


Abstract
Insight into the three-dimensional architecture of RNA is essential for understanding its cellular functions. However, even the classic transfer RNA structure contains features that are overlooked by existing bioinformatics tools. Here we present DSSR (Dissecting the Spatial Structure of RNA), an integrated and automated tool for analyzing and annotating RNA tertiary structures. The software identifies canonical and noncanonical base pairs, including those with modified nucleotides, in any tautomeric or protonation state. DSSR detects higher-order coplanar base associations, termed multiplets. It finds arrays of stacked pairs, classifies them by base-pair identity and backbone connectivity, and distinguishes a stem of covalently connected canonical pairs from a helix of stacked pairs of arbitrary type/linkage. DSSR identifies coaxial stacking of multiple stems within a single helix and lists isolated canonical pairs that lie outside of a stem. The program characterizes ‘closed’ loops of various types (hairpin, bulge, internal, and junction loops) and pseudoknots of arbitrary complexity. Notably, DSSR employs isolated pairs and the ends of stems, whether pseudoknotted or not, to define junction loops. This new, inclusive definition provides a novel perspective on the spatial organization of RNA. Tests on all nucleic acid structures in the Protein Data Bank confirm the efficiency and robustness of the software, and applications to representative RNA molecules illustrate its unique features. DSSR and related materials are freely available at http://x3dna.org/.
INTRODUCTION
The three-dimensional (3D) folding of RNA shows striking parallels to that of proteins. On the other hand, RNA is distinct from proteins due to its more flexible backbone and the wide variety of observed base-pairing motifs (1). In massive assemblies such as the ribosome, RNA displays a bewildering complexity that overwhelms our abilities to comprehend its organization (2). Even small RNA molecules (such as tRNA, riboswitches, and ribozymes) can fold into complex tertiary structures. Deciphering the information provided by the growing library of solved RNA structures and relating this information to biological function constitute two of the challenges of modern structural biology. For example, the design of RNA-based nanostructures relies on well-characterized small structural motifs (3).
Discoveries of new RNA folds and functions have stimulated interest in the development of technologies that can make sense of the complex spatial arrangements of these molecules. Fundamental RNA structural features are currently characterized by a plethora of computer programs and databases specialized in the identification of paired bases (4–8), A-form double helices (6,7), loops of various types (including multi-branched junction loops) (9–11), and pseudoknots (12,13). Use of one program often requires the output of another. For example, pseudoknot detection (12,14) requires a listing of canonical base pairs. Moreover, some of the programs do not consider modified nucleotides (4,8) and others ignore pseudoknots when finding junction loops (10,11).
The analysis of RNA 3D structure presents challenges not usually encountered in the characterization of DNA and protein structures, including: (i) a large number of chemically modified nucleotides; (ii) the presence of both canonical (Watson-Crick or G–U wobble) and noncanonical base pairs; (iii) the coaxial stacking and higher-order hydrogen-bonded, coplanar associations (multiplets) of base pairs; (iv) the formation of pseudoknots; (v) the heterogeneity of loops, including junction loops; (vi) a mix of structural motifs; and (vii) the RNA-specific interactions of the 2′-hydroxyl group. DSSR (Dissecting the Spatial Structure of RNA) is a computational tool that resolves all of these issues in a single self-contained program. The software consolidates, refines, and extends the functionality of the 3DNA suite of programs (6,7) for RNA structural analysis (15). DSSR is built upon our extensive experience in supporting 3DNA, growing knowledge of RNA structures, and refined programming skills.
The key features of DSSR are illustrated in Figure 1 and include: (i) recognition of nucleotides, both standard and modified, based on atom names and base planarity; (ii) detection of hydrogen-bonded base pairs regardless of tautomeric or protonation state, using embedded standard reference frames and simple geometric criteria; (iii) identification of higher-order, coplanar base associations (by searching horizontally in the base-pair plane for further hydrogen-bonding interactions); (iv) classification of arrays of stacked base pairs into helices (based on vertical exploration of stacking interactions regardless of backbone connectivity or base-pair type); (v) identification of stems of stacked and covalently connected canonical base pairs; (vi) grouping of coaxially stacked stems within helices; (vii) pinpointing isolated canonical base pairs outside of stems; (viii) identification of ‘closed’ hairpin, bulge, internal, and junction loops; (ix) detection and (optional) removal of pseudoknots of arbitrary complexity; (x) inclusion of isolated base pairs and pseudoknotted stems in loop identification; (xi) recognition of k-turns, U-turns, A-minor motifs, G-tetrads, ribose zippers, kissing loops, capping interactions, and sugar-phosphate backbone interactions; and (xii) characterization and classification of base-pair spatial arrangements, backbone conformations, and helical orientations. Importantly, with these combined features, DSSR has no match in its breadth and depth of functionality for nucleic acid structural analysis. For instance, no other program that we know of can characterize the classic tRNAPhe structure (with 14 modified nucleotides) as thoroughly as DSSR (Figure 2, Supplementary Figures S1 and S2, and Supplementary Sample Output).
Figure 1.
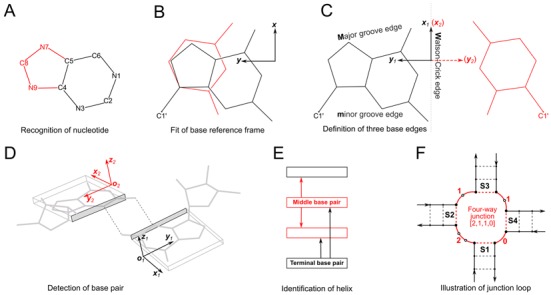
Summary of steps used to identify nucleic acid structural components. (A) Nucleotides are recognized using standard atom names and base planarity. A base is taken as a pyrimidine (six-membered ring) unless it possesses one of three purine atoms (red). (B) Bases are assigned a standard reference frame independent of sequence: purines and pyrimidines (red) are symmetrically placed with respect to the sugar. (C) The standard base frame is derived from an idealized Watson-Crick base pair, where the x1, y1-axes of the sequence base align with the x2-, y2-axes of its complement (red) and define three base edges (Watson-Crick, minor groove, major groove). (D) Base pairs are identified from the distance and coplanarity of base rings (highlighted by rectangular blocks with embedded reference frames and shaded minor-groove edges) and the occurrence of at least one hydrogen bond (dashed lines). (E) Helices are defined by base-stacking interactions. Whereas the two nearest neighbors of a terminal pair (black) lie on one side of the pair, those of a middle pair (red) lie on opposite sides. (F) Closed loops are delineated by the ends of stems (or isolated base pairs) and specified by the lengths of consecutive connecting loop segments. Here, the four-way junction (S1 to S4) is denoted [2,1,1,0] in terms of the connecting, unpaired loop nucleotides (white circles) running clockwise from S1 to S4. Arrows point from the 5′ to 3′ direction along each strand and dashed lines represent stem pairs.
Figure 2.
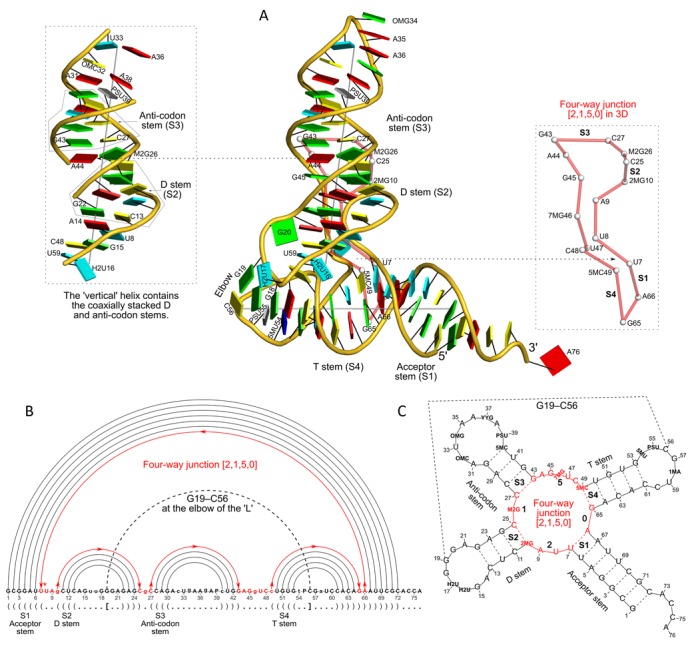
DSSR captures well-known features and provides a new perspective on the classic yeast tRNAPhe structure (PDB id: 1ehz (49)). (A) The software automatically detects the four stems and the two helices that form the L-shaped molecule, depicted here in cartoon-block representation (center). Whereas the helices may include all types of base pairs and backbone breaks, the stems comprise only canonical pairs with continuous backbones. Note the coaxial stacking of the D and anti-codon stems and the noncanonical features of the composite helix (represented by a gray line, left). The red ‘circle’, overlaid on the central image and detailed to the right, reveals the 3D pathway along the [2,1,5,0] four-way junction loop. (B) The dot-bracket notation derived by DSSR serves as input for the depicted linear (arc) representation of secondary structure. The bases comprising the four-way junction loop (red) run in sequential order from U7 (*) following the arrows to the right and returning along the outer A66→U7 arc. The pseudoknotted G19–C56 pair (with matched []) is noted by the dashed arc. (C) Both the four-way junction (red) and the three hairpin loops follow ‘circular’ routes within the traditional cloverleaf representation of tRNA. Here the 14 modified nucleotides are represented by three-letter codes. The 3D images were created using PyMOL (A, red; C, yellow; G, green; T, blue; U, cyan; pseudouridine P, gray), the 2D diagrams using VARNA, and the annotations using Inkscape.
Following the initial release of DSSR on the 3DNA Forum in early 2013, its rapid adoption has allowed us to refine and extend the software based on user feedback. Early versions of DSSR have been cited in annotations of hydrogen bonds in the crystal structure of the bacterial Alu domain of the signal recognition particle (16), the automated identification and classification of RNA base pairs by the RNApdbee webserver (12), and the characterization of RNA secondary structural features from crystal structures of the large ribosomal subunit (17) and the whole ribosome (18) of human mitochondria. DSSR is now a mature, actively supported software product, readily applicable to real-world nucleic acid structural analysis. The application of DSSR to a wide variety of nucleic acid structures (Table 1) underscores its robust performance and highlights its unique features.
Table 1. Summary of structural features identified by DSSR (in default settings) for ten representative RNA molecules.
| PDB id | Nucleotidesa | Pairsb | Multiplets | Helices | Stems | Hairpins | iloopsc | Junctions | Pseudoknotd | Timee | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yeast tRNAPhe | 1ehz (49) | 76 (14) | 34 (21) | 4 | 2 | 4 | 3 | 0 (0) | 1 | 1 | <1 s |
| Viral tRNA mimic | 4p5j (34) | 84 (1) | 37 (27) | 4 | 2 | 5 | 5 | 0 (0) | 1 | 1 | <1 s |
| Twister ribozyme | 4rge (46) (chain A) | 56 (0) | 31 (20) | 4 | 2 | 6 | 2 | 0 (0) | 1 | 2 | <1 s |
| SAM-I riboswitch | 2gis (48) | 95 (1) | 47 (30) | 8 | 3 | 7 | 3 | 3 (1) | 2 | 1 | <1 s |
| Cas9-sgRNA-DNA | 4oo8 (50) | 117 (0) | 49 (43) | 0 | 5 | 6 | 4 | 1 (1) | 0 | 0 | <1 s |
| Group I intron | 1gid (38) (chain A) | 158 (0) | 82 (48) | 14 | 4 | 10 | 3 | 4 (4) | 1 | 0 | <1 s |
| Group II intron | 3bwp (43) | 349 (0) | 159 (104) | 12 | 10 | 23 | 6 | 9 (1) | 2 | 4 | ∼1 s |
| Large ribosomal subunit | 1s72 (44) | 2876 (5) | 1459 (811) | 242 | 86 | 179 | 68 | 67 (36) | 36 | 3 | <1 min |
| E. coli 70S ribosomef,g | 5afi (45) | 4801 (53) | 2383 (1332) | 325 | 134 | 297 | 116 | 126 (66) | 54 | 2 | <3 min |
| S. cerevisiae 80S ribosomeg | 4u4o (47) | 10 398 (0) | 4927 (2705) | 572 | 317 | 636 | 231 | 348 (139) | 120 | 4 | ∼15 min |
aTotal number of nucleotides, with modified ones in parentheses. Low numbers of modified nucleotides may reflect limited resolution of the experiments.
bTotal number of base pairs, with canonical Watson-Crick and G–U wobble pairs in parentheses.
cInternal loops, with bulges in parentheses.
dOrder of pseudoknot of highest complexity.
eRuntime on a MacBook Air (Middle 2011) with 1.8 GHz Intel Core i7 and 4GB 1333 MHz DDR3.
fHigh-resolution cryo-electron microscopy (cryo-EM) structure at 2.9 Å.
gThese two structures are in PDBx/mmCIF format, due to their large size.
MATERIALS AND METHODS
Data sources
Nucleic-acid-containing structures were downloaded from the Protein Data Bank (PDB) (19) and updated weekly. Each release of DSSR was checked against all these structures, with the current version 1.2.8 validated on the PDB release as of June 12, 2015. Searches for motifs were performed on release 1.89 (December 5, 2014) of the non-redundant RNA crystal structures at 3.0-Å or better resolution (NR3A-dataset) curated by Leontis and Zirbel (20). The 3D images were created using PyMOL version 1.7.4.0 (http://pymol.org; the PyMOL Molecular Graphics System, Schrödinger, LLC), the 2D diagrams using VARNA (21) version 3.9, and the annotations using Inkscape version 0.48 (https://inkscape.org). The base rectangular block representation follows the style of Calladine et al. (22), with purines having dimensions of 4.5 Å (width, groove edges) by 4.5 Å (depth, side edges) by 0.5 Å (height) and pyrimidines of 3.0 Å × 4.5 Å × 0.5 Å, as in 3DNA (6,7). Blocks of these sizes approximately encompass all atoms of the bases, including the exocyclic atoms.
Identification of nucleotides
DSSR uses the atomic coordinates and standard names of base-ring atoms to identify a nucleotide (Figure 1A). All known nucleotides share a common six-membered pyrimidine ring, with atoms named consecutively (N1, C2, N3, C4, C5, C6), and purines include three additional atoms (N7, C8, N9). A least-squares fitting procedure matches atoms in a residue to those in a reference purine with nine ring atoms. A nucleotide is identified if a residue contains at least three base ring atoms and the root-mean-square deviation (rmsd) of the fit falls below a user-definable cutoff. Since base rings are rigid, the rmsd is normally <0.1 Å. To account for experimental error and special non-planar cases, such as 5,6-dihydrouridine (H2U) in yeast tRNAPhe (Figure 2), the default rmsd cutoff is set to 0.28 Å. The algorithm detects regular and modified nucleotides (in base, sugar, or phosphate; Table 1) and it is applicable to biopolymer chains as well as isolated ligands (e.g. SAM in the SAM-I riboswitch, Figure 5).
Figure 5.
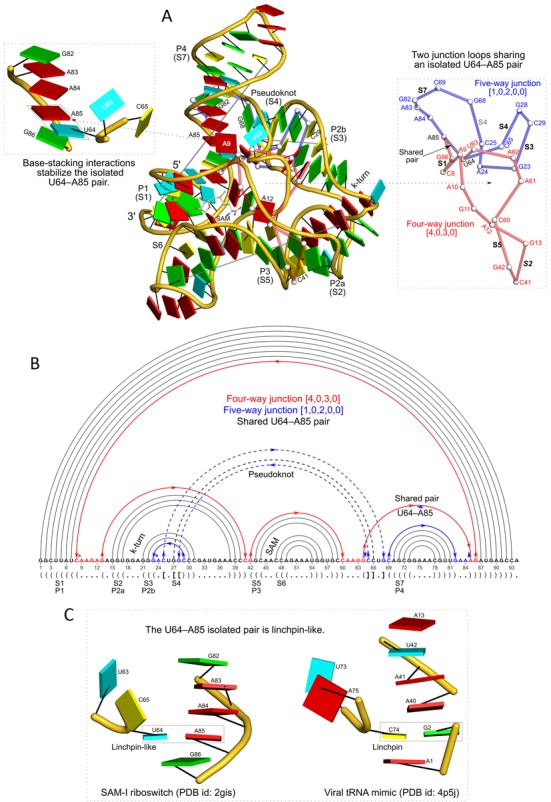
DSSR pinpoints a linchpin-like U64–A85 pair that is shared by a four-way and a five-way junction loop in the S-adenosyl methionine I riboswitch (PDB id: 2gis (48)). (A) DSSR identifies two junction loops (right): a [4,0,3,0] four-way junction loop (red) and a [1,0,2,0,0] five-way junction loop (blue), which share a common side, i.e. the isolated U64–A85 pair (left). (B) The linear secondary structure diagram, annotated with DSSR-derived dot-bracket notation, depicts the pathways of the two junction loops. The four-way loop runs from C8 (*), follows the red arrows to the right, and returns along the outer G86→C8 arc. The five-way loop starts at G23 (*), moves to the right following the blue arrows along two arcs (C25→G68 and C69→G82), and returns to the start along three arcs (A85→U64, C65→G28, C29→G23). Note that the shared U64–A85 arc is traversed twice, from left to right along the four-way junction loop, and right to left along the five-way junction loop. (C) The U64–A85 pair is stabilized by base-stacking interactions in a way strikingly similar to the G2–C74 linchpin pair in the viral tRNA mimic (see Figure 3), and may also be regarded as a ‘linchpin’. These two images take advantage of unique visualization features within 3DNA/DSSR, including the capability to orient different molecules into a common frame (here, the frames of the linchpin pairs with the minor-groove edges facing the viewer) and to represent bases as color-coded rectangular blocks.
As of June 12, 2015, DSSR detected over 630 different types of modified nucleotides in the PDB. In the derived base sequence, DSSR uses a one-letter shorthand for each identified nucleotide: upper case A, C, G, U and T for standard RNA and DNA bases, and lower case letters for modified nucleotides mapped to their canonical counterparts (e.g. ‘c’ for 5-methylcytidine, 5MC; Figure 2 and Supplementary Sample Output). Note that pseudouridine (PSU) is shortened to ‘P’, due to its special C1′–C5 glycosidic linkage (Figure 2).
Fitting of local base reference frames
Once a nucleotide has been identified, a local reference frame is used to specify the base in 3D space. Following 3DNA (6,7), DSSR employs the standard base reference frame (23) (Figure 1B and C) and performs a least-squares fit of the atomic coordinates for each nucleotide in the analyzed structure against a corresponding base in its standard frame (23). The fit uniquely defines the position and orientation (i.e. the reference frame) of each base in the structure, and the three axes are orthonormal by definition (Figure 1D). Moreover, the standard base frame of the purines and pyrimidines is symmetrically placed with respect to the sugar (Figure 1B) and thus independent of base identity. The frame also contains intuitive geometric features to define the three base edges (Watson-Crick, minor groove, major groove, Figure 1C).
Identification of hydrogen bonds and base-stacking interactions
To identify hydrogen bonds, DSSR implements a geometric approach based on the relative spatial positions of nitrogen (N) and oxygen (O) atoms. The method starts by enumerating all N/O atom pairs that are within a certain distance cutoff (default to 4.0 Å), and then filters each through a series of heuristic criteria (including hydrogen-bonding donor/acceptor properties, angles with neighboring atoms, and planarity with respect to the bases). Specifically, it uses the mutual shortest distance between atom pairs to avoid spurious hydrogen bonds (Supplementary Figures S1–S4 and S7) and to detect unconventional donor/acceptor combinations (e.g. the N3 to N3 hydrogen bond in the hemi-protonated cytosine–cytosine base pair in the i-motif (24), Supplementary Figure S2E).
To quantify base-stacking interactions, DSSR employs the shared overlap area of the two bases, projected onto the ‘mean’ plane (6) defined by the average z-axis of the reference frames. This quantitative measure is intuitive, easy to visualize, and follows the spirit of existing base-stacking diagrams of DNA structures (25).
Identification of base pairs
DSSR characterizes a pair geometrically, based on the local base reference frames (Figure 1D) and the following five key criteria (with default values in parentheses): (i) the distance between the two origins (≤15 Å); (ii) the vertical separation between the base planes (≤2.5 Å); (iii) the angle between the base normal vectors (≤65°); (iv) the absence of stacking between the two bases; and (v) the presence of at least one hydrogen bond involving a base atom. The default cutoff values are based on extensive tests in real-world applications (6,7), and work well even for distorted structures. As long as two interacting bases fulfill the above criteria, they are designated as a pair. This method is able to identify all pairs that actually exist in a given structure, either canonical (Watson-Crick or G–U wobble) or noncanonical. The latter pairs may include normal or modified nucleotides, regardless of tautomeric or protonation state (as in the cytosine-cytosine pair, Supplementary Figure S2E) (26,27).
The M–N versus M+N relative base orientations
Due to molecular asymmetry (28,29), each base has two unique faces that can be easily distinguished using the direction of the z-axis of the standard base reference frame (Figure 1B and C). When two bases (M and N) in a pair have opposite faces, the scalar product of their z-axes is negative, so the pair is denoted M–N (with a minus sign). The canonical Watson-Crick and wobble G–U pairs belong to this category. Conversely, if the M and N bases in a pair share the same face, the scalar product of their z-axes is positive, and the pair is denoted M+N (with a plus sign). The well-known Hoogsteen A+U pair belongs to this class. An M–N pair becomes M+N if either M or N (but not both) is flipped (30), and vice versa. For example, the reverse Watson-Crick pairs are M+N, and the reverse Hoogsteen pair (Supplementary Figure S4) is M–N.
Six base-pair parameters
As in 3DNA (6,7), DSSR takes advantage of the six standard base-pair parameters—three translations (Shear, Stretch, Stagger) and three rotations (Buckle, Propeller, Opening)—to quantify the relative spatial position and orientation of any two interacting bases rigorously. Among the six parameters, only Shear, Stretch, and Opening are critical for characterizing different types of pairs. Buckle, Propeller and Stagger, on the other hand, describe the nonplanarity of a given pair (6). By virtue of the definition of the standard base reference frame, Shear, Stretch, and Opening are all close to zero for Watson-Crick pairs. Moreover, every other type of pair has a set of characteristic parameters. For example, the wobble G–U pair is characterized by an average Shear of –2.2 Å, and the Hoogsteen A+U pair is distinguished by a Stretch of approximately –3.5 Å and an Opening of near 66°.
Common names of base pairs
DSSR assigns names of common pairs (30,31) based on sequence identity and characteristic parameters. The list includes Watson-Crick A–U and G–C, wobble G–U, sheared G–A, Hoogsteen A+U, and reverse Hoogsteen A–U, among others. With common names, the stretches of canonical base pairs within a structure are immediately obvious, which helps in visually pinpointing double-helical stem regions.
Classification of base pairs
DSSR classifies base pairs by two commonly used nomenclatures: the 28 hydrogen-bonding types from Saenger (31) and the 12 basic geometric classes of Leontis-Westhof (LW) (32). Additionally, DSSR introduces a new classification scheme that defines three base-centric interacting edges (Watson-Crick, minor groove, major groove; Figure 1C) and takes consideration of the two relative base orientations (‘+’ and ‘–’, see above). These geometrically defined base edges retain the simplicity and usefulness of the LW method, and eliminate the ambiguities associated with the LW RNA-specific ‘sugar’ edge that ties the base minor-groove edge to the ribose sugar 2′-hydroxyl group. Details about the new base-pair classification scheme will be reported elsewhere.
Higher-order coplanar base associations (multiplets)
DSSR defines multiplets as three or more bases associated in a coplanar geometry via a network of hydrogen-bonding interactions. Multiplets are identified through inter-connected base pairs, filtered by pair-wise stacking interactions and vertical separations to ensure overall coplanarity (Supplementary Figures S1, S3, S4 and S7). The abundant A-minor motifs (33) (types I and II, Supplementary Figures S3, S4 and S7) are base triplets, the smallest multiplet. The G-tetrad motif, where four guanines are associated via four pairs in a square planar geometry, is another special case of a multiplet.
Helices, stems, coaxial stacking, and isolated canonical pairs
DSSR defines a helix by base-stacking interactions, regardless of pairing type (canonical or otherwise) or backbone connectivity (continuous or broken, Figures 1E and 2A). By definition, a helix contains at least two base pairs. A pair may belong to only one helix. When a base is involved in multiple pairs, the pair geometrically closest to a Watson-Crick interaction is selected for helix formation. For example, in the triplet of a type I A-minor motif, the receptor pair (normally a G–C) would be chosen. A stem is a helix consisting of only canonical pairs with continuous backbones. Coaxial stacking occurs when more than one stem exists in a single helix, where neighboring stems can stack either directly above one another or on either side of one or more noncanonical pairs (Figure 2A). An isolated pair is a canonical Watson-Crick or G–U wobble pair not belonging to any stem. Isolated pairs are common in RNA structures (Figures 2, 3, 5, 6), and may play a critical role in stabilizing various folds (e.g. the linchpin G–C pair in the viral tRNA mimic (34), Figure 3).
Figure 3.
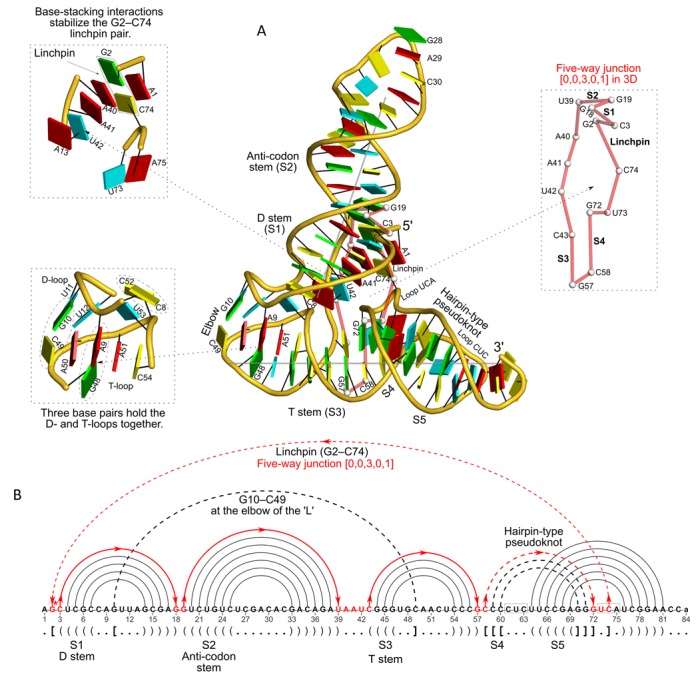
DSSR reveals the striking global similarity and distinct local variations between the tRNA mimic from turnip yellow mosaic virus (PDB id: 4p5j (34)) and yeast tRNAPhe. (A) The viral tRNA mimic assumes an overall L-shaped tertiary structure (center) composed of two helices (gray lines). DSSR uncovers a [0,0,3,0,1] five-way junction loop (right) enabled by the hairpin-type pseudoknot at the 3′-end of the molecule and the G2–C74 linchpin pair. This critical linchpin is unique to the tRNA mimic, where it is stabilized by extensive base-stacking interactions (upper-left). The lower-left inset emphasizes the intricate interactions between the D- and T-loops in the mimic, including the three base pairs (within dashed ellipses) and the unique base triplet at the elbow (Supplementary Figure S3A). (B) The linear secondary structure diagram generated with the DSSR-derived dot-bracket notation shows the sequential location of the bases comprising the linchpin pair, the five-way junction loop (red), the G10–C49 pair at the elbow, and the hairpin-type pseudoknot. Note that the dashed arcs connecting the so-called first-order pseudoknotted pairs (indicated by matched []) do not cross each other along the linear sequence. The numbering of residues used here follows that in the PDB file, which is offset by two nucleotides from that given in the original publication (e.g. the G2–C74 linchpin is termed G4–C76 there).
Figure 6.
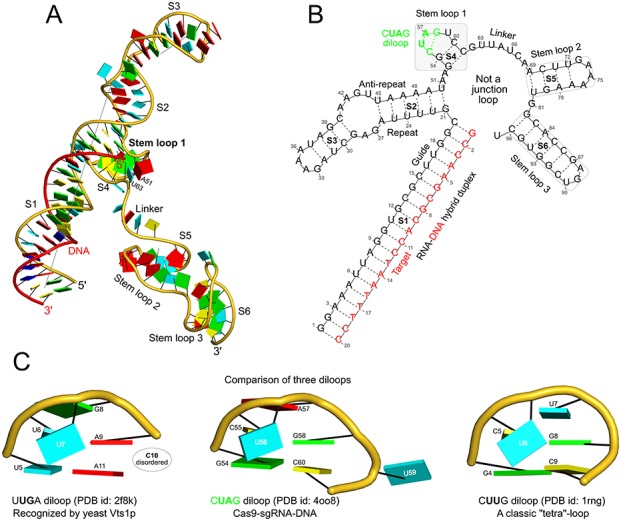
DSSR applies to RNA-DNA hybrid structures, such as the CRISPR Cas9-sgRNA-DNA ternary complex (chains B and C, PDB id: 4oo8 (50)). (A) The software identifies five helices (depicted by gray lines) and six stems (annotated) in the structure. The longest helix includes the RNA-DNA hybrid duplex (S1, depicted by intertwined gold-red backbone tubes) and the repeat:anti-repeat RNA stem (S2). (B) The secondary structure diagram, derived using DSSR, shows that the hybrid structure does not form a ‘closed’ junction loop. DSSR classifies the CUAG hairpin loop as a diloop (instead of a tetraloop) because the C and G form a Watson-Crick pair that closes the loop, leaving only a two-nucleotide (UA) loop segment. (C) Comparison of the CUAG diloop (center) with the UUGA diloop from a yeast Vts1p-RNA hairpin complex (referred to in the literature as part of a pentaloop (62), left) shows the remarkable similarity between the two loops despite the large difference in their base sequences. The CUAG diloop also shares common features with the NMR solution structure of the classic CUUG diloop (63) (often called a tetraloop, right), including the flipped out second position U and the stacking of the closing C–G pair over a neighboring G–C pair. The diloops differ, however, in terms of the inter-pair twist angle at the GpC dinucleotide step. These three images are oriented in the frames of the purines stacked above the terminal nucleotides (A9, left; G58, middle; G8, right) with the minor-groove edges facing the viewer.
Stems and isolated pairs delineate secondary structure components, including various loops (see below). Canonical pairs (either in stems or when isolated) create pseudoknots when they cross each other along the linear base sequence (Figures 2–5). DSSR treats isolated pairs as a special case of stems in identifying loops and characterizing pseudoknots. For each identified helix (or stem), DSSR derives a least-squares fitted linear helical axis (25) (Figures 2–6), useful for schematic representation (7) and quantifying the geometric relationship between two helices (e.g. the inter-helix angle). DSSR also calculates a comprehensive set of helical parameters (e.g. twist angles) (6) for dinucleotide steps in a helix/stem.
Algorithmically, DSSR searches vertically in the neighborhood of selected pairs for stacking interactions with other pairs (Figure 1E). The two nearest neighbors of a terminal pair lie on the same side of the pair whilst those of a middle pair sit on opposite sides. Starting from one end, a helix/stem can be assembled one pair at a time until the other end is reached. The same algorithm can be applied to identify continuous base stacks (Figures 3 and 5), by using bases instead of pairs as the assembly unit. Circular RNA or DNA molecules are treated as special cases and properly handled.
Pseudoknot detection and removal
DSSR characterizes pseudoknots in an expanded dot-bracket notation (dbn), using matched symbols ([], {}, <>) and letters (upper/lower case) for successively higher-order pseudoknotted pairs. Thus, a first-order pseudoknot can be fully differentiated using matched [] (Figures 2, 3, 5), a second-order by [] and {} (Figure 4), and so on. The program adapts the elimination-gain heuristics of Smit et al. (14) and works iteratively to derive the dbn for consecutively higher-order pseudoknots. DSSR also has functionality for removing pseudoknotted pairs (changing [], {}, etc. to dot in dbn) to produce a fully nested structure (Figure 4B and C). As noted above, DSSR employs only canonical base pairs (either in stems or when isolated) in defining RNA secondary structures (35,36) and characterizing pseudoknots (13).
Figure 4.
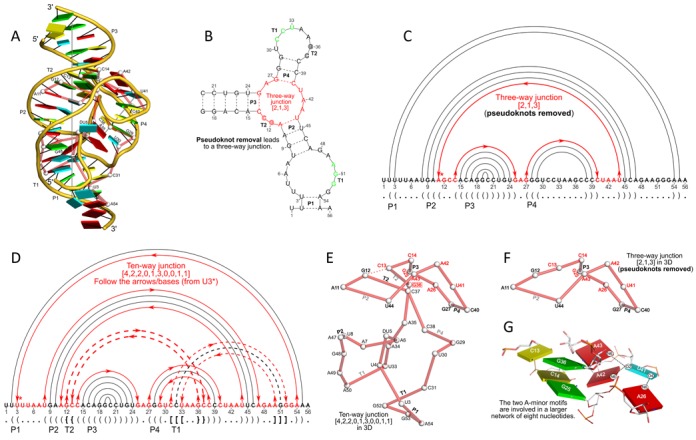
DSSR discloses complexity in the folding of the env22 twister ribozyme not apparent in the two-armed tertiary structure (chain A, PDB id: 4rge (46)). (A) The software automatically detects the long helical arm with five coaxially stacked stems and the short single-stemmed arm of the molecule. Failing to account for the pseudoknots within the structure leads to a characterization of the molecule very different from its real organization. When pseudoknots are omitted, the RNA appears to form a simplified [2,1,3] three-way junction as shown in both planar (B) and linear (C) secondary structure diagrams. In reality, the DSSR-derived dot-bracket notation points to a double-pseudoknotted structure (D) with two types of brackets distinguishing the pseudoknotted pairs (matched [] and {}), and uncovers a novel [4,2,2,0,1,3,0,0,1,1] ten-way junction loop (D and E). The junction, which can be traced by following the arrows along the red arcs and bases (starting from U3, marked with *) in D, contains both ends of four of the six stems and follows a supercoiled pathway in 3D (Supplementary Figure S5). In contrast, without consideration of pseudoknots (F), the junction forms a simple relaxed circle (Supplementary Figure S5). DSSR also detects three previously ignored base pairs that help to anchor the consecutive A-minor motifs reported in the literature (46) (G). U41 pairs with A42 and A43 through bifurcated hydrogen bonding, as well as with A26 (Supplementary Figure S4C, D). Moreover, U41 and A42 constitute a UpA dinucleotide platform, and in combination with G25 and A26, create a unique network of eight interacting nucleotides (G). All eight nucleotides are involved in the ten-way junction loop (labeled red in (E)).
Loop identification and classification
DSSR delineates loops using the terminal base pairs of stems and the bridging nucleotides (Figure 1F). Depending on the number of stems involved, loops are classified into three categories: a hairpin loop is delimited by one stem, an internal/bulge loop by two stems, and a junction (multi-branched) loop by three or more stems. In DSSR, a loop forms a ‘closed’ circle with any two sequential nucleotides connected either by a phosphodiester linkage or a canonical base pair, and is specified by the lengths of consecutive bridging-nucleotide segments (Figures 1F and 2C). For example, the [2,1,5,0] four-way junction loop in tRNAPhe (Figure 2C) contains two bridging nucleotides between stems S1 and S2, one between S2 and S3, five between S3 and S4, and zero between S4 and S1.
The well-known GNRA (N for A/C/G/U, and R for A/G) tetraloop has four nucleotides in its single loop segment; if the closing pair is considered, however, the loop contains a total of six nucleotides. In contrast, the so-called CUUG tetraloop is termed a diloop in DSSR since the C and G form a closing canonical pair, leaving only UU in the loop segment. Similarly, a noncanonical pair in an otherwise continuous helix composed of canonical pairs signifies a [1,1] internal loop and is not considered as pseudoknotted (Figure 5B). Both ends of pseudoknotted stems may be involved in the same junction loop, creating intricate topologies (Figures 4 and 5). Pseudoknot removal leads to simplified loops, which help to reveal basic secondary structure features at the expense of missing the precise folding topology (Figure 4).
Representation of secondary structure
DSSR produces RNA secondary structures in three commonly used file formats—ViennaRNA package dbn (36), Mfold connect table (.ct) (35), and CRW bpseq (37)—that can be fed directly into visualization tools such as VARNA (21). DSSR employs an extended dbn to account for pseudoknots (see above) and chain breaks or multiple chains (Yann Ponty, personal communication). The DSSR-derived .ct file contains the actual PDB sequential number of each nucleotide, and allows for the representation of multiple molecules.
Other functionality
DSSR provides detailed listings of continuous base stacks, non-pairing interactions (between two nucleotides not involved in a base pair), and interactions involving phosphate groups; the software also detects ribose zipper motifs (38), types I and II A-minor motifs (33), U-turns of the UNR-type (39) and the GNRA-type (40), kissing loops, and local (i.e. non-composite) k-turns (41) and calculates a comprehensive set of commonly used sugar-phosphate backbone parameters.
Implementation and software availability
DSSR was implemented in ANSI C as a stand-alone command-line program. It is self-contained and the binaries for common operating systems are tiny (<1mb), without runtime dependencies on third-party libraries. DSSR is distributed in compiled form (for Mac OS X, Linux and Windows), with an extensive manual. User questions are promptly addressed on the public 3DNA Forum. A simple web interface to DSSR has been implemented, making its major functionality easily accessible. DSSR has also been integrated into the Jmol (42) molecular graphic visualization program (Robert Hanson, personal communication; details of the Jmol-DSSR integration will be reported elsewhere). Since its initial release in early 2013, DSSR has been continuously refined based on user feedback, and is currently at a stable version (1.2.8). The software and related resources are freely available at the 3DNA homepage: http://x3dna.org/.
RESULTS
DSSR readily analyzes any RNA structure in PDB or PDBx/mmCIF format. Table 1 summarizes the results of running DSSR (using default settings) on ten representative RNA-containing structures: a tRNA and its mimic, a ribozyme, a riboswitch, an RNA-DNA hybrid duplex from the CRISPR-Cas9 complex, group I and group II (43) introns, a large ribosomal subunit (44), and the entire Escherichia coli (45) and Saccharomyces cerevisiae ribosomes. The size of the RNA components in these structures ranges from 56 nucleotides for the env22 twister ribozyme (46) (PDB id: 4rge) to 10,398 nucleotides for the yeast 80S ribosome (47) (PDB id: 4u4o, in mmCIF format). DSSR runs almost instantaneously on a contemporary laptop computer, except for the analyses of very large ribosomal RNA structures.
The number of base pairs in these ten RNA molecules is roughly half the number of nucleotides. Approximately 60% of the identified pairs are canonical; the remaining 40% are noncanonical. DSSR identifies all nucleotides within each structure, including those that are chemically modified. Other automatically detected structural features include multiplets, helices/stems, various ‘closed’ loops (hairpin, internal/bulge and junction), and pseudoknots (Table 1).
In the following sections, we use four functionally important noncoding RNA molecules (tRNAPhe, a viral tRNA mimic, a twister ribozyme and a SAM-I riboswitch) (34,46,48–49) as well as the CRISPR Cas9-sgRNA-DNA ternary complex (50) to illustrate salient features of the program. In each and every case, DSSR not only characterizes key features reported in the literature but also reveals significant new findings undetected previously.
Yeast phenylalanine tRNA
Starting from the 3D atomic coordinates of yeast tRNAPhe (PDB id: 1ehz (49)), DSSR identifies the four stems (acceptor, D, anti-codon, T), the three peripheral hairpin loops, and the central four-way junction that comprise its classic cloverleaf secondary structure (39) (Figure 2). Moreover, the program captures the organization of the L-shaped tertiary fold of the molecule through the pairwise coaxial stacking of adjacent stems: the acceptor and T stems constitute one arm of the overall structure, and the D and anti-codon stems the other. These two helices (represented by gray lines along the ‘best-fitted’ helical axes (25)) lie at an angle of 82° (Figure 2A). The ‘horizontal’ helix contains the acceptor and T stems in direct coaxial alignment, while the ‘vertical’ helix comprises the D and anti-codon stems, coaxially stacked on either side of the noncanonical M2G26–A44 pair. Whereas the helices identified by DSSR include all types of base pairs and may contain backbone breaks, the stems contain only canonical pairs with continuous backbones. The highly conserved Watson-Crick G19–C56 pair, located at the elbow of the L-shaped tertiary structure, connects the D and T loops via a kissing-loop interaction and creates a first-order pseudoknot (Figure 2B). DSSR denotes the elbow G–C pair as isolated since it does not belong to a stem, which requires at least two canonical pairs. The elbow G–C pair of tRNA plays an important role in its complex with the stem I domain of the T-box riboswitch, where its stacking against a base triplet of the riboswitch stabilizes the complex (51).
The procedure that DSSR employs to identify junction loops is exemplified in Figure 2B by the [2,1,5,0] four-way junction in tRNAPhe (shown in red). The loop starts at U7, follows along the bases sequentially to 2MG10, crosses over the 2MG10–C25 terminal pair of S2 (D stem) to C25, continues to pass through S3 (anti-codon stem) and S4 (T stem) to G65, and returns via the A66–U7 terminal pair of S1 (acceptor stem) to complete a ‘closed’ circle. DSSR introduces a novel schematic 3D representation of the junction loop (Figure 2A, right inset), in which nucleotides are simplified to nodes (white spheres coinciding with C1′ atoms) connected by virtual edges (red). The 3D coordinates of the nodes can be used to derive topological parameters that characterize the junction loops (52). Here, the [2,1,5,0] four-way junction forms a simple relaxed circle.
DSSR detects the four base triplets (Supplementary Figure S1) originally reported by Quigley and Rich (39) in tRNAPhe, including the two that contain modified nucleotides, (2MG10, C25, G45) and (C13, G22, 7MG46). The software further reveals the extensive base-stacking interactions within the molecule: only four nucleotides (H2U17, G20, U47, A76) do not participate in intra-molecular stacks. In addition, DSSR identifies the three base pairs that hold the D-loop (C13 to G22) and T-loop (G53 to C61) in place (Figure 2 and Supplementary Figure S2): G19–C56 at the elbow, G18+PSU55 within the ‘horizontal’ side of the L, and H2U16+U59 on the ‘vertical’ side. To the best of our knowledge, the latter pair has never been mentioned in the literature although it closely matches the C8+C52 pair in the viral tRNA mimic (Supplementary Figure S2; see also below). The three inter-loop base pairs, combined with the interdigitated stacking of bases from the loops, stabilize the overall L-shaped fold of tRNA (39,53).
Transfer RNA contains the most diverse set of modified nucleotides found in nature (54), with more than 100 variants in the RNA Modification Database (55). The modifications tend to occur in the loops, where they form crucial tertiary pairing and stacking interactions. These extensive modifications pose a challenge to RNA bioinformatics software since the majority of programs cannot consistently handle non-standard nucleotides (4,8). DSSR solves this problem by treating standard and modified nucleotides in a uniform framework (Figure 1). For the yeast tRNAPhe structure (PDB id: 1ehz), the software identifies a total of 76 nucleotides including all 14 modified ones (Figure 2C).
The viral tRNA-like structure
A single RNA sequence can play multiple functional roles by adopting different tertiary structures. A prototype for this is the tRNA-like structure from the 3′-end of turnip yellow mosaic virus, recently solved at 2.0-Å resolution by Colussi et al. (PDB id: 4p5j) (34). As noted by the authors, the RNA adopts a shape that mimics tRNA, but it uses a very different set of intra-molecular interactions to achieve this shape. This structure provides an excellent example of how DSSR deciphers global similarity in spite of the many local differences between the two molecules (Figure 3).
DSSR identifies the two helices comprising the L-shape in the tRNA mimic (Figure 3), just as it does in tRNAPhe (Figure 2). However, the tRNA mimic contains five stems instead of the four observed in tRNAPhe. The ‘vertical’ helix consists of the D- and anti-codon stems as in tRNA, but the ‘horizontal’ helix is comprised of the T-stem and two other stems involved in the hairpin-type pseudoknot at the 3′-end of the structure (Figure 3A). Although the tRNA mimic and tRNAPhe adopt similar overall folds, the two helices in the mimic lie at a different angle (71°) from that in tRNAPhe (82°). As in tRNAPhe, the mimic also contains an isolated G–C pair at the elbow and two additional noncanonical pairs holding the D-loop and T-loop together (Figure 3 and Supplementary Figure S2). Two continuous base stacks stabilize these base pairs (Figure 3A). Moreover, the presumably hemi-protonated C+C pair (24) (as in the i-motif (56)) matches the similarly positioned and previously unnoticed H2U16+U59 pair in tRNAPhe. Note that DSSR uses a geometric approach to identify the otherwise acceptor-acceptor N3 to N3 hydrogen bond in the C+C pair (Supplementary Figure S2). The software identifies any existing base pair, regardless of tautomeric form or protonation state (26,27).
The G–C linchpin is an isolated and pseudoknotted pair, specific to the tRNA mimic (Figure 3A, upper-left inset). It plays a critical role in the fold and function of the mimic and is stabilized by extensive base-stacking interactions (Figure 3A). By taking isolated canonical pairs and pseudoknotted stems into consideration, DSSR identifies a [0,0,3,0,1] five-way junction loop. This previously unreported structural feature, supported by the linchpin and the hairpin-type pseudoknot at the 3′-end of the molecule, plays a role similar to that of the [2,1,5,0] four-way junction in tRNAPhe, holding the two arms of the L in place. Four base triplets also occur in the mimic (Supplementary Figure S3). While the triplets in tRNAPhe all lie near the D stem, three of those in the mimic cluster near the hairpin-type pseudoknot (Supplementary Figure S3B-D). The fourth triplet in the mimic involves a G+U platform (57) and the elbow G–C pair (Supplementary Figure S3A).
The env22 twister ribozyme
The twister ribozyme belongs to a class of small self-cleaving nucleolytic ribozymes recently discovered through bioinformatic analysis (58). The subsequently determined crystal structures of the env22 (46) and Oryza sativa (59) twister ribozymes show similar global folds, involving the coaxial stacking of five helical stems and the formation of a second-order pseudoknot. Here we use the env22 twister ribozyme (46) (chain A, PDB id: 4rge) to illustrate how DSSR captures these features and reveals a surprisingly intricate ten-way junction in the small, compact RNA molecule (Figure 4). The software automatically identifies the two reported helices (46) (Figure 4A): a longer helix with five stems (P1–T1–P2–T2–P3) and a shorter helix with a single stem (P4). Moreover, the program confirms the compactness of the twister ribozyme in terms of the stacking of all but one of the nucleotides (DU5 near the DU5-A6 cleavage site). DSSR also correctly characterizes the double pseudoknots (Figure 4D) and detects the four base triplets reported in the literature (46) (Supplementary Figure S4). The output further reveals the expansion of two consecutive A-minor motifs into higher-order base multiplets through U41 (Figure 4G). This uracil simultaneously forms three single hydrogen-bonded base pairs: U41+A42 (a dinucleotide platform), U41+A43, and U41+A26. In each case, the N6 atom of an adenine forms a hydrogen bond with one of the uracil carbonyl groups. A survey of the NR3A-dataset (20) shows no examples of similar interactions in other types of RNA molecules.
Strikingly, by taking the pseudoknotted stems into consideration, DSSR uncovers a novel [4,2,2,0,1,3,0,0,1,1] ten-way junction loop that includes 34 of the 56 nucleotides (Figure 4D and E). The ten-way junction arises from the six-stem structure by traversing both ends of the four stems involved in pseudoknots. This intricate loop follows a supercoiled pathway, with a linking number of 3 (Supplementary Figure S5) (52). Removal of the two pseudoknotted stems (T1 and T2) leads to an over-simplified three-way junction loop, consisting of only 12 nucleotides (Figure 4B, C and F). Moreover, this three-way junction is no longer supercoiled, but forms a simple relaxed circle (Supplementary Figure S5). We know of no other RNA structural analysis tool that can automatically delineate the two helices in the twister ribozyme or characterize the ten-way junction loop.
The SAM-I riboswitch
Riboswitches are cis-acting genetic elements that bind to specific metabolites to regulate gene expression at the level of transcription or translation. S-Adenosyl-Methionine (SAM) riboswitches are the most common type, and the SAM-I family is among the best characterized (60). We use the prototypical SAM-I riboswitch from Thermoanaerobacter tengcongensis (48) (PDB id: 2gis) for further illustration of the definitions of helices, loops and pseudoknots used in DSSR, and show how the software can treat isolated ligands, like SAM, as modified nucleotides in detecting base pairs and multiplets (Figure 5).
By default, DSSR includes isolated pairs and pseudoknotted stems in defining various loops, as shown above for the viral tRNA mimic and the twister ribozyme (Figures 3 and 4). When applied in the same manner to the SAM-I riboswitch, DSSR detects two junction loops: a [4,0,3,0] four-way junction and a [1,0,2,0,0] five-way junction (Figure 5A). Moreover, these two junction loops share the isolated U64–A85 pair, which is held in place by base-stacking interactions strikingly similar to those of the G–C linchpin in the viral tRNA mimic (Figure 5C). The linchpin-like U64–A85 pair in the SAM-I riboswitch is further stabilized by the formation of a base triplet with A24 (Supplementary Figure S7F). If the isolated pairs and pseudoknotted stem are not taken into account, the structure appears to form a [6,1,8,3] four-way junction loop as originally reported (48). Alternatively, exclusion of the isolated pairs but inclusion of the pseudoknotted stem reveals a [6,1,4,0,3,2,3] seven-way junction loop. Thus, DSSR provides a new perspective on junction loops, which play critical roles in RNA folding and biological functions. The isolated U64–A85 pair additionally demonstrates that linchpin-like motifs may serve as a general stabilizing factor in RNA folding.
DSSR identifies three helices in the SAM-I riboswitch instead of the two previously reported (48). The helix containing the k-turn is broken into two pieces (61), due to a lack of stacking interactions around the kink (Supplementary Figure S6). It is also worth noting that DSSR uses only canonical pairs in characterizing pseudoknots. While the noncanonical U26–U67 pair was previously taken as part of a four-pair pseudoknot (48), DSSR denotes it as a [1,1] internal loop (Figure 5B). DSSR detects a total of eight base triplets (Supplementary Figure S7), including one where SAM interacts with A45 and U57. In addition to its involvement in this base triplet, SAM also forms other hydrogen-bonding (with G11, G58 and C59) and stacking (with C47) interactions.
The Cas9-sgRNA-DNA ternary complex
Application of DSSR to the crystal structure of the Streptococcus pyogenes CRISPR-associated endonuclease Cas9 in complex with its single guide RNA (sgRNA) and the DNA that the protein has cleaved (50) (PDB id: 4oo8) illustrates the capability of the program to treat RNA-DNA hybrid complexes (Figure 6 and Supplementary Figure S8). The software detects five helices and six stems in the structure. The guide RNA:target DNA hybrid stem (S1) and the repeat:anti-repeat RNA stem (S2) stack coaxially along the longest helix (26 base pairs). Each of the four remaining helices contains a single stem. The three stems (S1, S2 and S4) previously reported to form a three-way junction loop (50) do not form the ‘closed’ circle required by DSSR for such a loop. While the stems are indeed in close spatial proximity (near stem loop 1, Figure 6A), the DNA target strand (S1) and the 3′-end of sgRNA S4 (stem loop 1) are not connected. Thus, DSSR does not detect such a three-way junction loop, or the overall five-way junction loop suggested by the secondary structure diagram (Figure 6B and Supplementary Figure S8).
In addition to the two GAAA tetraloops (denoted as [4] hairpin loops in the DSSR output) and one AGU triloop (termed a [3] hairpin loop), DSSR also identifies a UA diloop (labeled as a [2] hairpin loop) that is closed by a Watson-Crick C–G pair (here referred to as a CUAG diloop with inclusion of the closing base pair in the name). The CUAG diloop (C55 to G58) lies in stem loop 1, a region recognized by the REC lobe of Cas9 and critical for the function of the protein (50). The flip-out of U59 (to make extensive interactions with the Cas9 protein) places the closing C55–G58 pair directly over the G54–C60 pair. As shown in Figure 6C, the CUAG diloop is strikingly similar to the UUGA diloop, which is recognized by the sequence-specific RNA binding sterile alpha motif domain of yeast post-transcriptional regulator Vts1p and referred to in the literature as part of a pentaloop (62). The structural features of the CUAG diloop may thus be related to the critical function that stem loop 1 plays in the Cas9 system, along lines similar to the key role played by the UUGA diloop in protein–RNA recognition.
DSSR also classifies the so-called CUUG tetraloop (63) as a diloop because of the closing Watson-Crick C–G pair and its distinction from a proper GNRA tetraloop. The CUUG fold, however, differs from the CUAG fold in terms of the twisting of the closing C–G pair and the G–C pair that stacks next to it (Figure 6C). While the CUUG diloop (63) has been well characterized along with the GNRA and UNCG tetraloops (64), we find no literature references to the CUAG (or more generally, CURG) diloop. A survey of the NR3A-dataset (20) for all diloops gives a total of 15 hits (Supplementary Figure S9), which can be categorized into five groups by base sequence: GGUC, with the second position G flipped away from the closing pair; CARG, with the second position A extruded into the minor-groove side of the closing pair; CUUG, with structural variations in the three crystallographic examples and differences from their NMR solution counterpart; CUAG, with all four instances found in Cas9 complexes (50,65); and UUKA (K for G/U), with irregular shapes.
DISCUSSION
DSSR uncovers a broad range of RNA structural information in a consistent, easily accessible framework. Starting from a 3D atomic coordinate file in either PDB or PDBx/mmCIF format, the software automatically identifies nucleotides, including those that are chemically modified. By employing a standard base reference frame (23) and simple geometric criteria, the program characterizes all existing base pairs, including noncanonical associations. DSSR detects multiplets (base triplets or even higher-order coplanar arrangements) by searching horizontally in the plane of the paired bases for further hydrogen bonding interactions. The program finds helices by exploring vertically in the neighborhood of selected pairs for stacking interactions, regardless of pairing type or backbone connectivity. DSSR defines a stem as a special type of helix made up of canonical pairs with continuous backbones, and describes coaxial stacking by the presence of two or more stems within a single helix. The program pinpoints isolated canonical pairs that lie outside of a stem and employs both stems and isolated pairs to delineate ‘closed’ loops of various types and to characterize pseudoknots of high complexity.
A rigorous comparison (66) between the diverse functionality of DSSR and each of the many existing RNA bioinformatics tools with which it overlaps is beyond the scope of the present work. Suffice it to say that DSSR performs individual tasks in unique ways, greatly extending yet still remaining consistent with our earlier treatment of double-helical DNA and RNA structures (6,7). Importantly, when the many features are combined, DSSR possesses, to the best of our knowledge, a much broader set of functionality for nucleic acid structural analysis than any existing method. Using the classic yeast tRNAPhe structure (39,49) as an example, DSSR detects 14 modified nucleotides, four base triplets (39), two helices corresponding to the L-shaped tertiary structure, four stems and three hairpin loops matching the cloverleaf secondary structure, and a [2,1,5,0] four-way junction loop, among other structural features (Table 1, Figure 2, Supplementary Figures S1 and S2, and Supplementary Sample Output.) We know of no other widely used structural analysis tools (4–5,8) with equivalent functionality.
DSSR differentiates a ‘helix’ from a ‘stem’, terms often used interchangeably in the literature (along with words like ‘arm’ or ‘paired region’) to describe a double-helical fragment. The helix/stem distinction introduced here leads naturally to a definition of coaxial stacking, another widely used concept. By definition, the loops identified in DSSR are ‘closed’, with successive nucleotides connected by either a covalent phosphodiester linkage or a canonical base pair. The program introduces a consistent notation for various loops, leading to the characterization of largely ignored diloops (Figure 6C and Supplementary Figure S9). Distinct from common practice (10,11), DSSR allows for pseudoknotted pairs in the delineation of junction loops (Figures 3–5), providing a novel perspective on RNA folding. The program is unique in pinpointing isolated canonical pairs, which are prevalent (Figures 2, 3, 5, 6) and perform critical roles in RNA folding and function (as demonstrated by the G–C linchpin pair of the viral tRNA mimic (34)). This new capability, to detect isolated canonical pairs, may allow the RNA community to uncover the wider range of roles potentially played by such residues.
Following standard conventions (13,35–36), DSSR by default uses only canonical pairs to identify stems and to characterize pseudoknots. Nevertheless, the program contains provisions for including noncanonical pairs in an extended definition of stems. DSSR-derived secondary structures are written in three commonly used formats (dot-bracket notation, connect table, bpseq) that can be easily connected to visualization tools such as VARNA (21), or to other algorithms for pseudoknot removal (14).
In summary, DSSR is an integrated computational tool, designed from the bottom up to streamline the analysis of RNA 3D structures. The program automatically characterizes nucleotides, base pairs, multiplets, pseudoknots, loops, stems, and coaxially stacked helices. By taking isolated canonical pairs and pseudoknotted stems into account, DSSR uncovers novel, intricate junction loops overlooked until now in the literature, even in small RNA molecules. Overall, DSSR has a combined set of functionalities well beyond the scope of any known specialized resources. The software is efficient and robust due to extensive tests against all nucleic-acid-containing structures in the PDB and continued refinements based on user feedback. DSSR can potentially serve as a cornerstone for RNA structural bioinformatics and will benefit a broad range of possible applications. For example, more complete knowledge of the building blocks of RNA and the spatial arrangements of these components in known high-resolution structures can help to distill the role of RNA tertiary organization in biological function, to predict the folding of long RNA molecules from nucleotide sequence, and to facilitate the design of RNA-binding ligands and the engineering of new supramolecular RNA materials.
Supplementary Material
Acknowledgments
We would like to thank Pascal Auffinger, Andrew Colasanti, Jan Hajic, Robert Hanson, Cathy Lawson, Marc Parisien and the user community for adopting earlier releases of DSSR and providing us with feedback. We thank Andrew Colasanti, Mauricio Esguerra, Jan Hajic, Cathy Lawson, Yin Yin Lu, John Trent and Huanwang Yang for critically reading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01GM096889 to X.J.L., R01HG003008 to H.J.B. and R01GM034809 to W.K.O.]. Funding for open access charge: National Institutes of Health [R01GM096889].
Conflict of interest statement. None declared.
REFERENCES
- 1.Moore P.B. Structural motifs in RNA. Annu. Rev. Biochem. 1999;68:287–300. doi: 10.1146/annurev.biochem.68.1.287. [DOI] [PubMed] [Google Scholar]
- 2.Noller H.F. RNA structure: reading the ribosome. Science. 2005;309:1508–1514. doi: 10.1126/science.1111771. [DOI] [PubMed] [Google Scholar]
- 3.Chworos A., Severcan I., Koyfman A.Y., Weinkam P., Oroudjev E., Hansma H.G., Jaeger L. Building programmable jigsaw puzzles with RNA. Science. 2004;306:2068–2072. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- 4.Lemieux S., Major F. RNA canonical and non-canonical base pairing types: a recognition method and complete repertoire. Nucleic Acids Res. 2002;30:4250–4263. doi: 10.1093/nar/gkf540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H., Jossinet F., Leontis N., Chen L., Westbrook J., Berman H., Westhof E. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003;31:3450–3460. doi: 10.1093/nar/gkg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu X.J., Olson W.K. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu X.J., Olson W.K. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarver M., Zirbel C.L., Stombaugh J., Mokdad A., Leontis N.B. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol. 2008;56:215–252. doi: 10.1007/s00285-007-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lescoute A., Westhof E. Topology of three-way junctions in folded RNAs. RNA. 2006;12:83–93. doi: 10.1261/rna.2208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bindewald E., Hayes R., Yingling Y.G., Kasprzak W., Shapiro B.A. RNAJunction: a database of RNA junctions and kissing loops for three-dimensional structural analysis and nanodesign. Nucleic Acids Res. 2008;36:D392–397. doi: 10.1093/nar/gkm842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laing C., Schlick T. Analysis of four-way junctions in RNA structures. J. Mol. Biol. 2009;390:547–559. doi: 10.1016/j.jmb.2009.04.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antczak M., Zok T., Popenda M., Lukasiak P., Adamiak R.W., Blazewicz J., Szachniuk M. RNApdbee—-a webserver to derive secondary structures from pdb files of knotted and unknotted RNAs. Nucleic Acids Res. 2014;42:W368–W372. doi: 10.1093/nar/gku330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taufer M., Licon A., Araiza R., Mireles D., van Batenburg F.H., Gultyaev A.P., Leung M.Y. PseudoBase++: an extension of PseudoBase for easy searching, formatting and visualization of pseudoknots. Nucleic Acids Res. 2009;37:D127–D135. doi: 10.1093/nar/gkn806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smit S., Rother K., Heringa J., Knight R. From knotted to nested RNA structures: a variety of computational methods for pseudoknot removal. RNA. 2008;14:410–416. doi: 10.1261/rna.881308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olson W.K., Esguerra M., Xin Y., Lu X.J. New information content in RNA base pairing deduced from quantitative analysis of high-resolution structures. Methods. 2009;47:177–186. doi: 10.1016/j.ymeth.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kempf G., Wild K., Sinning I. Structure of the complete bacterial SRP Alu domain. Nucleic Acids Res. 2014;42:12284–12294. doi: 10.1093/nar/gku883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown A., Amunts A., Bai X.C., Sugimoto Y., Edwards P.C., Murshudov G., Scheres S.H., Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science. 2014;346:718–722. doi: 10.1126/science.1258026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amunts A., Brown A., Toots J., Scheres S.H., Ramakrishnan V. The structure of the human mitochondrial ribosome. Science. 2015;348:95–98. doi: 10.1126/science.aaa1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leontis N.B., Zirbel C.L. In: RNA 3D Structure Analysis and Prediction. Leontis NB, Westhof E, editors. Vol. 27. Berlin; Heidelberg: Springer; 2012. pp. 281–298. [Google Scholar]
- 21.Darty K., Denise A., Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calladine C.R., Drew H.R., Luisi B.F., Travers A.A. Understanding DNA: The Molecule and How it Works. 3rd edn. San Diego, CA: Academic Press; 2004. [Google Scholar]
- 23.Olson W.K., Bansal M., Burley S.K., Dickerson R.E., Gerstein M., Harvey S.C., Heinemann U., Lu X.J., Neidle S., Shakked Z., et al. A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 2001;313:229–237. doi: 10.1006/jmbi.2001.4987. [DOI] [PubMed] [Google Scholar]
- 24.Gehring K., Leroy J.L., Gueron M. A tetrameric DNA structure with protonated cytosine·cytosine base pairs. Nature. 1993;363:561–565. doi: 10.1038/363561a0. [DOI] [PubMed] [Google Scholar]
- 25.Lu X.J., El Hassan M.A., Hunter C.A. Structure and conformation of helical nucleic acids: analysis program (SCHNAaP) J. Mol. Biol. 1997;273:668–680. doi: 10.1006/jmbi.1997.1346. [DOI] [PubMed] [Google Scholar]
- 26.Kimsey I.J., Petzold K., Sathyamoorthy B., Stein Z.W., Al-Hashimi H.M. Visualizing transient Watson-Crick-like mispairs in DNA and RNA duplexes. Nature. 2015;519:315–320. doi: 10.1038/nature14227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh V., Fedeles B.I., Essigmann J.M. Role of tautomerism in RNA biochemistry. RNA. 2015;21:1–13. doi: 10.1261/rna.048371.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavery R., Zakrzewska K., Sun J.S., Harvey S.C. A comprehensive classification of nucleic acid structural families based on strand direction and base pairing. Nucleic Acids Res. 1992;20:5011–5016. doi: 10.1093/nar/20.19.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose I.A., Hanson K.R., Wilkinson K.D., Wimmer M.J. A suggestion for naming faces of ring compounds. Proc. Natl. Acad. Sci. U.S.A. 1980;77:2439–2441. doi: 10.1073/pnas.77.5.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burkard M.E., Turner D.H., Tinoco I. Jr. In: The RNA World. Gesteland RF, Cech TR, Atkins JF, editors. NY: Cold Spring Harbor Laboratory Press; 1999. pp. 675–680. [Google Scholar]
- 31.Saenger W. Principles of Nucleic Acid Structure. NY: Springer-Verlag; 1984. [Google Scholar]
- 32.Leontis N.B., Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nissen P., Ippolito J.A., Ban N., Moore P.B., Steitz T.A. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colussi T.M., Costantino D.A., Hammond J.A., Ruehle G.M., Nix J.C., Kieft J.S. The structural basis of transfer RNA mimicry and conformational plasticity by a viral RNA. Nature. 2014;511:366–369. doi: 10.1038/nature13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuker M., Mathews D.H., Turner D.H. In: RNA Biochemistry and Biotechnology. Barciszewski J, Clark BFC, editors. Vol. 90. Kluwer Academic Publishers; 1999. pp. 11–43. [Google Scholar]
- 36.Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cannone J.J., Subramanian S., Schnare M.N., Collett J.R., D'Souza L.M., Du Y., Feng B., Lin N., Madabusi L.V., Muller K.M., et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cate J.H., Gooding A.R., Podell E., Zhou K., Golden B.L., Kundrot C.E., Cech T.R., Doudna J.A. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 39.Quigley G.J., Rich A. Structural domains of transfer RNA molecules. Science. 1976;194:796–806. doi: 10.1126/science.790568. [DOI] [PubMed] [Google Scholar]
- 40.Jucker F.M., Pardi A. GNRA tetraloops make a U-turn. RNA. 1995;1:219–222. [PMC free article] [PubMed] [Google Scholar]
- 41.Klein D.J., Schmeing T.M., Moore P.B., Steitz T.A. The kink-turn: a new RNA secondary structure motif. EMBO J. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanson R.M. Jmol—-a paradigm shift in crystallographic visualization. J. Appl. Crystallogr. 2010;43:1250–1260. [Google Scholar]
- 43.Toor N., Keating K.S., Taylor S.D., Pyle A.M. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klein D.J., Moore P.B., Steitz T.A. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 2004;340:141–177. doi: 10.1016/j.jmb.2004.03.076. [DOI] [PubMed] [Google Scholar]
- 45.Fischer N., Neumann P., Konevega A.L., Bock L.V., Ficner R., Rodnina M.V., Stark H. Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature. 2015;520:567–570. doi: 10.1038/nature14275. [DOI] [PubMed] [Google Scholar]
- 46.Ren A., Kosutic M., Rajashankar K.R., Frener M., Santner T., Westhof E., Micura R., Patel D.J. In-line alignment and Mg2+ coordination at the cleavage site of the env22 twister ribozyme. Nat. Commun. 2014;5:5534. doi: 10.1038/ncomms6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garreau de Loubresse N., Prokhorova I., Holtkamp W., Rodnina M.V., Yusupova G., Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- 48.Montange R.K., Batey R.T. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441:1172–1175. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- 49.Shi H., Moore P.B. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J., Ferre-D'Amare A.R. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature. 2013;500:363–366. doi: 10.1038/nature12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clauvelin N., Olson W.K., Tobias I. Characterization of the geometry and topology of DNA pictured as a discrete collection of atoms. J. Chem. Theory Comput. 2012;8:1092–1107. doi: 10.1021/ct200657e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zagryadskaya E.I., Doyon F.R., Steinberg S.V. Importance of the reverse Hoogsteen base pair 54–58 for tRNA function. Nucleic Acids Res. 2003;31:3946–3953. doi: 10.1093/nar/gkg448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juhling F., Morl M., Hartmann R.K., Sprinzl M., Stadler P.F., Putz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Limbach P.A., Crain P.F., McCloskey J.A. Summary: the modified nucleosides of RNA. Nucleic Acids Res. 1994;22:2183–2196. doi: 10.1093/nar/22.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Day H.A., Pavlou P., Waller Z.A. i-Motif DNA: structure, stability and targeting with ligands. Bioorg. Med. Chem. 2014;22:4407–4418. doi: 10.1016/j.bmc.2014.05.047. [DOI] [PubMed] [Google Scholar]
- 57.Lu X.J., Olson W.K., Bussemaker H.J. The RNA backbone plays a crucial role in mediating the intrinsic stability of the GpU dinucleotide platform and the GpUpA/GpA miniduplex. Nucleic Acids Res. 2010;38:4868–4876. doi: 10.1093/nar/gkq155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roth A., Weinberg Z., Chen A.G., Kim P.B., Ames T.D., Breaker R.R. A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat. Chem. Biol. 2014;10:56–60. doi: 10.1038/nchembio.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y., Wilson T.J., McPhee S.A., Lilley D.M. Crystal structure and mechanistic investigation of the twister ribozyme. Nat. Chem. Biol. 2014;10:739–744. doi: 10.1038/nchembio.1587. [DOI] [PubMed] [Google Scholar]
- 60.Price I.R., Grigg J.C., Ke A. Common themes and differences in SAM recognition among SAM riboswitches. Biochim. Biophys. Acta. 2014;1839:931–938. doi: 10.1016/j.bbagrm.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lilley D.M. The structure and folding of kink turns in RNA. Wiley Interdiscip. Rev. RNA. 2012;3:797–805. doi: 10.1002/wrna.1136. [DOI] [PubMed] [Google Scholar]
- 62.Aviv T., Lin Z., Ben-Ari G., Smibert C.A., Sicheri F. Sequence-specific recognition of RNA hairpins by the SAM domain of Vts1p. Nat. Struct. Mol. Biol. 2006;13:168–176. doi: 10.1038/nsmb1053. [DOI] [PubMed] [Google Scholar]
- 63.Jucker F.M., Pardi A. Solution structure of the CUUG hairpin loop: a novel RNA tetraloop motif. Biochemistry. 1995;34:14416–14427. doi: 10.1021/bi00044a019. [DOI] [PubMed] [Google Scholar]
- 64.Woese C.R., Winker S., Gutell R.R. Architecture of ribosomal RNA: constraints on the sequence of ‘tetra-loops’. Proc. Natl. Acad. Sci. U.S.A. 1990;87:8467–8471. doi: 10.1073/pnas.87.21.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anders C., Niewoehner O., Duerst A., Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu X.J., Olson W.K. Resolving the discrepancies among nucleic acid conformational analyses. J. Mol. Biol. 1999;285:1563–1575. doi: 10.1006/jmbi.1998.2390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.