

Abstract
Cardiovascular disease, which is often driven by hypercholesterolemia and subsequent coronary atherosclerosis, is the number-one cause of morbidity and mortality in the United States. Based on long-term epidemiological studies, high-density lipoprotein cholesterol (HDL-C) levels are inversely correlated with risk for coronary artery disease (CAD). HDL-mediated reverse cholesterol transport (RCT) is responsible for cholesterol removal from the peripheral tissues and return to the liver for final elimination.1 In atherosclerosis, intraplaque angiogenesis promotes plaque growth and increases plaque vulnerability. Conceivably, the acceleration of RCT and disruption of intraplaque angiogenesis would inhibit atherosclerosis and reduce CAD. We have identified a protein called apoA-I binding protein (AIBP) that augments HDL functionality by accelerating cholesterol efflux. Furthermore, AIBP inhibits vascular endothelial growth factor receptor 2 activation in endothelial cells and limits angiogenesis.2 The following discusses the prospect of using AIBP as a novel therapeutic approach for the treatment of CAD.
Keywords: apoA-I binding protein, AIBP, angiogenesis, lymphangiogenesis, cholesterol efflux, reverse cholesterol transport

L. Zhu, B.S.

L. Fang, Ph.D.
Vasa Vasorum and Plaque Growth
Vasa vasorum are the microvessels that underlie the adventitial layer of large arteries or veins and provide oxygen and nutrients to these major vessels. Emerging evidence suggests that atherosclerotic plaque growth is closely associated with intraplaque angiogenesis, which occurs from vasa vasorum. Local administration of fibroblast growth factors (FGF), a potent angiogenic factor that promotes angiogenesis in the adventitia, enhanced the growth of atherosclerotic lesions in apoE-deficient mice.3 In rat models receiving injury to the arterial walls, administration of vascular endothelial growth factor (VEGF) exacerbated neointimal thickening, although VEGF alone was not sufficient to initiate the thickening.4 One important component that instigates intraplaque angiogenesis is inflammation, which causes oxidative stress. For example, transgenic mice—which ectopically express p22-phox, a major subunit of NAD(P)H oxidase in the smooth muscle cells—will generate increased oxidative stress in carotid lesions, further contributing to angiogenesis and plaque progression.5 It is posited that plaques behave like the tumor microenvironment in which inflammatory cells and/or other cells secret a plethora of angiogenic factors to induce angiogenesis. Due to hypoxia, the intraplaque microenvironment often results in vascular leakage, which further contributes to intraplaque hemorrhage, elevated lipid accumulation, and exacerbated inflammation. All of these effects result in the formation of vulnerable plaques. Interestingly, the perivascular microvessels also serve as niches for multipotent pericytes and endothelial progenitor cells. These cells play a critical role in augmenting intraplaque angiogenesis by providing endothelial cells (ECs) or supporting cells for angiogenic growth.6
Reverse Cholesterol Transport, Lymphatics, and CAD
Mounting evidence suggests that lymphatics play an essential role in lipid transport. Chylomicrons, a form of dietary lipids, are transported to the blood via intestinal lymphatic vessels.7 Similarly, the transport of high-density lipoproteins (HDL) from the peripheral tissues back to plasma depends on lymphatic vasculature but not on venous capillaries. It has been shown that there is roughly 344 mg of HDL transported in human peripheral lymph daily.8 Inactivation of one copy of VEGFR3 in mice diminishes lymphatics in the skin but retains a normal blood vasculature. Accordingly, these mice demonstrate attenuated ability to remove subcutaneous fat.9 Lymphatic vessels are found in the adventitia of arteries and are in close proximity to the blood vessels.10 Surgical disruption of lymphatic vessels, by using aortic transplantation as well as blocking lymphatic regrowth using VEGFR3-neutralizing antibody, led to a remarkable increase in aortic lipid accumulation.11
Lymphatic malfunction contributes to the pathogenesis of atherosclerosis, cardiovascular disease, and diabetes. The connection between atherosclerosis and impaired lymphatic functions has been observed for a long time.12,13 Experimental evidence has recently emerged suggesting that lymphatic vasculature provides a protective pathway for lipid and inflammatory cell removal from the arterial wall and peripheral tissues and thus alleviates the development of lipid deposition.11,14 It remains unclear whether lymphatic vessels play an active role, which is speculated to occur via SR-BI-mediated transendocytosis, or a passive role, where it functions merely as a collecting conduit.
HDL in the Regulation of Angiogenesis and Lymphangiogenesis
Numerous reports indicate that HDL associated bioactive lipids and HDL-evoked changes in cholesterol levels regulate angiogenesis and lymphangiogenesis. In ECs, cholesterol is mostly enriched in lipid rafts of the cell membrane, where it is essential for cell signaling and maintenance of regular cellular functions such as migration and proliferation.15 Manipulation of cholesterol levels in ECs to decrease lipid rafts would interfere with membrane receptor-like VEGF receptor 2 (VEGFR-2) signaling and subsequently impair angiogenesis. Notably, other molecules involved in angiogenesis such as FGFR, ICAM-1, and VCAM-1 also localize to membrane lipid rafts and may be affected.16,17
Sphingosine-1-phosphate (S1P), which docks on HDL via apoM, exerts an effect on both angiogenesis and lymphangiogenesis.18 The effects of HDL-associated S1P on angiogenesis seem to be context-dependent. For example, S1P on HDL stimulates in vitro angiogenesis as well as ovarian angiogenesis in the follicular fluid via ERK, PKC, Akt, and eNOS activation.19 Endothelial lipase is shown to play a critical role in S1P-induced aortic ring neovascularization, presumably by releasing free S1P from HDL.20 In contrast, another study tested the effect of different concentrations of HDL on ECs and found a biphasic effect of HDL on endothelial progenitor cell (EPC)-mediated in vitro angiogenesis. High levels of HDL inhibit angiogenesis through stimulation of Rho-associated kinase (ROCK) and inhibition of the Akt and ERK pathways.21 In vivo, S1P receptor deficiency increases while S1P receptor activation decreases angiogenesis. Further, S1P signaling in ECs preserves vascular integrity by stabilizing adherent junctions between neighboring ECs and responds to laminar shear stress to transduce blood flow-regulated EC homeostasis.22,23
S1P promotes human lymphatic endothelial cell (LEC) tube formation and cell migration in vitro and facilitates growth of lymphatic vessels in the Matrigel plug in vivo.24 Mechanistically, it promotes lymphangiogenesis through stimulation of PLCg and calcium signaling. S1P levels are increased in breast cancer patients, in whom lymphatic vasculature is essential for tumor dissemination. Similarly, sphingosine kinase, a kinase that produces S1P, has increased expression levels and supports lymphangiogenesis in the tumor microenvironment.25
Despite many papers ascribing the actions of S1P to its receptor, HDL, at least in part, is involved in S1P-induced signaling. For example, SR-BI, the HDL receptor, is shown to regulate S1P-induced upregulation of NF-kB and cell adhesion molecules such as VCAM-1 and ICAM-1 via activation of PI3K and eNOS.26 Of note, the receptors for S1P, G protein-coupled receptors, localize and activate in lipid rafts.27 Whether targeted cholesterol efflux mediated by AIBP regulates the signals initiated from S1P is an intriguing question that merits further study.
Zebrafish: a Unique Animal Model for the Study of Atherosclerosis, Angiogenesis, and Lymphangiogenesis
Through our series of studies, we have established that zebrafish fed a high cholesterol diet (HCD) are a novel animal model to study the early atherosclerogenesis, when foam cell formation occurs. The advantages of using zebrafish include its optical transparency, easy genetic manipulation, generation of a large quantity of progeny per breeding, short life cycle, and rapid development of diseases. When fed an HCD similar to that used for mouse atherosclerosis studies (4% cholesterol weight for weight in food) for 2 months, adult zebrafish develop early healed lesions in the dorsal aorta.28 These lesions revealed lipid accumulation, macrophage retention, EC inflammation, and smooth muscle cell proliferation. All of these features are found in early human atherosclerotic lesions. Oxidation of low-density lipoprotein (LDL) renders it proinflammatory, and oxidized LDL (OxLDL) is believed to be one of the key culprits that promote atherogenesis. There is a 20-fold increase in OxLDL levels in zebrafish fed an HCD compared to control diet, and the absolute levels are much higher than that previously observed in humans.28 The OxLDL levels are measured using an ELISA assay based on E06, a monoclonal antibody against oxidized phospholipids developed by the Witztum lab.29 Consistent with this data, liquid chromatography tandem mass spectrometry (LC-MS/MS) measurement of the levels of oxidized cholesterol esters and phospholipids indicates a 10- to 70-fold increase, further supporting the use of zebrafish for studying atherosclerosis and lipid oxidation.30 The epitope recognized by E06 is also found on zebrafish HDL; this epitope is rarely found in human HDL. Yet there are reports on human apoA-I oxidation,31 so it is possible that the modified apoA-I is trapped in the intima and precludes its detection in human plasma.32
After the proof-of-concept studies, the zebrafish larvae were used for further analyses because of the aforementioned advantages. Feeding transgenic zebrafish larvae an HCD for 2 weeks also resulted in oxidation-specific epitopes (OSE) accumulation and vascular inflammation, which increase vascular leakage and macrophage recruitment.33 We created a transgenic zebrafish with conditional expression of EGFP-tagged single chain monoclonal antibody IK17, which targets malondialdehyde-modified epitopes, to monitor the spatiotemporal accumulation of OSE. Furthermore, the expression of IK17-EGFP reduced vascular lipid accumulation.33 Thus, from a pathogenesis point of view, the initial events of atherogenesis are conserved between zebrafish and humans.
Zebrafish angiogenesis proceeds in a similar manner to humans, and the genetic pathways governing angiogenesis such as VEGF and Notch are conserved in zebrafish. Several transgenic zebrafish lines such as EC-specific promoter driving fluorescent protein expression—including Tg(fli1:EGFP) and Tg(flk1:mcherry-RAS)—have been extensively used for zebrafish angiogenesis studies.34 These transgenic zebrafish permit a spatiotemporal and real-time examination of the development of in vivo angiogenesis for which fine-tuned mechanisms are delineated. Using the two transgenic zebrafish lines, we deciphered the role of AIBP in regulation of angiogenesis as discussed above in detail.
Zebrafish have also been employed to study lymphangiogenesis, although it appears that factors such as Prox1, Coup-TFII, and Sox18 that are essential for mouse lymphatic vessel growth seems to be dispensable for zebrafish lymphangiogenesis.35 Nonetheless, people have used different lymphatic reporter fish lines to study the functionality of the lymphatic system in zebrafish, and the key features are congruent with their mammal counterparts. For example, lymphatics in zebrafish are important for removal of subcutaneous fluids, and defects in lymphatics result in edema.36,37 The differential mechanism controlling zebrafish lymphatic vessel development may be adopted to treat lymphedema in humans.
AIBP-Regulated Cholesterol Efflux, Angiogenesis, and Atherosclerosis
Cholesterol is essential for normal cell function, from maintenance of structural integrity to biogenesis of hormones and ligands. However, excessive cholesterol is detrimental to cells. Cholesterol efflux enables the clearance of excess cholesterol from cells and is essential for maintenance of cholesterol homeostasis.38–40 Cholesterol efflux is carried out by cholesterol transporters such as ATP-binding cassette family members A1 (ABCA1) and G1 (ABCG1), and scavenger receptor class B member 1(SR-BI) in an ATP-dependent manner.41–45 ABCA1 resides on the cell membrane and transports cholesterol to lipid-poor apoA-I or nascent HDL, while ABCG1 is located in the cytosol and deploys cholesterol to HDL. Lack of ABCG1 facilitates ex vivo angiogenesis of a murine aortic ring. SR-BI is a receptor for HDL and mediates selective uptake of cholesterol esters on HDL.46 Interestingly, SR-BI favors cholesterol efflux from a phospholipid-rich environment.47 In addition, SR-BI is located in lipid rafts and was recently indicated as a sensor to regulate cholesterol homeostasis in the membrane.48 It is also reported to engage in active lymphatic transport of interstitial HDL.14
High levels of HDL may be beneficial because it carries excess cholesterol away from cells. HDL-C in epidemiological studies is inversely associated with CVD risk. Infusion of wild type apoA-I, apoA-I Milano, or its mimetic peptides into rabbits or mice mitigates atherosclerosis.49,50 Thus, different approaches have been applied to increase HDL-C levels such as CETP inhibitors and RVX-208, a compound that functions as an inhibitor for BET bromodomains to increase apoA-I protein synthesis.51,52 However, clinical trials at the Cleveland Clinic with RVX-208 failed to document an increase in apoA-I protein levels in humans compared to placebo control. So far, the majority of clinical trials aiming to reduce risks for CAD by increasing HDL-C levels have failed to show efficacy.53,54 Moreover, the genetic evidence supporting the cause-and-effect relationship between higher HDL-C levels and lower CVD risk is controversial.55 To address the discrepancy, several investigators proposed that augmenting the ability of cholesterol removal by HDL instead of increasing HDL-C levels would improve the outcome of CVD.56 Indeed, large-scale measurement of the ability of LDL-depleted plasma to facilitate cholesterol efflux confirmed lower risk for prevalent atherosclerotic burden and CAD in patients with higher cholesterol efflux capacity.57–59
We have recently reported that AIBP demonstrates a capability to enhance cholesterol efflux of HDL and therefore may fulfill the role of boosting HDL functionality (Figure 1).2 AIBP is a secreted protein discovered in a screen of proteins that interact with apoA-I.60 Human AIBP is abundantly expressed in a majority of tissues that govern secretions. Of particular interest, it is expressed in the vascular smooth muscle and heart muscle but not in skeletal muscle.61 The protein localization may imply its role in cardiovascular functions. In addition, AIBP was detected in human cerebrospinal fluid and urine but not in the plasma of healthy subjects.60 The human apoAIBP gene resides on the chromosome region associated with familial combined hyperlipidemia, which may cause premature CAD.62 Before our studies, there were no publications documenting the role of AIBP in lipid metabolism and cardiovascular disease.63 We found that an incubation of AIBP with HDL increased cholesterol efflux to HDL in ECs. The ability of AIBP to promote cholesterol efflux plateaus after 6 hours. One reason for this plateau may be the relatively small amount of AIBP used (the molar ratio of AIBP to HDL is 75). Since HDL is documented to trigger AIBP release from cells,60 it is possible that AIBP released from HDL-supplemented cells will catch up with the effect caused by trace amounts of AIBP. Another reason for the plateau may be due to an insufficient HDL cholesterol deposition pool for AIBP to carry out its function in an in vitro system. Under in vivo conditions, we do not expect such a problem because a much larger pool of HDL is present. We showed that this change of cholesterol efflux is a spatiotemporal-dependent event that occurs in development during zebrafish angiogenesis.2
Figure 1.

AIBP-mediated targeted cholesterol efflux from endothelial cells disrupts caveolae/lipid rafts and inhibits VEGFR2 signaling. VEGFA-stimulated VEGFR2 signaling induces the receptor dimerization in the caveolae/lipid rafts enriched with cholesterol. Enhanced removal of cholesterol by AIBP/HDL from the EC plasma membrane disrupts caveolae/lipid rafts, resulting in retarded VEGFR2 signaling and restricted angiogenesis. AIBP: apoA-I binding protein; VEGFR: vascular endothelial growth factor receptor; HDL: high-density lipoprotein; EC endothelial cells.
Most studies on cholesterol efflux associated with atherosclerosis were done in macrophages, so whether AIBP accelerates cholesterol efflux from these particular cells merits further investigation. Additionally, maintenance of cholesterol homeostasis in ECs appears critical for proper vascular function. Increased cholesterol levels in ECs can result in elevated levels of lipid rafts and their associated membrane receptor activation, leading to inflammation and/or hyperproliferation and angiogenesis.2,64 This should be avoided in the plaque microenvironment, where intraplaque angiogenesis may incur vulnerability and culminate in its rupture.65 It will be important to investigate if the enhancement effect of AIBP on cholesterol removal may improve the functionality of HDL in vivo and mitigate the development of atherosclerosis and CVD in at-risk patients.
AIBP documents a remarkable capability to restrict cell migration and angiogenesis in vitro in human ECs, ex vitro in mouse aortic rings, and in vivo in zebrafish.2 This inhibitory effect of AIBP depends on cholesterol transporter ABCG1, which is responsible for cholesterol removal in ECs.66 Accordingly, loss of ABCG1 abolishes the enhanced effect of AIBP on cholesterol efflux in ECs. Interestingly, neovascularization using mouse aortic ring assay demonstrates that ABCG1 deficiency increases microvessel sprouting, which is not retarded by AIBP. Consistently, the ability of AIBP to inhibit segmental artery angiogenesis is comprised in ABCA1A- and ABCG11-deficient zebrafish. In this scenario, it is likely that there are increased levels of lipid rafts, similar to in myeloid cells,38,67 in ECs lacking ABCG1, which in turn promotes VEGFR2 signaling and thus augments angiogenesis. An in vivo assessment of lipid rafts in zebrafish, known as membrane lipid order, which was measured using a lipophilic probe laurdan, reveals that there are more lipid rafts in tip cells than in stalk cells. This is in line with other findings as tip cells, characteristic of VEGFR2 activation, are known to be responsible for vascular sprouting and migration in zebrafish. Stalk cells, instead, follow the migration of tip cells and are believed to be quiescent, although recent studies indicate that tip and stalk cells may compete and alternate their cell fates due to synchronization of VEGFR and Notch signaling pathways during angiogenesis.68 The higher membrane order in tip cells can be lowered using cholesterol acceptor HDL, which has no effect on lipid raft abundance in the major blood vessel dorsal aorta (DA).2 Since vasculogenesis, which gives rise to the DA, has completed at this time,69 it is comprehensible that the cells may be quiescent in this setting. This will be of particular importance in using AIBP to treat CVD because it manifests no adverse effect on established vessels. Rather, it only targets newly formed vasculature that is detrimental for atherogenesis.
Future Prospective: Using AIBP to Treat Human CAD
We found that downregulation of AIBP gene expression increases while upregulation decreases lipid rafts in the presence of HDL. Concurrent with this result, enhanced neovascularization is observed in the aortic rings from ABCG1−/− mice.2 Administering recombinant apoA-I or genetically raising levels of apoA-I inhibit angiogenesis and tumor growth in mice.70 The blood vasculature and lymphatic vessels share many features and are functionally related. Lymphangiogenic sprouting initiates from venous ECs and intersegmental arteries provide cues for lymphangiogenesis.71 The potential role of AIBP in lymphangiogenesis may be worth investigating since lymphatics is regarded as a conduit for reverse cholesterol transport.
Considering that angiogenesis promotes atherosclerotic plaque growth and rupture, targeting AIBP may inhibit intraplaque angiogenesis and mitigate atherosclerotic plaque growth. Insights gained from zebrafish angiogenesis studies may be translated to targeting intraplaque neovascularization. We have made transgenic zebrafish with conditional expression of AIBP. Furthermore, taking advantage of the recently developed gene editing technology Crispr/Cas9,72 we can make an AIBP mutant zebrafish. Those AIBP loss- and gain-of-function zebrafish models will be of tremendous help in elucidating the role of AIBP in the early steps of atherogenesis. Inferred from studies of apoA-I mimetic peptides,73 there may be AIBP derivate peptides or mimetics that can replace AIBP function and are more amenable for therapeutic applications.
Since AIBP also promotes cholesterol removal from cells, it is possible that AIBP could “kill two birds with one stone” by both inhibiting intraplaque angiogenesis and accelerating cholesterol efflux in the plaque settings (Figure 2). This may inhibit plaque growth and result in conversion of a vulnerable plaque to a stable plaque that poses less threat to patients. The therapeutic potential of AIBP to boost HDL functionality may also underpin the direction of future drug discovery for cardiovascular disease.
Figure 2.
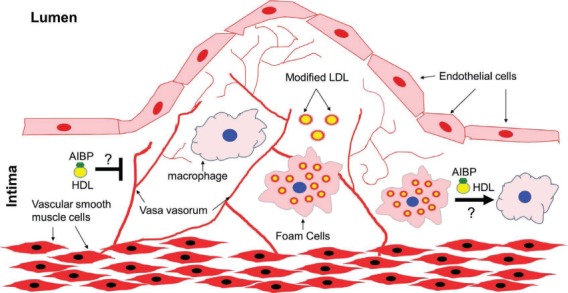
Presumptive protective role of AIBP in atherosclerosis. In atherosclerosis, LDL that is retained in the intima becomes proinflammatory modified LDL (e.g., OxLDL), which can be taken up by macrophages and become lipid-laden foam cells. AIBP, by accelerating cholesterol efflux, can reverse foam cell formation. In addition, AIBP may inhibit intraplaque angiogenesis and alleviate plaque growth. All these await further future studies. AIBP: apoA-I binding protein; LDL: low-density lipoprotein; HDL: high-density lipoprotein.
Acknowledgments
We thank Dina Schneider of the University of California, San Diego for critical reading of the manuscript.
Conflict of Interest Disclosure: Dr. Fang's research was supported by National Institutes of Health grant HL114734.
References
- 1.Glomset JA, Janssen ET, Kennedy R, Dobbins J. Role of plasma lecithin: cholesterol acyltransferase in the metabolism of high density lipoproteins. J Lipid Res. 1966 Sep;7(5):638–48. [PubMed] [Google Scholar]
- 2.Fang L, Choi SH, Baek JS et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature. 2013 Jun 6;498(7452):118–22. doi: 10.1038/nature12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka K, Nagata D, Hirata Y, Tabata Y, Nagai R, Sata M. Augmented angiogenesis in adventitia promotes growth of atherosclerotic plaque in apolipoprotein E-deficient mice. Atherosclerosis. 2011, Apr:215, 366–73. doi: 10.1016/j.atherosclerosis.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 4.Kwon HM, Sangiorgi G, Ritman EL et al. Adventitial vasa vasorum in balloon-injured coronary arteries: visualization and quantitation by a microscopic three-dimensional computed tomography technique. J Am Coll Cardiol. 1998 Dec;32:2072–9. doi: 10.1016/s0735-1097(98)00482-3. [DOI] [PubMed] [Google Scholar]
- 5.Khatri JJ, Johnson C, Magid R et al. Vascular oxidant stress enhances progression and angiogenesis of experimental atheroma. Circulation. 2004 Feb 3;109:520–5. doi: 10.1161/01.CIR.0000109698.70638.2B. [DOI] [PubMed] [Google Scholar]
- 6.Kawabe J, Hasebe N. Role of the vasa vasorum and vascular resident stem cells in atherosclerosis. Biomed Res Int. 2014;2014:701571. doi: 10.1155/2014/701571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tso P, Pitts V, Granger DN. Role of lymph flow in intestinal chylomicron transport. Am J Physiol. 1985 Jul;249(1 Pt 1):G21–8. doi: 10.1152/ajpgi.1985.249.1.G21. [DOI] [PubMed] [Google Scholar]
- 8.Nanjee MN, Cooke CJ, Olszewski WL, Miller NE. Lipid and apolipoprotein concentrations in prenodal leg lymph of fasted humans. Associations with plasma concentrations in normal subjects, lipoprotein lipase deficiency, and LCAT deficiency. J Lipid Res. 2000 Aug;41(8):1317–27. [PubMed] [Google Scholar]
- 9.Karkkainen MJ, Saaristo A, Jussila L et al. A model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci U S A. 2001 Oct 23;98(22):12677–82. doi: 10.1073/pnas.221449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kholová I, Dragneva G, Cermáková P et al. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest. 2011 May;41(5):487–97. doi: 10.1111/j.1365-2362.2010.02431.x. [DOI] [PubMed] [Google Scholar]
- 11.Martel C, Li W, Fulp B et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest. 2013 Apr;123(4):1571–9. doi: 10.1172/JCI63685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemole GM. The role of lymphstasis in atherogenesis. Ann Thorac Surg. 1981 Mar;31(3):290–3. doi: 10.1016/s0003-4975(10)60949-6. [DOI] [PubMed] [Google Scholar]
- 13.Miller AJ, DeBoer A, Palmer A. The role of the lymphatic system in coronary atherosclerosis. Med Hypotheses. 1992 Jan;37(1):31–6. doi: 10.1016/0306-9877(92)90009-2. [DOI] [PubMed] [Google Scholar]
- 14.Lim HY, Thiam CH, Yeo KP et al. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013 May 7;17(5):671–84. doi: 10.1016/j.cmet.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Lisanti MP, Scherer PE, Vidugiriene J et al. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol. 1994 Jul;126(1):111–26. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhainds D, Bourgeois P, Bourret G, Huard K, Falstrault L, Brissette L. Localization and regulation of SR-BI in membrane rafts of HepG2 cells. J Cell Sci. 2004 Jul 1;117(Pt 15):3095–105. doi: 10.1242/jcs.01182. [DOI] [PubMed] [Google Scholar]
- 17.van Buul JD, van Rijssel J, van Alphen FP, van Stalborch AM, Mul EP, Hordijk PL. ICAM-1 clustering on endothelial cells recruits VCAM-1. J Biomed Biotechnol. 2010;2010:120328. doi: 10.1155/2010/120328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christoffersen C, Obinata H, Kumaraswamy SB et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011 Jun 7;108(23):9613–8. doi: 10.1073/pnas.1103187108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miura S, Fujino M, Matsuo Y et al. High density lipoprotein-induced angiogenesis requires the activation of Ras/MAP kinase in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol. 2003 May 1;23(5):802–8. doi: 10.1161/01.ATV.0000066134.79956.58. [DOI] [PubMed] [Google Scholar]
- 20.Tatematsu S, Francis SA, Natarajan P et al. Endothelial lipase is a critical determinant of high-density lipoprotein-stimulated sphingosine 1-phosphate-dependent signaling in vascular endothelium. Arterioscler Thromb Vasc Biol. 2013 Aug;33(8):1788–94. doi: 10.1161/ATVBAHA.113.301300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang CY, Lin FY, Shih CM et al. Moderate to high concentrations of high-density lipoprotein from healthy subjects paradoxically impair human endothelial progenitor cells and related angiogenesis by activating Rho-associated kinase pathways. Arterioscler Thromb Vasc Biol. 2012 Oct;32(10):2405–17. doi: 10.1161/ATVBAHA.112.248617. [DOI] [PubMed] [Google Scholar]
- 22.Gaengel K, Niaudet C, Hagikura K et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell. 2012 Sep 11;23(3):587–99. doi: 10.1016/j.devcel.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Jung B, Obinata H, Galvani S et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell. 2012 Sep 11;23(3):600–10. doi: 10.1016/j.devcel.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon CM, Hong BS, Moon HG et al. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood. 2008 Aug 15;112(4):1129–38. doi: 10.1182/blood-2007-11-125203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagahashi M, Ramachandran S, Kim EY et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012 Feb 1;72(3):726–35. doi: 10.1158/0008-5472.CAN-11-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura T, Tomura H, Mogi C et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J Biol Chem. 2006 Dec 8;281(49):37457–67. doi: 10.1074/jbc.M605823200. [DOI] [PubMed] [Google Scholar]
- 27.Ephstein Y, Singleton PA, Chen W et al. Critical role of S1PR1 and integrin beta4 in HGF/c-Met-mediated increases in vascular integrity. J Biol Chem. 2013 Jan 25;288(4):2191–200. doi: 10.1074/jbc.M112.404780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoletov K, Fang L, Choi SH et al. Vascular lipid accumulation, lipoprotein oxidation, and macrophage lipid uptake in hypercholesterolemic zebrafish. Circ Res. 2009 Apr 24;104(8):952–60. doi: 10.1161/CIRCRESAHA.108.189803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hörkkö S, Bird DA, Miller E et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999 Jan;103(1):117–28. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang L, Harkewicz R, Hartvigsen K et al. Oxidized cholesteryl esters and phospholipids in zebrafish larvae fed a high cholesterol diet: macrophage binding and activation. J Biol Chem. 2010 Oct 15;285(42):32343–51. doi: 10.1074/jbc.M110.137257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borges CR, Rehder DS, Jensen S et al. Elevated plasma albumin and apolipoprotein A-I oxidation under suboptimal specimen storage conditions. Mol Cell Proteomics. 2014 Jul;13(7):1890–9. doi: 10.1074/mcp.M114.038455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, DiDonato JA, Levison BS et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med. 2014 Feb;20(2):193–203. doi: 10.1038/nm.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang L, Green SR, Baek JS et al. In vivo visualization and attenuation of oxidized lipid accumulation in hypercholesterolemic zebrafish. J Clin Invest. 2011 Dec;121(12):4861–9. doi: 10.1172/JCI57755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002 Aug 15;248(2):307–18. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 35.van Impel A, Zhao Z, Hermkens DM et al. Divergence of zebrafish and mouse lymphatic cell fate specification pathways. Development. 2014 Mar;141(6):1228–38. doi: 10.1242/dev.105031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hogan BM, Bos FL, Bussmann J et al. Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat Genet. 2009 Apr;41(4):396–8. doi: 10.1038/ng.321. [DOI] [PubMed] [Google Scholar]
- 37.Küchler AM, Gjini E, Peterson-Maduro J, Cancilla B, Wolburg H, Schulte-Merker S. Development of the zebrafish lymphatic system requires VEGFC signaling. Curr Biol. 2006 Jun 20;16(12):1244–8. doi: 10.1016/j.cub.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 38.Yvan-Charvet L, Pagler T, Gautier EL et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010 Jun 25;328(5986):1689–93. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armstrong AJ, Gebre AK, Parks JS, Hedrick CC. ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J Immunol. 2010 Jan 1;184(1):173–83. doi: 10.4049/jimmunol.0902372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bensinger SJ, Bradley MN, Joseph SB et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008 Jul 11;134(1):97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bodzioch M, Orsó E, Klucken J et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999 Aug;22(4):347–51. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 42.Rust S, Rosier M, Funke H et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999 Aug;22(4):352–5. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 43.Klucken J, Büchler C, Orsó E et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci U S A. 2000 Jan 18;97(2):817–22. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996 Jan 26;271(5248):518–20. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 45.Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem. 1994 Aug 19;269(33):21003–9. [PubMed] [Google Scholar]
- 46.Ji Y, Jian B, Wang N et al. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem. 1997 Aug 22;272(34):20982–5. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 47.Jian B, de la Llera-Moya M, Ji Y et al. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998 Mar 6;273(10):5599–606. doi: 10.1074/jbc.273.10.5599. [DOI] [PubMed] [Google Scholar]
- 48.Saddar S, Carriere V, Lee WR et al. Scavenger receptor class B type I is a plasma membrane cholesterol sensor. Circ Res. 2013 Jan 4;112(1):140–51. doi: 10.1161/CIRCRESAHA.112.280081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flynn R, Qian K, Tang C et al. Expression of apolipoprotein A-I in rabbit carotid endothelium protects against atherosclerosis. Mol Ther. 2011 Oct;19(10):1833–41. doi: 10.1038/mt.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lebherz C, Sanmiguel J, Wilson JM, Rader DJ. Gene transfer of wild-type apoA-I and apoA-I Milano reduce atherosclerosis to a similar extent. Cardiovasc Diabetol. 2007 May 2;6:15. doi: 10.1186/1475-2840-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLure KG, Gesner EM, Tsujikawa L et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS One. 2013 Dec 31;8(12):e83190. doi: 10.1371/journal.pone.0083190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003 Feb 1;23(2):160–7. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 53.AIM-HIGH Investigators. Boden WE, Probstfield JL et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011 Dec 15;365(24):2255–67. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz GG, Olsson AG, Abt M et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012 Nov 29;367(22):2089–99. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 55.Voight BF, Peloso GM, Orho-Melander M et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012 Aug 11;380(9841):572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012 Sep;18(9):1344–6. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 57.Khera AV, Cuchel M, de la Llera-Moya M et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011 Jan 13;364(2):127–35. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li XM, Tang WH, Mosior MK et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol. 2013 Jul;33(7):1696–705. doi: 10.1161/ATVBAHA.113.301373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rohatgi A, Khera A, Berry JD et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014 Dec 18;371:2383–93. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ritter M, Buechler C, Boettcher A et al. Cloning and characterization of a novel apolipoprotein A-I binding protein, AI-BP, secreted by cells of the kidney proximal tubules in response to HDL or ApoA-I. Genomics. 2002 May;79(5):693–702. doi: 10.1006/geno.2002.6761. [DOI] [PubMed] [Google Scholar]
- 61.Wilhelm M, Schlegl J, Hahne H et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014 May 29;509(7502):582–7. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 62.Bodnar JS, Chatterjee A, Castellani LW et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat Genet. 2002 Jan;30(1):110–6. doi: 10.1038/ng811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peloso GM, Demissie S, Collins D et al. Common genetic variation in multiple metabolic pathways influences susceptibility to low HDL-cholesterol and coronary heart disease. J Lipid Res. 2010 Dec;51(12):3524–32. doi: 10.1194/jlr.P008268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Setiadi H, McEver RP. Clustering endothelial E-selectin in clathrin-coated pits and lipid rafts enhances leukocyte adhesion under flow. Blood. 2008 Feb 15;111(4):1989–98. doi: 10.1182/blood-2007-09-113423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Virmani R, Kolodgie FD, Burke AP et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005 Oct;25(10):2054–61. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- 66.Terasaka N, Yu S, Yvan-Charvet L et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J Clin Invest. 2008 Nov;118(11):3701–13. doi: 10.1172/JCI35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whetzel AM, Sturek JM, Nagelin MH et al. ABCG1 deficiency in mice promotes endothelial activation and monocyte-endothelial interactions. Arterioscler Thromb Vasc Biol. 2010 Apr;30(4):809–17. doi: 10.1161/ATVBAHA.109.199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jakobsson L, Franco CA, Bentley K et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010 Oct;12(10):943–53. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 69.Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011 Aug 23;12(9):551–64. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zamanian-Daryoush M, Lindner D, Tallant TC et al. The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. J Biol Chem. 2013 Jul 19;288(29):21237–52. doi: 10.1074/jbc.M113.468967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011 Nov 7;17(11):1371–80. doi: 10.1038/nm.2545. [DOI] [PubMed] [Google Scholar]
- 72.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014 Jun 5;157(6):1262–78. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reddy ST, Navab M, Anantharamaiah GM, Fogelman AM. Apolipoprotein A-I mimetics. Curr Opin Lipidol. 2014 Aug;25(4):304–8. doi: 10.1097/MOL.0000000000000092. [DOI] [PMC free article] [PubMed] [Google Scholar]