


Abstract
The protein–DNA interactions between transcription factors and transcription factor binding sites are essential activities in gene regulation. To decipher the binding codes, it is a long-standing challenge to understand the binding mechanism across different transcription factor DNA binding families. Past computational learning studies usually focus on learning and predicting the DNA binding residues on protein side. Taking into account both sides (protein and DNA), we propose and describe a computational study for learning the specificity-determining residue-nucleotide interactions of different known DNA-binding domain families. The proposed learning models are compared to state-of-the-art models comprehensively, demonstrating its competitive learning performance. In addition, we describe and propose two applications which demonstrate how the learnt models can provide meaningful insights into protein–DNA interactions across different DNA binding families.
INTRODUCTION
Given a protein sequence, we are interested in which residues on the given protein sequence are important for protein functions. Identifying these residues would be very helpful in understanding the protein. Assuming functional residues are evolutionarily conserved, Casari et al. have proposed a high-dimensional projection method to identify different groups of residue conservation patterns from related species sequences to identify functional residues (1). Lichtarge et al. have proposed an evolutionary trace method (calculating conservations at different levels of a gene tree) to identify DNA-binding surfaces of the nuclear hormone receptors from homologous sequences (2).
In the context of DNA-binding proteins (3), each residue can be predicted and labeled as two class outputs (either DNA binding or neutral). Several approaches have been proposed to solve this classification problem: Ahmad et al. have implemented neural network approaches to predict DNA binding residues on amino acid sequences. Position specific scoring matrices generated by PSI-BLAST have been adopted in the first approach (4), whereas sequence composition and solvent accessibility information have also been adopted in another approach (5). Using three sequence features, a SVM model has been trained and tested by Wang et al. (6). Secondary structure element alignments have been incorporated into SVM models to perform predictions by Chu et al. (7). The electrostatic potential and curvature information of protein structures have been used and reported (8). Gaussian network models have been applied to model protein structures to perform predictions by Ozbek et al. (9). A random forest method using hybrid features has been adopted and reported by Wu et al. (10). An ensemble method has been proposed by Hwang et al. (11). A neighboring residue network based score has been proposed to improve the prediction by Miao and Westhof (12).
Exploiting the assumption that different DNA-binding specificities exist among paralogous sequences, Mirny and Gelfand have proposed a mutual information method to compute statistical significance for distinguishing specificity-determining residues from the other residues of bacterial transcription factors (13). For eukaryotes, Donald and Shakhnovich argued that sequence data were scarce. They proposed a single-linkage hierarchical clustering method on all the available homologous sequences based on sequence similarity (assuming the correlation between sequence similarities and functional similarities). They found that their method performed better than the previous method and the evolutionary trace method on the basic leucine zippers and nuclear receptors families (14).
Nonetheless, the past studies are usually devoted to learn and predict the DNA binding residues based on amino acid sequences only, ignoring their DNA counterpart sequences (15). This may be partly due to the high resolution protein–DNA binding data scarcity and the difficulty in handling the expanded search space. However, such bottlenecks have been alleviated in recent years. Specifically, the modern high throughput biotechnology can enable us to have sufficient data to look at both sides (protein and DNA sides) simultaneously. For example, Wong et al. have proposed a data mining framework to discover the protein motifs and DNA motifs in a coupled manner (16) as well as extension works to handle sequence degeneracy (17) and binding combinatorics (18). Mahony et al. have proposed incorporating mutual information into analyzing residue–nucleotide binding on aligned parts of DNA-binding domain (DBD) sequences of zinc finger, Homeodomain and bHLH DBD families (19). A recognition model to predict DNA motifs from Homeodomain protein sequences has also been proposed (20).
In this work, we aim at analyzing and building learning models to learn pair-wise specificity-determining residue–nucleotide interactions for different DNA-binding families.
MATERIALS AND METHODS
Collecting DBD family data
It has been found that protein–DNA binding interactions are diverse in binding modes (21,22). Different DNA motifs could be bound by the same DNA-binding protein. Therefore, we have tried to collect as much interaction data as possible to capture those diverse binding mechanisms. We have selected the latest protein–DNA binding interaction database, CISBP (23), which is the most comprehensive database to the best of our knowledge. We have collected the entire experimentally verified pairs of human DBD sequences and the corresponding DNA motif matrices from CISBP (v0.71) (23). For each DBD sequence, it is possible to have multiple motif matrices measured by different biotechnologies (e.g. ChIP-Exo, ChIP-Seq, ChIP-Chip, PBM and SELEX). To be unbiased for each DBD sequence, STAMP is used to combine them into a consensus motif matrix with the default setting (24). After that, we have searched and limited our study to the human DNA-binding domain families which have at least 10 pairs in CISBP (v0.71) as shown in Supplementary Table S1.
For each of those DNA-binding domain families, we used MUSCLE (25) and STAMP (19) to align the DBD sequences and DNA motif matrices, respectively (with the default setting). The resultant alignment sequence logos can be found in Supplementary Figures S2 and S3. We construct a Spearman rank correlation heat map for the pair-wise residue–nucleotide co-variations between the aligned DBD amino acid sequences and the corresponding DNA motif matrices for each DBD family as shown in Step 5 of Supplementary Figure S1. Its implementation details can be found in Supplementary Materials.
Nonetheless, such a co-variation analysis has several limitations; for instance, if a position is well conserved, that position should be somewhat functionally important in protein–DNA binding. Nonetheless, it cannot be captured by this kind of co-variation analysis since the position is not varied at all. Furthermore, the physicochemical properties (e.g. residue polarity) have not been taken into account. To address the issues, we proceed to collect the entire available protein–DNA binding complex structures and build inference models to take all those factors into account for learning pair-wise residue–nucleotide interactions across different DBD families.
Collecting protein–DNA complex structures
To train (or build) learning models for different DBD families, we collected the entire protein–DNA complex structures from RCSB PDB in May 2013. CD-HIT (with the default setting) is used to remove sequence redundancy among the entire protein chains in the structures (26), resulting in a set of 833 protein chains as well as a set of 1469 protein–DNA binding pairs.
Among them, we identify and extract the annotated DBD sequences of the DBD families listed in Supplementary Table S1. The total numbers of DBD sequences extracted from PDB are summarized in Supplementary Table S2. In addition, we also extract the DNA-binding site sequences in the same protein–DNA complex structures. After reverse complements are identified and incorporated, we have obtained the binding pairs of DBD sequences and DNA sequences for different DBD families. The statistics are tabulated in Supplementary Table S2. We followed the common definition of previous studies that a residue is called a DNA-binding residue if any of its atoms fall within a cutoff distance of 3.5 Å from any of DNA molecules’ atoms in at least one known protein–DNA binding complex (27,28). A residue–nucleotide pair is called a specificity-determining residue–nucleotide interaction pair if any of its residue's side-chain atoms fall within a cutoff distance of 3.5 Å from any of its nucleotide's base atoms in at least one known protein–DNA binding complex (16).
Training and testing procedures
Based on the CISBP data and PDB data, we can train and test models for learning and predicting the specificity-determining residue–nucleotide interactions for each DBD family. The overall approach is summarized in Figure 1, which can be divided into two phases: training and testing.
Figure 1.

Training and Testing Classification Models for Predicting Residue-Nucleotide Interactions on protein–DNA binding sequence pairs. Description can be found on the main text.
Training procedure
For the training procedure as shown in Figure 1, we adopt the protein–DNA binding sequence pairs from PDB and the corresponding DBD family sequence alignments in CISBP to build feature vectors as the inputs to train models. On the other hand, we adopt the structural information from PDB (i.e. three-dimensional residue–nucleotide binding information) as the gold-standard outputs to train models. (Steps A and B) For each DBD family (Domain X in this figure), we search through the PDB and obtain the known protein–DNA binding pairs from PDB for training models. (Step C) CD-HIT is run to remove protein sequence redundancy. (Step D) The structure of each training protein–DNA binding pair is measured to reveal the residue-nucleotide interactions. (Step E) The interaction information is isolated from the feature building steps later for sequence-only-input training. (Steps F, G and H) On the other hand, we also query the CISBP to retrieve the domain X family sequence alignments. (Step I) Each training protein–DNA binding sequence pair is aligned to the family sequence alignment. (Step J) Given the resultant alignment, feature vectors are calculated at each possible interaction position pair. (Steps K and L) Given the feature vectors as well as the measured structural binding labels at each possible interaction position, standard classification techniques are used to train an inference model for domain X (i.e. a random forest classification model with 100 decision trees using 30 random features (from the WEKA software) is used in this study).
Testing procedure
For the testing procedure as shown in Figure 1, we are not given any training protein–DNA binding sequence pair with known structural interactions. Instead, we are just given an input pair of protein sequence and DNA sequence with known DBD (domain X in Figure 1) as well as CISBP (However, in practice, we also get that input sequence pair from PDB and discard its structural information which is reserved for validation in later steps). The model testing part is to use the corresponding trained model to predict the possible residue–nucleotide interactions on the input pair. (Steps 1 and 2) The protein sequence of the input protein–DNA binding sequence pair is scanned by Pfam to predict which DBD domain that the protein sequence belongs to (Domain X in this example). (Steps 3, 4 and 5) CISBP is then queried to retrieve the domain X family alignment. (Step 6) The input protein–DNA binding sequence pair is aligned to its family (domain X in Figure 1) alignment. (Step 7) Feature vectors are built from the resultant alignment at each possible interaction position of the input protein–DNA binding sequence pair. (Step 8) The feature vectors are inputted into the trained model of domain X. (Step 9) The trained model predicts which possible interaction is truly a protein–DNA binding interaction.
Building feature vectors
Given an input pair of protein sequence and DNA sequence, we build a feature vector at each possible residue–nucleotide interaction. Mathematically, if the input protein sequence is of length laa and the input DNA sequence is of length ldna, the total number of possible residue–nucleotide interaction is laa × ldna, resulting in laa × ldna feature vectors. Based on the feature vectors, we use the trained model to predict which possible residue–nucleotide interaction (feature vector) is actually binding (positive class).
For each possible residue–nucleotide interaction feature vector, we compute several features which are essential for the binding prediction. The features are listed in Supplementary Table S3. In summary, we calculate the physicochemical properties (e.g. residue polarity), sequence context (e.g. the 2nd preceding residue), sequence co-variation (e.g. the correlation between the current residue and the current nucleotide in the family alignment from CISBP), conservation (e.g. entropy of the current residue's aligned position in the family alignment from CISBP), positional information (e.g. the aligned position of the current nucleotide in the family alignment from CISBP) and mixed features (e.g. the average residue mass at the aligned position of the current residue in the family alignment from CISBP).
Running time
Both of the training and testing procedures are of polynomial time complexity; details of which can be found in Supplementary Materials. In practice, the whole training and testing procedure are implemented in Java programming language. The computing equipment is a dedicated Intel Xeon W3550 server with 12 GB memory. For the protein side prediction benchmarking, the feature building and evaluation procedure took 1.5 h and 2 h, respectively. For the prediction benchmarking on both sides (protein and DNA), the feature building and evaluation procedure took 4 h and 9 h, respectively. If we take into account the other methods’ benchmarking, the overall computing time took about 1 week.
RESULTS
To validate the learning approach outlined in Figure 1 we tested it using leave-one-out cross-validation: for each DBD family, we leave out one pair of DBD sequence-DNA sequence from PDB for model testing and apply the rest for model training. It is repeated until all pairs have been left out once; for instance, we have 22 pairs of DBD Sequence-DNA Sequence Pairs from the Homeodomain DBD family in PDB. In the first round, the 1st pair is held out and the 2nd–22nd pairs are used for training. In the second round, the 2nd pair is held out and the 1st, 3rd–22nd pairs are used for training. The procedure is repeated until all pairs have been held out once.
Predicting on protein side only
To compare our prediction approach to the other proposed methods, we need to first reduce our prediction problem back to the classic problem, i.e. predicting DNA-binding residues on input protein sequences. We first train and test our method only on protein sequences first, ignoring the DNA sequences. The entire features which are linked to DNA in Supplementary Table S3 are discarded. Only the protein features are used for training and testing the learning models (denoted as ‘ours’ on figures). On the other hand, we have also re-run our approach using both the protein features and the discarded DNA features. For each residue position, the maximal prediction score is taken for all observed nucleotides on the corresponding DNA side (denoted as ‘ours-both’ on figures).
Feature ranking on protein features
We have ranked the protein features on the protein sequences of the PDB data collected using information gain as tabulated in Supplementary Table S4. The top feature is the entropy of the current residue's aligned position in the family alignment using polarity symbols (polarity_entropy). Comparing to the polarity of the current residue (polarity, ranked 25th), this top feature is significantly ranked higher than the polarity feature. It may indicate that the conservation of polarity is a key determining factor for predicting DNA binding residues on protein sequences. The second top feature is the average pH of the current residue's aligned position in the family alignment (avg_Ph). Similar to the previous case, its pH feature (pH) is ranked (11th) lower than itself because the pH evolutionary conservation in its DBD family alignment is more informative than the pH observed at the current residue. The third top feature is the occurring fraction of non-gap residues at the current residue's aligned position in the family alignment (aa_obsCount), it indicates that the evolutionary presence of the current residue's aligned position in the family alignment can be used to predict the DNA-binding residues on DBD protein sequences. Last but not least, the fourth top feature is the aligned position of the current residue (aaMSAind). It can be observed that our proposed method can adopt that feature to take advantage of the sequence position information of each family-wise-aligned DNA-binding sequence.
Comparing to sequence-based methods
For each DBD family, we have written network scripts to send the testing DBD sequences to the BindN web-server, BindN+ web-server and DISIS web-server for obtaining their predictions with the default settings suggested. Briefly, BindN is a support vector machine classifier using physicochemical sequence features (6). BindN+ is an extension of BindN which also takes in account the evolutionary information (29). DISIS is also a support vector machine classifier which considers evolutionary information, predicted secondary structural information and the neighboring residue information (28). The Receiver Operating Characteristic (ROC) and precision-recall (PRC) curves for the entire DBD families are plotted and shown in Figure 2 and Supplementary Figure S4. It can be observed that our proposed method using protein-only features (ours) and that using both-protein–DNA features (ours-both) have a competitive edge over the other sequence-based methods at low false positive rates.
Figure 2.
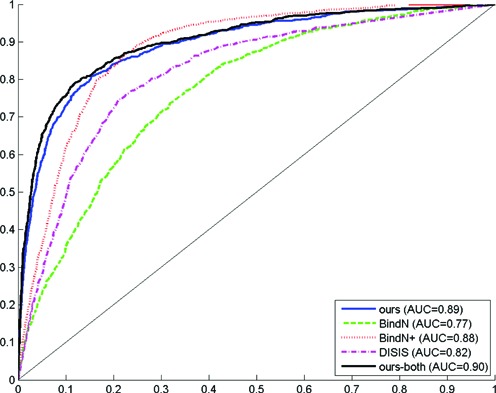
Receiver Operating Characteristic (ROC) curves for our proposed methods (in Blue and Black), BindN (in Green), BindN+(in Red) and DISIS (in Violet) on the entire DBD families.
Comparing to structural methods
Although it is unfair for our proposed methods to be compared to the structural method since our methods are not given any structural information (e.g. three-dimensional coordinates of atoms) during model testing (p.s. three-dimensional information has been adopted as class labels during model training), it may still be interesting to check how well our proposed methods can be compared to the structural methods for model testing. Thus we have also written network scripts to send the structural information of each testing DBD sequence to the DBD-Hunter web-server and DISPLAR web-server with the default settings suggested. Briefly, DBD-Hunter is a structural template matching method using statistical potential (30) while DISPLAR is a neural network method taking into account the evolutionary information, the solvent accessibilities of each residue, as well as the spatial neighbors (31). Since DBD-Hunter is a template matching method, we seek to calculate how often a residue is predicted as DNA-binding among the templates suggested by DBD-Hunter. We denote DBDhunter(k) as the DBD-Hunter program only using the top k templates ranked by TM scores while DBDhunter is denoted as the DBD-Hunter program using the entire templates suggested. On the other hand, DISPLAR just outputs discrete results without any confidence number. Thus we assigned 1 and 0 to the residue predicted as DNA-binding and Not-DNA-binding by DISPLAR. 0.5 is assigned to the residue which cannot be predicted by DISPLAR. The ROC and PRC curves for the entire DBD families are also plotted and shown in Figure 3 and Supplementary Figure S5.
Figure 3.
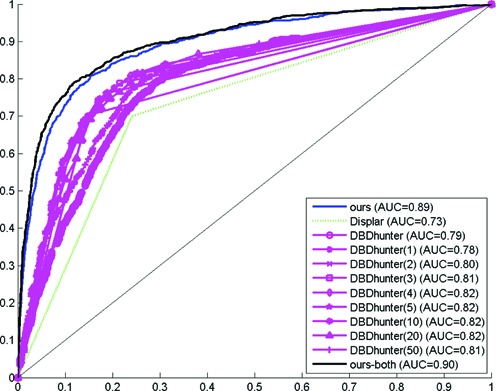
ROC curves for our proposed method (in Blue and Black), DBD-Hunter (in Violet), DISPLAR (in Green) on the entire DBD families.
It can be observed that our proposed methods have a competitive edge over the structural methods even though our proposed methods are not given any structural information (i.e. spatial information) during model testing. Nonetheless, we note that such a performance degradation of DBD-Hunter may be due to the fact that its scoring system is not consistent across different DBD families. In other words, if a residue is predicted as DNA-binding in half of the templates in the Homeodomain DBD family, its prediction confidence is not necessarily equivalent to that of a residue predicted as DNA-binding in half of the templates in the bHLH DBD family. Thus, it may be beneficial for DBD-Hunter to compare its results with the others on individual DBD family data sets, although the sequence-based methods do not suffer from this issue. As illustrative examples, we depict the ROC and PRC curves for the top DBD families (i.e. bHLH and Homeodomain) in Supplementary Figures S6, S7, S8 and S9.
Interestingly, it can be observed that the performance of DBD-Hunter is improved if we limit the input data to a specific DBD family. Furthermore, even after we have allowed DBD-Hunter to use various number of templates, it can be observed that our proposed methods are still comparable to the DBD-Hunter which is the best available structural method for predicting DNA-binding interactions to our knowledge, although our proposed methods are not given any structural information during model testing (p.s. three-dimensional information has been adopted as class labels during model training). We attribute the good performance to three major reasons (i) comprehensive training data from CISBP and PDB (ii) elaborated input feature building, and (iii) state-of-the-art ensemble classification model (i.e. Random Forest which is scale-free and efficient to train).
Predicting on both protein and DNA sides
Having demonstrated our proposed methods are competitive among the state-of-the-art methods on the classic problem, we proceed to verify our approach on the new problem: Given a pair of known DBD protein sequence and DNA sequence, we seek to learn and predict the specificity-determining residue–nucleotide interactions between the known DBD protein sequence and DNA sequence.
Feature ranking on all features
We have ranked all features on the PDB data collected using information gain as tabulated in Supplementary Table S5. It is not surprising that the top feature is the entropy of the current residue's aligned position in the family alignment (aa_entropy) because DNA-binding residues are supposed to undergo negative selection during its evolutionary history, resulting in a strong conservation signal (e.g. low entropy). The 2nd–6th features are similar to what we have discussed in the previous feature ranking section. The most interesting observation is that, among the top 10 features, only 2 features account for both sides (i.e. aa_nt (the current residue and nucleotide pair) and aa_presence_total (the sum of the correlations between the current residue's aligned position column profile and the entire nucleotide column profiles in family alignment)), indicating that the residue–nucleotide binding pairs are largely determined by the protein information. Surprisingly, the mutual information feature (MI) is only ranked at the middle of the list (44th), reflecting that MI-based studies may not have enough distinguishing power for learning. To have a broad view on such observation, we have calculated the total and average information gains of different types of features as shown in Figure 4. Again, we can observe that most of the information gains are given by the protein features which average information gain is higher than the other feature types.
Figure 4.

(A) Total information gain distribution of different feature types. (B) Average information gains of different feature types. It can be observed that protein features constitutes most of the information gains.
Performance on different DBD families
We have used the leave-one-out cross-validation approach aforementioned to test our approach. The results are depicted in Figure 5 and Supplementary Figure S10.
Figure 5.
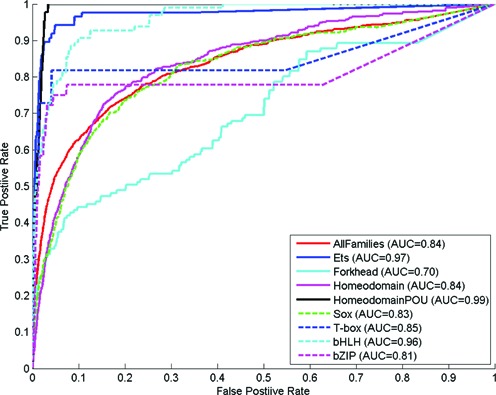
Receiver Operating Characteristic (ROC) curves for our proposed method on the DBD family data. Each line corresponds to a DBD family.
It can be observed that the performance of our proposed method varies on different families. It performed very well for the ETS family. On the other hand, it performed the worst for the Forkhead family.
The ETS family is a cancer-related domain which is highly conserved (32). To illustrate our predictions on ETS family, we have selected the crystal structure of the protein–DNA complex of human PDEF ETS domain bound to the prostate specific antigen regulatory site (PDB code: 1YO5) as an example. Our top five predictions are highlighted in colors in Figure 6. We can observe that our method is capable of predicting the binding cores of PDEF. The only false positive is the residue–nucleotide pair which are very proximal to each other (4.06 Å) but cannot exceed the 3.5 Å threshold.
Figure 6.
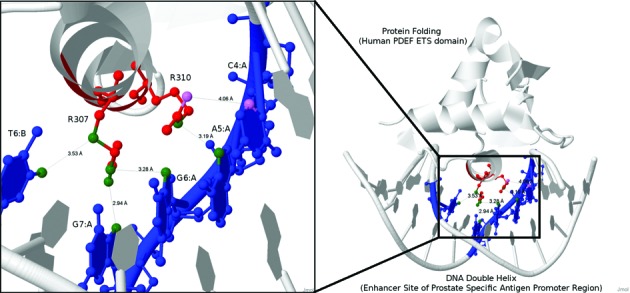
Crystal structure of the protein–DNA complex of human PDEF ETS domain bound to the prostate specific antigen regulatory site (PDB code: 1YO5). Our top five predictions are highlighted in colors. The red and blue molecules denote the DNA binding residues and nucleotides predicted by our method, respectively. In particular, the binding atoms within our predicted DNA binding residue–nucleotide pairs are highlighted in green and violet, indicating true positives and false positives, respectively.
In contrast, the Forkhead family is a transcription factor family known to be involved in early developmental decisions of cell fates during embryogenesis (33). Especially, it has just been recently characterized that the Forkhead family has diverged into different subfamilies with different DNA-binding specificities (34). As such, it is not surprising that our proposed method cannot work well for the Forkhead family data which has not been divided into different sub-families very well under the existing annotation system.
Predictions at low false positive rates
In practice, we are especially interested in the prediction accuracies at low false positive rates. Thus, we have also examined and plotted the previous benchmark ROC curves at low false positive rates as shown in Figure 7. It can be observed that our method can still show its own competitive edges over the other methods. In particular, different performance is observed on different DNA-binding families because of different training data set availabilities and binding mechanism complexities; for instance, our approach performs very well on the cancer-related DNA-binding family (ETS) because its sequences are highly conserved, resulting in high-quality training and thus testing performance (AUC=0.97).
Figure 7.
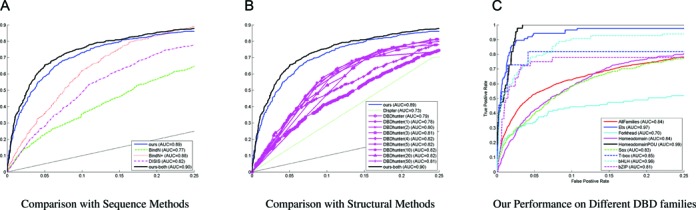
(A and B) Performance comparison of our method with the other methods at low false positive rates. (C) Performance comparison of our method on different families for the proposed problem at low false positive rates.
Using different classification models
On the other hand, we are also interested in the learning performance of Random Forest (discriminative classifier), comparing to the other classification methods. We have re-run the previous computational experiments using Naive Bayes (generative classifier) and Adaboost (ensemble classifier) as depicted in Supplementary Figures S11 and S12, respectively. Comparing their curves (Supplementary Figures S11 and S12) with the original curves of Random Forest (Figures 2, Supplementary Figure S4, 3, Supplementary Figure S5, 5 and Supplementary Figure S10), it can be observed that Random Forest is the best option among the three classification models for this study.
APPLICATIONS
Connecting to recognition model
Combined with the existing recognition models to predict DNA motifs (e.g. PreMoTF (20)), our proposed method can pave a new direction in protein–DNA binding prediction: Given a protein sequence of known DBD domain, we could predict its DNA motif using its corresponding domain recognition model. After that, the proposed method here can be used to predict the interacting pairs of residues and nucleotides between the protein and the predicted DNA motif.
To demonstrate the concept, we have selected the NMR solution structure of the Homeodomain of Pitx2 in complex with a TAATCC DNA binding site (PDB code: 2LKX) as an example. First, we ignore the bound DNA sequence as well as the three-dimensional structural information. Only the protein sequence is submitted to PreMoTF. PreMoTF then predicts and outputs a DNA motif matrix which is believed to be bound by the protein sequence. The predicted DNA motif matrix can be found in Figure 8. We scan the bound DNA sequence using the predicted DNA motif matrix and locate the DNA motif position. Our proposed method is then applied to predict the specificity-determining residue–nucleotide binding pairs between the protein sequence and the DNA motif. After that, we map our sequence-based predictions back to its actual three-dimensional structure as shown in Figure 8. It can be observed that our proposed method combined with PreMoTF can predict the residue–nucleotide binding pairs accurately.
Figure 8.
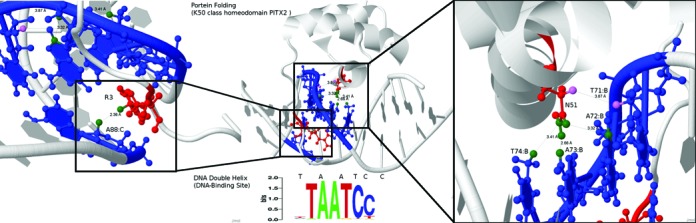
NMR solution structure of the Homeodomain of Pitx2 in complex with a TAATCC DNA binding site (PDB code: 2LKX). Our top five predictions are highlighted in colors. The red and blue molecules denote the DNA binding residues and nucleotides predicted by our method, respectively. In particular, the binding atoms within our predicted DNA binding residue–nucleotide pairs are highlighted in green and violet, indicating true positives and false positives, respectively. The DNA motif position predicted by PreMoTF is indicated by the sequence logo.
DNA motif recognition
Since our method takes a binding pair of known DBD protein sequence and DNA sequence as an input, we can enumerate the entire possible DNA sequences given a protein sequence to observe which DNA sequence has its nucleotides predicted to be bound by the protein more frequently than the others. As a result, we can obtain a predicted score for each possible DNA sequence given a protein. Such a predicted score can be used as a delegate of protein–DNA binding affinity for ranking the entire possible DNA sequences. It is similar to the Protein Binding Microarray (PBM) technology but in silico. If we further post-process the predicted scores to build a DNA motif matrix, it is also similar to the DNA motif recognition model described in the previous section. Nonetheless, this application, similar to PBM, can have a higher resolution map of DNA motifs (score for each possible k-mer) than DNA motif matrix models which assume positional independence.
As an illustrative example, we have selected the bHLH DBD domain protein sequence of the transcription factor E2-alpha (UniProt code: P21677; UniPROBE code: Tcfe2a). With its bHLH domain protein sequence fixed, we generate the entire possible DNA 8-mers, resulting in 65536 (48) binding pairs of the bHLH domain protein sequence and DNA 8-mer. For each binding pair, we feed it into the prediction model trained on bHLH DBD binding pairs as described in the previous section. The maximum of the prediction model score is taken as the predicted score for each binding pair. Thus, we obtain a predicted score for each possible DNA 8-mer from its corresponding binding pair. Consistent with the previous study by Zhao and Stormo (35), we pick the top 25 scoring 8-mers and compare them with the previous study's top 25 8-mers with the highest median binding intensities measured by PBM (36). It can be observed that our top 25 scoring 8-mers share similar patterns CANNTG with those measured by PBM as shown in Supplementary Table S6. As a summary, we also build a sequence logo from our top 25 scoring 8-mers as shown in Figure 9. It can be observed that our sequence logo is quite similar to the logos measured and reported by the previous PBM study as shown in Supplementary Figure S13 (36).
Figure 9.
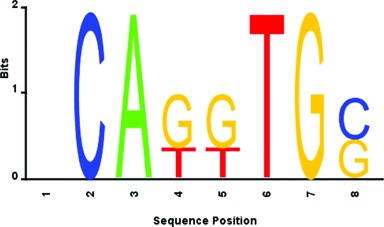
Sequence logo obtained by our prediction enumeration on the bHLH DBD domain of the transcription factor E2-alpha (UniProt code: P21677, UniPROBE code: Tcfe2a).
DISCUSSION
In this study, we have proposed and described a computational study on learning specificity-determining residue–nucleotide interactions. Our proposed solution is to take advantage of the vast amount of protein–DNA binding sequence pairs from CISBP to learn inference models on PDB data for different DNA-binding families.
To study its learning performance, we have conducted comprehensive analysis and case studies. (i) We adapted our proposed methods to the classic problem (predicting DNA binding residues on protein side only), on which we have compared our methods with the state-of-the-art methods. The results reveal that our methods are competitive among the sequence-based methods at low false positive rates. Furthermore, our method shows comparable results with DBDhunter which is still the best available structural method for the classic problem. In addition, the proposed approach using both DNA and protein features performs better than that using protein features alone. (ii) Having demonstrated our method competitiveness, we proceed to estimate the proposed method's performance on the new problem (predicting specificity-determining residue–nucleotide pairs). It can be observed that the proposed method performs differently across different DBD families. In particular, it works well for POU and ETS DBD families (AUC = 0.99 and AUC = 0.97, respectively). (iii) To shed light on that, we have studied our predictions on the PDEF protein. From the crystal structure of the protein–DNA complex of human PDEF ETS domain bound to the prostate specific antigen regulatory site (PDB code: 1YO5), we observe that our top predictions can reflect the experimentally verified residue–nucleotide binding pairs. (iv) To apply the proposed method, we have also discussed and implemented two additional potential applications. For the first application, we have run PreMoTF to predict a DNA motif from a Homeodomain protein sequence and used our proposed method to predict residue–nucleotide binding pairs between the Homeodomain DBD protein sequence and the predicted DNA motif instance. For the second application, we have used our proposed method to predict and rank which DNA 8-mer is bound by a bHLH DBD protein sequence in silico.
In light of the above, we believe our proposed computational study is capable of learning specificity-determining residue–nucleotide binding pairs at competitive levels (AUC = 0.70–0.99). In the future, we can foresee that our proposed approach will become very useful as massive protein–DNA interaction data are being generated using next generation sequencing technology.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank the anonymous reviewers, Anthony Booner, Rui Kuang, Gary Bader and John Parkinson for their comments. The authors would also like to thank Bulyk Lab for making their PBM data publicly available. The authors would also like to thank Timothy R. Hughes and his lab members for making their CISBP database publicly available. Amazon Web Service (AWS) in Education Research Grant is acknowledged by Ka-Chun Wong.
FUNDING
City University of Hong Kong [Project no. 7200444/CS to K.W.]; Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery [327612 to Z.Z.]. Funding for open access charge: Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery [327612 to Z.Z.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Casari G., Sander C., Valencia A. A method to predict functional residues in proteins. Nat. Struct. Biol. 1995;2:171–178. doi: 10.1038/nsb0295-171. [DOI] [PubMed] [Google Scholar]
- 2.Lichtarge O., Bourne H.R., Cohen F.E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 3.Wong K.C., Li Y., Peng C., Zhang Z. SignalSpider: probabilistic pattern discovery on multiple normalized ChIP-Seq signal profiles. Bioinformatics. 2015;31:17–24. doi: 10.1093/bioinformatics/btu604. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad S., Sarai A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinformatics. 2005;6:33. doi: 10.1186/1471-2105-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmad S., Gromiha M.M., Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. doi: 10.1093/bioinformatics/btg432. [DOI] [PubMed] [Google Scholar]
- 6.Wang L., Brown S.J. Prediction of DNA-binding residues from sequence features. J. Bioinform. Comput. Biol. 2006;4:1141–1158. doi: 10.1142/s0219720006002387. [DOI] [PubMed] [Google Scholar]
- 7.Chu W.Y., Huang Y.F., Huang C.C., Cheng Y.S., Huang C.K., Oyang Y.J. ProteDNA: a sequence-based predictor of sequence-specific DNA-binding residues in transcription factors. Nucleic Acids Res. 2009;37:396–401. doi: 10.1093/nar/gkp449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuchiya Y., Kinoshita K., Nakamura H. PreDs: a server for predicting dsDNA-binding site on protein molecular surfaces. Bioinformatics. 2005;21:1721–1723. doi: 10.1093/bioinformatics/bti232. [DOI] [PubMed] [Google Scholar]
- 9.Ozbek P., Soner S., Erman B., Haliloglu T. DNABINDPROT: fluctuation-based predictor of DNA-binding residues within a network of interacting residues. Nucleic Acids Res. 2010;38:W417–W423. doi: 10.1093/nar/gkq396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J., Liu H., Duan X., Ding Y., Wu H., Bai Y., Sun X. Prediction of DNA-binding residues in proteins from amino acid sequences using a random forest model with a hybrid feature. Bioinformatics. 2009;25:30–35. doi: 10.1093/bioinformatics/btn583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hwang S., Gou Z., Kuznetsov I.B. DP-Bind: a web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics. 2007;23:634–636. doi: 10.1093/bioinformatics/btl672. [DOI] [PubMed] [Google Scholar]
- 12.Miao Z., Westhof E. Prediction of nucleic acid binding probability in proteins: a neighboring residue network based score. Nucleic Acids Res. 2015;43:5340–5351. doi: 10.1093/nar/gkv446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirny L.A., Gelfand M.S. Using orthologous and paralogous proteins to identify specificity-determining residues in bacterial transcription factors. J. Mol. Biol. 2002;321:7–20. doi: 10.1016/s0022-2836(02)00587-9. [DOI] [PubMed] [Google Scholar]
- 14.Donald J.E., Shakhnovich E.I. Predicting specificity-determining residues in two large eukaryotic transcription factor families. Nucleic Acids Res. 2005;33:4455–4465. doi: 10.1093/nar/gki755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong K.C., Chan T.M., Peng C., Li Y., Zhang Z. DNA motif elucidation using belief propagation. Nucleic Acids Res. 2013;41:e153. doi: 10.1093/nar/gkt574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung K.S., Wong K.C., Chan T.M., Wong M.H., Lee K.H., Lau C.K., Tsui S.K. Discovering protein-DNA binding sequence patterns using association rule mining. Nucleic Acids Res. 2010;38:6324–6337. doi: 10.1093/nar/gkq500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan T.M., Wong K.C., Lee K.H., Wong M.H., Lau C.K., Tsui S.K., Leung K.S. Discovering approximate-associated sequence patterns for protein-DNA interactions. Bioinformatics. 2011;27:471–478. doi: 10.1093/bioinformatics/btq682. [DOI] [PubMed] [Google Scholar]
- 18.Wong K.C., Peng C., Wong M.H., Leung K.S. Generalizing and learning protein-DNA binding sequence representations by an evolutionary algorithm. Soft Comput. 2011;15:1631–1642. [Google Scholar]
- 19.Mahony S., Auron P.E., Benos P.V. Inferring protein-DNA dependencies using motif alignments and mutual information. Bioinformatics. 2007;23:297–304. doi: 10.1093/bioinformatics/btm215. [DOI] [PubMed] [Google Scholar]
- 20.Christensen R.G., Enuameh M.S., Noyes M.B., Brodsky M.H., Wolfe S.A., Stormo G.D. Recognition models to predict DNA-binding specificities of homeodomain proteins. Bioinformatics. 2012;28:i84–i89. doi: 10.1093/bioinformatics/bts202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Narasimhan K., Lambert S.A., Yang A.W., Riddell J., Mnaimneh S., Zheng H., Albu M., Najafabadi H.S., Reece-Hoyes J.S., Fuxman Bass J.I., et al. Mapping and analysis of Caenorhabditis elegans transcription factor sequence specificities. Elife. 2015;4 doi: 10.7554/eLife.06967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hume M.A., Barrera L.A., Gisselbrecht S.S., Bulyk M.L. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2014;43:D117–D122. doi: 10.1093/nar/gku1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahony S., Benos P.V. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007;35:W253–W258. doi: 10.1093/nar/gkm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 27.Ahmad S., Gromiha M.M., Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. doi: 10.1093/bioinformatics/btg432. [DOI] [PubMed] [Google Scholar]
- 28.Ofran Y., Mysore V., Rost B. Prediction of DNA-binding residues from sequence. Bioinformatics. 2007;23:i347–i353. doi: 10.1093/bioinformatics/btm174. [DOI] [PubMed] [Google Scholar]
- 29.Wang L., Huang C., Yang M.Q., Yang J.Y. BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst. Biol. 2010;4 Suppl 1:S3. doi: 10.1186/1752-0509-4-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao M., Skolnick J. DBD-Hunter: a knowledge-based method for the prediction of DNA-protein interactions. Nucleic Acids Res. 2008;36:3978–3992. doi: 10.1093/nar/gkn332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tjong H., Zhou H.X. DISPLAR: an accurate method for predicting DNA-binding sites on protein surfaces. Nucleic Acids Res. 2007;35:1465–1477. doi: 10.1093/nar/gkm008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomlins S.A., Laxman B., Dhanasekaran S.M., Helgeson B.E., Cao X., Morris D.S., Menon A., Jing X., Cao Q., Han B., et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 33.Hacker U., Grossniklaus U., Gehring W.J., Jackle H. Developmentally regulated Drosophila gene family encoding the fork head domain. Proc. Natl. Acad. Sci. U.S.A. 1992;89:8754–8758. doi: 10.1073/pnas.89.18.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa S., Gisselbrecht S.S., Rogers J.M., Hartl D.L., Bulyk M.L. DNA-binding specificity changes in the evolution of forkhead transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2013;110:12349–12354. doi: 10.1073/pnas.1310430110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y., Stormo G.D. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nat. Biotechnol. 2011;29:480–483. doi: 10.1038/nbt.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robasky K., Bulyk M.L. UniPROBE, update 2011: expanded content and search tools in the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2011;39:D124–D128. doi: 10.1093/nar/gkq992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.