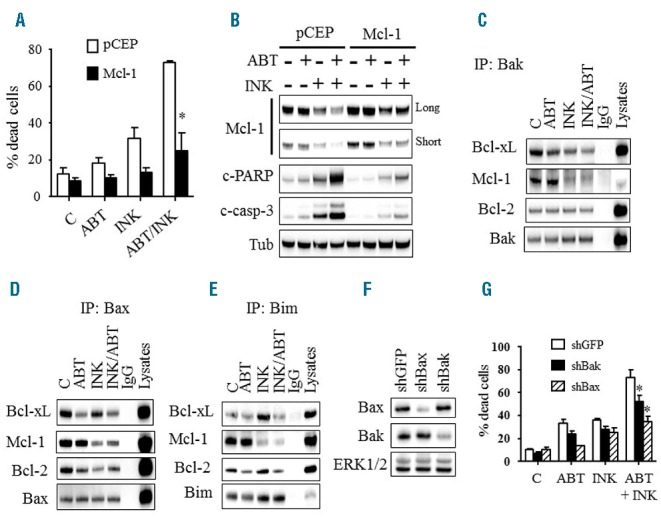
Functional role of Mcl-1, Bax, and Bak in INK128/ABT-737-mediated cell death. A) U937 cells ectopically expressing Mcl-1 and their empty vector control pCEP cells were exposed to INK128 (200 nM) ± ABT-737 (500 nM) for 24 hrs, after which cell death was assessed using the Annexin V/PI staining assay. Error Bars: SD of 3 independent experiments; *P<0.01. Alternatively, protein lysates were prepared and subjected to Western blot analysis (B). (C–E) U937 cells were treated with INK128 (200 nM) ± ABT-737 (500 nM) for 4 hrs after which cells were lysed, Bak (C), Bax (D), or Bim (E) were immuno-precipitated, and the immuno-precipitates were subjected to immunoblotting. F) Western blot analysis in U937 cells in which Bax or Bak was knocked down using shRNA. G) These cells were exposed to INK128 (200 nM) ± ABT-737 (500 nM) for 24 hrs, after which cell death was monitored using the Annexin V/PI staining assay. Error Bars: SD of 3 independent experiments; *P<0.01 for shBax and P<0.05 for shBak.
Parallel immunoprecipitation analysis revealed that INK128 ± ABT-737 markedly diminished Bak, Bax and Bim binding to Mcl-1, presumably reflecting diminished Mcl-1 protein levels (Figures 6C–E). ABT-737 or INK128 decreased Bak binding to Bcl-xL but not to Bcl-2, an effect that was enhanced by combined treatment (Figure 6C). However, combined treatment markedly diminished Bax binding to both Bcl-2 and Bcl-xL, (Figure 6D). Notably, a decline in Bax protein bound to Bcl-2 or Bcl-xL was also observed in cells exposed to either agent alone (Figure 6D). Finally, while INK128 increased Bim binding to Bcl-2 and Bcl-xL, similar to our previous findings with PI3K/mTOR inhibitors,19 ABT-737 decreased Bim binding to Bcl-2 but not to Bcl-xL (Figure 6E). Combined treatment led to diminished Bim/Bcl-2 binding and abrogation of INK128-mediated increases in Bim/Bcl-xL binding (Figure 6E).
The role of Bax and Bak in cell death mediated by combined treatment with INK128 and ABT-737, was assessed in Bax or Bak knockdown cells (Figure 6F). Notably, these cells were significantly less sensitive to INK128/ABT-737 compared to non-targeted shRNA controls, reflected by diminished Annexin V/PI positivity (Figure 6G). Similar results were obtained in MV4-11 cells (data not shown). Consistent with the notion that Bax, Bak, and Bim play important roles in INK128/ABT-737 lethality, cells ectopically expressing Bcl-2 or Bcl-xL, which, like Mcl-1 sequester Bax, Bak, and Bim,19,26 were significantly less susceptible to INK128/ABT-737 (Online Supplementary Figure S3C,D). Together, these findings suggest that Bax, Bak, and Bim release from Bcl-2/Bcl-xL (by ABT-737 or INK128) and Mcl-1 (by INK128) contributes to INK128/ABT-737 anti-leukemic effects.
AKT inhibition enhances rapamycin/ABT-737 lethality associated with Mcl-1 down-regulation
To gain insights into the disparate interactions of INK128 and rapamycin with ABT-737, key proteins were monitored in U937 and MV4-11 cells. In contrast to INK128, which sharply dephosphorylated AKT and down-regulated Mcl-1 protein levels, rapamycin failed to diminish AKT phosphorylation or Mcl-1 protein expression, including at concentrations significantly higher than achievable plasma levels24 (Figure 7A). Instead, increased AKT phosphorylation occurred in both U937 and MV4-11 cells following a 4- or 8- hour exposure to rapamycin, consistent with previous reports11 (Figure 7A and Online Supplementary Figure S4A,B). INK128 was more effective than rapamycin in diminishing 4EBP1 phosphorylation as previously reported in other cell types27 (Figure 7A). Furthermore, co-administration of a selective AKT inhibitor (AKT inhibitor VIII) with rapamycin/ABT-737 suppressed cell growth and viability (Figure 7B) associated with decreased Mcl-1 protein levels (Figure 7C), comparable to findings with INK128/ABT-737. Similar results were obtained in MV4-11 cells (Online Supplementary Figure S4C), arguing that more pronounced AKT inactivation and Mcl-1 down-regulation contribute to the enhanced potentiation of ABT-737 anti-leukemic activity by INK128 compared to rapamycin.
Figure 7.
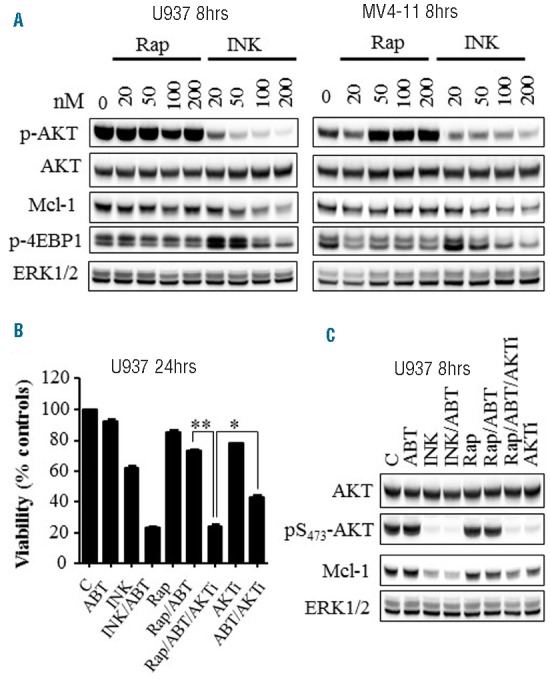
Inhibition of AKT enhances rapamycin/ABT-737 lethality in association with Mcl-1 down-regulation. A) U937 or MV4-11 cells were exposed to the designated concentrations of rapamycin or INK128 for 8 hrs, after which cells were lysed and protein lysates were subjected to Western blot analysis. B–C) Cell growth and viability (B) or Western blot analysis (C) performed in U937 cells following exposure to the indicated agents for 24 hrs or 8 hrs, respectively. The concentrations used in this study were: 500 nM ABT-737; 200 nM INK; 20 nM rapamycin; and 2 μM AKTi (AKT inhibitor VIII). **P<0.001; *P<0.02.
Co-administration of INK128/ABT-737 markedly decreases tumor growth in vivo and prolongs survival in a systemic leukemia xenograft model
To determine whether INK128/ABT-737 co-treatment exhibited anti-leukemic activity in vivo, NOD/SCID-gamma mice bearing systemic MV4-11-derived xenografts were employed. Combined INK128 (0.5 mg/kg) and ABT-737 (80 mg/kg) treatment significantly reduced leukemia growth (Figure 8A) and strikingly prolonged mouse survival. Moreover, on day 108 after treatment initiation, 50% of the mice treated with the combination remained alive with healthy appearances, whereas all of the mice from vehicle or single agent groups had died by day 57 (Figure 8B). Western blot analysis performed on subcutaneous tumor tissues excised from mice 4 hours post-treatment revealed that INK128 ± ABT-737 markedly diminished 4EBP1 phosphorylation and decreased Mcl-1 protein (Figure 8C), analogous to in vitro results. Notably, the effects of combined treatment or INK128 alone on these proteins were similar, as shown by densitometry (Online Supplementary Figure S4D). Significantly, combined treatment did not induce weight loss (Online Supplementary Figure S5) or other toxicities (e.g., fur loss, behavioral changes etc., data not shown). Together, these findings indicate that co-administration of INK128 and the BH3-mimetic ABT-737 markedly reduces in vivo leukemia growth associated with 4EBP1 dephosphorylation and Mcl-1 down-regulation, and significantly prolongs the survival of mice bearing systemic leukemia.
Figure 8.
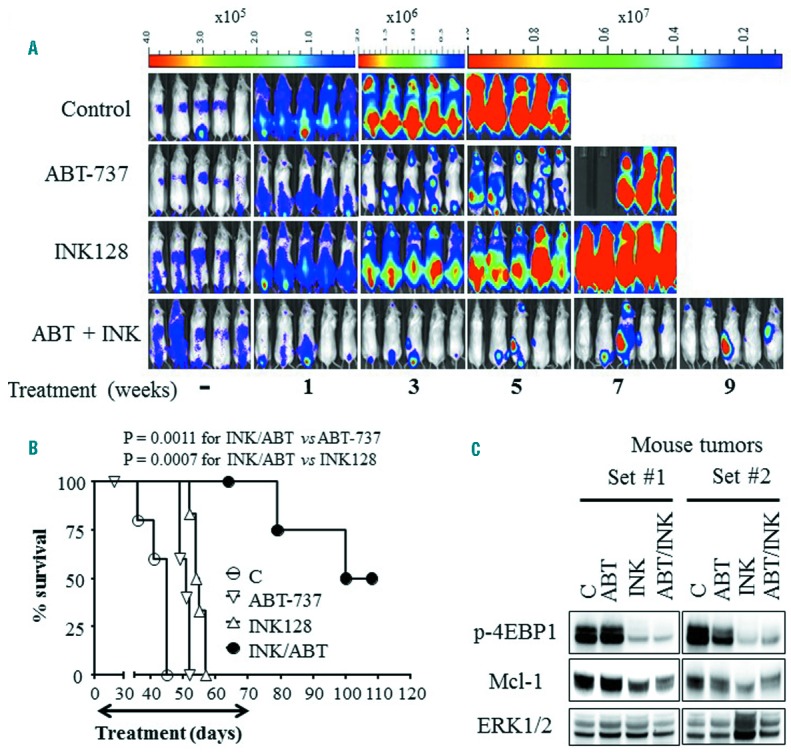
Co-administration of INK128 and ABT-737 exhibits potent in vivo anti-leukemia activity. (A) NOD/SCID-gamma mice were inoculated via tail-vein with MV4-11 cells expressing luciferase. Five days later, mice were treated with INK128 (0.5 mg/kg) ± ABT-737 (80 mg/kg) and imaged using the IVIS 200 system (A), and survival was analyzed using Kaplan-Meier survival plots (B). Studies involved 5–6 mice per condition; the survival of mice treated with the combination was significantly prolonged compared to mice treated with single agents (P=0.0011, and P=0.0007 for combination vs. ABT-737 or INK128 respectively, log-rank test). Treatment was discontinued on day 70. On day 108, 2 mice were still alive with healthy appearances; on the same day, the experiment was terminated. C) Nude mice were injected subcutaneously with MV4-11 cells. Once the tumors reached 1 cm in diameter, mice were treated with INK128 (0.5 mg/kg) ± ABT-737 (80 mg/kg). 4 hrs later, tumors were excised, lysed and analyzed by Western blot analysis.
Discussion
Susceptibility of various tumor types, including AML, to BH3-mimetics like ABT-737 is regulated by Mcl-1 expression.28,29 This has prompted combination strategies to down-regulate Mcl-1, including CDK inhibitors,30 inhibitors of translation,31 and deubiquitinase inhibition.32 Ourselves and others have reported that PI3K/AKT/mTOR pathway inhibition down-regulates Mcl-1 expression19 and increases BH3-mimetic lethality.19,33,34 Rapamycin is an approved selective mTORC1 inhibitor with a relatively low toxicity profile,24 and in pre-clinical studies targets primitive leukemia progenitors.9 In contrast, INK128 is a novel, clinically relevant ATP-competitive dual TORC1/TORC2 inhibitor that potently inhibits ser473 AKT phosphorylation, a process requiring TORC2 activation.35 INK128 is effective against B-lymphoblastic leukemia cells,36 but AML activity has not been explored. Furthermore, dual TORC1/TORC2 inhibition by AZD8055 enhances ABT-737 lethality in malignant epithelial cells,37,38 but this strategy has not been investigated in hematologic malignancies, including AML. Herein we report that the novel dual TORC1/TORC2 inhibitor INK128 potently inhibits AKT activation and triggers Mcl-1 down-regulation in cultured and primary AML cells, sharply increasing BH3 mimetic susceptibility.
The capacity of INK128 to potentiate ABT-737 anti-leukemic effects was greater than that of rapamycin. Notably, disabling Bcl-2 and Bcl-xL pharmacologically by ABT-737 or genetically by tet-inducible shRNA knockdown rendered AML cells exquisitely sensitive to INK128 but not rapamycin, suggesting that combined mTORC1 and mTORC2 inhibition is required for effective interactions. The superiority of INK128 in potentiating BH3-mimetic anti-leukemic activity could reflect multiple factors. For example, rapamycin may be a less potent inhibitor of protein translation, as suggested by its diminished inhibition of 4EBP1 phosphorylation in AML cells, consistent with results in other malignant hematopoietic cells.27,36 Second, in contrast to INK128, rapamycin failed to down-regulate Mcl-1 efficiently, an action that we and others have shown to play a key role in ABT-737 sensitivity.19,28,29 Failure of rapamycin to down-regulate Mcl-1 may reflect ineffective inhibition of mRNA translation39 or other mechanisms e.g., increased Mcl-1 stability through activation of AKT and inactivation of GSK3.40 Third, rapamycin activates AKT through feedback mechanisms involving p70S6K inactivation and IRS1 activation,11 raising the apoptotic threshold. Consistently, rapamycin activated AKT and did not significantly induce Mcl-1 down-regulation. However, the addition of AKT inhibitor VIII to rapamycin/ABT-737 sharply increased apoptosis in association with pronounced AKT inactivation and Mcl-1 down-regulation, as observed with INK128/ABT-737. These findings suggest that rapamycin-mediated AKT activation, and its failure to down-regulate Mcl-1 or dephosphorylate 4EBP1, may contribute to minimal rapamycin/ABT-737 interactions. In addition to Mcl-1, a variety of AKT downstream targets might play a role in conferring resistance to BH3-mimetics, including 4EBP1, GSK3 and Bim, among others.19,28,29
Translation of the short-lived protein Mcl-1 is regulated by mTORC1.41 The observation that INK128 significantly down-regulated Mcl-1, but rapamycin exerted only modest effects, suggests that factors other than mTORC1 contribute to the marked Mcl-1 down-regulation, at least in some cell types. These findings are consistent with recent reports42 describing Mcl-1 down-regulation in mantle cell lymphoma cells by dual mTORC1/TORC2 inhibitors, but not mTORC1 inhibitors alone. However, rapamycin did not induce significant 4EBP1 dephosphorylation, which may lead to only partial 4EBP1/eIF4E complex dissociation, limiting effects on cap-dependent mRNA translation. Of note, previous studies have demonstrated a prominent role for eIF4E in controlling Mcl-1 translation.43 It is also possible that mTORC2 inhibition potentiates Mcl-1 down-regulation through AKT inactivation, GSK3 activation, and MCL-1 degradation.40 The observation that enforced Mcl-1 expression markedly diminished INK128/ABT-737-mediated apoptosis indicate that Mcl-1 down-regulation contributes functionally to cell death, consistent with previous findings from our group and others that Mcl-1 plays a critical role in determining ABT-737 sensitivity.19 Significantly, Mcl-1 down-regulation was similar following exposure to INK128 alone or ABT-737/INK128, whereas cell death was considerably more pronounced with combined treatment. This suggests that Mcl-1 down-regulation is not solely responsible for the observed cell death, but may cooperate with Bcl-2 and Bcl-xL inhibition. However, the observation that similar interactions occurred with ABT-199, which inhibits Bcl-2 but not Bcl-xL,16 suggest that Bcl-2 inhibition may be particularly important in some AML cells.
Recent studies have highlighted interactions between anti- and pro-apoptotic Bcl-2 proteins in cell fate.19,44 Combined INK128/ABT-737 treatment not only diminished Mcl-1 protein levels, but also markedly reduced Bax, Bak, and Bim binding to Mcl-1 and Bcl-xL. Combined treatment also significantly decreased Bax/Bcl-2 and Bim/Bcl-2 binding, collectively leading to increased free Bax, Bak, and Bim, culminating in apoptosis. This interpretation is supported by evidence that Bax or Bak knockdown, or ectopic expression of Mcl-1, Bcl-2, or Bcl-xL, significantly diminished INK128/ABT-737 lethality. Notably, INK128 decreased Bak binding to Bcl-xL and Bax binding to Bcl-2 and Bcl-xL, consistent with our previous findings with PI3K inhibitors.19 The mechanism underlying this phenomenon could reflect the observed increase in Bim binding to Bcl-2 and Bcl-xL which would displace Bax and Bak from these anti-apoptotic proteins. Together, these findings support a model wherein INK128/ABT-737 co-administration induces multiple perturbations that cooperatively induce cell death, including AKT inactivation, 4EBP1 dephosphorylation, Mcl-1 down-regulation, and Bax, Bak, and Bim release from all the major pro-apoptotic Bcl-2 members e.g., Bcl-2, Bcl-xL, and Mcl-1.
Combined INK128/ABT-737 treatment significantly increased cell death in primary blast specimens isolated from multiple AML patients. These samples exhibited diverse genetic aberrations including mutations in FLT3 (FLT3-ITD or FLT3-D835H), Kras, IDH2 (R140Q, R172K), and NPM1 (p.W288fs*12). Significant heterogeneity occurred in primary AML cell responses to this regimen, as with AML lines, in that some specimens responded to very low ABT-737 concentrations (e.g., 7.5 – 10 nM) while others required significantly higher concentrations (e.g., 500 nM), although the latter were equivalent to pharmacologically achievable concentrations of the clinically relevant ABT-263. These observations are consistent with previous reports from our and other groups.19,23 The molecular basis for this heterogeneity is unknown, but may stem from intrinsic disparities in Bcl-2 family protein expression as reflected by BH3-profiling.45 In this regard, AML cells with defined genetic backgrounds e.g., MLL translocation or IDH1/2 mutations, are highly sensitive to Bcl-2 inhibition.46,47 Differential sensitivity of cell lines and primary specimens to INK128 also occurred, perhaps reflecting dependence of a particular leukemic cell on the mTOR pathway and/or activation of compensatory survival pathways.
Interestingly, ABT-737/INK128 regimen activity was, if anything, more pronounced in CD34+/CD38−/CD123+ populations enriched in leukemia progenitor cells than in bulk blast populations. These findings are consistent with evidence that primitive blast progenitors may be particularly susceptible to Bcl-2/Bcl-xL inhibitors such as ABT-737,28 or to rapalogs9 when administered individually.
Despite pronounced resistance of Ba/F3 leukemia cells expressing FLT3-ITD to the Bcl-2 antagonist ABT-737, these cells were very sensitive to combined ABT-737/INK128 exposure. Furthermore, FLT3-ITD F691L mutants exhibiting marked resistance to FLT3 inhibitors e.g., sorafenib or quizartinib were also susceptible to combined treatment. The ability of INK128/ABT-737 to kill leukemia cells regardless of upstream FLT3 status may reflect disruption of critical cell survival factors downstream of this genetic aberration. In this regard, recent studies suggest that Mcl-1 represents a critical survival factor for FLT3-ITD leukemias.48 It is noteworthy that the INK128/ABT-737 regimen effectively killed leukemia cells in the presence of a protective microenvironment e.g., HS5 cells, suggesting that this regimen might circumvent bone marrow stromal cell protective effects, known to play an important role in leukemia cell survival. Microenvironmental factors have also been postulated to contribute to leukemic cell resistance to various therapies, including cytotoxic agents.49 Moreover, the PI3K/mTOR inhibitor BEZ235 combined with the HDACI panobinostat circumvented stromal cell resistance of diffuse large B-cell lymphoma cells through a mechanism involving Mcl-1 down-regulation and Bim up-regulation.50 Studies are currently underway to determine whether such mechanisms are operative in the present setting. Whatever the mechanism, the ability of the ABT-737/INK128 regimen to trigger cell death and/or diminish survival in AML cells co-cultured with stromal cells argues that this strategy may be able to bypass survival signals conferred by the microenvironment.
In vivo studies employing a mouse model bearing a systemic U937 xenograft exhibiting tet-inducible Bcl-2 and Bcl-xL dual knockdown demonstrated that disabling Bcl-2 and Bcl-xL markedly increased INK128 anti-leukemic activity and significantly prolonged survival. Furthermore INK128/ABT-737 co-administration significantly reduced leukemia growth and prolonged survival in a systemic xenograft mouse model without exerting significant toxicity. Such findings are concordant with evidence that both of these agents preferentially target neoplastic cells, and particularly leukemia stem cells.9,28 Significantly, several INK128/ABT-737 regimen actions observed in vitro were recapitulated in a subcutaneous xenograft model, including 4EBP1 dephosphorylation, and Mcl-1 down-regulation. The relative lack of toxicity in vivo is compatible with the observation that the INK128/ABT-737 regimen was minimally toxic to normal CD34+ cells at exposures that induced marked cell death in their leukemic counterparts. Taken together, these findings demonstrate that combined treatment with INK128 and BH3-mimetics such as ABT-737 robustly kills diverse myeloid leukemia cells in vitro as well as in vivo through a mechanism involving Mcl-1 down-regulation and Bax/Bak activation. They also raise the possibility that combining a dual mTORC1/mTORC2 inhibitor such as INK128 with a BH3-mimetic such as ABT-263 or ABT-199 may be particularly effective in the setting of AML.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by CA167708, CA142509, and Leukemia and Lymphoma Society of America award #6472-15.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Manzoli L, McCubrey JA. Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opin Investig Drugs. 2009; 18(9):1333–1349. [DOI] [PubMed] [Google Scholar]
- 2.Sujobert P, Bardet V, Cornillet-Lefebvre P, et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005;106(3):1063–1066. [DOI] [PubMed] [Google Scholar]
- 3.Chapuis N, Tamburini J, Cornillet-Lefebvre P, et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica. 2010;95(3):415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8(3):179–183. [DOI] [PubMed] [Google Scholar]
- 5.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwitkowski VE, Prowell TM, Ibrahim A, et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist. 2010;15(4):428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapuis N, Tamburini J, Green AS, et al. Perspectives on inhibiting mTOR as a future treatment strategy for hematological malignancies. Leukemia. 2010;24(10):1686–1699. [DOI] [PubMed] [Google Scholar]
- 9.Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005;106(13):4261–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shangary S, Johnson DE. Recent advances in the development of anticancer agents targeting cell death inhibitors in the Bcl-2 protein family. Leukemia. 2003;17(8):1470–1481. [DOI] [PubMed] [Google Scholar]
- 13.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30(18):3667–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. [DOI] [PubMed] [Google Scholar]
- 15.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. [DOI] [PubMed] [Google Scholar]
- 16.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. [DOI] [PubMed] [Google Scholar]
- 17.Ng SY, Davids MS. Selective Bcl-2 inhibition to treat chronic lymphocytic leukemia and non-Hodgkin lymphoma. Clin Adv Hematol Oncol. 2014;12(4):224–229. [PubMed] [Google Scholar]
- 18.Konopleva M, Polleya DA, Potluri J, et al. A Phase 2 Study of ABT-199 (GDC-0199) in Patients with Acute Myelogenous Leukemia (AML). Blood. 2014;124(21):118. [Google Scholar]
- 19.Rahmani M, Aust MM, Attkisson E, Williams DC, Jr, Ferreira-Gonzalez A, Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013; 73(4):1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahmani M, Dai Y, Grant S. The histone deacetylase inhibitor sodium butyrate interacts synergistically with phorbol myristate acetate (PMA) to induce mitochondrial damage and apoptosis in human myeloid leukemia cells through a tumor necrosis factor-alpha-mediated process. Exp Cell Res. 2002;277(1):31–47. [DOI] [PubMed] [Google Scholar]
- 21.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280(42):35217–35227. [DOI] [PubMed] [Google Scholar]
- 22.Infante JR, Tabernero J, Burris HA, et al. A phase I, open label, dose escalation study of an oral mammalian target of rapamycin inhibitor INK128 administered once daily in patients with advanced malignancies. Cancer Res. 2012;72(8 Suppl):Abstract nr 5588. [Google Scholar]
- 23.Zhang W, Ruvolo VR, Gao C, et al. Evaluation of apoptosis induction by concomitant inhibition of MEK, mTOR, and Bcl-2 in human acute myelogenous leukemia cells. Mol Cancer Ther. 2014; 13(7):1848–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jimeno A, Rudek MA, Kulesza P, et al. Pharmacodynamic-guided modified continuous reassessment method-based, dose-finding study of rapamycin in adult patients with solid tumors. J Clin Oncol. 2008;26(25):4172–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan R, Hogdal LJ, Benito JM, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4(3):362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willis SN, Chen L, Dewson G, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005; 19(11):1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maiso P, Liu Y, Morgan B, et al. Defining the role of TORC1/2 in multiple myeloma. Blood. 2011;118(26):6860–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006; 10(5):375–388. [DOI] [PubMed] [Google Scholar]
- 29.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67(2):782–791. [DOI] [PubMed] [Google Scholar]
- 31.Zhang W, Konopleva M, Ruvolo VR, et al. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia. 2008; 22(4):808–818. [DOI] [PubMed] [Google Scholar]
- 32.Schwickart M, Huang X, Lill JR, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463(7277):103–107. [DOI] [PubMed] [Google Scholar]
- 33.Jin L, Tabe Y, Kojima K, et al. PI3K inhibitor GDC-0941 enhances apoptotic effects of BH-3 mimetic ABT-737 in AML cells in the hypoxic bone marrow microenvironment. J Mol Med. 2013;91(12):1383–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaillant F, Merino D, Lee L, et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell. 2013;24(1):120–129. [DOI] [PubMed] [Google Scholar]
- 35.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. [DOI] [PubMed] [Google Scholar]
- 36.Janes MR, Vu C, Mallya S, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2013; 27(3):586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faber AC, Coffee EM, Costa C, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by sup pressing MCL-1. Cancer Discov. 2014;4(1): 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Preuss E, Hugle M, Reimann R, Schlecht M, Fulda S. Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J Biol Chem. 2013; 288(49):35287–35296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tamburini J, Green AS, Bardet V, et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood. 2009; 114(8):1618–1627. [DOI] [PubMed] [Google Scholar]
- 40.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21(6):749–760. [DOI] [PubMed] [Google Scholar]
- 41.Mills JR, Hippo Y, Robert F, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci USA. 2008;105(31):10853–10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller A, Zang C, Chumduri C, Dorken B, Daniel PT, Scholz CW. Concurrent inhibition of PI3K and mTORC1/mTORC2 overcomes resistance to rapamycin induced apoptosis by down-regulation of Mcl-1 in mantle cell lymphoma. Int J Cancer. 2013; 133(8):1813–1824. [DOI] [PubMed] [Google Scholar]
- 43.Hsieh AC, Costa M, Zollo O, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17(3):249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morales AA, Kurtoglu M, Matulis SM, et al. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood. 2011;118(5):1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12(2):171–185. [DOI] [PubMed] [Google Scholar]
- 46.Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015; 21(2):178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niu X, Wang G, Wang Y, et al. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia. 2014;28(7):1557–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasper S, Breitenbuecher F, Heidel F, et al. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J. 2012;2(3):e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2014; 25(4);198–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rahmani M, Aust MM, Benson EC, Wallace L, Friedberg J, Grant S. PI3K/mTOR inhibition markedly potentiates HDAC inhibitor activity in NHL cells through BIM- and MCL-1-dependent mechanisms in vitro and in vivo. Clin Cancer Res. 2014;20(18):4849–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]