


Abstract
Selenocysteine (Sec) is found in the catalytic centers of many selenoproteins and plays important roles in living organisms. Malfunctions of selenoproteins lead to various human disorders including cancer. Known as the 21st amino acid, the biosynthesis of Sec involves unusual pathways consisting of several stages. While the later stages of the pathways are well elucidated, the molecular basis of the first stage—the serylation of Sec-specific tRNA (tRNASec) catalyzed by seryl-tRNA synthetase (SerRS)—is unclear. Here we present two cocrystal structures of human SerRS bound with tRNASec in different stoichiometry and confirm the formation of both complexes in solution by various characterization techniques. We discovered that the enzyme mainly recognizes the backbone of the long variable arm of tRNASec with few base-specific contacts. The N-terminal coiled-coil region works like a long-range lever to precisely direct tRNA 3′ end to the other protein subunit for aminoacylation in a conformation-dependent manner. Restraints of the flexibility of the coiled-coil greatly reduce serylation efficiencies. Lastly, modeling studies suggest that the local differences present in the D- and T-regions as well as the characteristic U20:G19:C56 base triple in tRNASec may allow SerRS to distinguish tRNASec from closely related tRNASer substrate.
INTRODUCTION
Aminoacyl-tRNA synthetases (aaRSs) catalyze the attachment of specific amino acids to the 3′ end of cognate tRNA molecules for protein synthesis. The reaction occurs as a two-step process, in which an enzyme-bound amino acid is first condensed with ATP to form a molecule of aminoacyl adenylate, and it is subsequently transferred to the 3′ terminal adenosine ribose of tRNA (1). Each of the 20 common amino acids corresponds to a specific aaRS in general. However, the 21st amino acid selenocysteine (Sec) lacks a proprietary aaRS in all three domains of life (2,3). Sec-tRNASec is synthesized by the conversion of serine through a multistep process in a Sec-specific tRNA (tRNASec)-dependent manner. All selenoproteins are selenium-dependent enzymes, generally with Sec at their active sites, and many of them are essential for organismal viability (4,5). In Sec biosynthesis, tRNASec is first aminoacylated with serine by seryl-tRNA synthetase (SerRS) to produce Ser-tRNASec. The following step is species-dependent: in bacteria, Sec synthetase (SelA) converts Ser-tRNASec to Sec-tRNASec in a single-step reaction; in contrast, archaea and eukaryotes carry on the synthesis through an intermediate step where the serine moiety is phosphorylated by O-phosphoseryl-tRNA kinase (PSTK) to produce O-phosphoseryl-tRNASec (Sep-tRNASec) (6), which in turn becomes the substrate of O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthetase (SepSecS) to generate Sec-tRNASec (7) (Supplementary Figure S1).
Dimeric SerRS is a class II aaRS (8–11), whose catalytic aminoacylation domain (AD) is formed by a seven-stranded antiparallel β-sheet and contains three conserved motifs. In all organisms except for methanogens, SerRSs also possess a characteristic N-terminal tRNA-binding domain (TBD), which is composed of two long α-helices (coiled-coil region), protruding away from the AD. Compared to lower organisms, vertebrate SerRSs also display a ∼30-residue C-terminal extension named the UNE-S domain, and two other insertion domains named insertion I and insertion II, respectively (12). Besides the well-characterized aminoacylation functions, mammalian SerRSs play a non-canonical role in the process of vascular development. This alternative function requires the UNE-S domain (12–14) and is independent of the aminoacylation activity (15).
The cocrystal structure of T. thermophilus SerRS (TtSerRS) complexed with tRNASer (TttRNASer) was determined to 2.9 Å more than two decades ago (PDB code 1SER) (16). By contrast, cocrystal structures of tRNASec have not been available until recently. In the heterologous complex formed between A. aeolicus tRNASec and M. kandleri SerRS, the N-terminal domain of the non-canonical SerRS interacts with the extra arm stem and the outer corner of tRNASec (17). Addtionally, the crystal structure of A. aeolicus SelA complexed with tRNASec shows that the N-terminal domain of the protein interacts with the D-arm of tRNA, and a large cleft between two protein subunits accommodates the 3′ terminal region of Ser-tRNASec (18). The crystal structure of archaeal PSTK in complex with tRNASec indicates that PSTK distinguishes Ser-tRNASec from Ser-tRNASer by recognizing the characteristic D-arm of the former (19). The crystal structure of human tRNASec in complex with SepSecS shows that SepSecS employs a primordial tRNA-dependent catalytic mechanism where the enzyme makes contacts with the acceptor-, TψC- and variable arms of Ser-tRNASec (20). Lastly, the structure of hSerRS complxed with the Ser-AMP analog 5′-O-(N-(L-seryl)-sulfamoyl) adenosine (Ser-SA) shows that the binding of Ser-SA dramatically leverages the position of the TBD over a 70 Å distance (PDB code 4L87). Interestingly, this leverage is specific to higher eukaryotes but is not seen in bacterial, archaeal and lower eukaryotic SerRSs (21).
All these complex structures provide great insights into the important steps in the Sec biosynthesis pathway. But to date, no structural information is available for the serylation step in terms of the interaction mode between the enzyme and the tRNA substrate, i.e. the serylation details of the Sec biosynthesis are still missing (Supplementary Figure S1). Here we report two crystal structures of human SerRS (hSerRS)-tRNASec complex in different stoichiometry. We also demonstrate that both complexes are present in solution as observed in crystals. The structures address the recognition mechanism of tRNA substrates and allow for better understanding the dual substrate specificity of SerRS for both tRNASec and tRNASer.
MATERIALS AND METHODS
Cloning, expression and purification of hSerRS and mutants
The wild-type (WT) full-length hSerRS gene SARS (GenBank Accession No. BAA95602) was amplified from cDNA library of human 293T cell, which contains 514 residues. It was cloned into the expression vector pET-20b (+) vector (Novagen) with the restriction sites NheI and XhoI. The E156T/R157S, E447K and other mutants were generated by the quikchange method (Stratagene) with the WT plasmid as the template. All the primers used in this study were listed in Supplementary Table S1.
The plasmids of WT SerRS and mutants were transformed into the E. coli strain BL21 (DE3) cells for overexpression. An overnight WT culture was grown in Luria–Bertani broth containing 50 μg/ml ampicillin. A 2 l fresh culture medium was inoculated with 10 ml of the overnight culture. When A600 reached 0.6–0.8 at 37°C, the expression of SerRS was induced by 0.2 mM isopropyl β-D-thiogalactopyranoside (IPTG) and was kept shaking overnight at 25°C. The E. coli cells were then harvested by centrifugation at 4500 rpm for 20 min and resuspended in pre-cooled nickel-nitrilotriacetic acid (Ni-NTA) buffer A (20 mM Tris-HCl (pH 8.0), 250 mM NaCl, 10 mM imidazole, 1 mM β-mercaptoethanol (β-ME) and 1 mM PMSF. The cells were disrupted by ultrasonication and the supernatant was obtained by centrifugation at 14 000 rpm for 1 h at 4°C. The supernatant was then applied to Ni-NTA affinity column (Qiagen), which was previously equilibrated with Ni-NTA buffer A. The target protein was eluted with Ni-NTA buffer B (20 mM Tris-HCl (pH 8.0), 250 mM NaCl, 250 mM imidazole, 1 mM β-ME and 1 mM PMSF). The SerRS-containing fractions were pooled, dialyzed in a buffer containing 20 mM Tris-HCl (pH 8.0), 50 mM NaCl and 1 mM DTT. SerRS was further purified by anion exchange chromatography on a Q-HP column (GE Healthcare) with a NaCl gradient, and SerRS was eluted at ∼250 mM NaCl. The final separation was performed by size-exclusion chromatography with a Superdex 200 column (10/30), and the protein was eluted with 20 mM HEPES (pH 7.0), 75 mM NaCl and 1 mM DTT. The pure protein was collected and concentrated to 4 mg/ml. The aliquoted protein was flash-frozen and stored at −80°C until further use. For mutants to be tested with aminoacylation activity assays, 5% glycerol was added to the concentrated protein before it was frozen. Additionally, for the C46S and C117S mutants, no reducing reagent was added during the process of protein extraction and purification. All the purified mutants displayed a symmetrical peak on the size-exclusion column, suggesting that they were well folded.
In-vitro transcription of tRNA substrate
Synthetic oligos corresponding to the T7 promoter plus tRNASec-encoding sequences were ligated into the pUC19 vector with the restriction sites HindIII and XbaI. The transcription template was produced by PCR amplification of the ligated DNA fragment. The PCR product was phenol-extracted and precipitated with 95% ethanol after storage at −80°C for 2 h. The dry pellet of DNA after precipitation was redissolved in DEPC-treated water to a concentration ∼400 μg/ml. The in vitro transcription was carried out for 4 h at 37°C in a mixture containing 2.5 mM of each NTP,20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 20 mM MgCl2, 5 mM DTT, 1 mM spermidine and 0.3 μM T7 RNA polymerase. The transcribed tRNA was purified by 12% denaturing Urea-PAGE gel, extracted, and was then ethanol-precipitated. The pellet was washed and redissolved in a buffer containing 10 mM sodium arsenate (pH 6.5) to a concentration 5 mg/ml. tRNA was annealed by heating to 65°C and allowed to cool to room temperature after addition of 10 mM MgCl2. The annealed RNA was stored at −80°C for further use. Other tRNASec mutants were prepared using the same protocol and all the tRNA sequences used in this study were listed in Supplementary Table S2.
Crystallization, data collection and structure determination
High-throughput crystallization screen was performed using the Mosquito liquid transfer robot (TTP Labtech) and the sitting-drop vapor diffusion method. In order to obtain the best crystals possible for structure determination, we introduced point mutations to both the protein and tRNA constructs for crystallization. These mutations greatly enhanced the diffraction quality of the cocrystals and finally led to the crystallization of the two complex structures. For crystallization of the 2:1 complex, E447K was mixed with tRNASec(G-C) at a molar ratio of 2:1.2 in a buffer containing 2 mM serine, 2 mM AMPPNP, 5 mM β-ME and 5 mM MgCl2. Cocrystals of the 2:1 complex were found in 12% PEG 3350, 0.2 M NaCl and 0.1 M Tris-HCl (pH 7.5). After optimization, the best crystals were produced in a reservoir solution with 18% PEG 3350, 0.1 M NaCl, 0.1 M Tris-HCl (pH 8.0) and 0.1 M sodium malonate (pH 7.0). The 2:2 complex was prepared by mixing the E156T/R157S double mutant with tRNASec(G-G) at a molar ratio of 1:2.4, and MgCl2 was omitted. All crystals were grown at 25°C and the fully-grown crystals were soaked for 2 min in a cryoprotective solution containing all the components of the reservoir solution plus 20% (v/v) ethylene glycol. The soaked crystals were mounted on a nylon loop and were flash-frozen in liquid nitrogen.
Data were collected from frozen crystals at −173°C using Beamline 17U (BL17U) at the Shanghai Synchrontron Radiation Facility (SSRF, Shanghai, P.R.China). The two data sets were processed with the program HKL2000 (22). The E447K cocrystals belong to space group P21212 and diffract to a resolution of 3.55 Å while the E156T/R157S cocrystals belong to P21 and diffract to 4.0 Å resolution. The structures of the complexes were solved by molecular replacement (MR) using PHENIX (23). Both the coordinates of the ligand-bound SerRS (PDB code 4L87 (21) and the coordinates of tRNASec (PDB code 3A3A (24)) were used as the search models and both components were searched simultaneously. The initial models generated by MR for the E156T/R157S cocrystals contain a protein dimer and two tRNA molecules in the asymmetric unit while the E447K cocrystals contain a protein dimer and one tRNA molecule. The initial models of both complexes had large conformational changes in the TBD and were manually built with the program COOT (25). The models were then fed to the refinement program PHENIX.REFINE (26). Multiple cycles of refinement with NCS implemented for the AD within the dimer (N153-A514) were carried out and alternated with model rebuilding. The final Rfree and R-factors for the E156T/R157S-tRNASec-serine-AMPPNP complex were 0.3220 and 0.2736, while for the E447K-tRNASec-serine-AMPPNP complex were 0.3140 and 0.2530, respectively (Supplementary Table S3).
Aminoacylation assay
The reaction was performed at 25°C, in a buffer containing 150 mM HEPES (pH 7.5), 20 mM KCl, 20 μM L-serine, 4 mM MgCl2, 2 mM DTT, 4 mM ATP, 15 μM annealed human tRNASec or tRNASer(UGA) and 2 μM [3H]-L-serine (PerkinElmer, 22 Ci/mmol). 200 nM SerRS or mutants were added to initiate the reaction. Aliquots of the reaction were transferred to filter papers pre-soaked with 5% TCA, and washed three times with 5% ice-cold TCA and twice with 95% ethanol. The radioactivity of the acid-insoluble fractions was quantified by liquid scintillation counting. For variants to be used in the oxidation-reduction experiments, no reducing agent was added during the purification process. After the protein purification, the oxidation reaction was carried out by incubation with 1 mM GSSG for 1 h while the reduction reaction was carried out by treatment with 10 mM DTT for 12 h after dialyzing away the extra GSSG.
Electrophoretic mobility shift assay (EMSA)
The binding reactions between tRNA and SerRS were incubated on ice for 30 min in 5 μl buffer containing 20 mM HEPES (pH 7.0), 75 mM NaCl, 2 mM serine, 2 mM AMPPNP, 5 mM β-ME and 5 mM MgCl2. Two identical sets of binding reactions were set up at molar ratios of 2:0.3, 2:0.6, 2:1.2, 2:2.4 and 2:4.8 (SerRS/tRNA). 8.53 μM E447K was mixed with increasing different amounts of tRNASec(G-C) or 5.15 μM tRNASec(G-C) was mixed with decreasing amounts of SerRS according to the ratios shown above. Samples of each set were mixed with 5 μl 0.5X TBE buffer (pH 8.3) containing 30% glycerol and 20 μl sample was loaded onto a 4% polyacrylamide gel separately. Electrophoresis was performed at 4°C for 1.5 h at 80 V after pre-running the gel for 30 min, with 0.5X TBE buffer as the running buffer. The two gels were run in parallel and were stained either with ethidium bromide or commassie brilliant blue.
Size-exclusion chromatography analysis
The size-exclusion chromatography analysis was performed on a Superdex 200 column (10/30), pre-equilibrated with 20 mM HEPES (pH 7.0), 75 mM NaCl and 1 mM DTT. Samples were prepared in the same fashion as in the EMSA assay at the molar ratios of 2:0.3, 2:0.6, 2:1.2 and 2:2.4. The protein concentration was fixed at 50 μM. 100 μl complex of each sample was incubated for 30 min at 4°C before being injected into the column at a flow rate of 0.5 ml/min. The elution profile was monitored at 254 and 280 nm, and fractions of each peak were collected for gel analysis.
Dynamic light scattering (DLS) measurements
DLS measurements were carried out with a photogoniometer (plate reader, Wyatt Technology). The protein concentration was fixed throughout at 4.5 μM and the complexes were formed at molar ratios ranging from 2:0.9 to 2:1.4, with fine increments of RNA. Samples were subjected to a 2000 rpm centrifugation for 15 s to remove large particles or air bubbles before readout in the temperature-controlled DynaPro plate reader (Wyatt Technology). Each sample was run in triplicates and each well was measured 10 times, with 1 s acquisition time.
RESULTS
The overall structure of the 2:1 and 2:2 complexes
The full-length hSerRS protein was used for crystallization and contains 520 residues, including a C-terminal 6×His tag (Figure 1A). In the process of crystal optimization, we first changed the C2 base in the original tRNASec sequence to a G. The base opposite to this G was either changed to a complementary base C or left unchanged. The two resulting tRNA molecules, designated tRNASec(G-C) and tRNASec(G-G) respectively, were used in separate cocrystallization trials (Figure 1B). Activity assays show that the WT enzyme retains at least 2/3 activity toward the tRNASec(G-G) substrate (Supplementary Figure S2). We next generated two rounds of mutagenesis on the protein with the purpose to generate diffraction crystals. The first round of mutagenesis was mainly based on the crystal structure of the TtSerRS-TttRNASer complex (PDB code 1SER) (16) and we mutated many non-conserved residues in close range to the negatively tRNA molecule to positively charged lysine or arginine residues, in order to form stronger electrostatic interactions between hSerRS and tRNA. This allows us to obtain some low-resolution cocrystal structures (worse than 4.0 Å). We subsequently analyzed the crystal packing patterns of these structures, and performed the second round of mutagenesis to create possible specific interactions (namely hydrogen bonds or salt bridges) between the symmetry molecules. We tested as many as 34 mutants, among which the E447K and E156T/R157S mutants gave the best results after extensive screening and led to two forms of cocrystals: the E447K-tRNASec(G-C) complex diffracted to 3.55 Å, and there were a hSerRS dimer and one tRNA molecule in the asymmetric unit (named the 2:1 complex hereafter); the E156T/R157S-tRNASec(G-G) complex diffracted to 4.0 Å and there were a hSerRS dimer and two tRNA molecules in the asymmetric unit (named the 2:2 complex).
Figure 1.
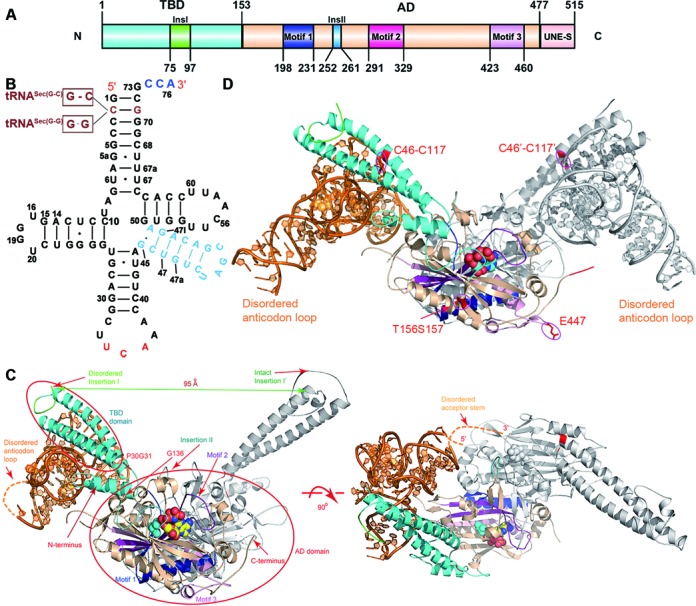
The overall view of the hSerRS-tRNASec complexes. (A) The domain architecture of hSerRS. The TBD and the AD are colored cyan and wheat, respectively. The three signature motifs 1–3 are colored blue, purple and light pink, respectively. Insertion I is green and Insertion II is light blue. (B) The secondary structure of tRNA molecules used in this study. The 3′ terminal CCA tail is colored blue, the anticodon triplet and the variable arm are colored red and light blue, respectively. tRNASec(G-C) and tRNASec(G-G) (depicted brown) are mutant tRNAs with alterations to the second base pair (the C2-G71 base pair) in the original tRNASec sequence. The short lines represent the Watson–Crick base pairs and the dots represent non-Watson–Crick base pairs. (C) Two orthogonal views of the 2:1 complex in ribbon rendition. The active site AMPPNP and serine are shown as spheres. The domain color scheme of monomer 1 is as in Figure 1A and monomer 2 is gray. tRNA is in orange. P30G31 and G136 are marked red and indicated by the red arrows. (D) The view of the 2:2 complex in ribbon rendition. The C46–C117 disulfide bonds on the two monomers are circled and the positions of the E447 and T156S157 residues (mutated from the E156R157 dipeptide) are also indicated.
In the 2:1 complex, protein subunit 1 (the tRNA-bound subunit) of the hSerRS dimer is visible from V2-I476 except for residues G65-L86 and K414-K415; subunit 2 (the free subunit) is free from internal disorder from V2-P475. The tRNA acceptor end (G1-C4, C68-A76) and the anticodon loop (G30-U43) are not resolved (Figure 1C and Supplementary Figure S3A). The core region of tRNA comes into contacts with the coiled-coil region of subunit 1 while the acceptor end points to the catalytic region of subunit 2. In the 2:2 complex structure, residues V2-E72, L85-Y410, M417-E479 are visible for protein subunit 1; subunit 2 is ordered from V2-K69, E82-D88 and A92-A474 (Figure 1D and Supplementary Figure S3B). Both tRNA chains resolve more residues and only U34-C35 and G73-A76 of chain B, and U34-C35 and C72-A76 of chain D are missing. The structure closely resembles the 2:1 complex structure in most of the region. Like the apoenzyme, the UNE-S domains in both complexes are invisible.
hSerRS–tRNASec interactions
Due to its higher diffraction resolution, we will focus on the 2:1 complex crystal structure to discuss the protein–tRNA interactions. The interactions are mainly formed between coiled-coil region of the synthetase and the 16-nt variable arm, the D- and TψC loops of tRNASec. The tRNASec-facing side in the first long helix of the TBD is mainly comprised of charged or polar residues (referred to as helix 1, Figure 2A and Supplementary Figure S4). The OD2 atom of the highly conserved residue D51 is only 3.06 Å from the N2 atom of G47a and also within 4 Å distance from the O2′ atom of the ribose in A47k and the O2 atom of C47j (3.96 and 3.93 Å, respectively). To assess the importance of the critical residues, we created a series of mutants and performed activity assays. We first discovered that 200 nM WT hSerRS keeps a linear initial velocity for up to 15 min under specified reaction conditions (inset in Figure 2B). For the mutants, we only measured the activities at 2- and 5-min time points to ensure the time points that we chose were well within the linear range. Compared to WT, the activity of the D51A mutant is undetectable (Figure 2A, B and Supplementary Figure S5). Semiconserved N54 can donate a hydrogen bond to the O2′ atom of the ribose in A47k while the invariant N58 is capable of forming two hydrogen bonds with the non-bridging oxygens of the phosphate in G47l as well as that of A47k. Similarly, D51 and R47 are near A47k for interactions with the sugar ring, while S61 is also likely to donate a hydrogen bond to the N3 atom of U20. The substitutions of these residues to alanines reduced aminoacylation activities to original 1/3–1/2. Furthermore, two highly conserved residues, R9 and R44 possibly form salt bridges with the phosphate oxygens of U47b and C47c (Figure 2C). Mutations of these two residues to alanines reduced the activities by more than 6- and 50-fold, respectively, suggesting their important roles in tRNA recognition. Interestingly, E447K displayed even slightly higher activity than that of WT enzyme, whereas the double-mutant E156T/R157S retained only ∼10% activity of WT for reasons that are not clear (Figure 2B). Finally, K104 and R107, two basic residues at the beginning of the second long helix (α4 in Supplementary Figure S6, referred to as helix 2) in the TBD, are in close distance from the sugar ring and the phosphate oxygen of C56, respectively, and may form contacts from the back of tRNA elbow (Figure 2D). Their alanine substitutions decreased the activities to ∼1/3 of WT (Figure 2B). One should note that at 3.55 Å resolution, the specific interactions can not always be determined with absolute certainty. But in combination with the results from the sequence alignment and activity assays, we can fairly assess the contribution of individual residues to tRNA recognition and aminoacylation (Figure 2E).
Figure 2.
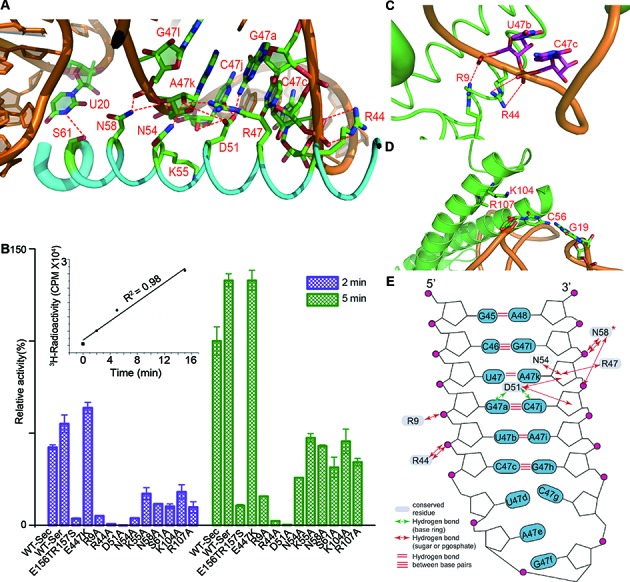
Substrate recognition by hSerRS. (A) The interactions of helix 1 with the minor groove of the variable region. Interactions within 4 Å distance are indicated by red dotted lines. (B) The time course of serylation activity assays of the mutants that are involved in WT tRNASec recognition. Two sets of data are shown, representing the measurements at 2- (purple) and 5-min time points (green), respectively. The activity of WT hSerRS with WT tRNASec at the 5-min time point is regarded as 100%, and the activity of each mutant with WT tRNASec is compared to that of WT hSerRS. The readings at time point zero are used as blanks and the error bars are calculated from three measurements. The time course of assay of the WT enzyme is shown in the inset. (C) The interactions of R9 and R44 with tRNA residues U47b and C47c at the variable region. Note that the well-resolved side chain of R9 forms a hydrogen bond with the U47b phosphate oxygen. (D) The interactions of two positively charged residues K104 and R107 from helix 2, with the G19-C56 base pair. The R107 side chain is invisible in the electron density map and is only modeled to Cβ. (E) A diagram of the protein-tRNA interactions in the variable region. Hydrogen bond distances smaller than 4.0 Å are indicated by red, double-headed arrows. N58 is an absolutely conserved residue (marked with a star).
Structural changes during formation of the complex
The structural changes induced by enzyme-substrate binding in tRNASec are small. tRNASec retains the basic shape of the free form of human tRNASec (PDB code 3A3A)(24). They both resemble that of mouse tRNASec (PDB code 3RG5)(27), with only minor deviations in the variable and the anticodon region (Figure 3A). In terms of hSerRS, the orientations of the TBDs are quite different, as revealed by the superposition of hSerRS structures of the apoenzyme (PDB code 3VBB)(12), Ser-SA-bound complex (PDB code 4L87)(21) and tRNA-bound complex, respectively (the 2:1 complex, Figure 3B). tRNA induces similar conformational changes in the TBD domain as does Ser-SA, which is significantly different from the apo form.
Figure 3.
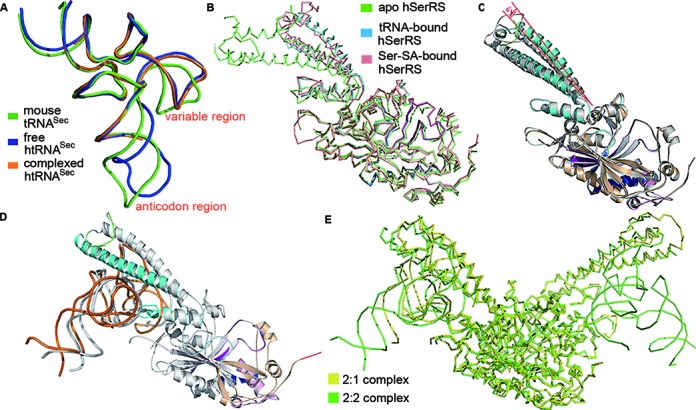
Structural changes induced by tRNA binding. (A) The structural superposition of the free human tRNASec (blue, PDB 3A3A), complexed human tRNASec (orange) and the free mouse tRNASec (green, PDB 3RG5). (B) The backbone alignment of the free hSerRS (green, PDB 3VBB), Ser-SA-bound hSerRS (salmon, PDB 4L87) and tRNA-bound hSerRS in the 2:1 complex (blue). (C) The overlay of the two monomers of the 2:1 complex. There is a 6°-angle rotation in the helical TBD due to the binding of tRNA, while the ADs superimpose well. (D) The overlay of the two monomers of the 2:2 complex. One subunit is in color (tRNA in orange) as in Figure 1A while the other is in gray. (E) The backbone overlay of the ADs of the 2:2 and 2:1 complexes. hSerRS in the 2:2 complex is colored green while in the 2:1 complex is yellow.
Comparison of the conformation of the protein monomers in the 2:1 complex suggests that two subunits are also quite different in the TBD regions: first, subunit 1 is missing ∼20 residues in the insertion I region while its counterpart in the other monomer is intact. The disorder is most likely caused by clashes with the incoming tRNA molecule. Second, the two α4-helices in the TBDs rotate an angle of ∼6° from one to another, and their RMSD is relatively large (1.4 Å over 112 Cαs) (Figure 3C). The ADs of the two subunits are similar, with a RMSD of 0.8 Å over 320 Cαs. In the 2:2 complex, the two subunits are more similar to each other, but larger differences are also observed in the TBDs. Additionally, the two tRNA molecules in the 2:2 complex are not in the exact same positions relative to their TBDs. By rotating 180° around the 2-fold axis of the dimer, we can nearly superimpose the protein molecules (Figure 3D). However, the acceptor stem of chain B is closer to the TBD of the other subunit, suggesting a tighter binding of chain B than chain D. Additionally, the anticodon regions of tRNAs are at large variations, which is probably due to the fewer contacts with the enzyme in these regions.
Lastly, the structures of two complexes also exhibit subtle differences. The tRNA position in the 2:2 complex is closer to the active site than that in the 2:1 complex although the TBD domains in the two complexes align well (Figure 3E). These structural differences of the protein and RNA molecules between the two complexes or within the same complex, may reflect the intrinsic flexibility and constant adjustments of the TBD to external events like the binding of tRNA substrate.
Flexibility of TBD during serylation
To test how the flexibility of the TBD affects catalytic efficiencies of hSerRS, we introduced the two mutations at the hinge region of the coiled-coil region: P30G31Y (the mutation of the P30G31 dipeptide to a single tyrosine) and G136V (Figure 4A). These two mutations are intended to make the residues bulkier and hence to restrict the movements of the helical domain. Notably, the P30G31Y mutant almost eliminated serylation while the other is only about 15% as active as that of WT (Figure 4B).
Figure 4.
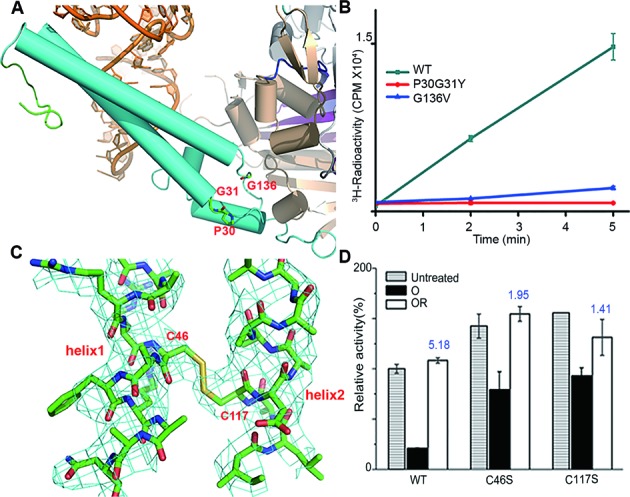
The structural flexibility of the TBD and evaluation of its importance to activities. (A) The residues P30G31 and G136 at the hinge region. Helices of the protein are represented by cylinders. (B) The time course of serylation activity assays of P30G31Y and G136V measured at the 2-, and 5-min time points. The experiment is performed in quadruplicate. (C) The C46-C117 disulfide bond of chain is shown in 2Fo-Fc maps. The map is contoured at 1σ. (D) Evaluation of the oxidation/reduction effects on WT, C46S and C117S mutants by activity assays. Untreated samples are represented by the bars with horizontal stripes; the oxidized samples are represented by the black solid bars and indicated by ‘O’, while samples treated by oxidation followed by reduction are represented by the white solid bars and indicated by ‘OR’. The numbers on top of the white bars indicate the folds of activity gain, calculated by dividing the ‘OR’ sample activity by the ‘O’ sample activity. The activity of untreated WT hSerRS at the 5-min time point is 100% and the readings at time point zero of the variants are used as blanks. The error bars are calculated from three measurements.
Interestingly, an intra-subunit disulfide bond between C46 (from helix 1 of the TBD) and C117 (from helix 2 of the TBD) is discovered in each monomer of the 2:2 complex (Figure 4C). The difference map shows obvious continuous positive electron density when the two cysteines are truncated to Cβs (Supplementary Figure S7). During crystallization, pre-added dithiothreitol (DTT) might have lost the reducing power during prolonged exposure to the air and leads to the formation of a disulfide bond between C46 and C117. Since the disulfide bridge may greatly constrain the relative movements of the two helices in the TBD and thus bundles them together, we suspected that this unforeseen linkage might play a role in aminoacylation. Since the formation of the disulfide bond could be easily controlled by oxidation with oxidized glutathione (GSSG), and by reduction with DTT, we could switch on and off the disulfide bridge and examine its effects by activity assays.We first checked the activity loss by oxidizing the WT enzyme with 1 mM GSSG, which only retained ∼16% activity of the untreated enzyme. But the enzyme after the oxidation followed by 10 mM DTT reduction fully recovered its activity (Figure 4D). The WT activity gain (defined as the ratio of WT activity with reduction to that with oxidation) is ∼5.2-fold. In comparison, after the same oxidation-reduction cycle, the activity gain of the C46S mutant is only 1.95-fold. A similar trend is also exhibited by the C117S mutant, but to an even lesser extent (1.4-fold). On the other hand, the untreated C46S and C117S mutants exhibit even higher serylation activity than that of WT, suggesting that WT enzyme has been already partially oxidized during the course of protein purification (Figure 4D). Therefore, both cysteine-to-serine mutants are less affected in serylation by the oxidation-reduction cycle treatment as reflected by the activity gains, due to their inability to form the disulfide bridge. These results confirmed our hypothesis and indicated that free movements of the TBD are important to the aminoacylation functions of hSerRS, presumably because the movements induced by the binding of tRNA at the TBD are coupled to subsequent binding of the acceptor end of tRNA at the active site.
Characterization of the complex formation in solution
We observed two types of SerRS-tRNA complexes in crystalline state, and we then tested whether we could obtain the same result in solution. We first checked the elution profile of the complexes formed at various protein/RNA molar ratios on an analytical Superdex 200 column (10/30, GE healthcare). E447K and tRNASec were mixed in the presence of excessive AMPPNP, in molar ratios ranging from 2:0.3 to 2:4.8. tRNASec amount was increased gradually while the E447K amount was kept constant (Figure 5A). Starting from the 2:0.3 ratio, an extra, faster-moving peak at 11.96 ml forms, indicating the formation of the 2:1 complex. Increase of the ratio to 2:0.6 greatly promoted the peak formation of another complex that eluted at 11.72 ml (the 2:2 complex). As a result, two peaks are obvious although they are still partially overlapping. From ratios 2:1.2 to 2:2.4, two better-resolved, symmetrical peaks are generated. Additionally, the 2:2 complex species dominates over the 2:1 complex. Each fraction of the peaks was analyzed by silver staining of the SDS-PAGE gel. The intensities of the protein and RNA bands increase with RNA, indicating the formation of larger amounts of the complexes (Supplementary Figure S8).
Figure 5.
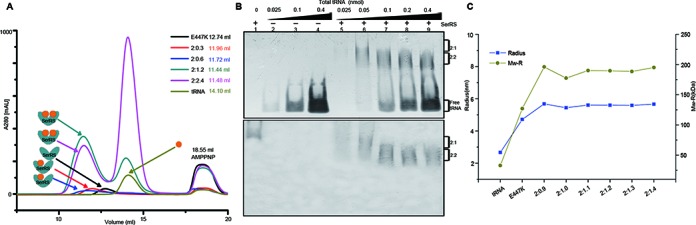
Characterization of solution behavior of hSerRS. (A) The chromatograms of free tRNA, the E447K mutant and complexes formed at different molar ratios on a Superdex 200 column (10/30). The species eluted from the column are represented by the solid circles, while the orange circles stand for tRNA molecule and the dark cyan V-shaped symbol for the hSerRS dimer. The retention volumes of each eluted species (indicated by the arrows) are given in the top right corner. (B) EMSA of hSerRS-tRNA complexes with a constant amount of protein (167 pmol) and increasing amounts of tRNA (molar ratios of protein to RNA ranging from 2:0.3 to 2:4.8). The two identical sets of samples are stained by ethidium bromide (upper panel) and commassie blue (lower panel) after gel electrophoresis. (C) The DLS analyses of hSerRS-tRNA complexes. The left vertical axis indicates the particle sizes of the species while the right vertical axis indicates the molecular weights of the major species in solution estimated by DLS. The horizontal axis indicates different molar ratios of protein to RNA.
Next, we performed EMSA to evaluate the contribution of the two types of complexes. By keeping the protein amount constant while increasing RNA, we saw the formation of the 2:1 complex at ratios of 2:0.3–2:0.6. Starting from 2:1.2, we observed a new, faster-migrating band (Figure 5B), suggesting the binding of the second tRNA molecule. Additionally, comparison of the unbound RNA in the complex samples with the control samples on the left side indicates that less amounts of free tRNA was present in the complexes (lane 5 versus 2, 7 versus 3 and 9 versus 4). This trend holds true for molar ratios up to 2:4.8.
Last, DLS was also used to characterize the behavior of the enzyme. The major particle size of SerRS alone is around 4.5–5.1 nm, corresponding to a protein size of 115–154 kDa. This calculation is marginally larger than the hSerRS dimer (∼120 kDa) but is understandable, considering the elongated shape of the hSerRS dimer. The size of tRNASec alone is around 2.5–3.0 nm or ∼30 kDa, which is close to the theoretical value. Interestingly, we saw an increase of the major particle radius accordingly as well as a constant increase in molecular weight ∼70–80 kDa (roughly the size of two tRNASec molecules) with the increment of tRNA component all the way up to the 2:1.4 ratio (Figure 5C). We however did not obtain meaningful data for complex formation under the ratio range of 2:0.3–2:0.6 (the range where the 2:1 complex forms), where protein aggregation occurred and interfered with the sensitive DLS measurements. Taken together, our studies indicate that both stable complexes are authentic in solution and the formation of the 2:2 complex in solution starts from the molar ratio of 2:0.9. We later performed the same set of experiments with the WT enzyme and found that it displayed very similar behavior to E447K in solution (data not shown).
DISCUSSION
tRNA recognition mode of SerRS
The dimeric hSerRS makes most of the contacts with the variable arm of tRNASec non-specifically using one monomer, and interacts with the acceptor end using the other. This binding mode is quite different from leucyl-tRNA synthetase (LeuRS) to tRNALeu, another tRNA with a long variable arm (28). Aside from the differences between the class I and II synthetases, LeuRS interacts with the elbow of tRNALeu as well as the D-arm. All the interactions come from one protein molecule, which justifies the monomeric form as the catalytic form for LeuRS. Additionally, our structure reveals that the hSerRS contacts the tRNA backbone and thus recognizes the tRNA shape rather than specific nucleotides. This binding mode is reminiscent of the crystal structure of TtSerRS in complex with TttRNASer, and the mutations of individual residues on TtSerRS barely cause significant consequences in aminoacylation (16). In the cocrystal structure of the unorthogonal complex formed between A. aeolicus tRNASec and M. kandleri SerRS (MkSerRS) (17), the orientation of the variable loop including the U20:G19:C56 base triple from A. aeolicus tRNASec is similar to that of htRNASec, and the backbones of two tRNA molecules superpose well (Supplementary Figure S9). However, A. aeolicus tRNASec is almost completely resolved and contains a 9-bp variable arm, in contrast to the 5-bp arm present in htRNASec. Additionally, being a non-canonical SerRS, MkSerRS forms six-stranded β-sheet and four α-helices tRNA-binding module, instead of the coiled-coil observed in the typical SerRSs. Nevertheless, MkSerRS interacts with tRNASec using this module in a similar fashion to our complex structure, by contacting mainly with the phosphate groups and ribose moieties of nucleotides in variable region as well as the U20:G19:C56 base triple in tRNASec. The involvement of tRNA serylation by the idiosyncratic N-terminal domain in MkSerRS was in agreement with the biochemical studies conducted by Jaric et al. (29). Based on a docking model, they tested the activities of mutants within the N-terminal region, and proved their involvement in binding and serylation of its cognate substrate tRNASer. Three positively charged residues, Arg76, Lys79 and Arg94 within the N-terminal domain were found to have a pronounced effect on the ability of the enzyme to serylate cognate tRNA.
The flexibility of the TBD is essential to aminoacylation. The translational and rotational movements of this region are governed by charged interactions, hydrogen bonds as well as the C46-C117 disulfide bridge. As revealed by the alignment in the Supplementary Figure S6, C46 is conserved only in vertebrate SerRSs while C117 is not conserved. Therefore, the C46–C117 covalent bond is probably a species-specific factor. Intrasubunit disulfide bonds formed upon oxidative stress have been reported to play a crucial role in cellular defense systems against a variety of stress challenges (30–34). For example, in the brain type creatine kinase, disulfide formation between the residues C74 and C254 can serve as a cellular defense mechanism against intracellular oxidative stress (34). Although currently we are not clear about the physiological relevance of the disulfide bond, one possibility is that free radicals lead to the formation of the disulfide bridge on hSerRS in damaged cells, and serylation efficiencies may be affected, which in turn triggers downstream signaling pathways to counteract the harmful effects. Furthermore, we found that mutations at the hinge region P30G31 and G136 (conserved residues in vertebrates) are highly detrimental to aminoacylation activities. Taken together, we speculate that the structural flexibility is the intrinsic nature of the TBD, which is adapted to serylation.
Dual substrate specificity
The idiosyncratic feature of tRNASer and tRNASec is their long variable arms (35,36), which are the most important identity element of these two closely related tRNA molecules. Their ability to interact with the TBD allows SerRS to distinguish them from normal tRNAs, which usually contain a 3–4 nt variable loop.
However, previous studies have demonstrated that hSerRS serylates the tRNASec substrate with only 1/10 efficiency of the tRNASer substrate (37). Therefore, the enzyme is still able to distinguish tRNASec from tRNASer during the charging process, which is a problem of interest. The TtSerRS-TttRNASer complex structure reveals that the coiled-coil region from one subunit of SerRS establishes contacts with the variable arm and the TψC loop of tRNASer, whereas the active site of the other subunit interacts with the acceptor stem. Here, our cocrystal structure may offer an explanation. Modeling studies by superposition of the SerRS from our complex with the TtSerRS-TttRNASer complex suggest that hSerRS could adjust the orientation of the coiled-coil region to respond to similar cognate tRNA substrates (Figure 6). However in tRNASec, G19 and U20 in the D-loop and C56 in the T-loop form a characteristic base triple, U20:G19:C56 (24,38). In tRNASer, the G19:C56 base pair is conserved (16) (Supplementary Figure S10). In the tertiary interaction U20:G19:C56, U20 is bulged out and becomes accessible to the solvent (Figure 6). There is only partial electron density for the U20 base ring but its backbone is clearly visible. The position of U20 is unique, because the corresponding base U20 in tRNAPhe or C20 in TttRNASer points to the inside of these tRNA molecules. When in contact with the enzyme, the N3 atom of U20 is only 3.8 Å from OG of highly conserved S61. In addition to the stem-loop pattern differences in the D- and T-regions (6 bp + 4 nt in D-region and 4 bp in the T-arm in tRNASec versus 4 bp + 8 nt in D-region and 5 bp in the T-arm in tRNASer), this unique base triple present in the elbow region may form the structural basis for SerRS to recognize tRNASec and thus distinguish two highly similar tRNA structures. The ability of hSerRS to distinguish two tRNA substrates is reasonable and is consistent with the needs of living species. There are only about 25 Sec-containing enzymes in human but practically every human protein contains serine. Serylation of tRNASer by hSerRS is in much higher demands than serylation of tRNASec. Higher serylation efficiency of the former ensures that Ser-tRNASer is in an ample supply when serine is needed for protein synthesis. Similarly, other enzymes in the rest of the Sec biosynthetic route also make contacts with the D-loop of tRNASec (18–20), which can be readily distinguished from tRNASer due to either the characteristic 4 nt D-loop in tRNASec, or the differences of the base triple in the elbow region. Therefore, this structural feature may serve as important identity elements not only for the serylation of tRNASec, but also for the following steps in Sec generation.
Figure 6.
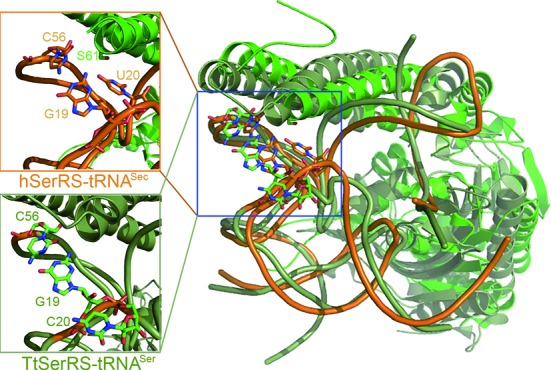
The mechanism by SerRS to distinguish two similar tRNA substrates. The structure overlay of human tRNASec (orange) with TttRNASer (deep teal) both of which are in complex with their cognate synthetases. Only one subunit of the enzyme in each complex is shown. The insets show the close-up details of the tertiary structure at the elbow region of each tRNA, viewed from the same angle from that in the original. Note that U20 in human tRNASec is accessible to the solvent whereas C20 in TttRNASer points to the inside of the core region (‘buried’).
Stoichiometry
Crystallographic and solution studies suggested that SerRSs from both T. thermophilus and E. coli can simultaneously bind to two tRNA molecules (16,39–41). In addition, binding behavior characterization of yeast SerRS by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-MS) has shown that yeast SerRS can also bind to tRNASer in both 2:1 and 2:2 stoichiometry (42). EMSA experiments of the native and crosslinked SerRS-tRNASer complex, as well as the pyrophosphate exchange assay suggest that two tRNASer binding sites on this enzyme are not equivalent, consistent with previous calculation (43,44). Recently, French et al. have shown that the SepSecS tetramer is able to bind to one or two tRNASec molecules (45), but the binding preference of hSerRS to tRNASec is not clear. In this study, we obtained two types of complexes of crystals in different stoichiometry to rule out the possibility of crystallization artifacts. We went on further through size-exclusion chromatography studies, EMSA and DLS analyses to demonstrate that hSerRS remains the ability to form the same types of complexes in solution. Whether these complexes are both biologically relevant is not the focus of our research and is also beyond the scope of this study.
Consequences for Sec biosynthesis
PSTK accesses the tRNASec acceptor stem from the major groove and makes contacts with the D-arm using the C-terminal three-helix bundle. By superposition of the two tRNASec molecules in their complex forms, we found that the catalytic domain in the two structures partially overlap. Interestingly, their RNA-recognition elements (the coiled-coil region of SerRS and the three-helix bundle domain of PSTK), complement each other well (Supplementary Figure S11A). Together these tRNA-recognition domains form a ring structure and wrap around the tRNA substrate. This raises the possibility that SerRS, PSTK and tRNA form a transient ternary complex during the substrate channeling process. We could speculate that upon the synthesis of Ser-tRNASec, PSTK approaches tRNA from the D-arm, and competes SerRS off the substrate by shoveling against SerRS with its catalytic domain. SerRS gradually loses ‘grip’ on the acceptor end and eventually becomes released from tRNA (Supplementary Figure S11B).
ACCESSION NUMBERS
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession codes 4RQF and 4RQE for the 2:1 and 2:2 complexes, respectively.
Supplementary Material
Acknowledgments
We thank L Kang for helpful discussion and SSRF for access to the BL17U beamline.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Sciences Foundation of China [31100579]; Guangdong Innovative Research Team Program NO. 2011Y038. Funding for open access charge: National Sciences Foundation of China [31100579], Guangdong Innovative Research Team Program NO. 2011Y038.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ibba M., Söll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 2.Commans S., Böck A. Selenocysteine inserting tRNAs: an overview. FEMS Microbiol. Rev. 1999;23:335–351. doi: 10.1111/j.1574-6976.1999.tb00403.x. [DOI] [PubMed] [Google Scholar]
- 3.Leinfelder W., Zehelein E., Mandrand-Berthelot M.A., Böck A. Gene for a novel tRNA species that accepts L-serine and cotranslationally inserts selenocysteine. Nature. 1988;331:723–725. doi: 10.1038/331723a0. [DOI] [PubMed] [Google Scholar]
- 4.Kryukov G.V., Castellano S., Novoselov S.V., Lobanov A.V., Zehtab O., Guigo R., Gladyshev V.N. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 5.Rayman M.P. The importance of selenium to human health. Lancet. 2000;356:233–241. doi: 10.1016/S0140-6736(00)02490-9. [DOI] [PubMed] [Google Scholar]
- 6.Carlson B.A., Xu X.M., Kryukov G.V., Rao M., Berry M.J., Gladyshev V.N., Hatfield D.L. Identification and characterization of phosphoseryl-tRNASer/Sec kinase. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12848–12853. doi: 10.1073/pnas.0402636101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu X.M., Carlson B.A., Mix H., Zhang Y., Saira K., Glass R.S., Berry M.J., Gladyshev V.N., Hatfield D.L. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol. 2007;5:e4. doi: 10.1371/journal.pbio.0050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cusack S., Berthet-Colominas C., Hartlein M., Nassar N., Leberman R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature. 1990;347:249–255. doi: 10.1038/347249a0. [DOI] [PubMed] [Google Scholar]
- 9.Ribas de Pouplana L., Schimmel P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001;104:191–193. doi: 10.1016/s0092-8674(01)00204-5. [DOI] [PubMed] [Google Scholar]
- 10.Schimmel P., Ribas de Pouplana L. Formation of two classes of tRNA synthetases in relation to editing functions and genetic code. Cold Spring Harb. Symp. Quant. Biol. 2001;66:161–166. doi: 10.1101/sqb.2001.66.161. [DOI] [PubMed] [Google Scholar]
- 11.Wu X.Q., Gross H.J. The long extra arms of human tRNASer/Sec and tRNASer function as major identify elements for serylation in an orientation-dependent, but not sequence-specific manner. Nucleic Acids Res. 1993;21:5589–5594. doi: 10.1093/nar/21.24.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu X., Shi Y., Zhang H.M., Swindell E.C., Marshall A.G., Guo M., Kishi S., Yang X.L. Unique domain appended to vertebrate tRNA synthetase is essential for vascular development. Nat. Commun. 2012;3:681. doi: 10.1038/ncomms1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo M., Schimmel P. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013;9:145–153. doi: 10.1038/nchembio.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo M., Yang X.L., Schimmel P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010;11:668–674. doi: 10.1038/nrm2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukui H., Hanaoka R., Kawahara A. Noncanonical activity of seryl-tRNA synthetase is involved in vascular development. Circ. Res. 2009;104:1253–1259. doi: 10.1161/CIRCRESAHA.108.191189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biou V., Yaremchuk A., Tukalo M., Cusack S. The 2.9 Å crystal structure of T. thermophilus seryl-tRNA synthetase complexed with tRNASer. Science. 1994;263:1404–1410. doi: 10.1126/science.8128220. [DOI] [PubMed] [Google Scholar]
- 17.Itoh Y., Sekine S., Suetsugu S., Yokoyama S. Tertiary structure of bacterial selenocysteine tRNA. Nucleic Acids Res. 2013;41:6729–6738. doi: 10.1093/nar/gkt321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itoh Y., Brocker M.J., Sekine S., Hammond G., Suetsugu S., Söll D., Yokoyama S. Decameric SelA-tRNASec ring structure reveals mechanism of bacterial selenocysteine formation. Science. 2013;340:75–78. doi: 10.1126/science.1229521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiba S., Itoh Y., Sekine S., Yokoyama S. Structural basis for the major role of O-phosphoseryl-tRNA kinase in the UGA-specific encoding of selenocysteine. Mol. Cell. 2010;39:410–420. doi: 10.1016/j.molcel.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Palioura S., Sherrer R.L., Steitz T.A., Söll D., Simonović M. The human SepSecS-tRNASec complex reveals the mechanism of selenocysteine formation. Science. 2009;325:321–325. doi: 10.1126/science.1173755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X., Shi Y., Yang X.L. Crystal structure of human Seryl-tRNA synthetase and Ser-SA complex reveals a molecular lever specific to higher eukaryotes. Structure. 2013;21:2078–2086. doi: 10.1016/j.str.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh Y., Chiba S., Sekine S., Yokoyama S. Crystal structure of human selenocysteine tRNA. Nucleic Acids Res. 2009;37:6259–6268. doi: 10.1093/nar/gkp648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganichkin O.M., Anedchenko E.A., Wahl M.C. Crystal structure analysis reveals functional flexibility in the selenocysteine-specific tRNA from mouse. PloS One. 2011;6:e20032. doi: 10.1371/journal.pone.0020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palencia A., Crepin T., Vu M.T., Lincecum T.L. Jr, Martinis S.A., Cusack S. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2012;19:677–684. doi: 10.1038/nsmb.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaric J., Bilokapic S., Lesjak S., Crnkovic A., Ban N., Weygand-Durasevic I. Identification of amino acids in the N-terminal domain of atypical methanogenic-type seryl-tRNA synthetase critical for tRNA recognition. J. Biol. Chem. 2009;284:30643–30651. doi: 10.1074/jbc.M109.044099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghezzi P., Bonetto V., Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxid. Redox Signal. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 31.Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 32.Li W., Zhang J., An W. The conserved CXXC motif of hepatic stimulator substance is essential for its role in mitochondrial protection in H2O2-induced cell apoptosis. FEBS Lett. 2010;584:3929–3935. doi: 10.1016/j.febslet.2010.08.043. [DOI] [PubMed] [Google Scholar]
- 33.Wei P.C., Hsieh Y.H., Su M.I., Jiang X., Hsu P.H., Lo W.T., Weng J.Y., Jeng Y.M., Wang J.M., Chen P.L., et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell. 2012;48:747–759. doi: 10.1016/j.molcel.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X.H., Chen Z., Gao Y.S., Yan Y.B., Zhang F., Meng F.G., Zhou H.M. Generation of the oxidized form protects human brain type creatine kinase against cystine-induced inactivation. Int. J. Biol. Macromol. 2011;48:239–242. doi: 10.1016/j.ijbiomac.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 35.Himeno H., Hasegawa T., Ueda T., Watanabe K., Shimizu M. Conversion of aminoacylation specificity from tRNATyr to tRNASer in vitro. Nucleic Acids Res. 1990;18:6815–6819. doi: 10.1093/nar/18.23.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sampson J.R., Saks M.E. Contributions of discrete tRNASer domains to aminoacylation by E.coli seryl-tRNA synthetase: a kinetic analysis using model RNA substrates. Nucleic Acids Res. 1993;21:4467–4475. doi: 10.1093/nar/21.19.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amberg R., Mizutani T., Wu X.Q., Gross H.J. Selenocysteine synthesis in mammalia: an identity switch from tRNASer to tRNASec. J. Mol. Biol. 1996;263:8–19. doi: 10.1006/jmbi.1996.0552. [DOI] [PubMed] [Google Scholar]
- 38.Juhling F., Morl M., Hartmann R.K., Sprinzl M., Stadler P.F., Putz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borel F., Vincent C., Leberman R., Hartlein M. Seryl-tRNA synthetase from Escherichia coli: implication of its N-terminal domain in aminoacylation activity and specificity. Nucleic Acids Res. 1994;22:2963–2969. doi: 10.1093/nar/22.15.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price S., Cusack S., Borel F., Berthet-Colominas C., Leberman R. Crystallization of the seryl-tRNA synthetase: tRNASer complex of Escherichia coli. FEBS Lett. 1993;324:167–170. doi: 10.1016/0014-5793(93)81386-e. [DOI] [PubMed] [Google Scholar]
- 41.Yaremchuk A.D., Tukalo M.A., Krikliviy I., Malchenko N., Biou V., Berthet-Colominas C., Cusack S. A new crystal form of the complex between seryl-tRNA synthetase and tRNASer from Thermus thermophilus that diffracts to 2.8 Å resolution. FEBS Lett. 1992;310:157–161. doi: 10.1016/0014-5793(92)81319-h. [DOI] [PubMed] [Google Scholar]
- 42.Gruic-Sovulj I., Lüdemann H.-C., Hillenkamp F., Weygand-Durasevic I., Kucan Z., Peter-Katalinic J. Detection of noncovalent tRNA· aminoacyl-tRNA synthetase complexes by matrix-assisted laser desorption/Ionization mass spectrometry. J. Biol. Chem. 1997;272:32084–32091. doi: 10.1074/jbc.272.51.32084. [DOI] [PubMed] [Google Scholar]
- 43.Krauss G., Pingoud A., Boehme D., Riesner D., Peters F., Maass G. Equivalent and non-equivalent binding sites for tRNA on aminoacyl-tRNA synthetases. Eur. J. Biochem. 1975;55:517–529. doi: 10.1111/j.1432-1033.1975.tb02189.x. [DOI] [PubMed] [Google Scholar]
- 44.Rigler R., Pachmann U., Hirsch R., Zachau H.G. On the interaction of seryl-tRNA synthetase with tRNASer. Eur. J. Biochem. 1976;65:307–315. doi: 10.1111/j.1432-1033.1976.tb10418.x. [DOI] [PubMed] [Google Scholar]
- 45.French R.L., Gupta N., Copeland P.R., Simonović M. Structural asymmetry of the terminal catalytic complex in selenocysteine synthesis. J. Biol. Chem. 2014;289:28783–28794. doi: 10.1074/jbc.M114.597955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.