

Abstract
The technological development of induced pluripotent stem cells (iPSCs) has overcome many of the limitations of adult and embryonic stem cells. We have found that activation of innate immunity signaling is necessary for this process, as it facilitates epigenetic plasticity in cells by a process called transflammation. More recently, we have discovered that transflammation also facilitates the transdifferentiation of cells directly from one somatic cell type to another. This insight may lead to a promising therapeutic pathway that avoids reverting cells all the way back to pluripotency before achieving a cell type of interest. While there is much therapeutic promise to transflammation and transdifferentiation, there is also evidence that transdifferentiation plays a role in some pathological conditions, including atherosclerosis. Ultimately, better understanding of transflammation will facilitate the development of regenerative therapies.
Keywords: induced pluripotent stem cells, transdifferentiation, innate immunity, epigenetic plasticity, transflammation

J. P. Connell, Ph.D.
Introduction
Cardiovascular diseases, including ischemic heart disease, heart failure, stroke, and peripheral arterial disease, are responsible for one out of every three deaths in the United States.1 Most of this morbidity is associated with structural and functional impairment of the vasculature that impairs perfusion and causes end-organ damage. Current therapies for cardiovascular disease are imperfect in that they primarily ameliorate disease but do not reverse it. For example, ischemic heart disease causes scarring and reduced cardiac function that may be improved by vasodilator and beta-blocker therapy, but reversal of heart failure may require an orthotopic heart transplant.2 Such an approach is frequently complicated by acute or chronic rejection, and the immunosuppressive therapy required by these individuals can cause dyslipidemia and hypertension, side effects that promote the progression of vascular disease. Thus, current cardiovascular therapies do not achieve the promise of regenerative medicine, the goal of which is to replace or restore damaged tissues and organs.
One of the major hurdles to the development of regenerative medicine therapies, particularly for cardiovascular applications, is the identification of suitable cell sources. An ideal cell source would be autologous to avoid the issue of immune rejection as well as the ethical issues surrounding embryonic stem cells. Adult stem cells are an autologous source for regeneration. For example, mesenchymal stromal cells can be isolated from bone marrow or adipose tissue, expanded in vitro, and then administered to an ischemic tissue. These cells secrete angiogenic cytokines and immunomodulatory factors that can increase capillary density, improve perfusion, reduce inflammation, and promote healing. However, in patients with chronic disease, these adult stem cells are reduced in number and in function. Allogeneic mesenchymal stromal cells from healthy individuals can be employed and theoretically may be beneficial based on their secretion of paracrine factors. However, the residence time of such cells in the host tissue is limited, as is their proliferative capacity. Advances in the generation of induced pluripotent stem cells, as well as in the transdifferentiation of somatic cells, may be able to overcome these limitations and expand the potential of regenerative medicine.
Inducing Pluripotency
In 2006, Takahashi and Yamanaka first reported the use of a few defined factors to induce pluripotency in murine fibroblasts.3 Generation of human-induced pluripotent stem cells (iPSCs) was reported the next year.4 Yamanaka won the Nobel Prize in Physiology or Medicine in November 2012 for his discovery that overexpression of four transcription factors (Oct4, Sox2, KLF4, and cMyc) could induce somatic cells to form pluripotent stem cells.5 Pluripotent stem cells are capable of almost limitless selfrenewal and can differentiate into any somatic cell type. This pivotal step of transitioning cells from a differentiated state back to pluripotency has opened the door for new avenues of personalized medicine. For example, in a patient with a poorly understood neurological disease, one can produce iPSCs from an easily accessible cell, such as a skin fibroblast. These fibroblast-derived iPSCs can then be differentiated into neurons. To the extent that the neurological disease has a genetic basis, the iPSC-derived neurons should recapitulate the neurological pathobiology to provide for a “disease-in-a-dish” model. Having understood the pathobiology of the disease, one can also set up high-throughput screens for small molecules that may correct the pathobiology. For example, if the iPSC-derived neurons are found to have abnormal electrophysiology due to an overactive ion channel, one can develop an assay based upon ion channel activity and then screen a small molecule library for a drug that corrects the abnormality. In this way, iPSC-derived cells have accelerated the elucidation of disease pathobiology and the discovery of therapeutic molecules.
Finally, the iPSC technology provides a renewable source of any cell type for regenerative medicine applications. Because they can be expanded indefinitely, it is possible to generate the number of cells required to have a therapeutic effect. Moreover, as they can be differentiated into any cell type, iPSC-derived cells should have broad clinical application; in fact, preclinical studies have demonstrated that iPSC-derived cells show therapeutic promise for many indications. These cells may be genetically modified to correct a congenital abnormality or to express a therapeutic protein. Furthermore, they may be combined with bioengineered materials, biologicals, and/or small molecules to enhance their restoration of organ structure and function.6
Generating iPSCs remains technically challenging as it requires the activation of a complex network of genes affecting pluripotency. Many improvements have been made to the induction methods to accelerate the process, reduce the number of transcriptional factors needed, and express the Yamanaka factors without integrating foreign DNA into the host genome.7 However, there remain challenges to overcome. One concern is the ramifications of an incomplete differentiation of a batch of iPSC-derived therapeutic cells; for example, if some rare iPSCs are still present in the therapeutic cell product, these pluripotent cells could form a teratoma in the host. Another concern is the quality of the therapeutic cell product. Throughout the process of pluripotency induction, passaging, and differentiation into therapeutic cells, genetic abnormalities may be introduced. Such abnormalities may be due in part to the induction of pluripotency and to the prolonged time in cell culture. A greater understanding of the process of iPSC generation is required to enhance the quality of iPSCs. Finally, the process of differentiation into the therapeutic cell product requires careful characterization of the cells to document the fidelity of the differentiation. For some cell types (such as iPSC-derived cardiomyocytes), differentiation in vitro to a mature cell type that fully replicates the desired phenotype has not yet been fully achieved.
Role of Innate Immunity in Cellular Reprogramming
For iPSC technology to achieve its promise, it is vital to understand more about the mechanisms of cellular reprogramming. We recently discovered that the retroviral vectors used to transport the Yamanaka factors into the cell play a critical role in the reprogramming process.8 Essentially, the Yamanaka factors alone (as cell-permeant peptides) are not effective in generating pluripotency.9–12 However, when the Yamanaka factors are delivered in the form of modified messenger RNA (mmRNA) or encoded in a retroviral vector, the simultaneous activation of innate immune signaling promotes reprogramming. As described in more detail below, we discovered that the activation of innate immune signaling caused global changes in epigenetic modifiers so as to increase epigenetic plasticity, thereby facilitating the action of the Yamanaka factors through a process termed “transflammation.”
Pathogens, such as viruses or bacteria, activate innate immunity through toll-like receptors (TLRs),13 which recognize pathogen-associated molecular patterns that may be associated with viral proteins, lipopolysaccharides, DNA, or RNA.14–16 Toll-like receptor signaling may be mediated by an intracellular adaptor known as the myeloid differentiation primary response gene (MyD), in which case it is MyD88-dependent.17 Among the TLRs, only the TLR3 pathway acts fully independently of MyD88.18,19 We found that when we used retroviral vectors or mmRNA encoding the Yamanaka factors, inhibition of MyD88 had no effect on pluripotency induction. By contrast, the generation of iPSCs using the Yamanaka approach was markedly reduced in cells where TLR3 or its adaptor TRIF (toll/IL-1 receptor domain-containing adaptor-inducing interferon-β) were knocked down genetically,8 therefore TLR3 activation was necessary for the efficient generation of iPSCs. Further evidence for the involvement of the TLR3 signaling pathway was obtained using polyinosinicpolycytidylic acid (poly I:C), a synthetic analog of double-stranded RNA recognized specifically by TLR3.20 This TLR3 agonist enhanced the generation of iPSCs when used in combination with cell permeant proteins for the Yamanaka factors.8
Histone acetylation is associated with an open chromatin state that makes DNA accessible for transcription and allows for gene expression. In contrast, histone deacetylation is associated with a closed chromatin state. We have learned that activation of innate immunity through TLR3 causes epigenetic changes that favor reprogramming. Global changes in epigenetic modifiers that occur after TLR3 activation include down-regulation of the histone deacetylase family and up-regulation of histone acetyltransferases.8 The effect of TLR3 activation on epigenetic plasticity is mediated by NF-κB,8 a transcriptional effector of TLR3 activation21,22 that interacts with proteins containing histone acetyltransferase domains (p300 and CBP) to positively regulate expression of the target genes.23,24 Additionally, activation of IRF3 by the TLR3 pathway is necessary for efficient reprogramming.8
Though TLR3 and its subsequent signaling cascade are certainly involved in transflammation (Figure 1), our unpublished evidence indicates that activation of other receptors, such as TLR4 and RIG-I, may also promote epigenetic plasticity.8,25–27
Figure 1.
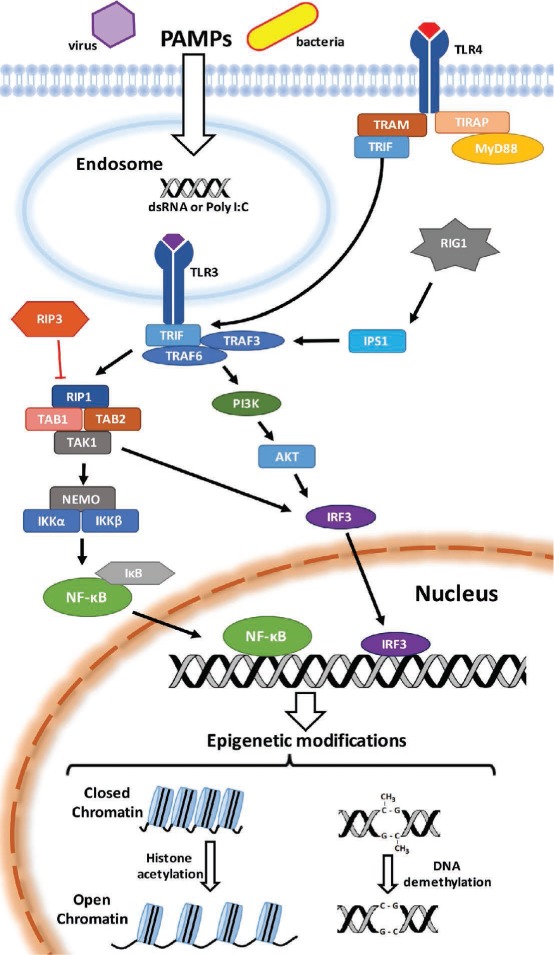
Pathogen-associated molecular patterns (PAMPs) are recognized by toll-like receptors (TLRs), including TLR3 and TLR4, on the cell surface or in endosomes. Stimulation of receptors activates innate immunity signaling, including mobilization of NF-κB and IRF-3, resulting in epigenetic modifications that change cellular plasticity. These modifications may include DNA demethylation or chromatin modifications due to an up-regulation of histone acetyltransferases and down-regulation of histone deacetylases. PAMPS: pathogen associated molecular patterns; TLR3: toll-like receptor 3; TLR4: toll-like receptor 4; TRAM: TRIF-related adaptor molecule; TIRAP: toll-interleukin 1 receptor (TIR) domain-containing adapter protein; TRIF: TIR-domain-containing adapter-inducing interferon-β; MyD88: myeloid differentiation primary response gene 88; RIG1: retinoic acid inducible gene I; IPS1: IFNβ-promoter stimulator-1; TRAF3: tumor necrosis factor (TNF) receptor-associated factor 3; TRAF6: tumor necrosis factor (TNF) receptor-associated factor 6; PI3K: phosphoinositide 3-kinase; AKT: Ak strain transforming; IRF3: interferon regulatory factor 3; RIP3: receptor-interacting protein kinase 3; RIP1: receptor-interacting protein kinase 1; TAK1: transforming growth factor β activated kinase 1; TAB1: TAK1-binding protein 1; TAB2: TAK1-binding protein 2; NEMO: NF-κB essential modulator; IKKa: inhibitor of nuclear factor kappa-B kinase subunit alpha; IKKβ: inhibitor of nuclear factor kappa-B kinase subunit beta; IκB: inhibitor of kappa B; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells.
Therapeutic Transdifferentiation
A greater understanding of cellular reprogramming may lead to improved protocols that enhance the efficiency, yield, and quality of iPSCs. Nevertheless, there remain a number of technical hurdles for the clinical application of iPSC-derived cells. One of the most daunting is how to deliver such cells. The application, dose, duration, frequency, and method (e.g., intramuscular injection) are all variable factors that must be understood. Administering the cells in a matrix that preserves cell viability and function may be a preferred approach, but it would be difficult to replicate the complexity of normal tissue architecture, with its microvasculature, innervation and intricate cellular associations. Another approach that avoids the complexities of cell delivery is to therapeutically transdifferentiate resident cells in the tissue to directly facilitate regeneration.28 For example, with an ischemic injury to the myocardium, one may apply a therapeutic transdifferentiation strategy that transforms a cardiac fibroblast into a cardiomyocyte to reduce scar formation and improve ventricular function. Using a transdifferentiation strategy in situ would avoid the complications of cell delivery, take advantage of the existing tissue architecture and resident cells, and avoid the concerns of iPSC-derived cells, including the risk of teratoma formation and undesirable genetic alterations.
Overexpression of lineage-specific transcription factors has been a successful strategy for transdifferentiating fibroblasts to neurons,29 cardiomyocytes,30 and endothelial cells.31,32 Indeed, proof-of-concept for in situ transdifferentiation has been obtained, and it appears that therapeutic transdifferentiation is a strategy within reach of clinical testing. However, most groups have used viral vectors to overexpress the transdifferentiation factors, which increases the risk of this therapeutic approach and complicates the regulatory strategy.
It occurred to us that innate immune signaling might also be important for the cellular reprogramming that occurs during transdifferentiation. In this case, it may be possible to first activate innate immunity using the FDA-approved TLR3 agonist poly I:C to enhance epigenetic plasticity, and then to provide “outside-in” signaling to obtain the desired phenotype. To that end, we have developed a method to transdifferentiate fibroblasts into endothelial cells using only growth factor proteins and small molecules in combination with poly I:C to induce transflammation and allow for cellular reprogramming.33 Fibroblasts were plated on gelatin-coated dishes and treated with poly I:C (30 ng/ml) for 1 week while cultured in an induction medium (Dulbecco's modified Eagle's medium with 7.5% fetal bovine serum and 7.5% knockout serum replacement). After 7 days in this condition, the cells were switched to a transdifferentiation medium supplemented with growth factors known to promote an endothelial lineage: basic fibroblast growth factor, vascular endothelial growth factor, and bone morphogenetic protein-4. Additional small molecules were later added to the culture medium to further enhance endothelial specification during maintenance (8-Br-cAMP, an agonist of cAMP-dependent protein kinase) or growth and monolayer formation during expansion (SB431542, a specific transforming growth factor-β receptor inhibitor).33
These induced endothelial cells (iECs) had the typical cobblestone morphology expected of endothelial cells and expressed endothelial markers CD31, VE-cadherin, and von Willebrand factor. Importantly, iECs had the functions expected of endothelial cells; they could incorporate acetylated low-density lipoprotein (LDL), form network structures on Matrigel®, and generate nitric oxide. When suspended in Matrigel and injected subcutaneously into immunodeficient mice, iECs were able to form capillary networks that integrated with the host vasculature as evidenced by the presence of red blood cells in the capillaries in the Matrigel plugs.33 Furthermore, in a mouse model of peripheral arterial disease, administration of iECs to the ischemic limb improved perfusion as assessed by laser Doppler spectroscopy.33 Consistent with these results, injections of iECs increased the capillary density and reduced clinical signs of ischemia (e.g., toe necrosis). The benefit of iECs was similar to that achieved with human microvascular endothelial cells and was superior to vehicle control.33
This study provides proof-of-concept for a non-viral strategy of therapeutic transdifferentiation. Ongoing studies are meant to simplify the strategy and to increase the yield and rate of transdifferentiation for in vivo application.
Clinical Perspectives
Our findings suggest that innate immune activation is necessary for cellular reprogramming. Activating innate immunity places cells into a state of epigenetic plasticity in which they are able to modify their cellular phenotype to meet the challenge of a pathogen or injury. This insight provides an opportunity to therapeutically manipulate an endogenous pathway for cellular transdifferentiation. One potential application of such therapeutic transdifferentiation would be immediately following myocardial infarction, when up to a billion cardiomyocytes may be lost.34 Following this tissue injury, the cardiac fibroblasts proliferate and migrate, deposit extracellular matrix in the infarcted and adjacent areas, and promote scar formation and adverse remodeling. If one could transdifferentiate a substantial number of these fibroblasts into endothelial cells, this might generate the microvasculature needed to supply the perfusion and provide the niche for generation of functional tissue. The desired effects would be a reduction in scar tissue, regeneration of cardiac tissue, improved ventricular function, and prevention of heart failure.
Pathologic Transdifferentiation
One concern regarding a strategy of therapeutic transdifferentiation stems from the fact that transdifferentiation plays a role in pathologic conditions, including atherosclerosis. Both monocyte-derived macrophages and vascular smooth muscle cells (SMCs) contribute to the development of atherosclerotic lesions.35,36 Recent evidence from Feil et al. demonstrates that vascular SMCs in atherosclerotic lesions may clonally expand and transdifferentiate into macrophage-like cells.37 The fate of medial SMCs during atherogenesis was tracked in hypercholesterolemic apolipoprotein E-deficient mice through SMC-specific tamoxifen-activated Cre recombinase and Cre reporter alleles, which allowed the originally labeled cells to be followed using β-galactosidase activity.37 Ten-week-old mice were treated with tamoxifen for 5 days, which resulted in the labeling of ~11% of SMCs in the aortic media, and this labeling remained in all subsequent daughter cells generated by the labeled SMCs. The fate of these cells was examined in 52-week-old mice. Few labeled cells were found in the atherosclerotic aortas, but occasionally large patches of labeled cells were found in atherosclerotic lesions. Some of these patches covered the entire intimal area of the plaque, indicating that the cells resulted from clonal expansion of a labeled SMC. The majority of the SMC-derived cells stained positive for MAC-2 and CD68, two markers commonly used to detect macrophages in plaque; however, they were either negative or had weak staining for smooth muscle α-actin compared to medial SMCs.37 This study is consistent with previous observations suggesting that SMCs may undergo a transformation into macrophage-like cells.37–43
This SMC-to-macrophage transdifferentiation may play a role in many other diseases, including hypertension, lung injury, and cancer;37 therefore, targeting this transdifferentiation may be a novel therapeutic pathway for treatment of such diseases. The contribution of pathological transdifferentiation to the progression of atherosclerosis raises some concerns regarding a therapeutic transdifferentiation strategy in patients with coronary artery disease. However, one recent study somewhat diminishes that concern as the onset of atherosclerosis is accelerated when TLR3 receptors are knocked out in the hypercholesterolemic apolipoprotein E−/− mouse. This finding suggests a protective role for TLR3 signaling in the vessel wall.44 Nevertheless, any strategy for therapeutic transdifferentiation is probably best constrained in space and time so as to reduce potential adverse effects.
Conclusion
Transflammation is a process that permits cells to respond to the challenge of pathogens or tissue damage. Pattern recognition receptors, such as the TLR3s, are stimulated by pathogens or tissue damage, activating cellular innate immunity. In addition to inducing the release of inflammatory cytokines, we have discovered that innate immune signaling causes global changes in epigenetic modifiers that induce epigenetic plasticity and enhance fluidity of cell phenotype. Understanding this process will facilitate cellular reprogramming for therapeutic applications. Recent work involving transdifferentiation of fibroblasts to iECs using small molecules to trigger transflammation and direct the cells towards endothelial phenotype may be useful for ischemic syndromes. Since transdifferentiation also occurs in pathologic conditions, a more comprehensive understanding of transflammation will be useful in developing strategies to reduce or prevent pathological transdifferentiation.
Conflict of Interest Disclosure: Dr. Cooke's research is funded by a grant from the National Institutes of Health (U01 HL100397, U01 HL099997, R01 AR063963, R21 AG044815).
References
- 1.Go AS, Mozaffarian D, Roger VL et al. Executive summary: Heart disease and stroke statistics--2014 update: A report from the american heart association. Circulation. 2014 Jan 21;129(3):399–410. doi: 10.1161/01.cir.0000442015.53336.12. [DOI] [PubMed] [Google Scholar]
- 2.Mancini D, Lietz K. Selection of cardiac transplantation candidates in 2010. Circulation. 2010 Jul 13;122(2):173–83. doi: 10.1161/CIRCULATIONAHA.109.858076. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006 Aug 25;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 4.Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007 Nov 30;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 5.Yamanaka S. The winding road to pluripotency (nobel lecture) Angew Chem Int Ed Engl. 2013 Dec 23;52(52):13900–9. doi: 10.1002/anie.201306721. [DOI] [PubMed] [Google Scholar]
- 6.Martins AM, Vunjak-Novakovic G, Reis RL. The current status of ips cells in cardiac research and their potential for tissue engineering and regenerative medicine. Stem Cell Rev. 2014 Apr;10(2):177–90. doi: 10.1007/s12015-013-9487-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leeper NJ, Hunter AL, Cooke JP. Stem cell therapy for vascular regeneration: Adult, embryonic, and induced pluripotent stem cells. Circulation. 2010 Aug 3;122(5):517–26. doi: 10.1161/CIRCULATIONAHA.109.881441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J, Sayed N, Hunter A et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012 Oct 26;151(3):547–58. doi: 10.1016/j.cell.2012.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho HJ, Lee CS, Kwon YW et al. Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood. 2010 Jul 22;116(3):386–95. doi: 10.1182/blood-2010-02-269589. [DOI] [PubMed] [Google Scholar]
- 10.Kim D, Kim CH, Moon JI et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009 Jun 5;4(6):472–6. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stadtfeld M, Hochedlinger K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010 Oct 15;24(20):2239–63. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou H, Wu S, Joo JY et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009 May 8;4(5):381–4. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. Ifn-regulatory factor 3-dependent gene expression is defective in tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):233–8. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004 Jul;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 15.Beutler B. Inferences, questions and possibilities in toll-like receptor signalling. Nature. 2004 Jul 8;430(6996):257–63. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 16.Kato H, Sato S, Yoneyama M et al. Cell type-specific involvement of rig-i in antiviral response. Immunity. 2005 Jul;23(1):19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat Immunol. 2010 May;11(5):373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 18.Adachi O, Kawai T, Takeda K et al. Targeted disruption of the myd88 gene results in loss of il-1-and il-18-mediated function. Immunity. 1998 Jul;9(1):143–50. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 19.Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol. 2006 May;117(5):979–87. doi: 10.1016/j.jaci.2006.02.023. quiz 88. [DOI] [PubMed] [Google Scholar]
- 20.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded rna and activation of nf-kappab by toll-like receptor 3. Nature. 2001 Oct 18;413(6857):732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 21.Hayden MS, Ghosh S. Signaling to nf-kappab. Genes Dev. 2004 Sep 15;18(18):2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 22.Hayden MS, West AP, Ghosh S. Nf-kappab and the immune response. Oncogene. 2006 Oct 30;25(51):6758–80. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 23.Farlik M, Reutterer B, Schindler C et al. Nonconventional initiation complex assembly by stat and nf-kappab transcription factors regulates nitric oxide synthase expression. Immunity. 2010 Jul 23;33(1):25–34. doi: 10.1016/j.immuni.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Verma IM. Nf-kappab regulation in the immune system. Nat Rev Immunol. 2002 Oct;2(10):725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 25.Cooke JP, Sayed N, Lee J, Wong WT. Innate immunity and epigenetic plasticity in cellular reprogramming. Curr Opin Genet Dev. 2014 Oct:2889–91. doi: 10.1016/j.gde.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawai T, Akira S. Toll-like receptor and rig-i-like receptor signaling. Ann N Y Acad Sci. 2008 Nov:11431–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 27.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr. Innate immunity induced by composition-dependent rig-i recognition of hepatitis c virus rna. Nature. 2008 Jul 24;454(7203):523–7. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooke JP. Therapeutic transdifferentiation: A novel approach for vascular disease. Circ Res. 2013 Mar 1;112(5):748–50. doi: 10.1161/CIRCRESAHA.113.301053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010 Feb 25;463(7284):1035–41. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ieda M, Fu JD, Delgado-Olguin P et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010 Aug 6;142(3):375–86. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ginsberg M, James D, Ding BS et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ets factors and tgfbeta suppression. Cell. 2012 Oct 26;151(3):559–75. doi: 10.1016/j.cell.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Margariti A, Winkler B, Karamariti E et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue-engineered vessels. Proc Natl Acad Sci U S A. 2012 Aug 21;109(34):13793–8. doi: 10.1073/pnas.1205526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sayed N, Wong WT, Ospino F et al. Transdifferentiation of human fibroblasts to endothelial cells: Role of innate immunity. Circulation. 2015 Jan 20;131(3):300–9. doi: 10.1161/CIRCULATIONAHA.113.007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansson EM, Lendahl U. Regenerative medicine for the treatment of heart disease. J Intern Med. 2013 Mar;273(3):235–45. doi: 10.1111/joim.12033. [DOI] [PubMed] [Google Scholar]
- 35.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011 May 19;473(7347):317–25. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 36.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011 Apr 29;145(3):341–55. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feil S, Fehrenbacher B, Lukowski R et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014 Sep 12;115(7):662–7. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 38.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014 Apr 15;129(15):1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 39.Andreeva ER, Pugach IM, Orekhov AN. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis. 1997 Nov;135(1):19–27. doi: 10.1016/s0021-9150(97)00136-6. [DOI] [PubMed] [Google Scholar]
- 40.Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in apoe knockout mice. Arterioscler Thromb Vasc Biol. 2006 Dec;26(12):2696–702. doi: 10.1161/01.ATV.0000247243.48542.9d. [DOI] [PubMed] [Google Scholar]
- 41.Feil S, Hofmann F, Feil R. Sm22alpha modulates vascular smooth muscle cell phenotype during atherogenesis. Circ Res. 2004 Apr 16;94(7):863–5. doi: 10.1161/01.RES.0000126417.38728.F6. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto K, Hirano K, Nozaki S et al. Expression of macrophage (mphi) scavenger receptor, cd36, in cultured human aortic smooth muscle cells in association with expression of peroxisome proliferator activated receptor-gamma, which regulates gain of mphi-like phenotype in vitro, and its implication in atherogenesis. Arterioscler Thromb Vasc Biol. 2000 Apr;20(4):1027–32. doi: 10.1161/01.atv.20.4.1027. [DOI] [PubMed] [Google Scholar]
- 43.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003 Nov 11;100(23):13531–6. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cole JE, Navin TJ, Cross AJ et al. Unexpected protective role for toll-like receptor 3 in the arterial wall. Proc Natl Acad Sci U S A. 2011 Feb 8;108(6):2372–7. doi: 10.1073/pnas.1018515108. [DOI] [PMC free article] [PubMed] [Google Scholar]