

Abstract
Endothelium-derived nitric oxide (eNO) is a multifunctional signaling molecule critically involved in the maintenance of metabolic and cardiovascular homeostasis. In addition to its role as a potent endogenous vasodilator, eNO suppresses key processes in vascular lesion formation and opposes atherogenesis. This review discusses eNO as an antiatherogenic molecule and highlights factors that influence its bioavailability and therapeutic approaches to restore or enhance its levels.
Keywords: nitric oxide, endothelium, endothelial nitric oxide synthase, asymmetric dimethylarginine, dimethylarginine dimethylaminohydrolase, atherosclerosis, vascular dysfunction, antiatherogenic

R. A. Sukhovershin, M.D., Ph.D.

G. Yepuri, Ph.D.

Y. T. Ghebremariam, Ph.D.
Introduction
Vascular endothelium is a mesodermal tissue that forms a monolayer of endothelial cells across the innermost surface of blood vessels.1 This monolayer serves as a semipermeable physical barrier between circulating blood and the remainder of the vessel lumen as well as other surrounding tissues in order to control vascular function including blood flow, contractility of blood vessels, and the transit of blood elements into and out of the bloodstream. A number of endogenous molecules are actively secreted by vascular endothelium. Among these bioactive molecules, endothelium-derived nitric oxide (eNO) is arguably the most potent and physiologically significant. Chemically, nitric oxide (NO) is a highly reactive molecule with an unpaired electron in its nitrogen atom. This chemical property is responsible for its short half-life (less than 30 seconds) and rapid oxidation into more stable products—nitrates (NO3) and nitrites (NO2). However, NO itself is efficient in activating vasoactive downstream targets including soluble guanylate cyclase (sGC) and cyclic guanosine monophosphate (cGMP) in order to transmit signals that regulate vascular tone, myocardial contractility, cell proliferation, platelet aggregation, superoxide production, expression of adhesion molecules, and endothelial-leukocyte interaction. Since eNO is essential in these physiological processes, suboptimal levels of eNO in circulation or in local tissues can contribute to the development and/or progression of cardiovascular diseases including atherosclerosis, the most common one. The current review provides an overview of eNO as an antiatherogenic molecule and includes factors that influence its bioavailability and therapeutic approaches to restore or enhance its levels.
Antiatherogenic Effects of Nitric Oxide
Atherosclerosis is a multifactorial disease involving interplay between genetics, environmental factors, and lifestyle choices. It is characterized by accumulation of oxidized low-density lipoproteins (oxLDL) in the vessel wall, which promotes infiltration of inflammatory cells, overproliferation of vascular smooth muscle cells (VSMCs), and accumulation of connective tissue components; collectively, these result in endothelial dysfunction characterized by marked impairment of eNO levels, increased adhesion of inflammatory cells to vessel wall, and development of fibrous atheromatous plaque.2
In the past two decades, a number of studies have investigated the role of NO in the development and progression of vascular inflammation and atherogenesis. Many of these studies demonstrate that eNO possesses an atheroprotective effect. For example, preclinical studies show that genetic deletion of endothelial NO synthase (eNOS) accelerates the progression of atherosclerosis in apolipoprotein E (ApoE)-deficient mice.3 Similarly, pharmacological inhibition of NOS promotes formation of fatty streaks in rabbits that are fed with cholesterolsupplemented diets.4 By contrast, increasing eNO levels using L-arginine (NO precursor) or synthetic NO donors has been shown to improve endothelial-dependent vasorelaxation and inhibit fatty streak formation in the same animal model.5,6 Meanwhile, modulation of the NOS pathway has been shown to influence the development of other atherosclerosis-related vascular diseases, including neointimal hyperplasia (thickening of blood vessels as a result of macrophage, connective tissue, and smooth muscle cell accumulation in the innermost layer of arterial wall) following balloon angioplasty or arterial vein grafting.7,8 In these studies, interventions aimed at increasing NO bioavailability (by arginine supplementation or eNOS-encoding gene therapy) suppressed neointimal thickening by restoring eNO production and improving vascular function.
Neointimal thickening is an underlying problem in a number of vascular diseases, including vasculopathy, restenosis, and atherosclerosis. The ability of eNO to attenuate or reverse these vascular diseases is a landmark discovery that should be effectively harnessed in translational studies aimed at treating different types of vascular disorders. In atherosclerosis, the attenuation of intimal thickening and fatty streak formation by NO is due to its ability to suppress lipid-induced adhesion of monocytes and aggregation of platelets to the endothelial wall. Studies have shown that dietary arginine reduces adhesiveness of monocytes to aortic endothelium isolated from hypercholesterolemic animals in an NO-dependent manner, whereas L-NAME, an inhibitor of NOS, disturbed NO production and promoted monocyte binding.9 Other in vitro studies have demonstrated that exogenous administration of NO dosedependently inhibited the ability of monocytes to interact with endothelial cells due, at least in part, to suppression of the gene expression of chemotactic factors that promote monocyte binding.10 In addition, eNO is known to suppress oxidant-sensitive proinflammatory transcription factors, such as nuclear factor κB (NFκB),11 and mitigate vascular oxidative stress by interacting with superoxide anion12 and directly inhibiting major pro-oxidant enzymes such as xanthine oxidase and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.13,14 Inactivation of superoxide and hydrogen peroxide by NO is due, in part, to peroxynitrite (OONO−) formation15 and subsequent interaction with thiol groups to form thionitrites, molecules capable of releasing NO.16 All of the above functionalities of eNO result in inhibition of reactive oxygen species (ROS) and attenuation of lipid peroxidation leading to an antiatherogenic effect.
Inhibition of VSMC proliferation and migration is another mechanism by which NO affects atherogenesis. Increased vascular inflammation due to progressive accumulation of oxidized lipoproteins promotes endothelial dysfunction in part by interrupting the permeability of the endothelium and allowing the infiltration of VSMCs and other cell types into the endothelial monolayer. This inward migration of VSMCs and inflammatory cells exacerbates an already dysfunctional endothelium, leading to hardening of the vessel wall and formation of lesions. The VSMCs continue to proliferate, migrate, and deposit collagen matrix in the inflamed vessels, processes that typically precede the development and rupture of atherosclerotic plaque. By contrast, eNO inhibits the proliferation and migration of VSMCs, thus resulting in normal vessel wall structure and function.
Endogenous Regulation of Nitric Oxide Levels
In endothelial cells, NO is constitutively produced by the enzyme endothelial NOS (eNOS). Another constitutive isoform, neuronal NOS (nNOS), is mainly expressed in neuronal cells to promote neurotransmission. Some studies have reported that nNOS is also expressed by endothelial cells, although its precise contribution to endothelium-derived NO is still unclear. The third isoform, inducible NOS (iNOS), is actively expressed by immune cells and not physiologically present in other cell types, including endothelial cells. However, in the setting of inflammation, endothelial cells express higher levels of iNOS. All the NOS isoforms utilize L-arginine as a substrate and require a number of cosubstrates (molecular oxygen, reduced NADPH) and cofactors (FAD, FMN, heme, and tetrahydrobiopterin; BH4) to produce bioactive NO. In addition, the NOS proteins need to be dimerized to make functional enzymes capable of catalyzing the oxidation of L-arginine.
An eNOS monomer consists of a reductase domain and an oxygenase domain. The former one transfers electrons from reduced NADPH through FAD and FMN to heme of the oxygenase domain of the second monomer. The transfer of electrons is activated by the binding of calcium (Ca2+)-dependent protein, calmodulin, to the conjugation site of the two domains. Once transferred, the electrons are used to activate molecular oxygen and oxidize L-arginine to equimolar quantities of L-citrulline and NO. Two monomers dimerize only in the presence of the cofactor heme and sufficient amounts of L-arginine and BH4 (BH4 serves as a donor of the second electron, while the first electron is transferred from the reductase domain).
One critical step in the endogenous regulation of eNOS activity is its interaction with calmodulin. This protein binds to eNOS in a reversible fashion that depends on the concentration of intracellular Ca2+. In resting cells, eNOS is localized in an inactive form within caveolae (an invagination of cytoplasmic membrane) bound to caveolin-1 protein. The enzymatic activity of eNOS is restored when Ca2+-loaded calmodulin displaces caveolin-1.17 This Ca2+-calmodulin-mediated regulation of eNOS activity is leveraged by several bioactive molecules that induce cytosolic Ca2+-influx to include bradykinin, acetylcholine, histamine, adenosine, and thrombin. Intriguingly, NO itself can also reversibly regulate eNOS enzymatic activity by forming a covalent adduct with target-specific sulfhydryl groups of the eNOS protein (known as “S-nitrosylation”).18
Phosphorylation of eNOS is another regulatory mechanism of its activity. The enzyme is subjected to phosphorylation at different sites that can either increase or suppress NO production.19 Stimuli that activate eNOS by phosphorylation include shear stress, insulin, bradykinin, vascular endothelial growth factor, estrogen, and platelet-derived lipid mediators. By contrast, under nonstimulated conditions, phosphorylation of threonine 495 residue of the eNOS protein appears to interfere with calmodulin binding and eNOS activation.20 Other stimuli that affect eNOS gene expression and/or stability of its mRNA include erythropoietin, hypoxia, oxLDL, and inflammatory stimuli.20
Inhibition of eNOS by its endogenous inhibitors asymmetrical dimethylarginine (ADMA) and monomethylarginine (MMA) is another important mechanism of regulating NO synthesis.21 These arginine derivatives (methylarginines) are produced in any nucleated cell due to post-translational modification of histone proteins, and they compete with arginine for the substrate-binding site in the NOS protein, thereby inhibiting NO production.22 This inhibition of eNOS activity by endogenous methylarginines is the basis for the “arginine paradox,” in which NO production is increased following exogenous supplementation of L-arginine despite sufficiently high intracellular concentration of L-arginine to saturate the NOS enzymes.23 Hence, some authors consider plasma L-arginine-to-ADMA ratio as a marker of endothelium-derived NO bioavailability.24
Under conditions in which arginine or BH4 is depleted or methylarginine levels are high (relative arginine deficiency), eNOS can produce superoxide instead of NO as a result of NADPH oxidase uncoupling from NO generation and inducing oxidative stress.25 This phenomenon, known as eNOS uncoupling, is involved in a number of pathological processes including atherosclerosis (Figure 1)
Figure 1.
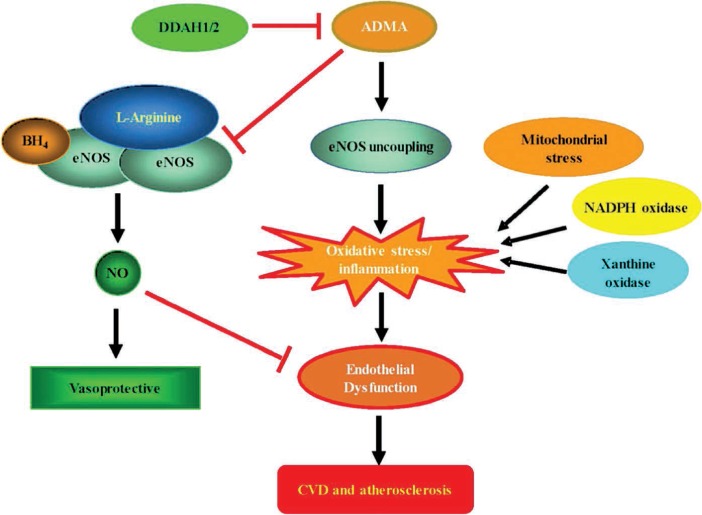
Pathophysiological role of endothelium-derived nitric oxide (eNO) in atherosclerosis. Endothelium-derived NO is an antiatherogenic molecule synthesized from the precursor L-arginine in the presence of cofactors, cosubstrates, and NO synthase (eNOS). NOS is competitively inhibited by asymmetrical dimethylarginine (ADMA), an endogenous substrate for dimethylarginine dimethylaminohydrolase (DDAH). Uncoupling of eNOS favors the generation of proinflammatory reactive oxygen and nitrogen species that contribute to endothelial dysfunction and development of cardiovascular disease (CVD), including atherosclerosis.
Mechanisms Leading to Reduced Nitric Oxide Bioavailability in Atherosclerosis
Several factors can influence NO availability in atherosclerosis. Some of these include reduced substrate (L-arginine) availability, suboptimal levels of cosubstrates and/or cofactors, eNOS uncoupling, loss-of-function polymorphism in the eNOS gene, elevated levels of methylarginines, plaque-mediated impairment in physiological shear stress, and flow disturbance or physical barriers such as accumulation of oxidized lipid particles (Figure 2). In brief, reduced L-arginine bioavailability may be due to reduced synthesis by the kidneys (e.g., in comorbid conditions where kidney disease exists alongside atherosclerosis), reduced transport into the plasma by the cationic amino acid transporters, or alternative metabolism of L-arginine by other enzymes including arginases. In addition, dysregulation of cofactors by oxidative stress may contribute to reduced NO bioavailability. Oxidative stress in the vessel wall might be exacerbated by cardiovascular risk factors such as hypercholesterolemia, diabetes, hypertension, or hyperhomocysteinemia, resulting in increased ROS production in the vessel wall.26 Reactive oxygen species are able to oxidize the eNOS cofactor BH4 and suppress expression of dihydrofolate reductase (an enzyme that recycles back oxidized BH2 to BH4), thereby depleting this cofactor and promoting eNOS uncoupling.27 Besides oxidizing BH4, superoxide ions directly scavenge NO, reducing its bioavailability and forming peroxynitrite. Peroxynitrite is able to oxidize BH4 and the substrate binding site of the eNOS protein, leading to reduced cofactor availability and further uncoupling of eNOS.28
Figure 2.
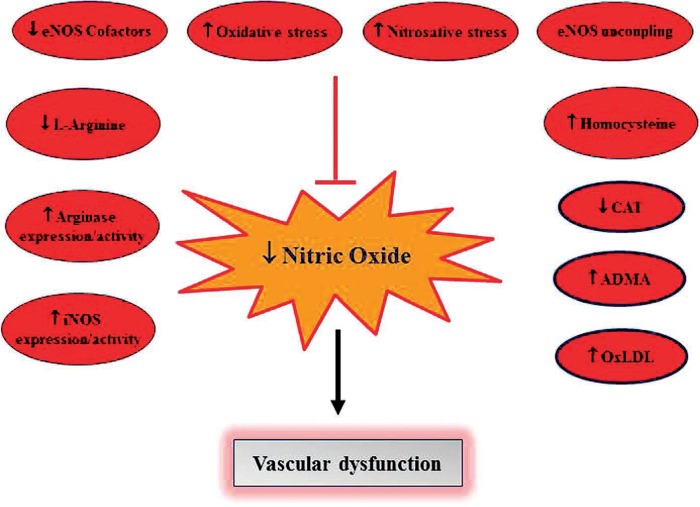
Endogenous factors that contribute to reduced nitric oxide (NO) bioavailability. Reduced NO directly contributes to vascular dysfunction and development of cardiovascular disease. iNOS: inducible NO synthase (NOS); eNOS: endothelial NOS; CAT: cationic amino acid transporter; ADMA: asymmetric dimethylarginine; OxLDL: oxidized low density lipoprotein.
Furthermore, accumulation of methylarginines—ADMA and MMA—due to increased synthesis and/or reduced metabolism is associated with reduced NO bioavailability, increased oxidative stress, development of vascular dysfunction, and atherosclerosis.29 Reduced metabolism of the methylarginines is mainly due to impairment in the enzyme that is chiefly responsible for their breakdown. Dimethylarginine dimethylaminohydrolase (DDAH), which exists in one of two isoforms in vascular cells, is physiologically responsible for the metabolism of MMA and ADMA into methylamine and citrulline (Figure 1). Unfortunately, DDAH is sensitive to oxidative stress, and its enzymatic activity is often impaired in cardiovascular disease including atherosclerosis.30 Consequently, elevated levels of circulating ADMA inhibits NO production and promotes superoxide production in the vessel wall, leading to eNOS uncoupling.31
Cholesterol and LDL were also reported to influence NO production. Interestingly, a low cholesterol concentration acutely increases NO synthesis, whereas high levels of cholesterol or LDL have the opposite effect.32,33 In addition, both native and oxLDL are capable of destabilizing eNOS mRNA and causing eNOS to uncouple.34,35 Moreover, oxLDL may also reduce arginine uptake by endothelial cells36 and increase the expression and activity of arginase, leading to reduced L-arginine bioavailability and impaired NOS signaling.37
Finally, the expression of iNOS, an enzyme with a rate constant 1,000-fold greater than eNOS, is upregulated in atherosclerotic plaques (predominantly in macrophages and VSMCs) and may contribute to eNOS uncoupling.38 Unlike eNOS, iNOS binds to calmodulin even at negligible Ca2+ concentrations and produces an extensive amount of superoxide anion (O2−) and NO that spontaneously react to form peroxynitrite anion (OONO−), a highly reactive and cytotoxic adduct. Since the supply of L-arginine is often depleted by iNOS, the level of eNO in the plaque area is anticipated to be low.
Strategies to Enhance Nitric Oxide Bioavailability
Given the multitude of factors that influence NO bioavailability in atherosclerosis, it is logical to target one or more of these processes to enhance NO levels in vivo. Some of the mechanisms by which existing drugs or future therapeutics may increase NO bioavailability include supplementation of L-arginine or NOreleasing molecules, restoration of cofactors and cosubstrates for eNOS, stabilization of eNOS mRNA or protein, increasing eNOS expression or activity, reducing synthesis or increasing metabolism of methylarginines, reducing oxidative stress, and/or enhancing the clearance of harmful lipoproteins (Figure 3). For example, along with reducing lipids, statins also reduce oxidative stress, stabilize eNOS expression, and increase its activity. Collectively, these effects might be incrementally responsible for slowing the progression of atherosclerosis and improving overall vascular health.39 In addition, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), the selective aldosterone antagonist eplerenone, and the direct renin inhibitor aliskiren all demonstrate favorable effects on NO bioavailability through a combination of the aforementioned mechanisms. Antioxidant supplementation can also be effective in scavenging ROS and preventing eNOS uncoupling.40 Furthermore, the use of folic acid, estrogen, and erythropoietin has been shown to increase BH4 bioavailability and improve endothelial function. Supplementation with L-arginine can also improve NO bioavailability but seems to be beneficial only when arginine is limited or ADMA levels are high.41 Meanwhile, the efficacy of NO donors in atherosclerosis has been evaluated and confirmed to restore NO levels and reduce LDL oxidation and lesion formation. However, their long-term therapeutic utility is complicated by the development of tolerance.42
Figure 3.
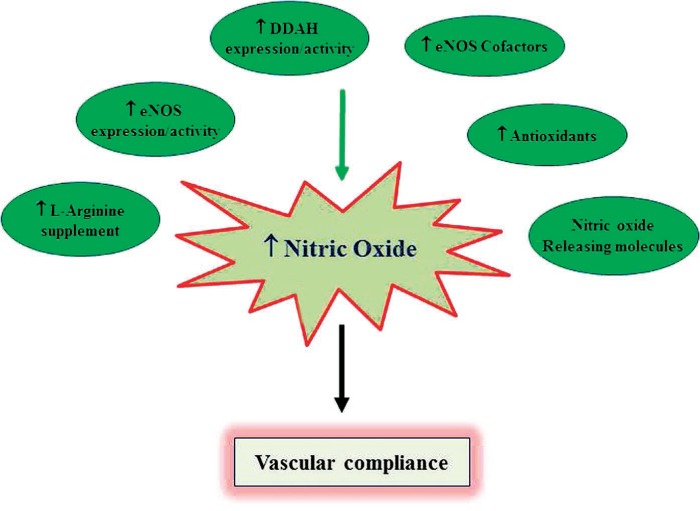
Factors that contribute to enhanced nitric oxide (NO) bioavailability. Increased NO directly contributes to vascular homeostasis and promotes cardiovascular health. eNOS: endothelial NO synthase (NOS); DDAH: dimethylarginine dimethylaminohydrolase.”
Finally, reducing the synthesis or increasing the metabolism of methylarginines is a plausible approach to enhance NO. The synthesis of ADMA (a more abundant molecule than MMA) may be controlled by regulating the expression of methyltransferase enzymes that are involved in the post-translational methylation of arginine residues in cellular proteins.43 On the other hand, ADMA metabolism can be increased by upregulating the expression or activity of DDAH. Intriguingly, cross-breeding of ApoE-deficient mice with DDAH transgenic animals has been shown to reduce plasma ADMA and attenuate atherosclerotic plaque formation,44 suggesting the potential efficacy of DDAH activating compounds in ameliorating the development and/or progression of atherosclerosis.
Conclusion
Endothelium-derived NO exerts a number of atheroprotective effects, and its bioavailability is tightly regulated by multiple fine-tuned mechanisms. The development and/or progression of atherosclerosis is associated with dysregulation of one or more of these mechanisms, resulting in the production of suboptimal levels of NO by the endothelium. As discussed above, there are several mechanisms by which endothelium-derived NO can be stimulated or exogenous NO can be supplemented. In recent years, some of these strategies have led to the development of promising NO-modulating agents that slow the progression of atherosclerosis. However, future studies should calibrate the effort and expand the strategies to not only treat atherosclerosis and its complications but also prevent its development. Preventing vascular inflammation and oxidative stress should be at the forefront of translational research aimed at reducing cardiovascular morbidity and mortality. In the meantime, we need to keep harnessing the cardiovascular benefits of physical exercise, which is known to stimulate coronary vasodilation due to enhancement of intracoronary blood flow and shear stress, to promote vascular health, and the consumption of green leafy vegetables, which are rich in the NO precursor molecules NO3 and NO2.45,46
Conflict of Interest Disclosure: The authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
References
- 1.Félétou M. The Endothelium: Part 1: Multiple functions of the endothelial cells-focus on endothelium-derived vasoactive mediators. San Rafael, CA: Morgan & Claypool Life Sciences; 2011. [PubMed] [Google Scholar]
- 2.Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006 Apr 18;47(8 Suppl):C7–12. doi: 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 3.Kuhlencordt PJ, Gyurko R, Han F et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001 Jul 24;104(4):448–54. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 4.Naruse K, Shimizu K, Muramatsu M et al. Long-term inhibition of NO synthesis promotes atherosclerosis in the hypercholesterolemic rabbit thoracic aorta. PGH2 does not contribute to impaired endothelium-dependent relaxation. Arterioscler Thromb. 1994 May;14(5):746–52. doi: 10.1161/01.atv.14.5.746. [DOI] [PubMed] [Google Scholar]
- 5.Cooke JP, Singer AH, Tsao P, Zera P, Rowan RA, Billingham ME. Antiatherogenic effects of L-arginine in the hypercholesterolemic rabbit. J Clin Invest. 1992 Sep;90(3):1168–72. doi: 10.1172/JCI115937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kojda G, Noack E. Effects of pentaerythrityl-tetranitrate and isosorbide-5-mononitrate in experimental atherosclerosis. Agents Actions Suppl. 1995;45:201–6. doi: 10.1007/978-3-0348-7346-8_29. [DOI] [PubMed] [Google Scholar]
- 7.von der Leyen HE, Gibbons GH, Morishita R et al. Gene therapy inhibiting neointimal vascular lesion: in vivo transfer of endothelial cell nitric oxide synthase gene. Proc Natl Acad Sci U S A. 1995 Feb 14;92(4):1137–41. doi: 10.1073/pnas.92.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis MG, Dalen H, Austerheim AM, Gulbrandsen TF, Svendsen E, Hagen PO. Suppression of intimal hyperplasia in experimental vein grafts by oral l-arginine supplementation and single ex vivo immersion in deferoxamine manganese. J Vasc Surg. 1996 Mar;23(3):410–20. doi: 10.1016/s0741-5214(96)80005-x. [DOI] [PubMed] [Google Scholar]
- 9.Tsao PS, McEvoy LM, Drexler H, Butcher EC, Cooke JP. Enhanced endothelial adhesiveness in hypercholesterolemia is attenuated by L-arginine. Circulation. 1994 May;89(5):2176–82. doi: 10.1161/01.cir.89.5.2176. [DOI] [PubMed] [Google Scholar]
- 10.Tsao PS, Wang B, Buitrago R, Shyy JY, Cooke JP. Nitric oxide regulates monocyte chemotactic protein-1. Circulation. 1997 Aug 5;96(3):934–40. doi: 10.1161/01.cir.96.3.934. [DOI] [PubMed] [Google Scholar]
- 11.De Caterina R, Libby P, Peng HB et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995 Jul;96(1):60–8. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Horke S, Förstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis. 2014 Nov;237(1):208–19. doi: 10.1016/j.atherosclerosis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Clancy RM, Leszczynska-Piziak J, Abramson SB. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest. 1992 Sep;90(3):1116–21. doi: 10.1172/JCI115929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukahori M, Ichimori K, Ishida H, Nakagawa H, Okino H. Nitric oxide reversibly suppresses xanthine oxidase activity. Free Radic Res. 1994 Sep;21(4):203–12. doi: 10.3109/10715769409056572. [DOI] [PubMed] [Google Scholar]
- 15.Struck AT, Hogg N, Thomas JP, Kalyanaraman B. Nitric oxide donor compounds inhibit the toxicity of oxidized low-density lipoprotein to endothelial cells. FEBS Lett. 1995 Mar 20;361(2–3):291–4. doi: 10.1016/0014-5793(95)00178-c. [DOI] [PubMed] [Google Scholar]
- 16.Stamler JS, Simon DI, Osborne JA et al. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A. 1992 Jan 1;89(1):444–8. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busse R, Mülsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990 Jun 4;265(1–2):133–6. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- 18.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2005 May 20;280(20):19888–94. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 19.Bauer PM, Fulton D, Boo YC et al. Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003 Apr 25;278(17):14841–9. doi: 10.1074/jbc.M211926200. [DOI] [PubMed] [Google Scholar]
- 20.Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010 May;459(6):793–806. doi: 10.1007/s00424-009-0767-7. [DOI] [PubMed] [Google Scholar]
- 21.Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol. 1990 Nov;101(3):746–52. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anthony S, Leiper J, Vallance P. Endogenous production of nitric oxide synthase inhibitors. Vasc Med. 2005 Jul;10(Suppl 1):S3–9. doi: 10.1191/1358863x05vm595oa. [DOI] [PubMed] [Google Scholar]
- 23.Böger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004 Oct;134(10 Suppl):2842S–2847S. doi: 10.1093/jn/134.10.2842S. discussion 2853S. [DOI] [PubMed] [Google Scholar]
- 24.Richir MC, van Lambalgen AA, Teerlink T et al. Low arginine/asymmetric dimethylarginine ratio deteriorates systemic hemodynamics and organ blood flow in a rat model. Crit Care Med. 2009 Jun;37(6):2010–7. doi: 10.1097/CCM.0b013e31819ffdaf. [DOI] [PubMed] [Google Scholar]
- 25.Vásquez-Vivar J, Kalyanaraman B, Martásek P et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998 Aug 4;95(16):9220–5. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Förstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008 Jun;5(6):338–49. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- 27.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2005 Jun 21;102(25):9056–61. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002 Mar;109(6):817–26. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sydow K, Münzel T. ADMA and oxidative stress. Atheroscler Suppl. 2003 Dec;4(4):41–51. doi: 10.1016/s1567-5688(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 30.Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007 Dec;293(6):H3227–45. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 31.Antoniades C, Shirodaria C, Leeson P et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J. 2009 May;30(9):1142–50. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- 32.Bialecki RA, Tulenko TN. Acute exposure to cholesterol increases arterial nitroprusside-and endothelium-mediated relaxation. Am J Physiol. 1993 Jan;264(1 Pt 1):C32–9. doi: 10.1152/ajpcell.1993.264.1.C32. [DOI] [PubMed] [Google Scholar]
- 33.Deliconstantinos G, Villiotou V, Stavrides JC. Modulation of particulate nitric oxide synthase activity and peroxynitrite synthesis in cholesterol enriched endothelial cell membranes. Biochem Pharmacol. 1995 May 26;49(11):1589–600. doi: 10.1016/0006-2952(95)00094-g. [DOI] [PubMed] [Google Scholar]
- 34.Liao JK, Shin WS, Lee WY, Clark SL. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J Biol Chem. 1995 Jan 6;270(1):319–24. doi: 10.1074/jbc.270.1.319. [DOI] [PubMed] [Google Scholar]
- 35.Pritchard KA, Jr, Groszek L, Smalley DM et al. Native low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res. 1995 Sep;77(3):510–8. doi: 10.1161/01.res.77.3.510. [DOI] [PubMed] [Google Scholar]
- 36.Vergnani L, Hatrik S, Ricci F et al. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production : key role of L-arginine availability. Circulation. 2000 Mar 21;101(11):1261–6. doi: 10.1161/01.cir.101.11.1261. [DOI] [PubMed] [Google Scholar]
- 37.Ryoo S, Lemmon CA, Soucy KG et al. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res. 2006 Oct 27;99(9):951–60. doi: 10.1161/01.RES.0000247034.24662.b4. [DOI] [PubMed] [Google Scholar]
- 38.Depre C, Havaux X, Renkin J, Vanoverschelde JL, Wijns W. Expression of inducible nitric oxide synthase in human coronary atherosclerotic plaque. Cardiovasc Res. 1999 Feb;41(2):465–72. doi: 10.1016/s0008-6363(98)00304-6. [DOI] [PubMed] [Google Scholar]
- 39.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998 Mar 31;97(12):1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 40.Mosca L, Rubenfire M, Mandel C et al. Antioxidant nutrient supplementation reduces the susceptibility of low density lipoprotein to oxidation in patients with coronary artery disease. J Am Coll Cardiol. 1997 Aug;30(2):392–9. doi: 10.1016/s0735-1097(97)00188-5. [DOI] [PubMed] [Google Scholar]
- 41.Cooke JP, Tsao PS. Arginine: a new therapy for atherosclerosis? Circulation. 1997 Jan 21;95(2):311–2. doi: 10.1161/01.cir.95.2.311. [DOI] [PubMed] [Google Scholar]
- 42.Herman AG, Moncada S. Therapeutic potential of nitric oxide donors in the prevention and treatment of atherosclerosis. Eur Heart J. 2005 Oct;26(19):1945–55. doi: 10.1093/eurheartj/ehi333. [DOI] [PubMed] [Google Scholar]
- 43.Böger RH, Sydow K, Borlak J et al. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S-adenosylmethionine-dependent methyltransferases. Circ Res. 2000 Jul 21;87(2):99–105. doi: 10.1161/01.res.87.2.99. [DOI] [PubMed] [Google Scholar]
- 44.Jacobi J, Maas R, Cardounel AJ et al. Dimethylarginine dimethylaminohydrolase overexpression ameliorates atherosclerosis in apolipoprotein E-deficient mice by lowering asymmetric dimethylarginine. Am J Pathol. 2010 May;176(5):2559–70. doi: 10.2353/ajpath.2010.090614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niebauer J, Cooke JP. Cardiovascular effects of exercise: role of endothelial shear stress. J Am Coll Cardiol. 1996 Dec;28(7):1652–60. doi: 10.1016/S0735-1097(96)00393-2. [DOI] [PubMed] [Google Scholar]
- 46.Cooke JP, Ghebremariam YT. Dietary nitrate, nitric oxide, and restenosis. J Clin Invest. 2011 Apr;121(4):1258–60. doi: 10.1172/JCI57193. [DOI] [PMC free article] [PubMed] [Google Scholar]